[level-membership-for-neurology-category]
CHAPTER 68 INHERITED ATAXIAS
The inherited ataxias are a heterogeneous group of neurodegenerative syndromes with a vast array of clinical signs and symptoms, both neurological and systemic. The clinical spectrum is wide and may range from “pure” cerebellar signs to constellations that include spinal cord syndromes, peripheral nerve disease, cognitive impairment, cerebellar or supranuclear ophthalmological signs, psychiatric problems, and seizure disorders. Typically, inherited ataxias develop over years to decades; recessively inherited forms start in childhood and those dominantly inherited start in adulthood. Historically, these disorders were diagnosed with difficulty on the basis of clinical manifestations. Classification schemes, complicated by eponymous designations in the absence of genetically based information, further complicated the literature. Genetic advances, however, have aided in the classification and diagnosis of the inherited ataxias, identifying specific chromosomal defects or gene loci associated with a clinical phenotype in many instances.
BRIEF DESCRIPTION
The inherited ataxias are grouped (Table 68-1) by mode of inheritance: autosomal dominant, autosomal recessive, mitochondrial, and X-linked. More than 32 known autosomal dominant cerebellar ataxias (ADCAs) were known as of May 2006. The known ADCAs are designated as spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy (DRPLA), episodic ataxia types 1 and 2, human spastic ataxia, and ataxia caused by mutations in the gene encoding fibroblast growth factor 14.1 These ataxias characteristically manifest symptomatically in adulthood. However, the phenomenon of anticipation occurs frequently because (1) many ADCAs occur on the basis of trinucleotide repeat expansion mutations, (2) the length of trinucleotide repeats may be correlated with symptomatic age-related onset, and (3) trinucleotide repeats may undergo further expansion in subsequent generations.
TABLE 68-1 Examples of Inherited Ataxias and Their Mode of Inheritance
| Mode of Inheritance | Inherited Ataxias |
|---|---|
| Autosomal recessive | Friedreich’s ataxia, late-onset Friedreich’s ataxia, FARR, AVED, ARSACS, Cayman’s ataxia, abetalipoproteinemia, ataxia telangiectasia, AOA, SCAN, xeroderma pigmentosum, Cockayne’s syndrome, trichothiodystrophy, Joubert’s syndrome, Gillespie’s syndrome, Behr’s disease, Marinesco-Sjögren syndrome, and metabolic ataxias (urea cycle defects, aminoacidurias, peroxisomal disorders, disorders of pyruvate and lactate, Wilson’s disease, hyperammonemic ataxia, Niemann-Pick disease type C, sialidosis, Refsum’s disease, ceroid lipofuscinosis, leukodystrophies, cholestanolosis, and gangliosidosis) |
| Autosomal dominant | Spinocerebellar ataxias, DRPLA, episodic ataxia types 1 and 2, HSA, and FGF14 |
| X-linked | Sideroblastic anemia and spinocerebellar ataxia, cerebellar ataxia 2 syndrome, ataxia syndrome with extrapyramidal involvement, Arts syndrome, ataxia with tremor and cognitive decline, and Pelizaeus-Merzbacher allelic variant |
| Mitochondrial | Leukodystrophy; MELAS; MERRF; NARP; Leigh’s syndrome; HAM; syndrome of ataxia, cataract, and diabetes mellitus; coenzyme Q10 deficiency; COX10 deficiency; cytochrome c oxidase I and II deficiency; pyruvate dehydrogenase disorders; and syndrome of sideroblastic anemia and spinocerebellar ataxia |
ADCA, autosomal dominant cerebellar ataxia; AOA, ataxia with ocular motor apraxia; ARSACS, autosomal recessive spastic ataxia of Charlevoix-Saguenay; AVED, ataxia with vitamin E deficiency; COX10, cytochrome oxidase 10; DRPLA, dentatorubral-pallidoluysian atrophy; FARR, Friedreich’s ataxia with retained reflexes; FGF14, fibroblast growth factor 14 (mutation causing disease); HAM, hearing loss, ataxia, and myoclonus; HSA, human spastic ataxia; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke; MERRF, myoclonic epilepsy and ragged red fibers; NARP, neuropathy, ataxia, and retinitis pigmentosa; SCAN, spinocerebellar ataxia with axonal neuropathy.
The autosomal recessive cerebellar ataxias (ARCAs) are less common than ADCAs. The two most common ARCAs are Friedreich’s ataxia and ataxia telangiectasia. Other less common ARCAs include ataxia with vitamin E deficiency (AVED), autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), abetalipoproteinemia, Refsum’s disease, infantile-onset spinocerebellar ataxia, spinocerebellar ataxia with axonal neuropathy, Cayman’s ataxia, trichothiodystrophy, xeroderma pigmentosum, Cockayne’s syndrome, and ataxia with oculomotor apraxia.2,3 The ARCAs begin in infancy and early life.
Pathophysiological clues for many of the inherited ataxias are known. A few of the ARCAs have been genetically characterized with pathogenesis resulting from the loss of function of specific cellular proteins crucial in metabolic homeostasis, cell cycle, or DNA repair. For the ADCAs, the pathogenesis often appears related to the production of a toxic or harmful protein.2
EPIDEMIOLOGY AND RISK FACTORS
Estimates of the prevalence of the ADCAs are restricted to a few epidemiological studies hindered by founder effects in the isolated geographical populations studied. The population studies do indicate that the prevalence of the ADCA subtypes varies among ethnic and geographical populations.1 Although the prevalence is probably underestimated at three cases per 100,000 people, ADCAs are relatively uncommon disorders.4 In comparison, the prevalence of Huntington’s disease is 5 to 10 cases per 100,000 people. The most common ADCA worldwide is spinocerebellar ataxia type 3, followed by types 2 and 6, type 1, type 8, and type 7.5,6 The prevalence of DRPLA is 0.25% to 2% among patients with ADCA and is especially prevalent in Japan.7 The remaining subtypes are very rare.6
The exact prevalence of X-linked and mitochondrial inherited ataxias is unknown but believed to be extremely low. For example, the estimated prevalence of all mitochondrial disorders (with and/or without ataxia) is 11.5 cases per 100,000 individuals.8
The main risk factor for the inherited ataxias is a family history of ataxia. Spontaneous mutations are rarely identified.6 A patient with an ADCA, the proband, typically has a parent with the disease. However, this, too, is not a universal finding, because the mutant allele may have decreased penetrance in the parent, onset of the disease may be late in the parent but early in the proband secondary to anticipation, an affected parent may die before the disease manifests, the proband’s background may be unknown because of adoption or unknown lineage, or symptoms in a family member may go unrecognized.
