CHAPTER 82 INHERITED NEUROPATHIES
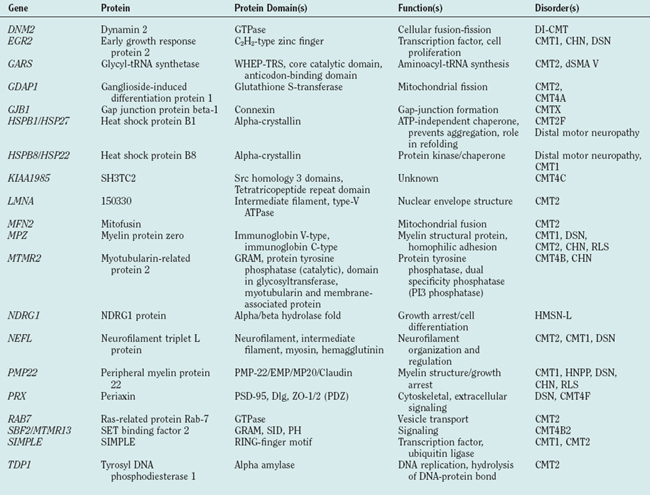
During the past decade, molecular genetics research has led to an exponential growth of knowledge of pathogenic genes and has suggested pathways involved in the pathomechanism of disease. In peripheral neuropathies, mutations or altered gene dosages in 25 genes cosegregated with disease, with each disease being due to a single gene in a given family. In vitro functional assays and experiments in animal models of specific genetic alterations elucidated the pathomechanisms by which specific genetic alterations cause disease and identified pathways involved in peripheral nerve biology (Table 82-1). As some of these mutant genes are found in significant numbers of patients with inherited peripheral neuropathy, molecular analysis plays a substantial role in establishing precise and secure etiological diagnoses.
DISEASE PHENOTYPES
Charcot-Marie-Tooth Disease (OMIM 118200, 118220)
The onset of clinical symptoms is in the first or second decade of life and includes progressive lower motor neuron-type weakness in a length-dependent manner. Weakness starts distally in the feet and progresses proximally in an ascending pattern. Early signs include tripping on uneven surfaces due to diminished dorsiflexion, difficulty walking on the heels, and tight heel cords. To compensate for the diminished ability to dorsiflex the foot, patients flex the hip with each step (steppage or equine-like gait). Neuropathic bony deformities develop, including pes cavus (high arched feet) and hammer toes. Early involvement of the peroneus group of muscles gives the legs a stork-like appearance. Patients also complain of leg cramps and lumbar pain after long walks. When the length-dependent progression reaches the length of the arm nerves, weakness and wasting of the intrinsic muscles of the hand appear. The thumb is seen to lie flat in the plane of the hand instead of opposing the other fingers. As a result, patients have difficulty opening jars, holding objects, writing, and buttoning. Muscle stretch reflexes disappear early in the ankles and later in the patella and upper limbs. Mild sensory loss to pain, temperature, or vibration in the legs is also noticed in some cases. Patients also complain of numbness and tingling in their feet and hands, but paresthesias are not as common as in acquired neuropathies. Restless leg syndrome, an irresistible urge to move the legs because of dysesthetic sensations when sitting or lying down, occurs in nearly 40% of patients with the axonal form.1 Neurophysiological studies establish the diagnosis of demyelinating or axonal CMT in most patients, but in a third type of CMT, the intermediate form, features of both demyelination and axonal loss coexist, and this mixed-type neuropathy directs attention to mutations in certain genes.
Hereditary Neuropathy with Liability to Pressure Palsies (HNPP, OMIM 162500)
The clinical phenotype is characterized by recurrent and transient nerve dysfunction at sites where the proximity of bony structures or muscle predisposes to nerve compression. Asymmetrical palsies occur after relatively minor compression or trauma of the peripheral nerves. This is a demyelinating neuropathy whose neuropathological hallmark is sausage-like thickening of myelin sheaths (tomacula). After repeated attacks, full reversal of neurological signs does not occur and the clinical picture becomes similar to CMT. Electrophysiological findings include mildly slowed nerve conduction velocity and conduction blocks,2 which sometimes lead to confusion with acquired neuropathy.
Dejerine-Sottas Neuropathy (DSN, OMIM 145900)
DSN is a clinical entity defined by early onset and developmental delay, followed by signs of lower motor neuron-type involvement, including hypotonia, weakness, and areflexia. Hypertrophic nerves can often be palpated. Neurophysiological studies reveal severe slowing of nerve conduction velocity (<10m/sec). Neuropathology reveals more pronounced demyelination and greater number of onion bulbs than in CMT. Cerebrospinal fluid proteins may be elevated. Most patients have significant disability.
Congenital Hypomyelinating Neuropathy (CHN, OMIM 605253)
As the name implies, CHN manifests at birth. However, it is often difficult to distinguish DSN from CHN clinically, as they both may manifest as floppy baby syndrome with absent tendon reflexes. The differential diagnosis between CHN and DSN rests on neuropathology and is based on the presence or absence of onion bulbs associated—in DSN—with absent or thin myelin sheets. CHN may manifest as arthrogryposis multiplex congenita.3
INHERITANCE PATTERN
CMT and related neuropathies exhibit all forms of mendelian inheritance—autosomal dominant, autosomal recessive, and X-linked. Autosomal dominant demyelinating CMT is the most frequent.4 Thirty-five linked loci (14 autosomal dominant, 12 autosomal recessive, and 3 X-linked; rarely mutations in a gene isolated as a dominant locus behave as recessive alleles in a given family) and 25 CMT-associated genes have been identified. HNPP and RLS show autosomal dominant inheritance, whereas CHN is autosomal recessive or sporadic. DSN shows both autosomal dominant and autosomal recessive inheritance. Genotype-phenotype correlation studies suggest that genetic heterogeneity, age-dependent penetrance, and variable expressivity are key characteristics of the hereditary motor-sensory neuropathies (HMSN). It is estimated that about one third of the mutations occur de novo5–7; thus absence of family history does not preclude genetic testing.
CLASSIFICATION
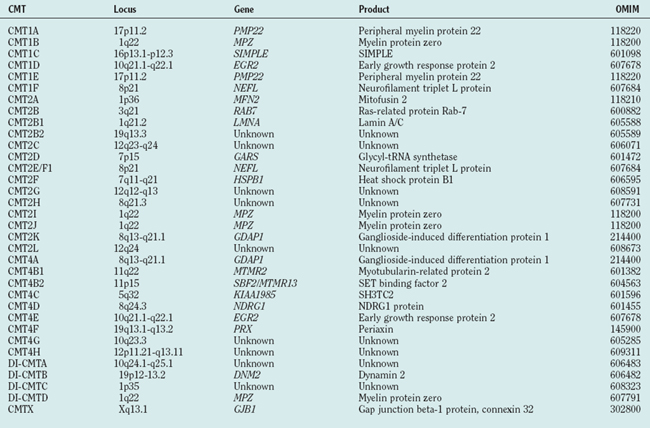
The classification system for CMT and related peripheral neuropathies is based on clinical phenotype, electrophysiology, and inheritance pattern (Table 82-2). This classification was based on large pedigrees, which were invaluable tools in identifying genes responsible for certain types of CMT. Loci identified from these families were named according to this classification as well. However, it became apparent that a substantial number of cases are sporadic and do not fit this classification. Genes associated with specific loci and disease types were found to be responsible for other types of CMT or with different inheritance patterns. Thus, this classification may need revision to make it both simpler and all inclusive. For a genetic overview of CMT, we divide the genes identified into functional groups, describe the clinical phenotypes associated with mutations in those genes, and briefly describe the presumptive pathomechanism deduced from in vitro and in vivo experiments.
