[level-membership-for-dermatology-category]
Inherited and Acquired Blistering Diseases
Anna L. Bruckner, Grainne M. O’Regan
Inherited blistering diseases
Epidermolysis bullosa
Epidermolysis bullosa (EB) is a family of rare, inherited disorders characterized by fragility of the skin and sometimes the mucosa in response to minor mechanical trauma. EB is caused by mutations in at least 14 genes1,2 that encode proteins of the basement membrane zone (BMZ) of the skin (Fig. 11.1). BMZ proteins are structural molecules involved in the adhesion of the epidermis and dermis. When an affected protein is absent or abnormal, adhesion strength is diminished and blistering occurs as a response to frictional stress. Data from the National Epidermolysis Bullosa Registry (NEBR) estimates the incidence of all forms of EB in the USA to be 20 cases/million live births.3
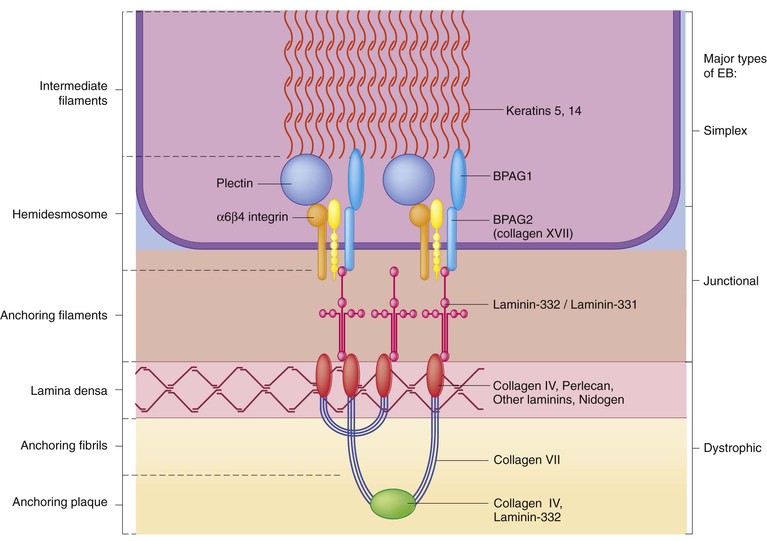
The classification of EB was revised in 2008 and utilizes the level of ultrastructural blistering as the primary categorical designation.1 The four major types of EB are: EB simplex (EBS) where cleavage occurs in the epidermis; junctional EB (JEB) with blistering through the BMZ; dystrophic EB (DEB) with cleavage arising in the superficial dermis, and Kindler syndrome where multiple splits can be seen. Each type is further divided into major and minor subtypes based on inheritance pattern, clinical findings, and the implicated molecular and genetic defects (Table 11.1).
TABLE 11.1
The classification of epidermolysis bullosa
| Major EB type | Major subtype | Minor subtype | Affected gene(s) (protein) | Typical inheritance |
| Simplex | Suprabasal | Lethal acantholytic EB | DSP (desmoplakin) | AR |
| Plakophilin deficiency (ectodermal dysplasia with skin fragility) | PKP1 (plakophilin) | AR | ||
| EBS superficialis | ? | ? | ||
| Basal | Localized (formerly Weber–Cockayne) | KRT5 (keratin 5), KRT14 (keratin 14) | AD | |
| Dowling–Meara | KRT5 (keratin 5), KRT14 (keratin 14) | AD | ||
| Other generalized (formerly Koebner) | KRT5 (keratin 5), KRT14 (keratin 14) | AD | ||
| With mottled pigmentation | KRT5 (keratin 5) | AD | ||
| With muscular dystrophy | PLEC1 (plectin) | AR | ||
| With pyloric atresia | PLEC1 (plectin), ITGA6, ITGB4 (α6β4 integrin) | AR | ||
| Autosomal recessive | KRT14 (keratin 14) | AR | ||
| Ogna | PLEC1 (plectin) | AD | ||
| Migratory circinate | KRT5 (keratin 5) | AD | ||
| Junctional | Herlitz | – | LAMA3, LAMB3, LAMC2 (laminin-332*) | AR |
| Other | Non-Herlitz, generalized | LAMA3, LAMB3, LAMC2 (laminin-332*), COL17A1 (type XVII collagen) | AR | |
| Non-Herlitz, localized | COL17A1 (type XVII collagen) | AR | ||
| With pyloric atresia | ITGA6, ITGB4 (α6β4 integrin) | AR | ||
| Inversa | LAMA3, LAMB3, LAMC2 (laminin-332*) | AR | ||
| Late onset | COL17A1 (type XVII collagen) | AR | ||
| LOC syndrome (Shabbir syndrome) | LAMA3 (laminin-332* α chain) | AR | ||
| Dystrophic | Dominant | Generalized | COL7A1 (type VII collagen) | AD |
| Acral | ||||
| Pretibial | ||||
| Pruriginosa | ||||
| Nails only | ||||
| Bullous dermolysis of the newborn | ||||
| Recessive | Severe generalized (former Hallopeau–Siemens) | COL7A1 (type VII collagen) | AR | |
| Generalized other (formerly non-Hallopeau–Siemens) | ||||
| Inversa | ||||
| Pretibial | ||||
| Pruriginosa | ||||
| Centripetalis | ||||
| Bullous dermolysis of the newborn | ||||
| Kindler syndrome | – | – | FERMT1 (kindlin-1) | AR |
AD, autosomal dominant; AR, autosomal recessive; LOC, laryngo-onycho-cutaneous; *Laminin-332 was formerly called laminin-5.