Mitochondrial disorders can result either from mutations in mitochondrial DNA that are maternally transmitted or from mutations in nuclear DNA that may follow either an autosomal dominant or an autosomal recessive pattern of inheritance.8
CLINICAL FEATURES
The Autosomal Recessive Cerebellar Ataxias
The ARCAs are rare, with the exceptions of Friedreich’s ataxia and ataxia telangiectasia. For simplification, they can be grouped etiologically (Table 68-2) as those resulting from potentially increased oxidative stress, those resulting from problems in DNA repair, and those caused by metabolic derangements. The more frequently encountered ARCAs are discussed in the following section; the features that may aid in determining diagnosis are highlighted.
TABLE 68-2 Pathophysiology of Autosomal Recessive Cerebellar Ataxias
| Pathophysiology | Disease |
|---|---|
| Oxidative stress | Friedreich’s ataxia, late-onset Friedreich’s ataxia, FARR, AVED, ARSACS, Cayman’s ataxia, and abetalipoproteinemia |
| DNA repair failure | Ataxia telangiectasia, AOA, SCAN, xeroderma pigmentosum, Cockayne’s syndrome, trichothiodystrophy |
| Metabolic causes | Urea cycle defects, aminoacidurias, peroxisomal disorders, disorders of pyruvate and lactate, Wilson’s disease, hyperammonemic ataxia, Niemann-Pick disease type C, sialidosis, Refsum’s disease, ceroid lipofuscinosis, leukodystrophies, cholestanolosis, and gangliosidosis |
| Congenital ataxia syndromes | Joubert’s syndrome, Gillespie’s syndrome, Behr’s disease, Marinesco-Sjögren syndrome |
AOA, ataxia with ocular motor apraxia; ARSACS, autosomal recessive ataxia of Charlevoix-Saguenay; AVED, ataxia with vitamin E deficiency; FARR, Friedreich’s ataxia with retained reflexes; SCAN, spinocerebellar ataxia with axonal neuropathy.
The ARCAs associated with oxidative stress include Friedreich’s ataxia and Friedreich’s ataxia–like syndromes, ataxias secondary to vitamin E deficiency, and Cayman’s ataxia. The most common of these is Friedreich’s ataxia. The classic clinical features include progressive gait and limb ataxia, dysarthria, absence of deep tendon reflexes, vibratory and proprioceptive sensory loss, and pyramidal weakness with a disease onset before 25 years of age.2 Symptomatic sensory loss typical of Friedreich’s ataxia helps to distinguish this ataxia from other spinocerebellar ataxias with severe reduction or loss of sensory action potentials without reduction of motor conduction velocities.9 About 25% of patients have an atypical manifestation after 25 years of age, called late-onset Friedreich’s ataxia, or with retained reflexes, known as Friedreich’s ataxia with retained reflexes, and/or slow disease progression. The skeletal, endocrine, and cardiovascular systems are affected; the disease manifests, respectively, as scoliosis and pes cavus, diabetes mellitus, and hypertrophic cardiomyopathy. Cardiomyopathy is a cardinal feature of Friedreich’s ataxia and detrimentally affects prognosis commonly, with early death secondary to heart failure or fatal arrhythmia. Optic neuropathy and sensorineural hearing may be present later in the disease. The pathogenesis of Friedreich’s ataxia involves a deficiency of a mitochondrial protein, frataxin, secondary to a guanine-adenine-adenine expansion. The genetic defect is believed to result in iron accumulation in mitochondria with oxygen-free radical production. The normal trinucleotide repeat range is estimated at 33 or fewer triplet repeats; pathological expansions range from 67 to 1000 triplets, with length inversely proportional to age at clinical onset, scoliosis, and cardiomyopathy.10 Unlike the ADCAs with trinucleotide repeats, Friedreich’s ataxia is not associated with anticipation. Patients with Friedreich’s ataxia become unable to walk within 11 years after disease onset and have a mean survival length of 36 years after onset of symptoms. Typically, more than two decades of life are spent in a debilitated motor state.11 To date, the size of the guanine-adenine-adenine pathological expansion has made development of transgenic animal models difficult.
AVED is a rare disorder with clinical manifestations similar to those of Friedreich’s ataxia. A patient with clinical features of Friedreich’s ataxia and negative genetic test results should be screened for AVED (with plasma vitamin E levels), especially because this syndrome is potentially treatable. Head titubation and dystonia are more common in AVED than in Friedreich’s ataxia; cardiomyopathy is a less frequent problem.2 The syndrome is caused by a defective transfer protein for α-tocopherol that prevents transfer of this most biologically active form of vitamin E to peripherally circulating lipoproteins.12 Oral vitamin E supplementation is the mainstay of treatment.
Cayman’s ataxia is an ARCA found in a population on Grand Cayman Island. It involves a mutation of the Caytaxin protein, found almost exclusively in the central nervous system, and is believed to bind a ligand with similar properties to vitamin E in the central nervous system.3 It is characterized by early-onset hypotonia, cerebellar ataxia, and psychomotor retardation.3
There are seven inherited ataxias caused by defective DNA repair: ataxia telangiectasia, ataxia with oculomotor apraxia, spinocerebellar ataxia with axonal neuropathy, ARSACS, xeroderma pigmentosum, Cockayne’s syndrome, and trichothiodystrophy. The most common of these is ataxia telangiectasia. Ataxia telangiectasia is typified by neurological, dermatological, and immunological symptoms starting in infancy or early childhood, with death in early adulthood. Neurological manifestations include cerebellar ataxia, slowed horizontal saccades, dystonic posturing, chorea, tics or jerks, dysphagia and choking, and peripheral neuropathy. The dermatological signs include oculocutaneous telangiectasia, premature graying of hair, and premature senile keratosis.12 Patients with ataxia telangiectasia have recurrent sinopulmonary infections secondary to derangement of cellular and humoral immunity (deficient immunoglobulin A levels). Malignancy and neoplasia are common, especially those involving hematological cells. The mutations associated with ataxia telangiectasia in the ataxia telangiectasia mutation gene lead to mutations in nuclear protein, which normally repairs DNA. Consequently, individuals with ataxia telangiectasia have a sensitivity to ionizing radiation—a diagnostic hallmark of ataxia telangiectasia.3 Almost all persons with ataxia telangiectasia have an elevated α-fetoprotein level; however, this is a nonspecific finding. Ataxia with oculomotor apraxia may be a separate syndrome. It lacks the immunological features and sensitivity to ionizing radiation seen in classic ataxia telangiectasia.2,13 Survival of patients with ataxia telangiectasia after age 30 is rare. ARSACS is a rare ARCA secondary to defective DNA repair found in parts of Quebec.
Xeroderma pigmentosum, Cockayne’s syndrome, and trichothiodystrophy are rare ARCAs characterized by extreme skin photosensitivity in combination with ataxia and possibly other neurological manifestations. Xeroderma pigmentosum is marked by the development of skin cancers, including squamous cell carcinomas and melanoma.12 Cockayne’s syndrome is associated with ataxia, skin manifestations, growth retardation, microcephaly and facial malformations, retinal and cochlear degeneration, and neuropathy.12 Trichothiodystrophy is associated with a mutation that produces a phenotype that overlaps with both xeroderma pigmentosum and trichothiodystrophy.