GENETICS
Genes Associated with Peripheral Nerve Structure
Peripheral Myelin Protein 22 (PMP22)
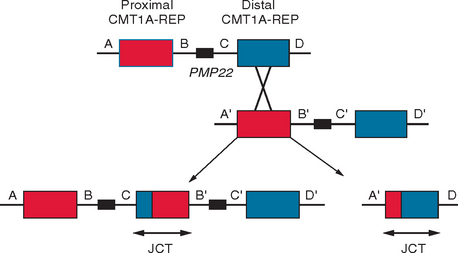
The first molecular event discovered as responsible for the majority of CMT was the duplication of the chromosomal segment harboring PMP22.8 This discovery introduced a novel molecular mechanism in human mutagenesis, nonallelic homologous recombination (Fig. 82-1), and defined a new group of disorders, the genomic disorders.9,10 The reciprocal molecular event, deletion—instead of duplication—of the same fragment was found in HNPP.11,12 This molecular event and the resulting diseases provided evidence for the presence of dosage-sensitive genes in the human genome.
Clinical phenotypes: An extra copy of PMP22, due to the CMT1A duplication, is associated with CMT18,13,14 and accounts for 70% of families with dominant CMT15,15 and 76% to 90% of sporadic CMT1.5,7 Reduced compound motor and sensory nerve action potentials correlate with clinical disability, whereas motor nerve conduction velocity does not.16 A prospective study of eight patients with CMT1A17 revealed that motor nerve conduction velocities and clinical motor examinations did not change significantly over a period of 22 years. The CMT1A duplication is also associated with neuropathy in patients with wide variations in clinical phenotypes, including DSN, RLS, calf hypertrophy, and scapuloperoneal atrophy or Davidenkow syndrome.4
Deletion of PMP22 leads to HNPP.11 In one study,18 50% of patients diagnosed with multifocal neuropathy had the 17p11.2 deletion associated with HNPP. Point mutations in PMP22 have been seen in CMT1, HNPP, DSN, and CHN phenotypes.19,20 As anticipated, loss of function mutations21 including frame-shift, nonsense, and splice site mutant alleles result in HNPP; analogous to the HNPP deletion, they effectively result in PMP22 haploinsufficiency.
Function: PMP22 is expressed in the peripheral nervous system, but its role is still unclear after 15 years of research. Most of the newly synthesized PMP22 is retained in the endoplasmic reticulum, where it is degraded.22 Only a small percentage of PMP22 is transported from the endoplasmic reticulum to the Golgi apparatus, where it undergoes complex glycosylation and becomes more stable. Axonal contact appears to stimulate the redistribution of PMP22 to the Schwann cell plasma membrane as myelination occurs.23 The ultrastructural pathology of the HNPP phenotype, tomacula, and reduced myelin compaction,24 suggests that PMP22 plays a structural role in myelin formation and/or maintenance.
Strategies aimed at normalizing PMP22 expression in transgenic mice have been encouraging25 and clinical trials are under way.
Myelin Protein Zero (MPZ)
Clinical phenotypes: About 85 to 90 different myelinopathy-associated MPZ mutations have been described (http://molgen-www.uia.ac.be/CMTMutations/). Most of them are associated with CMT1, but DSN and CMT2 phenotypes are also found, together with a few cases of CHN.26,27 A patient with a severe MPZ mutant allele27 presenting as a floppy baby taught us that innervation may be necessary for proper muscle differentiation and development. The original Roussy-Lévy family reported in 1926 has been shown to harbor a point mutation causing a missense amino acid substitution in the extracellular domain of MPZ.28
Function: MPZ is normally expressed exclusively by myelinating Schwann cells and accounts for 50% of the total PNS myelin protein.29 In vitro functional studies demonstrated that the MPZ truncating mutations associated with a severe form of peripheral neuropathy result in premature stop codons within the terminal or penultimate exons that escape nonsense-mediated decay and are stably translated into mutant proteins.30 However, a subset of these mutations, also escaping nonsense-mediated decay, resulted in a mild form of peripheral neuropathy. Further in vitro experiments demonstrated that the severity of disease phenotype depends on the amount of residual function of the mutant protein. Mutations altering the cytoplasmic domain and impairing adhesion act as null alleles. If the mutations disrupt the transmembrane domain, the mutant proteins are retained in the endoplasmic reticulum, undergo aggregation, and induce apoptosis.31
Genes Associated With Transport Through Myelin
Connexin 32 (Cx32)
Clinical phenotypes: Mutations in Cx32 account for nearly 10% of all CMT cases and are the second most frequent cause of CMT after PMP22 duplication. Over 250 different mutations have been described (http://molgen-www.uia.ac.be/CMTMutations/) throughout the entire Cx32 protein, which, unlike the PMP22 and MPZ mutations, are not concentrated in transmembrane or extracellular domains. These mutations behave in a dominant fashion and represent 90% of CMTX. Electrophysiological studies in patients with Cx32 mutations identified three patterns of neuropathy, axonal, demyelinating, and mixed.32,33
Function: The Cx32 (Gap junction B1; GJB1) gene encodes a gap junction protein containing four transmembrane domains. A connexon (hemichannel) consists of six connexin subunits and two connexons, one from each apposing membrane, which form a functional channel that allows rapid transport of ions and small molecules.34 Cx32 is expressed in myelinating Schwann cells and is localized to noncompact myelin in the paranode and Schmitt-Lanterman incisures, consistent with its role in providing a radial diffusion pathway between the adaxonal and perinuclear cytoplasm of the Schwann cell.35,36
Cx32-deficient mice mimic the human CMT1X phenotype37 with a slowly progressing demyelinating neuropathy. Enlarged periaxonal collars, abnormal noncompacted myelin domains, and axonal sprouts38 suggest that reflexive gap junctions may be required for myelin compaction or that Cx32 may play a structural role in myelin compaction. Mice lacking Cx32 show a distinct pattern of gene dysregulation in Schwann cells,39 indicating that Schwann cell homeostasis is critically dependent on the correct expression of Cx32.