Clinical features of epidermolysis bullosa
Friction-induced blisters and erosions are the cardinal cutaneous features of EB. The distribution and extent of blistering varies depending on the disease subtype. Some forms of EB, such as EBS and dominant DEB (DDEB), are stereotyped as mild and often localized, whereas blistering in recessive DEB (RDEB) and JEB is often severe and generalized. It is important to remember that these generalizations best apply to older infants and children in whom a ‘mature’ EB phenotype has developed. In contrast, neonates with any type of EB may present with widespread cutaneous blistering and erosions. Mucosal erosions and absent or dystrophic nails can also be seen in neonates with all forms of EB. Thus, diagnosing the specific type and subtype based on clinical findings alone can be difficult in the first weeks of life, and biopsies are indicated (see below). Certain EB subtypes are associated with extracutaneous manifestations and complications that can begin to present in infancy and early childhood.
Neonatal features
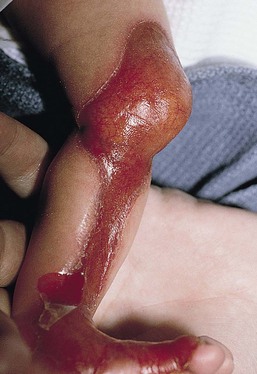
Neonates with EB may present at birth with large ulcers, usually on the lower extremities, called congenital localized absence of skin (CLAS) (Fig. 11.2). The edges of such ulcers are well-demarcated and the base is red and shiny. Bart and colleagues4 originally described the association of CLAS, mucosal blistering, and nail dystrophy and proposed that this triad represented a distinct syndrome, later termed Bart syndrome. Since that description, however, CLAS has been reported as a presenting sign in all types of EB, and Bart’s original kindred was further examined and found to have DDEB.5 CLAS likely results from intrauterine friction, such as the leg rubbing against the uterine wall, and is not specific for any one type of EB. The use of the term Bart syndrome is now discouraged.
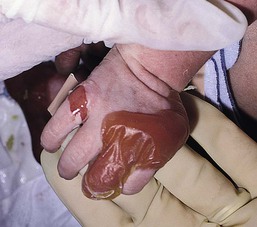
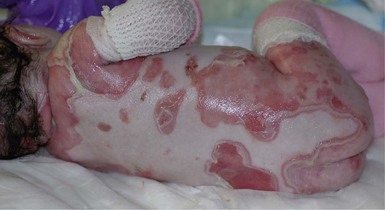
With or without CLAS, neonates with EB develop friction-induced blistering and erosions after birth. Skin changes may initially correlate with areas traumatized during birthing, such as the scalp and face in cases of vaginal birth. In many instances, blistering may be generalized. After birth, areas that are most subject to friction, such as the hands, diaper area, extensor extremities, and back, are most likely to blister (Fig. 11.3). Intact blisters are filled with serous or hemorrhagic fluid. In JEB and RDEB, the bullae can be quite large, and the pressure of fluid within the blister cavity can cause the lesion to extend (Fig. 11.4). More superficial blisters may rupture easily, leaving open erosions.