Metabolic derangements of many types cause neurodegenerative syndromes and ataxia (see Table 68-2). A complete review of these disorders is beyond the scope of this chapter. Refsum’s disease is one that is of importance because early diagnosis and intervention can prevent neurological manifestations. A rare disorder caused by accumulation of phytanic acid in body tissues, Refsum’s disease is a syndrome of cerebellar ataxia, sensorimotor polyneuropathy, and retinitis pigmentosa. The cerebrospinal fluid has a high protein concentration without pleocytosis.2 Treatment is based on dietary restriction of the fatty acid phytanic acid (Tables 68-3 and 68-4).
TABLE 68-3 Select Noncerebellar Clinical Findings in ARCA
| Neurological Signs (Other than Ataxia) | Disease |
|---|---|
| Retinopathy | Abetalipoproteinemia, Refsum’s disease, ARSACS, AVED |
| Optic atrophy | Friedreich’s ataxia, IOSCA |
| Ophthalmoplegia | IOSCA, AVED |
| Ocular apraxia | Ataxia telangiectasia, AOA |
| Hyporeflexia | Friedreich’s ataxia, AVED, abetalipoproteinemia |
| Hypotonia | Cayman’s ataxia |
| Spasticity | ARSACS |
| Peripheral neuropathy | Friedreich’s ataxia, AVED, ARSACS, SCAN |
| Proprioceptive loss | Friedreich’s ataxia, AVED, abetalipoproteinemia, PCRP |
| Muscle atrophy | Friedreich’s ataxia, AVED, abetalipoproteinemia |
| Pyramidal signs | Friedreich’s ataxia, AVED |
| Head titubation | AVED |
| Deafness | Friedreich’s ataxia, Refsum’s disease, IOSCA |
| Choreoathetosis | Ataxia telangiectasia, AOA, IOSCA |
| Mental retardation | Marinesco-Sjögren syndrome, AOA, coenzyme Q10 deficiency |
| Seizure | Coenzyme Q10 deficiency |
AOA, ataxia with ocular motor apraxia; ARSACS, autosomal recessive ataxia of Charlevoix-Saguenay; AVED, ataxia with vitamin E deficiency; IOSCA, infantile spinocerebellar ataxia; PCRP, posterior column ataxia with retinitis pigmentosa; SCAN, spinocerebellar ataxia with axonal neuropathy.
TABLE 68-4 Nonneurological Manifestations of Select Autosomal Recessive Cerebellar Ataxias
| Manifestation | Disease | Sign |
|---|---|---|
| Cardiovascular | Friedreich’s ataxia | Cardiomyopathy |
| AVED | Rare cardiomyopathy | |
| Pulmonary | Ataxia telangiectasia | Bronchiectasis secondary to chronic pulmonary infection |
| Gastrointestinal | Abetalipoproteinemia | Steatorrhea |
| Immunological/infectious | Ataxia telangiectasia | Immune deficiency with lymphopenia and low immunoglobulin levels with frequent sinopulmonary and cutaneous infections |
| Hematological/oncological | Ataxia telangiectasia | Lymphoreticular and germ cell malignancy in childhood and adenocarcinoma and solid tumors in adulthood |
| Abetalipoproteinemia | Acanthocytosis | |
| Xeroderma pigmentosum, trichothiodystrophy, and Cockayne’s syndrome | Skin cancers | |
| Musculoskeletal | Friedreich’s ataxia | Pes cavus and scoliosis |
| Cockayne’s syndrome and trichothiodystrophy | Short stature, microcephaly, and prognathism | |
| Dermatological | Ataxia telangiectasia | Ocular and cutaneous telangiectasias, premature senile keratosis, and premature graying hair |
| Xeroderma pigmentosum, trichothiodystrophy, and Cockayne’s syndrome | Skin photosensitivity and skin cancers | |
| Abetalipoproteinemia and AVED | Xanthelasmata and tendon xanthomas | |
| Endocrine | Friedreich’s ataxia | Diabetes mellitus |
| Ataxia telangiectasia | Hypogonadism |
AVED, ataxia with vitamin E deficiency.
The Autosomal Dominant Cerebellar Ataxias
The ADCAs are a clinically and genetically heterogeneous group of neurodegenerative disorders. Overlap exists between the ADCAs in manifestation to such a degree that genetic testing is required for definitive diagnosis. The following discussion, along with Table 68-5, highlights the clinical features of the six most common spinocerebellar ataxias, DRPLA, the episodic ataxias, human spastic ataxia, and fibroblast growth factor 14 mutation and are designed to narrow the differential diagnosis and number of tests required for diagnosis.1,14
TABLE 68-5 Selected Clinical Signs in Autosomal Dominant Cerebellar Ataxias2,17
| Clinical Signs (Other Than Ataxia) | Disease |
|---|---|
| Retinopathy | Spinocerebellar ataxia type 7 |
| Slow saccades | Spinocerebellar ataxia types 1, 2, 3, and 7 (rarely type 6) |
| Downbeat nystagmus | Spinocerebellar ataxia type 6 and episodic ataxia type 2 |
| Ophthalmoplegia | Spinocerebellar ataxia types 1, 2, and 3 |
| Upper motor neuron signs | Spinocerebellar ataxia types 1, 3, 7, and 12 (sometimes types 6, 8) |
| Extrapyramidal | Spinocerebellar ataxia types 2, 3, and 12 (parkinsonism) |
| DRPLA (chorea) | |
| Spinocerebellar ataxia type 3 (dystonia) | |
| Spinocerebellar ataxia types 2 and 14 and occasionally types 1, 3, 6, 7, and 19 (myoclonus) | |
| Spinocerebellar ataxia type 20 (dysphonia) | |
| FGF14 (dyskinesia) | |
| Spinocerebellar ataxia types 12, 16, and 19 and FGF14 (head or hand tremor) | |
| Spinocerebellar ataxia type 20 (palatal tremor) | |
| Cortical | Spinocerebellar ataxia types 13 and 21 and FGF14 (mental retardation) |
| Spinocerebellar ataxia types 10 and 7 and DRPLA (seizure) | |
| DRPLA and spinocerebellar ataxia type 17 (dementia) | |
| DRPLA and spinocerebellar ataxia type 17 (psychosis) | |
| Pontine signs | Spinocerebellar ataxia types 1, 2, and 3 |
| Fasciculations | Spinocerebellar ataxia type 3 |
| Myokymia | Episodic ataxia type 1 |
| Peripheral neuropathy | Spinocerebellar ataxia types 3, 4, 18, and 25 |
| Vertigo | Episodic ataxia type 2 |
DRPLA, dentatorubral-pallidoluysian atrophy; FGF14, fibroblast growth factor 14 (mutation causing disease).