Genes Associated With Axonal Transport
Neurofilament-Light (NEFL)
Clinical phenotypes: Mutations in NEFL have been identified in two independent families affected with autosomal dominant CMT2.40,41 Studies42,43 have identified additional mutations in NEFL among CMT and DSN cases. These patients had early onset, severe CMT or DSN, and moderate to severely reduced nerve conduction velocities.43
Function: NEFL encodes one of the three neurofilament subunits that are the major types of intermediate filaments found in neurons. In vitro functional studies of mutated neurofilament light chain showed defective assembly of intermediate filament networks, defective targeting of neurofilaments to processes, and altered intracellular distribution of mitochondria, suggesting defective axonal transport as the underlying pathomechanism.44
Transcription Factors Associated With Myelination
Early Growth Response 2 (EGR2)
Clinical phenotypes: Mutations in human EGR2 are found in patients with CMT1, DSN, and CHN.45,46 Patients with EGR2 mutations frequently have neuropathies affecting cranial nerves III, VII, and XII. Respiratory compromise is a common problem and requires careful monitoring.
Function: EGR2, also known as KROX20, encodes a Cys2His2 type zinc finger-containing protein. Most mutations occur in the zinc finger domain. Functional studies have shown that most EGR2 mutations affect the DNA binding and that the amount of residual binding correlates directly with disease severity.47 The same studies have shown that a mutation in the R1 domain of EGR2, which binds to the NAB corepressors and prevents their interaction with NAB proteins, leads to increased transcriptional activity of EGR2. Thus, failure to activate or inactivate downstream genes or deregulation of EGR2 activity could be a pathogenic mechanism.
Mouse EGR2 is implicated in the establishment of myelination and its subsequent expression is restricted to myelinating Schwann cells.48,49 Homozygous knockout mice for EGR2 show disruption in hindbrain segmentation50,51 and block of Schwann cells at an early stage of differentiation.52
Genes Associated with Signaling
Periaxin (PRX)
Clinical phenotype: Mutations in PRX are associated with autosomal recessive DSN and CMT4F.53–55 PRX mutations cause early onset but slowly progressive neuropathy with marked sensory component.56
Function: Alternative splicing of human PRX results in two forms: L-periaxin and S-periaxin.57 L-periaxin is first expressed in the nuclei of embryonic Schwann cells and then in the plasma membrane of myelinating Schwann cells.58 Its expression pattern in the rat sciatic nerve parallels the deposition of myelin.59 In mature myelin, periaxin is found in the cytoplasm-filled periaxonal regions of the sheath but is excluded from compact myelin. Mice disrupted for Prx develop PNS compact myelin that degenerates as the animals age,60 consistent with the role of periaxin in myelin stability. These mice are important models to study neuropathic pain in late onset demyelinating disease.
Myotubularin-Related Protein 2 (MTMR2)
Clinical phenotype: Mutations in MTMR2 cause a type of autosomal recessive CMT1 (CMT4B1) and CHN.61,62 CMT4B1 is characterized by focally folded myelin. The mutations are distributed throughout the open reading frame.
Function: MTMR2 encodes a dual specificity phosphatase. It also contains a GRAM domain, an SET-interacting domain, and a PDZ-binding domain. MTMR2 uses the lipid second messenger, phosphoinositol 3-phosphate, as a physiological substrate. The known63 disease-associated MTMR2 mutations show reduced phosphatase activity,64 indicating that the phosphatase activity of MTMR2 is crucial for its proper function in the peripheral nervous system.
SET Binding Factor 2 (SBF2) or Myotubularin-Related Protein 13 (MTMR13)
Clinical phenotype: A homozygous in-frame deletion encompassing exons 11 and 12 was detected in a consangious Turkish family.65 The Japanese family from one of the clinical reports of CMT and glaucoma66 was found to harbor a nonsense mutation in SBF2, which segregated with a phenotype characterized by markedly decreased MCV, myelin folding, and juvenile-onset glaucoma.67
N-myc Downstream Regulated Gene 1 (NDRG1)
Clinical phenotype: A homozygous C-to-T transition in exon 7 causing a nonsense allele (R148X) was identified in 60 individuals affected with hereditary motor and sensory neuropathy, Lom type (HMSNL).68 HMSNL is an autosomal recessive CMT1-type disorder with deafness and unusual neuropathological features.69
GDAP1
Clinical phenotype: Mutations in GDAP1 are associated with autosomal recessive axonal neuropathies70,71 and autosomal recessive CMT2 with vocal cord paresis and hoarseness.72 In one of these studies,71 the pathological allele (487C>T, Q163X) was observed in three unrelated Hispanic families that had the same haplotype, suggesting a founder mutation that probably arose in Spain and thereby entered the American Hispanic population.73
Function: GDAP1 encodes a ganglioside-induced differentiation protein originally isolated using a tetracycline-regulated expression system from differentiated Neuro2a cells.74 It contains a glutathione-S-transferase domain. It is expressed at high levels in the brain and spinal cord and at lower levels in human sural and mouse sciatic nerves.72 Recent studies suggest that GDAP1 regulates the mitochondrial network. Overexpression of GDAP1 induces fragmentation of mitochondria, while mitochondrial fission proteins (mitofusins 1 and 2 and Drp1) can counterbalance GDAP1-mediated fission. GDAP1-specific knockdown by RNAi results in tubular mitochondrial pathology. However, GDAP1 truncation mutations found in patients with CMT are not targeted to the mitochondria and have lost mitochondrial fragmentation activity in vitro.75
Genes Associated With Endosomes
RAB7
Clinical phenotype: Mutations in RAB7 have been associated with CMT2B, an axonopathy.76
Function: RAB7 encodes a GTP-binding protein, a member of the RAB family of small GTPases, which are important regulators of vesicular transport and are located in specific intracellular compartments. RAB7 has been localized to late endosomes and shown to be important in the late endocytic pathway.77
SIMPLE
Clinical phenotype: Mutations in SIMPLE may cause both demyelinating and axonal neuropathy.78,79
Function: This gene encodes an unglycosylated small integral membrane protein of the lysosome/late endosome.80 Bioinformatics suggest that SIMPLE may be a member of the RING-finger motif-containing subfamily of E3 ubiquitin ligases.