The blisters in EBS and JEB often heal without scarring, but macular hypo- or hyperpigmentation may be a transient change after blisters heal. DEB blisters heal with scarring that is often atrophic. Milia are most suggestive of DEB but can be seen in all forms of EB (Fig. 11.5). Granulation tissue, which is often described as ‘exuberant’ or ‘hyper-’ in JEB, can be seen in any EB erosion that has been slow to heal but is not often seen in the neonatal period.
Oral involvement is most often seen in neonates with JEB or DEB but can be seen in EBS as well. Open erosions or intact vesicles can occur on the lips, gums, and palate. These lesions probably result from the trauma of sucking and may result in pain with feeding. In any form of EB, trauma to the periungual skin can result in nails that are absent, dystrophic, or may be shed.
Blisters and erosions are at risk of becoming infected, signaled by the formation of crusts and foul-smelling or purulent drainage. Neonates with EB and extensive erosions are at risk for developing fluid and electrolyte abnormalities as well as sepsis. Neonates with EB and pyloric atresia may present with gastrointestinal obstruction at birth.6 In these cases, polyhydramnios and gastric distension may have been noted on prenatal ultrasound, and genitourinary strictures and obstruction may be present as well. Airway involvement is uncommon in neonates with EB, but can occur. Although it is most often associated with JEB-Herlitz (JEB-H), laryngeal involvement can occur in infants with certain forms of EBS and with RDEB.7 An early sign of blisters and erosions of the larynx is hoarseness, which can progress to stridor as airway obstruction worsens.
Specific epidermolysis bullosa subtypes
Suprabasal epidermolysis bullosa simplex
Lethal acantholytic EB (LAEB) is an extremely rare subtype of EB.8–10 Affected infants present at birth with rapidly progressive erosions with a positive Nikolsky’s sign. Intact vesicles and erosions are not seen. Other common features are complete alopecia, anonychia, and oral erosions. All reported cases have been fatal in the neonatal period. Histology shows suprabasal acantholysis, but immunofluorescence studies will be negative for pemphigus and the typical causes of EB (see below). Ultrastructural studies show that keratin filaments fail to connect properly to desmosomes. Loss of function mutations in DSP that lead to truncation of the desmoplakin protein are causative.
Plakophilin deficiency was initially described in 1997 as ectodermal dysplasia/skin fragility syndrome, by McGrath and colleagues.11 Children with this disorder develop spontaneous erosions and fissures, and the perioral area is commonly affected. Painful, fissured keratoderma, absent, sparse or wooly hair, nail dystrophy, and growth failure are other universal features.12 Biopsies of affected skin show acanthosis and hyperkeratosis and widened spaces between keratinocytes in the spinous layer. Desmosomes are small and poorly formed on electron microscopy. This is an autosomal recessive disorder caused by mutations in the PKP1 gene that encodes plakophilin.
Basal epidermolysis bullosa simplex
Basal EBS is the most common and often mildest form of EB. Most forms are transmitted in an autosomal dominant manner. Individuals with localized forms of EBS may not present for medical evaluation, thus precise estimates of the true prevalence of EBS are lacking. For example, the NEBR calculated the prevalence of EBS to be 4.60 cases/million, but also estimated that the registry probably captured only 10% of all individuals affected with EBS.3 Population-based data from Scotland show a prevalence of 28.6 cases/million in 1992.13
EBS, localized (EBS-loc, formerly Weber–Cockayne) is characterized by blisters of the hands and feet. Other than mild oral disease, extracutaneous involvement does not occur. Patients may first develop blisters at any age, including birth, but it is common for the first signs to present in the toddler years, or sometimes as late as adolescence, after a period of marked frictional stress.