Patients with spinocerebellar ataxia type 1 present with ataxia along with dysarthria and eventually develop bulbar signs such as atrophy of facial and masticatory muscles, perioral fasciculations, and severe dysphagia, leading to frequent aspiration.15 The mean age at onset of type 1 is 37, but it can occur from ages 4 to 74 years.16,17
Spinocerebellar ataxia type 2 is similar to types 1 and 3; however slowed saccades, postural and action tremors, myoclonus, and hyporeflexia are more commonly seen.1,18 The mean age at onset is 30.16
The most prevalent and perhaps the most variable ADCA is spinocerebellar ataxia type 3, or Machado-Joseph disease. Distinctive features in some patients include parkinsonism, restless legs syndrome and periodic leg movements in sleep, pseudoexophthalmos, faciolingual myokymia, and dystonia. A relatively specific sign in type 3 is the loss of temperature sensation, involving the limbs, trunk, and face. Three clinical forms of type 3 have been described. Type I disease is the most severe and is characterized by a young age at onset, prominent dystonia, rigidity, and bradykinesia. Ataxia is less prominent in type I. The most common form is type II, which is characterized by ataxia and upper motor neuron signs. Spastic paraparesis is a manifestation in some patients. Type III disease is typified by a pronounced peripheral neuropathy; it has a mean later age at onset, 42, and the age at onset is inversely correlated with severity of disease.16 Patients with spinocerebellar ataxia type 3 require walking aids after 10 to 15 years of disease duration.
Spinocerebellar ataxia type 6 is typified by “pure” cerebellar ataxia and late onset of disease; 60% of patients present after the age of 50 years.1 Other signs and symptoms can include diplopia, dysarthria, dysphagia, and horizontal and vertical nystagmus. The ataxia is slowly progressive, and life span is not shortened. Episodic ataxia type 2 and familial hemiplegic migraine are genetically related to type 6.
DRPLA, or Smith’s disease, is named for its characteristic pathology changes involving the dentate nucleus and external segment of the globus pallidus and their projections to the red and subthalamic nuclei, respectively. It is a rare disorder seen most often in Japan. The clinical features are variable but can include myoclonus, chorea, epilepsy, ataxia, and cognitive impairment.19 Age at onset is from childhood to late adulthood, with a mean age of 30 years.
Mutations in fibroblast growth factor 14 produce a rare ADCA with an early onset in childhood. Clinical features include ataxia, nystagmus, dysarthria, head and limb tremor, orofacial dyskinesias, reduced vibration in the legs, psychiatric symptoms of aggression and depression, and cognitive defects.17 Life span is not shortened in this disease.
Episodic ataxia types 1 and 2 are rare ADCAs associated with potassium channel and calcium channel dysfunction, respectively. Type 1 is characterized by paroxysmal spells of ataxia lasting seconds to minutes.20 Myokymia around the eyes, lips, or fingers is characteristic during or between episodes.12 Onset is usually in childhood or adolescence. Type 2 is typified by episodes of ataxia, nausea, and vertigo with interictal nystagmus, diplopia, and migraine. The episodes last hours to days and are brought on by stress, exercise, ethanol, phenytoin, and/or caffeine. Episodes can be aborted and prevented with acetazolamide. Type 2 is genetically related to familial hemiplegic migraine and spinocerebellar ataxia type 6. Other episodic ataxias have been recognized.
X-Linked Cerebellar Ataxias
Cerebellar ataxia has been described in families with an apparent X-linked inheritance (examples are listed in Table 68-1). These syndromes typically have associated clinical features, such as spasticity, mental retardation, deafness, dementia, or sideroblastic anemia.6 For a more complete description of the X-linked cerebellar ataxias, see the review by Evidente and associates.21
Mitochondrial Inherited Cerebellar Ataxias
Mitochondrial inherited cerebellar ataxia should be suspected if the ataxia is associated with additional features: neuropathy, myopathy, seizures, retinopathy, deafness, diabetes mellitus, cardiomyopathy, myoglobinuria, renal tubular acidosis, and/or short stature. Examples of mitochondrial inherited ataxias are listed in Table 68-1.
DIAGNOSTIC STRATEGY
If known, the ethnicity and geographic background of the patient should also be noted. It is well established that certain spinocerebellar ataxias are more prevalent in particular ethnic groups. Similarly, Friedreich’s ataxia is more common in white persons and has never been identified in sub-Saharan Africans, American Indians, Japanese persons, or Chinese persons. In the next section, Table 68-8 displays the inherited ataxias associated with particular racial groups.
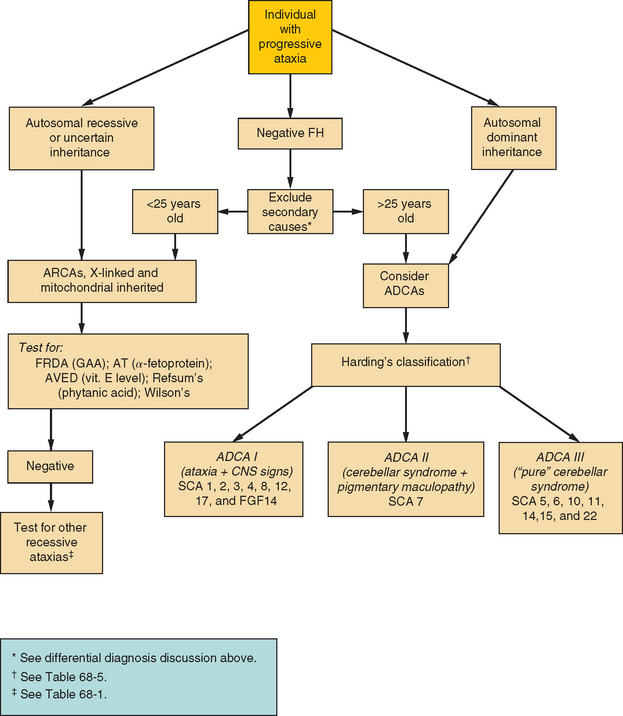
Although there is significant overlap between the phenotypes of inherited ataxias, clinical findings can aid in narrowing the differential diagnosis. If an ADCA is considered, the patient’s condition can be grouped into one of Harding’s clinical types represented in Table 68-6. These categories can be used for narrowing the differential diagnosis. Table 68-5 lists neurological signs other than ataxia that are associated with ADCAs, which may aid in genetic testing decisions. The ARCAs likewise have clinical characteristics both neurological and nonneurological that help distinguish them, such as telangiectasia in ataxia telangiectasia or hyporeflexia, cardiomyopathy, and diabetes mellitus in Friedreich’s ataxia. These are displayed in Tables 68-3 and 68-4. Figure 68-1 demonstrates a suggested diagnostic strategy.
| Harding’s Classification | Disease |
|---|---|
| ADCA I | Spinocerebellar ataxia types 1, 2, 3, 4, 8, 12, 17, and FGF14 |
| ADCA II | Spinocerebellar ataxia type 7 |
| ADCA III | Spinocerebellar ataxia types 5, 6, 10, 11, 14, 15, and 22 |
| Other | Spinocerebellar ataxia type 13 |
ADCA, autosomal dominant cerebellar ataxia. ADCA I, cerebellar syndrome + other central nervous system signs (pyramidal, extrapyramidal, ophthalmoplegia, and dementia); ADCA II, cerebellar syndrome + pigmentary maculopathy; ADCA III, “pure” cerebellar syndrome ± mild pyramidal signs; FGF14, fibroblast growth factor 14 (mutation causing disease).