Mitochondrial Gene
Mitofusin 2 (MFN2)
Clinical phenotype: Mutations in MFN2 were found in seven (19%) CMT2A pedigrees and in several sporadic cases.81 Most patients had moderately severe axonal neuropathy with onset in childhood. In a Japanese study, 8.6% of CMT2 and of unclassified patients had mutations in MFN2, and a recent study found that 23% of CMT2 families had mutations in this gene,82 making MFN2 the gene most commonly involved in CMT2.83
Function: Mitochondria are dynamic and highly motile organelles with frequent fusion and fission events. MFN2 is localized to the outer mitochondrial membrane, and—through fusion—regulates the architecture of the mitochondrial network. Although MFN2 is ubiquitously expressed, in the peripheral nerve the mitochondrial network has to be maintained long distances away from the cell body, which may explain the length-dependent axonal neuropathy developing in patients with MFN2 mutations.83
Chaperones
Heat-Shock Protein 27 (HSP27) and Heat-Shock Protein 22 (HSP22)
Clinical phenotype: Mutations in the small heatshock protein 27 were found in patients with distal motor neuropathy and in a family with CMT.84 In this family, distal motor neuropathy and CMT are allelic, suggesting that these two groups of disorders are intimately related. Although HSP22 mutations were originally found only in patients with distal motor neuropathy,84 recently—and not surprisingly—an HSP22 mutation was also identified in a family with CMT.85
Function: The pathomechanism of the neuropathy is less clear. In vitro data show that neuronal cells transfected with mutant HSP27 are less viable than cells transfected with the wild-type protein. When mutant HSP27 is cotransfected with NEFL, neurofilament assembly is altered. In a yeast two-hybrid system, HSP22 and HSP27 were found to interact.86
Genes Associated with DNA Single-Strand Break Repair
Tyrosyl DNA Phosphodiesterase 1 (TDP1)
Clinical phenotype: A familial homozygous TDP1 mutation has been associated with autosomal recessive spinocerebellar ataxia and axonal neuropathy (SCAN1).87 The phenotype caused by mutations in TDP1 does not quite fit the the CMT definition, as central nervous system involvement is also present. Rather, this clinical presentation belongs to a new group of disorders affecting—in various combinations—oculomotor praxis, the cerebellum, the spinal cord and the peripheral nerves, and all caused by alterations of the DNA repair pathways, with ataxia-oculomotor apraxia (AOA1 and AOA2).88,89
Function: TDP1 encodes a DNA repair enzyme that repairs both abortive single strand breaks created by topoisomerase190 and 3′-phosphoglycolated overhangs of DNA double-strand breaks.91 In the repair of single-strand breaks, TDP1 cleaves the covalent bond formed between the tyrosine moiety of TopoI and the 3′ end of the DNA, thus generating a 3′ end compatible with ligation.92 In the case of double-strand breaks, which leave 3′-phosphoglycolate overhangs, TDP1 removes the glycolate, leaving a 3′ phosphate, which becomes the substrate for ligation. In vitro functional studies revealed that mutations associated with the SCAN1 phenotype alter the sequestration of TDP1 into multiprotein single-strand break repair complexes that are catalitically inactive.93 Another set of in vitro functional studies showed that these mutations abolish the 3′-phosphoglycolate processing activity of the enzyme.94
Genes Associated with Other Peripheral Nerve System-Specific Functions
Lamin A/C (LMNA)
Clinical phenotype: One familial mutation in LMNA is associated with autosomal recessive CMT2 (CMT2B1).95 Other mutations in LMNA are associated with several different disorders, including Emery-Dreifuss muscular dystrophy (EDMD),96 limb-girdle muscular dystrophy,97 dilated cardiomyopathy,98 familial partial lipodystrophy,99 mandibuloacral dysplasia,100 and progeria.101,102 Thus, mutations in a single gene can cause different diseases affecting diverse tissues and organs, including neurons, muscles, cardiovascular and skeletal systems, and fat cells.
Function: LMNA encodes a structural protein with similarity to cytoplasmic intermediate filament proteins. Lamins are the major structural proteins of the nuclear lamina underlying the nuclear membrane. They appear to play a role in DNA replication, chromatin organization, spatial arrangements of nuclear pore complexes, nuclear growth, and anchorage of nuclear envelope proteins.103 Mice lacking Lmna develop to term with no overt abnormalities,104 but their postnatal growth is severely retarded and characterized by muscle weakness.
Glycyl tRNA Synthetase (GARS)
Clinical phenotype: Mutations in GARS have been found in patients with autosomal dominant CMT axonal neuropathy type 2, designated CMT2D. Distal spinal muscular atrophy type V (DSMAV) is an allelic disorder with a similar phenotype. The clinical picture of patients with GARS mutations differs from that of other axonal CMT2 types in that weakness and atrophy are more severe in the hands than in the feet and that sensory impairment is as frequent as motor involvement.105
Function: The human GARS protein is encoded by a 17-exon gene that spans about 40kb on chromosome 7p14 and is expressed ubiquitously. The four CMT2D/dSMA-V—associated mutations occured in conserved amino acids. GARS is a member of the family of aminoacyl tRNA synthetases responsible for charging tRNAs with their cognate amino acids. The functional holoenzyme exists as homodimer and contains three major functional domains: the WHEP-TRS domain for conjugation with other aminoacyl tRNA synthetases in enzyme complexes, the core catalytic domain for ligation; and the anticodon-binding domain for recognition of glycine-specific tRNAs.106
Dynamin 2 (DNM2)
Clinical phenotype: Mutations in DNM2 were found in three unrelated families with CMT originating from Australia, Belgium, and North America. Two additionally different mutations affecting the same amino acid, Lys558, segregated with CMT and neutropenia, a sign not previously associated with CMT neuropathies.107
Function: DNM2 belongs to the family of large GTPases and is part of the cellular fusion-fission apparatus. In vitro experiments showed that mutations of DNM2 substantially diminished binding of DNM2 to membranes by altering the conformation of the beta3/beta4 loop of the plecktrin homology domain.
GENETIC TESTING
Evidence-based data from 12 population-based studies from various ethnic backgrounds5,6,15,108–117 reported results on five genes/genomic rearrangements: PMP22 duplication/deletion; and on the following point mutations, MPZ, Cx32, and PMP22. The mutations of individual genes were uniformly distributed in the total population and in phenotypic subgroups. Applying a simple clinical classification (demyelinating versus axonal neuropathy) and considering the inheritance pattern111 improves markedly the diagnostic yield (Table 82-3).
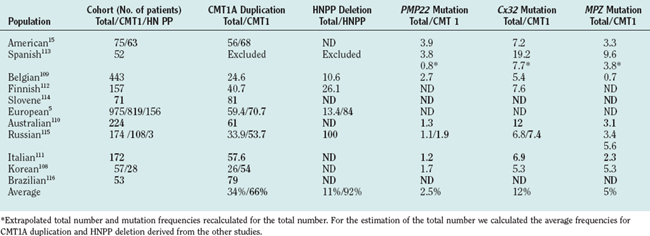
We used a cohort of 153 consecutive unrelated CMT cases collected before genetic testing became available in commercial laboratories to estimate the mutation frequencies for the genes that are not reported in the population-based studies. We tested 14 genes/genomic rearrangements (PMP22 dup/del, point mutations in Cx32, MPZ, PMP22, EGR2, PRX, NEFL, SOX10, SIMPLE, GDAP1, LMNA, TDP1, MTMR2) in this cohort. The frequencies of the five mutations screened in the population-based studies were similar to those reported, suggesting that estimates from this cohort are representative. At present, mutations in the genes for which population studies were not available seem to account for only a small minority (<1% to 2%) of patients with the CMT phenotype. Commercial laboratories report similar relative frequencies. Figure 82-2 illustrates the relative frequencies of genes whose mutations cause CMT1 or CMT2.