EBS, other generalized (EBS, gen-nonDM, formerly Koebner) presents at birth and is characterized by intraepidermal blistering in a generalized distribution. Oral involvement can be seen in infancy but improves with age. Atrophic scarring, dyspigmentation, and nail dystrophy may be seen. Extracutaneous involvement in this type of EB is uncommon. For both EBS-loc and EBS, gen-nonDM, life expectancy is normal and the overall prognosis is good. However, affected adults report that acral blistering can be painful and limits daily activities such as walking, thereby affecting quality of life.14,15
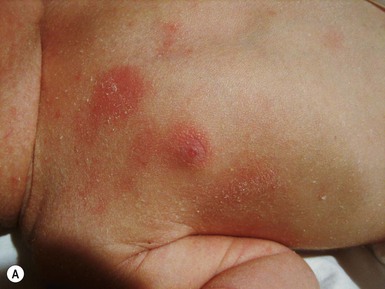
EBS Dowling–Meara (EBS-DM), on the other hand, is more severe and can even be fatal in the newborn period.14,16,17 Blistering presents at birth or within the first days of life. Skin involvement ranges from generalized to localization of blisters and erosions to areas of friction. A clinical clue to the diagnosis of EBS-DM is the occurrence of blisters in grouped or annular configurations (Fig. 11.6). This feature, however, is not always reliable and may not be present until after 1 year of age. Milia and atrophic scarring may occur, and nail dystrophy in which nails may be thickened, ridged, or shed, is fairly common. In childhood, a confluent palmoplantar keratoderma develops and becomes more prominent with age, persisting into adulthood. This hyperkeratosis can interfere with ambulation, and joint contractures may be a subsequent complication.14,17 Oral blistering is often present and varies in severity. Laryngeal involvement, presenting as hoarseness, has been reported, but unlike in JEB, does not signal a poor prognosis.18 Gastroesophageal reflux and constipation may also be seen as complications.19 The severity of EBS-DM generally improves over time, with less blistering in adolescence, and rare blistering in adulthood in many cases. Patients may also report reduced blistering during febrile illnesses.
EBS with mottled pigmentation (EBS-MP) is a rare subtype.20 It is characterized by acral, nonscarring blistering that presents in infancy. In addition, 2–5 mm hypo- and hyperpigmented macules occur in a reticulate pattern around the neck, axillary, and groin areas. This mottled pigmentation may be congenital or can develop later in infancy.
EBS with muscular dystrophy (EBS-MD), EBS with pyloric atresia (EBS-PA), and EBS, Ogna (EBS-Og) are all due to mutations in PLEC1, encoding plectin, although EBS-MD and EBS-PA are inherited recessively and EBS-Og is autosomal dominant.21 EBS-MD is a rare disorder that begins at or shortly after birth with mild generalized blistering and mucous membrane involvement. Other cutaneous findings include milia, atrophic scarring, and nail dystrophy.22 Nonmucosal findings can include respiratory involvement and dental enamel hypoplasia, leading to prominent caries. In most cases, progressive muscle weakness begins in adolescence or adulthood, although onset in infancy has been reported.23 The presentation of neonates with EB with pyloric atresia is discussed below.
Genetics and pathogenesis
EBS-DM, EBS-loc, and EBS, gen-nonDM are dominantly inherited, and mutations in KRT5 and KRT14 can be identified in about 75% of cases.24 Keratin-5 and -14 are complementary intermediate filaments expressed in basal keratinocytes. They are crucial components of the cytoskeleton involved in maintaining structural integrity. In addition, they function in the adhesion of these cells to the BMZ by attaching to the hemidesmosome via plectin (see Fig. 11.1).25 Mutations in these keratin genes lead to abnormal keratin intermediate filaments with reduced ability to withstand frictional stress, resulting in basal cell cytolysis histologically and in blistering clinically. Genotype–phenotype correlations for mutations KRT5 and KRT14 suggest that alterations in the highly conserved boundary domains of the α-helical rod domain lead to the Dowling–Meara phenotype, whereas mutations in the less conserved regions produce milder phenotypes.24
Most cases of EBS-MP are due to a specific point mutation in the nonhelical amino-terminal head domain of keratin-5,26 although a case with a mutation in KRT14 has been reported.27 Exactly how these mutations lead to disruption of keratin intermediate filaments or pigmentation is unclear.
Plectin, the affected protein in EBS-MD, EBS-PA, and EBS-Og anchors basal keratins to the hemidesmosome and is also expressed in the sarcolemma of muscle. The EBS-MD phenotype tends to correlate with mutations affecting the central rod domain of plectin, while EBS-PA is due to mutations outside this domain.28 The Ogna phenotype is due to a heterozygous missense mutation exerting a dominant negative effect.