From Harding AE: The clinical features and classification of the late onset autosomal dominant cerebellar ataxias: a study of 11 families, including descendants of “the Drew family of Walworth.” Brain 1982; 105:1-28.
EVALUATION, TESTS, AND LABORATORY FINDINGS
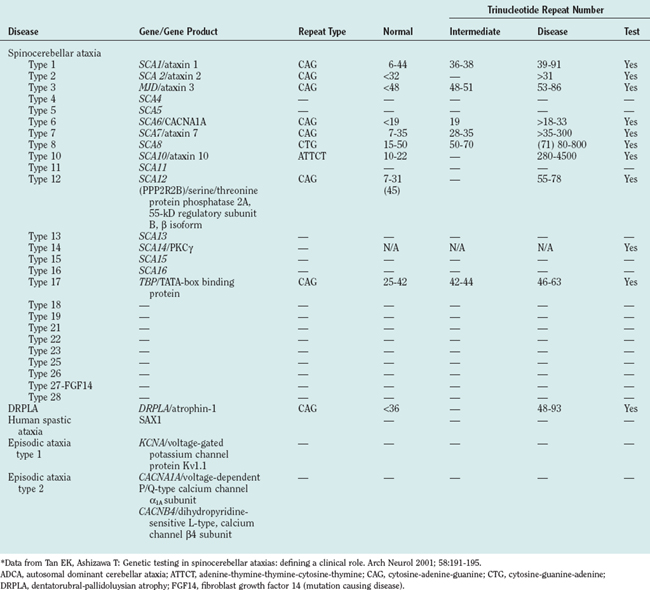
In patients with a clear inheritance pattern suggestive of a genetically derived ataxia, DNA testing should be the first step in establishing the diagnosis. Genetic testing is the most specific and definitive test for an inherited ataxia in which the specific genetic abnormality has been identified. It can also be the most cost-effective test if the direct genetic testing strategy can be directed by a thorough clinical examination, rather than by an expensive comprehensive inherited ataxia screen (several thousand U.S. dollars at most laboratories). Tables 68-7 and 68-8 list the inherited ataxias and their respective available genetic tests. A useful reference for current genetic tests and their availability is www.genetests.org. The ranges for the number of trinucleotide repeats for the ADCAs are listed in Table 68-8.

Interpreting test results can be difficult for the inherited ataxias associated with trinucleotide repeats. The presence of expansion repeats is tested by polymerase chain reaction or Southern blot techniques. For example, the test may reveal that only a single repeat length was detected. This usually indicates that the second allele is the same length and therefore cannot be distinguished.22 Another explanation is that the second allele repeat expansion was present but not detected, as can occur with polymerase chain reaction–based testing. The Southern blot should be employed in this instance because of its greater sensitivity. Interpretation of a test is also problematic when the length of the expansion repeat is on the lower border of the pathological range.22
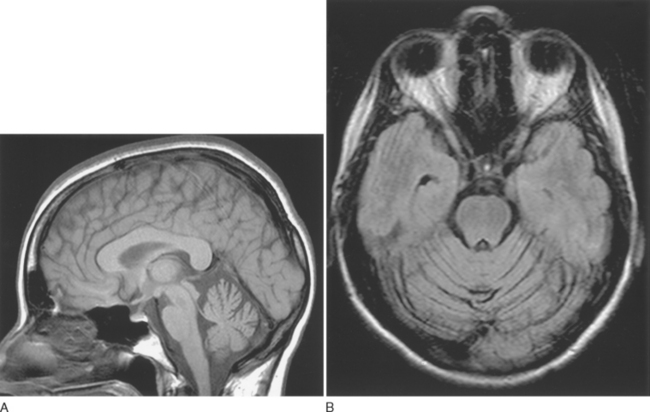
Magnetic resonance imaging findings such as cerebellar atrophy (Fig. 68-2), spinal atrophy, and olivopontocerebellar atrophy are nonspecific, as are abnormalities found on positron emission tomography studies in inherited ataxia. Similarly, no specific electrophysiological finding is associated with any one inherited ataxia; however, demonstrating a polyneuropathy in a patient may help narrow the differential diagnosis, because this is a finding common to some ataxias and infrequently encountered in others.
TREATMENT
Unfortunately, no specific treatment exists to halt or slow neuronal death in the inherited ataxias except for those secondary to vitamin E deficiency and/or other metabolic derangements (if recognized early in the course of illness). However, treatment strategies for certain ataxias are increasingly being studied. For instance, therapeutic strategies have been developed for Friedreich’s ataxia with the use of antioxidants (idebenone, coenzyme Q10, vitamin E, and mitoquinone), iron chelators (desferrioxamine, deferiprone, 2-pyridylcarboxaldehyde isonicotinoyl hydrazone analogs), glutathione peroxidase mimetics, pharmacological therapy to increase frataxin levels, and gene and cell therapies.10 No double-blind, placebo-controlled trials have demonstrated any benefit for the neurological manifestations of Friedreich’s ataxia. However, because patients may benefit from treatment of other medical problems related to the inherited ataxias (e.g., diabetes mellitus and cardiomyopathy in Friedreich’s ataxia), determining the diagnosis is important.
Because spinocerebellar ataxia type 6 is caused by a mutation in the α1A subunit of the voltage-gated neuronal calcium channel, calcium channel blockers and acetazolamide have been suggested as therapies because of their effectiveness in migraine prophylaxis and episodic ataxia type 2, respectively (two diseases with pathophysiology involving neuronal calcium flux). Ataxia was improved in the first open-label trial for spinocerebellar ataxia type 6 with the use of acetazolamide (250 to 500 mg/day) over 88 weeks.23 Parkinsonism associated with the spinocerebellar ataxias has been shown to improve with levodopa and dopamine agonists.24–26 Spinocerebellar ataxias with symptoms of dystonia and bradykinesia may respond to amantadine.27 Botulinum toxin injections may prove effective in cases with dystonia. Other symptoms, such as restless legs syndrome and periodic leg movements especially common in spinocerebellar ataxia type 3, can be treated with usual dopaminergic treatments.28,29 Table 68-9 lists clinical trials in inherited ataxias completed or ongoing as of January 2005. For a current list of clinical trials for inherited ataxias, refer to www.clinicaltrials.gov.
SUPPORTIVE CARE AND LONG-TERM MANAGEMENT
It is worthwhile to determine vitamin B12, folate, and thyroid status in all patients with ataxia, including those with inherited ataxia, because any patient may have a superimposed contribution of a readily treatable secondary cause of ataxia. For similar reasons, it is advisable for patients with ataxia to undergo magnetic resonance imaging and/or computed tomographic scanning of the head on at least one occasion.