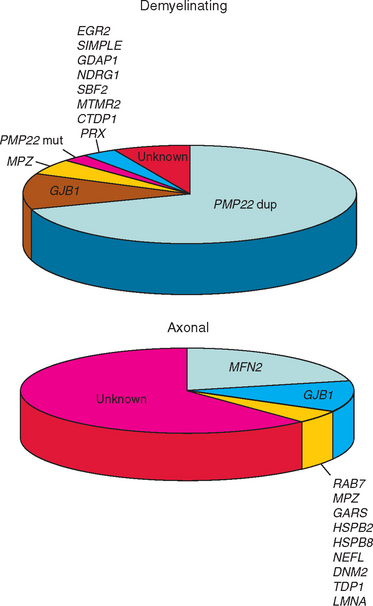
Duplication of a chromosomal segment harboring PMP22 (i.e., the CMT1A duplication)8 accounts for 43% of all CMT cases but for 70% of CMT1 patients. Thus, PMP22 duplication/deletion testing should be the initial step in demyelinating neuropathies of any severity.
Deletion of the same chromosomal segment results in HNPP.11 Although detection of deletion has a low yield in the total CMT population, deletion mutations are found in more than 90% of patients with HNPP. As HNPP occasionally can mimic multifocal neuropathy,18 a correct molecular diagnosis in this group can prevent unnecessary immunosuppressive therapy.
Population-based studies suggest that in patients with the CMT1 phenotype MPZ and PMP22 mutations are the next most common genetic errors, followed by mutations in rare genes.117 In the CMT2 group, Cx32 mutations are followed by MPZ mutations in frequency, but data, although not population based, suggest that MFN2 mutations may be common causes of CMT2.81,83
The high frequency of de novo mutations in duplication/deletion (37% to 90%)7,118 and in point mutations6 explains how genetic disease is commonly sporadic in presentation. The absence of a positive family history does not exclude CMT and related peripheral neuropathies. In fact, if a patient presents with chronic polyneuropathy without other signs or symptoms, and common systemic and treatable causes, such as diabetes, uremia, and nutritional deficiency, have been excluded, a genetic neuropathy is more likely than autoimmune or paraneoplastic neuropathy. A rational diagnostic approach is presented in Figure 82-3.
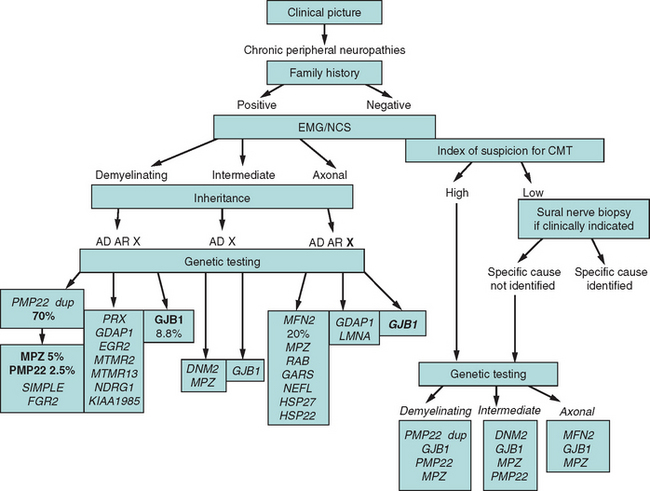
Finally, when performing genetic testing, one must consider the specific question posed and the likelihood that the result would affect medical management. PMP22 duplication and Cx32 mutation analysis establishes the molecular diagnosis in 65% of patients, but if patients with demyelinating neuropathy are tested as a group, the diagnostic yield increases to over 80%. A correct diagnosis identifies candidates for the clinical trials, families whose members are at risk for idiosyncratic drug reactions, and determines inheritance pattern, which may suffice to an adult with CMT and no reproductive plans.
MANAGEMENT
Treatment approaches to the hereditary sensorimotor neuropathies can be divided into preventive, symptomatic, and etiological. As CMT is a slowly progressive neurodegenerative disease, patients require periodic assessments. Physical therapy and occupational therapy are helpful in maintaining range of motion and function.119,120 Orthotic devices and assistive equipment can increase safety and function. In some instances, surgical interventions on the hands and feet become necessary.121,122
Symptomatic treatment may have a substantial impact on the quality of life. Maintenance of normal weight prevents strain on weak muscles and joints, prolongs ambulation, and prevents back pain. Nonsteroidal anti-inflammatory drugs may relieve low back or leg pain. Neuropathic pain can be treated with antiepileptic drugs (gabapentin, pregabalin, topiramate) or tricyclic antidepressants (amitriptyline).123,124 The tremor may respond to β blockers or primidone.125 Caffeine and nicotine can aggravate the fine intentional tremor and should be avoided. Neurotoxic drugs (http://www.charcot-marie-tooth.org/) and excessive alcohol should also be avoided. Even small doses of vincristine can produce devastating effects in CMT patients, and early detection of HMSN can avoid life-threatening vincristine neurotoxicity.126
Although etiological treatment is currently unavailable, therapeutic trials with a progesterone antagonist127 and with ascorbic acid127,128 have proved effective in animal models with a CMT-like neuropathy caused by the CMT1A duplication. Clinical trials in patients with the CMT1A duplication are under way.
GENETIC COUNSELING
An affected parent with autosomal dominant or X-linked dominant CMT1 or CMT2 has a 50% risk of having a child with the same mutation. Whether this child will be clinically affected sometime during his or her lifetime is not known because penetrance has not been determined in genetically well-defined patient populations. In general, only a few patients with autosomal dominant CMT1 or CMT2 have substantial difficulty walking before the age of 50 years, but almost all patients express some symptoms by the sixth decade of life.129 For fathers with X-linked dominant CMT, the risk of having an affected son is negligible but the risk of having an affected daughter is 100%. For mothers with X-linked dominant CMT, the risk of having an affected son or daughter is 50%.
In the absence of a molecular diagnosis, nerve conduction velocity slowing is detectable by age 2 to 5 years130,131 in patients with autosomal dominant CMT1. Therefore, if a young adult has normal nerve conduction velocities, the risk of developing autosomal dominant CMT1 is negligible, but if nerve conduction velocities are abnormal, the patient has at least a 90% risk of developing symptoms at some point in his or her life. Electrophysiological changes associated with autosomal dominant CMT2 develop with disease progression, such that only about one half of the patients can be identified by age 20 years.132
When unaffected parents have a child with CMT1 or CMT2, four possibilities exist: a de novo dominant mutation, autosomal recessive inheritance, X-linked inheritance, or nonpaternity. Distinction between these possibilities requires either the identification of the causative mutation(s) or identification of affected siblings. The identification of a de novo heterozygous presumed dominant mutation suggests a low recurrence risk for the parents; however, their risk is higher than that for the general population because of germline mosaicism.133 A proband with a heterozygous presumed dominant mutation has a 50% risk of having affected children. For autosomal recessive inheritance, the parental risk of an affected child is 25% because penetrance is nearly complete.
SUMMARY
Chance PF, Alderson MK, Leppig KA, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143-151.
Emerging pathways for hereditary axonopathies. J Mol Med. 2005 Aug 31.
Lupski JR, de Oca-Luna RM, Slaugenhaupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219-232.
Lupski JR, Garcia CA. Charcot-Marie-Tooth Peripheral Neuropathies and Related Disorders. In: The metabolic and molecular bases of inherited disease. McGraw-Hill; 2001.
Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003;4(9):714-726.