FUTURE CONSIDERATIONS
Therapeutic trials (see Table 68-9) may provide invaluable information concerning disease. Genetic research may also result in the discovery of gene modifiers that can be manipulated to delay the age at symptomatic onset or prevent neuronal destruction altogether. Likewise, delineating the pathophysiology responsible for the neurodegeneration caused by the ataxias may help provide not only therapies for the inherited ataxias but also information on the normal functioning of the cerebellum and its spinal tracts. Searches for biomarkers must be a priority, to determine prognosis before neuronal destruction and strategies for screening to identify asymptomatic individuals in whom definitive genetic testing is appropriate.
Di Donato S, Gellera C, Mariotti C. The complex and genetic classification of inherited ataxias. II. Autosomal recessive ataxias. Neurol Sci. 2001;22:219-228.
Harding AE. The clinical features and classification of the late onset autosomal dominant cerebellar ataxias: a study of 11 families, including descendants of “the Drew family of Walworth.”. Brain. 1982;105:1-28.
Rosa AL, Ashizawa T. Genetic ataxia. Neurol Clin North Am. 2002;20:727-757.
Schols L, Bauer P, Schmidt T, et al. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291-304.
Subramony SH, Currier RD. The classification of familial ataxias. In: Vinken PJ, Bruyn GW, Klawans HL, et al, editors. Handbook of Clinical Neurology, vol 60: Hereditary Neuropathies and Spinocerebellar Atrophies. Amsterdam: Elsevier Science; 1991:271-284.
1 Schols L, Bauer P, Schmidt T, et al. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3:291-304.
2 Di Donato S, Gellera C, Mariotti C. The complex and genetic classification of inherited ataxias. II. Autosomal recessive ataxias. Neurol Sci. 2001;22:219-228.
3 Taroni F, Di Donato. Pathways to motor incoordination: the inherited ataxias. Neuroscience. 2004;5:641-655.
4 Van de Warrenburg BPC, Sinke RJ, Verschuuren-Bemelmans CC, et al. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset analysis. Neurology. 2002;58:702-708.
5 Moseley ML, Benzow KA, Schut LJ, et al. Incidence of dominant spinocerebellar and Friedreich triplet repeats among 361 ataxia families. Neurology. 1998;51:1666-1671.
6 Bird TD. Hereditary ataxia overview. Available at: http://www.geneclinics.org/servlet/access?db=geneclinics&site-gt&id=8888891&key=U0ECBakjvMZq&gry=&fcn=y&fw=ig9-&filename=/profiles/ataxias/index.html. (accessed March 28, 2006).
7 Le Ber I, Camuzat A, Castelnovo G, et al. Prevalence of dentatorubral-pallidoluysian atrophy in a large series of white patients with cerebellar ataxia. Arch Neurol. 2003;60:1097-1099.
8 Chinnery PF. Mitochondrial disorders overview. Available at: http://www.geneclinics.org/servlet/access?id=8888892&key=mWTifFAAeBgi4&gry=INSERTGRY&fcn=y&fw=egAc&filename=/glossary/profiles/mt-overview/index.html. (accessed March 28, 2006).
9 Chokravarty A. Friedreich’s ataxia-yesterday, today and tomorrow. Neurol India. 2003;51:176-182.
10 Voncken M, Ioannou P, Delatycki MB. Friedreich ataxia—update on pathogenesis and possible therapies. Neurogenetics. 2004;5:1-8.
11 Klockgether T, Ludtke R, Kramer B, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain. 1998;121:589-600.
12 Rosa AL, Ashizawa T. Genetic ataxia. Neurol Clin North Am. 2002;20:727-757.
13 Aicarki J, Barbosa C, Andermann E, et al. Ataxia-ocular motor apraxia: a syndrome mimicking ataxia-telangiectasia. Ann Neurol. 1988;24:497-502.
14 Brusco A, Gellara C, Cagnoli C, et al. Molecular genetics of hereditary spinocerebellar ataxia: mutation analysis of spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian families. Arch Neurol. 2004;61:727-733.
15 Brandt VL, Zoghbi HY. Spinocerebellar ataxia type 1. Available at: http://www.geneclinics.org/servlet/access?id=8888892&key=2Jd7dxSHCvTiw&gry=INSERTGRY&fcn=y&fw=XQeU&filename=/glossary/profiles/sca1/index.html. (accessed March 28, 2006).
16 Klockgether T, Ludtke R, Kramer B, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain. 1998;121:589-600.
17 Hereditary ataxias: dominant. Available at: http://www.neuro.wustl.edu/neuromuscular/ataxia/domatax.html#sca1. (accessed March 28, 2006).
18 Pulst S-M. Spinocerebellar ataxia type 2. Available at: http://www.geneclinics.org/servlet/access?id=8888892&key=InWC1vXiLbpgH&gry=INSERTGRY&fcn=y&fw=ikyP&filename=/glossary/profiles/sca2/index.html. (accessed March 28, 2006).
19 Warner TT, Williams LD, Walker RWH, et al. A clinical and molecular genetic study of dentatorubropallidoluysian atrophy in four European families. Ann Neurol. 1995;37:452-459.
20 Albin RL. Dominant ataxias and Friedreich ataxia: an update. Curr Opin Neurol. 2003;16:507-514.
21 Evidente VGH, Gwinn-Hardy KA, Caviness JN, et al. Hereditary ataxias. Mayo Clinic Proc. 2000;75:475-490.
22 Margolis RL. The spinocerebellar ataxias: order emerges from chaos. Curr Neurol Neurosci Rep. 2002;2:447-456.
23 Yabe I, Sasaki H, Yamashita I, et al. Clinical trial of acetazolamide in SCA6, with assessment using the Ataxia Rating Scale and body stabilometry. Acta Neurol Scand. 2001;104:44-47.
24 Tuite PJ, Rogaeva EA, George-Hyslop PH, et al. Dopa-responsive parkinsonism phenotype of Machado-Joseph disease: confirmation of 14q CAG expansion. Ann Neurol. 2001;50:812-815.
25 Buhamann C, Bussopulos A, Oechsner M. Dopaminergic response in parkinsonian phenotype of Machado-Joseph disease. Mov Disord. 2003;18:219-221.
26 Furtado S, Farrer M, Tsuboi Y, et al. SCA-2 presenting as parkinsonism in an Alberta family: clinical, genetic, and PET findings. Neurology. 2002;59:1625-1627.
27 Woods BT, Schaumburg HH. Nigro-spino-dentatal degeneration with nuclear ophthalmoplegia: a unique and partially treatable clinico-pathological entity. J Neurol Sci. 1972;17:149-166.
28 Schols L, Haan J, Riess O, et al. Sleep disturbance in spinocerebellar ataxias: is the SCA3 mutation a cause of restless legs syndrome? Neurology. 1998;51:1603-1607.