1 Gemignani F, Marbini A, Di Giovanni G, et al. Charcot-Marie-Tooth disease type 2 with restless legs syndrome. Neurology. 1999;52:1064-1066.
2 Uncini A, Di Guglielmo G, Di Muzio A, et al. Differential electrophysiological features of neuropathies associated with 17p11.2 deletion and duplication. Muscle Nerve. 1995;18:628-635.
3 Boylan KB, Ferriero DM, Greco CM, et al. Congenital hypomyelination neuropathy with arthrogryposis multiplex congenita. Ann Neurol. 1992;31:337-340.
4 Lupski JR, Garcia CA. Charcot-Marie-Tooth peripheral neuropathies and related disorders. In: Scriver CR, Valle D, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 2001.
5 Nelis E, Van Broeckhoven C, De Jonghe P, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. 1996;4:25-33.
6 Boerkoel CF, Takashima H, Garcia CA, et al. Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann Neurol. 2002;51:190-201.
7 Hoogendijk JE, Hensels GW, Gabreels-Festen AA, et al. De-novo mutation in hereditary motor and sensory neuropathy type I. Lancet. 1992;339:1081-1082.
8 Lupski JR, de Oca-Luna RM, Slaugenhaupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219-232.
9 Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417-422.
10 Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74-82.
11 Chance PF, Alderson MK, Leppig KA, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143-151.
12 Chance PF, Abbas N, Lensch MW, et al. Two autosomal dominant neuropathies result from reciprocal DNA duplication/deletion of a region on chromosome 17. Hum Mol Genet. 1994;3:223-228.
13 Raeymaekers P, Timmerman V, Nelis E, et al. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group. Neuromuscul Disord. 1991;1:93-97.
14 Patel PI, Roa BB, Welcher AA, et al. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:159-165.
15 Wise CA, Garcia CA, Davis SN, et al. Molecular analyses of unrelated Charcot-Marie-Tooth (CMT) disease patients suggest a high frequency of the CMTIA duplication. Am J Hum Genet. 1993;53:853-863.
16 Krajewski KM, Lewis RA, Fuerst DR, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123(Pt 7):1516-1527.
17 Killian JM, Tiwari PS, Jacobson S, et al. Longitudinal studies of the duplication form of Charcot-Marie-Tooth polyneuropathy. Muscle Nerve. 1996;19:74-78.
18 Tyson J, Malcolm S, Thomas PK, et al. Deletions of chromosome 17p11.2 in multifocal neuropathies. Ann Neurol. 1996;39:180-186.
19 Valentijn LJ, Bolhuis PA, Zorn I, et al. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth disease type 1A. Nat Genet. 1992;1:166-170.
20 Simonati A, Fabrizi GM, Pasquinelli A, et al. Congenital hypomyelination neuropathy with Ser72Leu substitution in PMP22. Neuromuscul Disord. 1999;9:257-261.
21 Nicholson GA, Valentijn LJ, Cherryson AK, et al. A frame shift mutation in the PMP22 gene in hereditary neuropathy with liability to pressure palsies. Nat Genet. 1994;6:263-266.
22 Sancho S, Magyar JP, Aguzzi A, et al. Distal axonopathy in peripheral nerves of PMP22-mutant mice. Brain. 1999;122(Pt 8):1563-1577.
23 Pareek S, Suter U, Snipes GJ, et al. Detection and processing of peripheral myelin protein PMP22 in cultured Schwann cells. J Biol Chem. 1993;268:10372-10379.
24 Yoshikawa H, Dyck PJ. Uncompacted inner myelin lamellae in inherited tendency to pressure palsy. J Neuropathol Exp Neurol. 1991;50:649-657.
25 Perea J, Robertson A, Tolmachova T, et al. Induced myelination and demyelination in a conditional mouse model of Charcot-Marie-Tooth disease type 1A. Hum Mol Genet. 2001;10:1007-1018.
26 Warner LE, Hilz MJ, Appel SH, et al. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron. 1996;17:451-460.
27 Szigeti K, Saifi GM, Armstrong D, et al. Disturbance of muscle fiber differentiation in congenital hypomyelinating neuropathy caused by a novel myelin protein zero mutation. Ann Neurol. 2003;54:398-402.
28 Plante-Bordeneuve V, Guiochon-Mantel A, Lacroix C, et al. The Roussy-Lévy family: from the original description to the gene. Ann Neurol. 1999;46:770-773.
29 Lemke G. Unwrapping the genes of myelin. Neuron. 1988;1:535-543.
30 Inoue K, Khajavi M, Ohyama T, et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36:361-369.
31 Khajavi M, Inoue K, Wiszniewski W, et al. Curcumin treatment abrogates endoplasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am J Hum Genet. 2005;77:841-850.
32 Dubourg O, Tardieu S, Birouk N, et al. Clinical, electrophysiological and molecular genetic characteristics of 93 patients with X-linked Charcot-Marie-Tooth disease. Brain. 2001;124:1958-1967.
33 Tabaraud F, Lagrange E, Sindou P, et al. Demyelinating X-linked Charcot-Marie-Tooth disease: unusual electrophysiological findings. Muscle Nerve. 1999;22:1442-1447.
34 Bruzzone R, Ressot C. Connexins, gap junctions and cell-cell signalling in the nervous system. Eur J Neurosci. 1997;9:1-6.
35 Neuhaus IM, Bone L, Wang S, et al. The human connexin32 gene is transcribed from two tissue-specific promoters. Biosci Rep. 1996;16:239-248.
36 Bergoffen J, Scherer SS, Wang S, et al. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039-2042.
37 Anzini P, Neuberg DH, Schachner M, et al. Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J Neurosci. 1997;17:4545-4551.
38 Kobsar I, Maurer M, Ott T, et al. Macrophage-related demyelination in peripheral nerves of mice deficient in the gap junction protein connexin 32. Neurosci Lett. 2002;320:17-20.
39 Nicholson SM, Gomes D, de Nechaud B, et al. Altered gene expression in Schwann cells of connexin32 knockout animals. J Neurosci Res. 2001;66:23-36.
40 Mersiyanova IV, Perepelov AV, Polyakov AV, et al. A new variant of Charcot-Marie-Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am J Hum Genet. 2000;67:37-46.
41 De Jonghe P, Mersivanova I, Nelis E, et al. Further evidence that neurofilament light chain gene mutations can cause Charcot-Marie-Tooth disease type 2E. Ann Neurol. 2001;49:245-249.
42 Yoshihara T, Yamamoto M, Hattori N, et al. Identification of novel sequence variants in the neurofilament-light gene in a Japanese population: analysis of Charcot-Marie-Tooth disease patients and normal individuals. J Peripher Nerv Syst. 2002;7:221-224.
43 Jordanova A, De Jonghe P, Boerkoel CF, et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain. 2003;126:590-597.
44 Perez-Olle R, Jones ST, Liem RK. Phenotypic analysis of neurofilament light gene mutations linked to Charcot-Marie-Tooth disease in cell culture models. Hum Mol Genet. 2004;13:2207-2220.
45 Warner LE, Mancias P, Butler IJ, et al. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet. 1998;18:382-384.
46 Timmerman V, De Jonghe P, Ceuterick C, et al. Novel missense mutation in the early growth response 2 gene associated with Dejerine-Sottas syndrome phenotype. Neurology. 1999;52:1827-1832.
47 Warner LE, Svaren J, Milbrandt J, et al. Functional consequences of mutations in the early growth response 2 gene (EGR2) correlate with severity of human myelinopathies. Hum Mol Genet. 1999;8:1245-1251.
48 Zorick TS, Syroid DE, Arroyo E, et al. The transcription factors SCIP and Krox-20 mark distinct stages and cell fates in Schwann cell differentiation. Mol Cell Neurosci. 1996;8:129-145.
49 Kioussi C, Gruss P. Making of a Schwann. Trends Genet. 1996;12:84-86.
50 Swiatek PJ, Gridley T. Perinatal lethality and defects in hindbrain development in mice homozygous for a targeted mutation of the zinc finger gene Krox20. Genes Dev. 1993;7:2071-2084.
51 Schneider-Maunoury S, Topilko P, Seitandou T, et al. Disruption of Krox-20 results in alteration of rhombomeres 3 and 5 in the developing hindbrain. Cell. 1993;75:1199-1214.
52 Topilko P, Schneider-Maunoury S, Levi G, et al. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796-799.
53 Boerkoel CF, Takashima H, Stankiewicz P, et al. Periaxin mutations cause recessive Dejerine-Sottas neuropathy. Am J Hum Genet. 2001;68:325-333.
54 Guilbot A, Williams A, Ravise N, et al. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Hum Mol Genet. 2001;10:415-421.
55 Takashima H, Boerkoel CF, De Jonghe P, et al. Periaxin mutations cause a broad spectrum of demyelinating neuropathies. Ann Neurol. 2002;51:709-715.
56 Kijima K, Numakura C, Shirahata E, et al. Periaxin mutation causes early-onset but slow-progressive Charcot-Marie-Tooth disease. J Hum Genet. 2004;49:376-379.
57 Dytrych L, Sherman DL, Gillespie CS, et al. Two PDZ domain proteins encoded by the murine periaxin gene are the result of alternative intron retention and are differentially targeted in Schwann cells. J Biol Chem. 1998;273:5794-5800.
58 Sherman DL, Brophy PJ. A tripartite nuclear localization signal in the PDZ-domain protein L-periaxin. J Biol Chem. 2000;275:4537-4540.
59 Gillespie CS, Sherman DL, Blair GE, et al. Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron. 1994;12:497-508.
60 Gillespie CS, Sherman DL, Fleetwood-Walker SM, et al. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron. 2000;26:523-531.
61 Bolino A, Muglia M, Conforti FL, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. 2000;25:17-19.
62 Bolino A, Lonie LJ, Zimmer M, et al. Denaturing high-performance liquid chromatography of the myotubularin-related 2 gene (MTMR2) in unrelated patients with Charcot-Marie-Tooth disease suggests a low frequency of mutation in inherited neuropathy. Neurogenetics. 2001;3:107-109.
63 Kim SA, Taylor GS, Torgersen KM, et al. Myotubularin and MTMR2, phosphatidylinositol 3-phosphatases mutated in myotubular myopathy and type 4B Charcot-Marie-Tooth disease. J Biol Chem. 2002;277:4526-4531.
64 Berger P, Bonneick S, Willi S, et al. Loss of phosphatase activity in myotubularin-related protein 2 is associated with Charcot-Marie-Tooth disease type 4B1. Hum Mol Genet. 2002;11:1569-1579.
65 Senderek J, Bergmann C, Weber S, et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003;12:349-356.
66 Kiwaki T, Umehara F, Takashima H, et al. Hereditary motor and sensory neuropathy with myelin folding and juvenile onset glaucoma. Neurology. 2000;55:392-397.
67 Hirano R, Takashima H, Umehara F, et al. SET binding factor 2 (SBF2) mutation causes CMT4B with juvenile onset glaucoma. Neurology. 2004;63:577-580.
68 Kalaydjieva L, Gresham D, Gooding R, et al. N-myc downstream-regulated gene 1 is mutated in hereditary motor and sensory neuropathy-Lom. Am J Hum Genet. 2000;67:47-58.
69 Kalaydjieva L, Hallmayer J, Chandler D, et al. Gene mapping in Gypsies identifies a novel demyelinating neuropathy on chromosome 8q24. Nat Genet. 1996;14:214-217.
70 Baxter RV, Ben Othmane K, Rochelle JM, et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 2002;30:21-22.
71 Boerkoel CF, Takashima H, Nakagawa M, et al. CMT4A: identification of a Hispanic GDAP1 founder mutation. Ann Neurol. 2003;53:400-405.
72 Cuesta A, Pedrola L, Sevilla T, et al. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat Genet. 2002;30:22-25.
73 Claramunt R, Pedrola L, Sevilla T, et al. Genetics of Charcot-Marie-Tooth disease type 4A: mutations, inheritance, phenotypic variability, and founder effect. J Med Genet. 2005;42:358-365.
74 Liu H, Nakagawa T, Kanematsu T, et al. Isolation of 10 differentially expressed cDNAs in differentiated Neuro2a cells induced through controlled expression of the GD3 synthase gene. J Neurochem. 1999;72:1781-1790.
75 Niemann A, Ruegg M, La Padula V, et al. Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network: new implications for Charcot-Marie-Tooth disease. J Cell Biol. 2005;170:1067-1078.
76 Verhoeven K, De Jonghe P, Coen K, et al. Mutations in the small GTPase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. 2003;72:722-727.
77 Vitelli R, Chiariello M, Lattero D, et al. Molecular cloning and expression analysis of the human Rab7 GTPase complementary deoxyribonucleic acid. Biochem Biophys Res Commun. 1996;229:887-890.
78 Saifi GM, Szigeti K, Wiszniewski W, et al: SIMPLE mutations in Charcot-Marie-Tooth disease and the potential role of its protein product in protein degradation. Human Mutations 2005; in press.
79 Street VA, Goldy JD, Golden AS, et al. Mapping of Charcot-Marie-Tooth disease type 1C to chromosome 16p identifies a novel locus for demyelinating neuropathies. Am J Hum Genet. 2002;70:244-250.
80 Moriwaki Y, Begum NA, Kobayashi M, et al. Mycobacterium bovis Bacillus Calmette-Guerin and its cell wall complex induce a novel lysosomal membrane protein, SIMPLE, that bridges the missing link between lipopolysaccharide and p53-inducible gene, LITAF(PIG7), and estrogen-inducible gene, EET-1. J Biol Chem. 2001;276:23065-23076.
81 Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449-451.
82 Lawson VH, Graham BV, Flanigan KM. Clinical and electro-physiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. 2005:197-204.
83 Kijima K, Numakura C, Izumino H, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot-Marie-Tooth neuropathy type 2A. Hum Genet. 2004.
84 Evgrafov OV, Mersiyanova I, Irobi J, et al. Mutant small heatshock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36:602-606.