29 Abele M, Burk K, Laccone F, et al. Restless legs syndrome in spinocerebellar 1,2, and 3. J Neurol. 2001;248:311-314.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
CHAPTER 68 INHERITED ATAXIAS
The inherited ataxias are a heterogeneous group of neurodegenerative syndromes with a vast array of clinical signs and symptoms, both neurological and systemic. The clinical spectrum is wide and may range from “pure” cerebellar signs to constellations that include spinal cord syndromes, peripheral nerve disease, cognitive impairment, cerebellar or supranuclear ophthalmological signs, psychiatric problems, and seizure disorders. Typically, inherited ataxias develop over years to decades; recessively inherited forms start in childhood and those dominantly inherited start in adulthood. Historically, these disorders were diagnosed with difficulty on the basis of clinical manifestations. Classification schemes, complicated by eponymous designations in the absence of genetically based information, further complicated the literature. Genetic advances, however, have aided in the classification and diagnosis of the inherited ataxias, identifying specific chromosomal defects or gene loci associated with a clinical phenotype in many instances.
BRIEF DESCRIPTION
The inherited ataxias are grouped (Table 68-1) by mode of inheritance: autosomal dominant, autosomal recessive, mitochondrial, and X-linked. More than 32 known autosomal dominant cerebellar ataxias (ADCAs) were known as of May 2006. The known ADCAs are designated as spinocerebellar ataxias, dentatorubral-pallidoluysian atrophy (DRPLA), episodic ataxia types 1 and 2, human spastic ataxia, and ataxia caused by mutations in the gene encoding fibroblast growth factor 14.1 These ataxias characteristically manifest symptomatically in adulthood. However, the phenomenon of anticipation occurs frequently because (1) many ADCAs occur on the basis of trinucleotide repeat expansion mutations, (2) the length of trinucleotide repeats may be correlated with symptomatic age-related onset, and (3) trinucleotide repeats may undergo further expansion in subsequent generations.
TABLE 68-1 Examples of Inherited Ataxias and Their Mode of Inheritance
| Mode of Inheritance | Inherited Ataxias |
|---|---|
| Autosomal recessive | Friedreich’s ataxia, late-onset Friedreich’s ataxia, FARR, AVED, ARSACS, Cayman’s ataxia, abetalipoproteinemia, ataxia telangiectasia, AOA, SCAN, xeroderma pigmentosum, Cockayne’s syndrome, trichothiodystrophy, Joubert’s syndrome, Gillespie’s syndrome, Behr’s disease, Marinesco-Sjögren syndrome, and metabolic ataxias (urea cycle defects, aminoacidurias, peroxisomal disorders, disorders of pyruvate and lactate, Wilson’s disease, hyperammonemic ataxia, Niemann-Pick disease type C, sialidosis, Refsum’s disease, ceroid lipofuscinosis, leukodystrophies, cholestanolosis, and gangliosidosis) |
| Autosomal dominant | Spinocerebellar ataxias, DRPLA, episodic ataxia types 1 and 2, HSA, and FGF14 |
| X-linked | Sideroblastic anemia and spinocerebellar ataxia, cerebellar ataxia 2 syndrome, ataxia syndrome with extrapyramidal involvement, Arts syndrome, ataxia with tremor and cognitive decline, and Pelizaeus-Merzbacher allelic variant |
| Mitochondrial | Leukodystrophy; MELAS; MERRF; NARP; Leigh’s syndrome; HAM; syndrome of ataxia, cataract, and diabetes mellitus; coenzyme Q10 deficiency; COX10 deficiency; cytochrome c oxidase I and II deficiency; pyruvate dehydrogenase disorders; and syndrome of sideroblastic anemia and spinocerebellar ataxia |
ADCA, autosomal dominant cerebellar ataxia; AOA, ataxia with ocular motor apraxia; ARSACS, autosomal recessive spastic ataxia of Charlevoix-Saguenay; AVED, ataxia with vitamin E deficiency; COX10, cytochrome oxidase 10; DRPLA, dentatorubral-pallidoluysian atrophy; FARR, Friedreich’s ataxia with retained reflexes; FGF14, fibroblast growth factor 14 (mutation causing disease); HAM, hearing loss, ataxia, and myoclonus; HSA, human spastic ataxia; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke; MERRF, myoclonic epilepsy and ragged red fibers; NARP, neuropathy, ataxia, and retinitis pigmentosa; SCAN, spinocerebellar ataxia with axonal neuropathy.
The autosomal recessive cerebellar ataxias (ARCAs) are less common than ADCAs. The two most common ARCAs are Friedreich’s ataxia and ataxia telangiectasia. Other less common ARCAs include ataxia with vitamin E deficiency (AVED), autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS), abetalipoproteinemia, Refsum’s disease, infantile-onset spinocerebellar ataxia, spinocerebellar ataxia with axonal neuropathy, Cayman’s ataxia, trichothiodystrophy, xeroderma pigmentosum, Cockayne’s syndrome, and ataxia with oculomotor apraxia.2,3 The ARCAs begin in infancy and early life.
Pathophysiological clues for many of the inherited ataxias are known. A few of the ARCAs have been genetically characterized with pathogenesis resulting from the loss of function of specific cellular proteins crucial in metabolic homeostasis, cell cycle, or DNA repair. For the ADCAs, the pathogenesis often appears related to the production of a toxic or harmful protein.2
EPIDEMIOLOGY AND RISK FACTORS
Estimates of the prevalence of the ADCAs are restricted to a few epidemiological studies hindered by founder effects in the isolated geographical populations studied. The population studies do indicate that the prevalence of the ADCA subtypes varies among ethnic and geographical populations.1 Although the prevalence is probably underestimated at three cases per 100,000 people, ADCAs are relatively uncommon disorders.4 In comparison, the prevalence of Huntington’s disease is 5 to 10 cases per 100,000 people. The most common ADCA worldwide is spinocerebellar ataxia type 3, followed by types 2 and 6, type 1, type 8, and type 7.5,6 The prevalence of DRPLA is 0.25% to 2% among patients with ADCA and is especially prevalent in Japan.7 The remaining subtypes are very rare.6
The exact prevalence of X-linked and mitochondrial inherited ataxias is unknown but believed to be extremely low. For example, the estimated prevalence of all mitochondrial disorders (with and/or without ataxia) is 11.5 cases per 100,000 individuals.8
The main risk factor for the inherited ataxias is a family history of ataxia. Spontaneous mutations are rarely identified.6 A patient with an ADCA, the proband, typically has a parent with the disease. However, this, too, is not a universal finding, because the mutant allele may have decreased penetrance in the parent, onset of the disease may be late in the parent but early in the proband secondary to anticipation, an affected parent may die before the disease manifests, the proband’s background may be unknown because of adoption or unknown lineage, or symptoms in a family member may go unrecognized.