85 Tang BS, Zhao GH, Luo W, et al. Small heatshock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum Genet. 2004.
86 Benndorf R, Sun X, Gilmont RR, et al. HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 ((3D)HSP27). J Biol Chem. 2001;276:26753-26761.
87 Takashima H, Boerkoel CF, John J, et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32:267-272.
88 Moreira MC, Barbot C, Tachi N, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. 29(2):189–93. Nat Genet. 2001;29:189-193.
89 Moreira MC, Klur S, Watanabe M, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225-227.
90 Pouliot JJ, Yao KC, Robertson CA, et al. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552-555.
91 Inamdar KV, Pouliot JJ, Zhou T, et al. Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J Biol Chem. 2002;277:27162-27168.
92 Interthal H, Pouliot JJ, Champoux JJ. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc Natl Acad Sci U S A. 2001;98:12009-12014.
93 El-Khamisy SF, Saifi GM, Weinfeld M, et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108-113.
94 Zhou T, L J.W., Tatavarthi H, et al. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1). Nucleic Acids Res. 2005;33:289-297.
95 De Sandre-Giovannoli A, Chaouch M, Kozlov S, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726-736.
96 Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285-288.
97 Muchir A, Bonne G, van der Kooi AJ, et al. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet. 2000;9:1453-1459.
98 Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715-1724.
99 Cao H, Hegele RA. Nuclear lamin A/C R482Q mutation in canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet. 2000;9:109-112.
100 Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71:426-431.
101 Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293-298.
102 Mounkes LC, Kozlov S, Hernandez L, et al. A progeroid syndrome in mice is caused by defects in A-type lamins. Nature. 2003;423:298-301.
103 Stuurman N, Heins S, Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol. 1998;122:42-66.
104 Sullivan T, Escalante-Alcalde D, Bhatt H, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913-920.
105 Antonellis A, Ellsworth RE, Sambuughin N, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72:1293-1299.
106 Freist W, Logan DT, Gauss DH. Glycyl-tRNA synthetase. Biol Chem Hoppe Seyler. 1996;377:343-356.
107 Zuchner S, Noureddine M, Kennerson M, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet. 2005.
108 Choi BO, Lee MS, Shin SH, et al. Mutational analysis of PMP22, MPZ, GJB1, EGR2 and NEFL in Korean Charcot-Marie-Tooth neuropathy patients. Hum Mutat. 2004;24:185-186.
109 Janssen EA, Kemp S, Hensels GW, et al. Connexin32 gene mutations in X-linked dominant Charcot-Marie-Tooth disease (CMTX1). Hum Genet. 1997;99:501-505.
110 Nicholson GA. Mutation testing in Charcot-Marie-Tooth neuropathy. Ann N Y Acad Sci. 1999;883:383-388.
111 Mostacciuolo ML, Righetti E, Zortea M, et al. Charcot-Marie-Tooth disease type I and related demyelinating neuropathies: Mutation analysis in a large cohort of Italian families. Hum Mutat. 2001;18:32-41.
112 Silander K, Meretoja P, Juvonen V, et al. Spectrum of mutations in Finnish patients with Charcot-Marie-Tooth disease and related neuropathies. Hum Mutat. 1998;12:59-68.
113 Bort S, Nelis E, Timmerman V, et al. Mutational analysis of the MPZ, PMP22 and Cx32 genes in patients of Spanish ancestry with Charcot-Marie-Tooth disease and hereditary neuropathy with liability to pressure palsies. Hum Genet. 1997;99:746-754.
114 Leonardis L, Zidar J, Ekici A, et al. Autosomal dominant Charcot-Marie-Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies: detection of the recombination in Slovene patients and exclusion of the potentially recessive Thr118Met PMP22 point mutation. Int J Mol Med. 1998;1:495-501.
115 Mersiyanova IV, Ismailov SM, Polyakov AV, et al. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat. 2000;15:340-347.
116 Marques WJ, Freitas MR, Nascimento OJM, et al. 17p duplicated Charcot-Marie-Tooth 1A. J. Neurol. 2005;252:972-979.
117 Szigeti K, Garcia C, Lupski J. Charcot-Marie-Tooth disease and related hereditary polyneuropathies: molecular diagnostics determine aspects of medical management. Genetics in Medicine. 2005;8:86-92.
118 Warner LE, Roa BB, Lupski JR. Absence of PMP22 coding region mutations in CMT1A duplication patients: further evidence supporting gene dosage as a mechanism for Charcot-Marie-Tooth disease type 1A. Hum Mutat. 1996;8:362-365.
119 Lindeman E, Spaans F, Reulen J, et al. Progressive resistance training in neuromuscular patients. Effects on force and surface EMG. J Electromyogr Kinesiol. 1999;9:379-384.
120 Njegovan ME, Leonard EI, Joseph FB. Rehabilitation medicine approach to Charcot-Marie-Tooth disease. Clin Podiatr Med Surg. 1997;14:99-116.
121 Mann DC, Hsu JD. Triple arthrodesis in the treatment of fixed cavovarus deformity in adolescent patients with Charcot-Marie-Tooth disease. Foot Ankle. 1992;13:1-6.
122 Guyton GP, Mann RA. The pathogenesis and surgical management of foot deformity in Charcot-Marie-Tooth disease. Foot Ankle Clin. 2000;5:317-326.
123 Backonja MM. Use of anticonvulsants for treatment of neuropathic pain. Neurology. 2002;59:S14-S17.
124 Bissar-Tadmouri N, Parman Y, Boutrand L, et al. Mutational analysis and genotype/phenotype correlation in Turkish Charcot-Marie-Tooth Type 1 and HNPP patients. Clin Genet. 2000;58:396-402.
125 Koller WC, Hristova A, Brin M. Pharmacologic treatment of essential tremor. Neurology. 2000;54:S30-S38.
126 Naumann R, Mohm J, Reuner U, et al. Early recognition of hereditary motor and sensory neuropathy type 1 can avoid life-threatening vincristine neurotoxicity. Br J Haematol. 2001;115:323-325.
127 Sereda MW, Meyer zu Horste G, Suter U, et al. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med. 2003;9:1533-1537.
128 Passage E, Norreel JC, Noack-Fraissignes P, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10:396-401.
129 Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103:259-280.
130 Kaku DA, Parry GJ, Malamut R, et al. Uniform slowing of conduction velocities in Charcot-Marie-Tooth polyneuropathy type 1. Neurology. 1993;43:2664-2667.
131 Kaku DA, Parry GJ, Malamut R, et al. Nerve conduction studies in Charcot-Marie-Tooth polyneuropathy associated with a segmental duplication of chromosome 17. Neurology. 1993;43:1806-1808.
132 Harding AE, Thomas PK. Autosomal recessive forms of hereditary motor and sensory neuropathy. J Neurol Neurosurg Psychiatry. 1980;43:669-678.
133 Takashima H, Nakagawa M, Kanzaki A, et al. Germline mosaicism of MPZ gene in Dejerine-Sottas syndrome (HMSN III) associated with hereditary stomatocytosis. Neuromuscul Disord. 1999;9:232-238.