Mitochondrial disorders can result either from mutations in mitochondrial DNA that are maternally transmitted or from mutations in nuclear DNA that may follow either an autosomal dominant or an autosomal recessive pattern of inheritance.8
CLINICAL FEATURES
The Autosomal Recessive Cerebellar Ataxias
The ARCAs are rare, with the exceptions of Friedreich’s ataxia and ataxia telangiectasia. For simplification, they can be grouped etiologically (Table 68-2) as those resulting from potentially increased oxidative stress, those resulting from problems in DNA repair, and those caused by metabolic derangements. The more frequently encountered ARCAs are discussed in the following section; the features that may aid in determining diagnosis are highlighted.
TABLE 68-2 Pathophysiology of Autosomal Recessive Cerebellar Ataxias
| Pathophysiology | Disease |
|---|---|
| Oxidative stress | Friedreich’s ataxia, late-onset Friedreich’s ataxia, FARR, AVED, ARSACS, Cayman’s ataxia, and abetalipoproteinemia |
| DNA repair failure | Ataxia telangiectasia, AOA, SCAN, xeroderma pigmentosum, Cockayne’s syndrome, trichothiodystrophy |
| Metabolic causes | Urea cycle defects, aminoacidurias, peroxisomal disorders, disorders of pyruvate and lactate, Wilson’s disease, hyperammonemic ataxia, Niemann-Pick disease type C, sialidosis, Refsum’s disease, ceroid lipofuscinosis, leukodystrophies, cholestanolosis, and gangliosidosis |
| Congenital ataxia syndromes | Joubert’s syndrome, Gillespie’s syndrome, Behr’s disease, Marinesco-Sjögren syndrome |
AOA, ataxia with ocular motor apraxia; ARSACS, autosomal recessive ataxia of Charlevoix-Saguenay; AVED, ataxia with vitamin E deficiency; FARR, Friedreich’s ataxia with retained reflexes; SCAN, spinocerebellar ataxia with axonal neuropathy.
The ARCAs associated with oxidative stress include Friedreich’s ataxia and Friedreich’s ataxia–like syndromes, ataxias secondary to vitamin E deficiency, and Cayman’s ataxia. The most common of these is Friedreich’s ataxia. The classic clinical features include progressive gait and limb ataxia, dysarthria, absence of deep tendon reflexes, vibratory and proprioceptive sensory loss, and pyramidal weakness with a disease onset before 25 years of age.2 Symptomatic sensory loss typical of Friedreich’s ataxia helps to distinguish this ataxia from other spinocerebellar ataxias with severe reduction or loss of sensory action potentials without reduction of motor conduction velocities.9 About 25% of patients have an atypical manifestation after 25 years of age, called late-onset Friedreich’s ataxia, or with retained reflexes, known as Friedreich’s ataxia with retained reflexes, and/or slow disease progression. The skeletal, endocrine, and cardiovascular systems are affected; the disease manifests, respectively, as scoliosis and pes cavus, diabetes mellitus, and hypertrophic cardiomyopathy. Cardiomyopathy is a cardinal feature of Friedreich’s ataxia and detrimentally affects prognosis commonly, with early death secondary to heart failure or fatal arrhythmia. Optic neuropathy and sensorineural hearing may be present later in the disease. The pathogenesis of Friedreich’s ataxia involves a deficiency of a mitochondrial protein, frataxin, secondary to a guanine-adenine-adenine expansion. The genetic defect is believed to result in iron accumulation in mitochondria with oxygen-free radical production. The normal trinucleotide repeat range is estimated at 33 or fewer triplet repeats; pathological expansions range from 67 to 1000 triplets, with length inversely proportional to age at clinical onset, scoliosis, and cardiomyopathy.10 Unlike the ADCAs with trinucleotide repeats, Friedreich’s ataxia is not associated with anticipation. Patients with Friedreich’s ataxia become unable to walk within 11 years after disease onset and have a mean survival length of 36 years after onset of symptoms. Typically, more than two decades of life are spent in a debilitated motor state.11 To date, the size of the guanine-adenine-adenine pathological expansion has made development of transgenic animal models difficult.
AVED is a rare disorder with clinical manifestations similar to those of Friedreich’s ataxia. A patient with clinical features of Friedreich’s ataxia and negative genetic test results should be screened for AVED (with plasma vitamin E levels), especially because this syndrome is potentially treatable. Head titubation and dystonia are more common in AVED than in Friedreich’s ataxia; cardiomyopathy is a less frequent problem.2 The syndrome is caused by a defective transfer protein for α-tocopherol that prevents transfer of this most biologically active form of vitamin E to peripherally circulating lipoproteins.12 Oral vitamin E supplementation is the mainstay of treatment.
Cayman’s ataxia is an ARCA found in a population on Grand Cayman Island. It involves a mutation of the Caytaxin protein, found almost exclusively in the central nervous system, and is believed to bind a ligand with similar properties to vitamin E in the central nervous system.3 It is characterized by early-onset hypotonia, cerebellar ataxia, and psychomotor retardation.3
There are seven inherited ataxias caused by defective DNA repair: ataxia telangiectasia, ataxia with oculomotor apraxia, spinocerebellar ataxia with axonal neuropathy, ARSACS, xeroderma pigmentosum, Cockayne’s syndrome, and trichothiodystrophy. The most common of these is ataxia telangiectasia. Ataxia telangiectasia is typified by neurological, dermatological, and immunological symptoms starting in infancy or early childhood, with death in early adulthood. Neurological manifestations include cerebellar ataxia, slowed horizontal saccades, dystonic posturing, chorea, tics or jerks, dysphagia and choking, and peripheral neuropathy. The dermatological signs include oculocutaneous telangiectasia, premature graying of hair, and premature senile keratosis.12 Patients with ataxia telangiectasia have recurrent sinopulmonary infections secondary to derangement of cellular and humoral immunity (deficient immunoglobulin A levels). Malignancy and neoplasia are common, especially those involving hematological cells. The mutations associated with ataxia telangiectasia in the ataxia telangiectasia mutation gene lead to mutations in nuclear protein, which normally repairs DNA. Consequently, individuals with ataxia telangiectasia have a sensitivity to ionizing radiation—a diagnostic hallmark of ataxia telangiectasia.3 Almost all persons with ataxia telangiectasia have an elevated α-fetoprotein level; however, this is a nonspecific finding. Ataxia with oculomotor apraxia may be a separate syndrome. It lacks the immunological features and sensitivity to ionizing radiation seen in classic ataxia telangiectasia.2,13 Survival of patients with ataxia telangiectasia after age 30 is rare. ARSACS is a rare ARCA secondary to defective DNA repair found in parts of Quebec.
Xeroderma pigmentosum, Cockayne’s syndrome, and trichothiodystrophy are rare ARCAs characterized by extreme skin photosensitivity in combination with ataxia and possibly other neurological manifestations. Xeroderma pigmentosum is marked by the development of skin cancers, including squamous cell carcinomas and melanoma.12
[/not-level-membership-for-neurology-category]