Junctional epidermolysis bullosa
Junctional epidermolysis bullosa (JEB) is the least common type of EB. Data from the NEBR suggests an incidence of two cases/million live births.3 Inheritance of all forms of JEB is autosomal recessive, and a spectrum of clinical phenotypes can be seen. The JEB–Herlitz (JEB-H) and JEB with pyloric atresia (JEB-PA) subtypes, however, are often incompatible with survival beyond infancy.
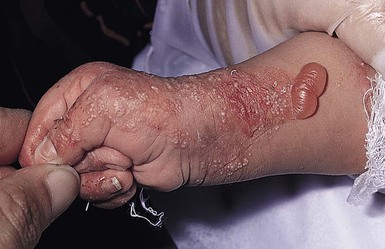
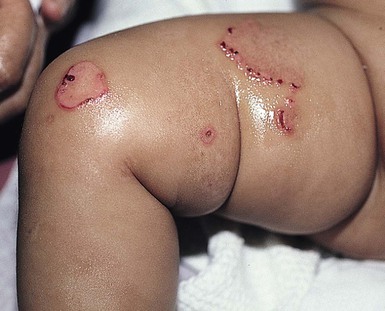
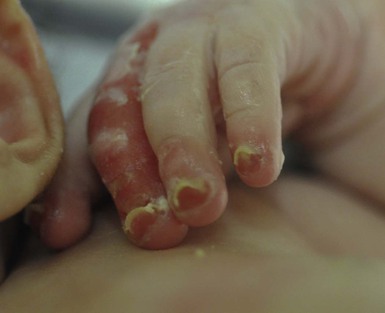
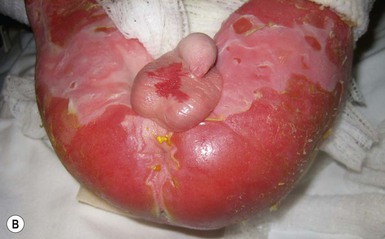
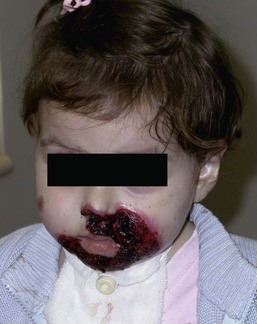
JEB-H is characterized by generalized skin and mucosal erosions with a propensity for wounds to form exuberant granulation tissue. Other associated findings include nail dystrophy (Fig. 11.7), dental enamel hypoplasia, and involvement of the respiratory epithelium. Blistering and erosions are seen at, or shortly after, birth (Fig. 11.8A), although the amount of blistering is not predictive of outcome, as infants with little skin involvement can do poorly. Severe involvement of the back and buttocks is common but not pathognomonic (Fig. 11.8B). In neonates who survive into infancy, the development of exuberant or hypergranulation tissue within erosions is a finding with high specificity for JEB-H. The central face, especially the periorificial areas (Fig. 11.9), periungual skin (paronychial inflammation), and nape of the neck are commonly affected. Absent or dystrophic nails or nail shedding are often present. Ocular erosions can also occur.7 Oral blistering and erosions are often present in the neonatal period and can make feeding difficult. The laryngeal and respiratory epithelia may also be affected, typically first presenting as a weak or hoarse cry.29 Stridor suggests worsening airway obstruction, which can be fatal. Involvement of the gastrointestinal and urinary epithelia can also occur. Anemia, probably due to a combination of iron deficiency and chronic inflammation, is common. Patients with JEB-H typically die in the first few years of life due to failure to thrive, sepsis, pneumonia or respiratory failure. Data from the NEBR reported a cumulative risk of death of ~45% in the first year of life,30 while the Dutch EB registry found that over 23 years, all 22 subjects with JEB-H passed away in the first 3 years, with the average age of death being 5.8 months.31
JEB-non-Herlitz, generalized (JEB-nH gen) has features similar to JEB-H, although it is generally less severe and its overall prognosis tends to be better.32 In the neonatal period, however, clinical signs and pathologic analysis may not fully distinguish between JEB-H and JEB-nH gen, and a final diagnosis may be based on long-term outcome and/or results of genetic mutation analysis. Generalized blistering and mucous membrane involvement are seen in the neonatal period, and both improve as the child ages. Healing with granulation tissue is less common and less pronounced than in JEB-H, but can occur. Cutaneous lesions may heal with atrophy and pigmentary alterations. In hair-bearing areas, progressive alopecia may result, although this finding is most pronounced in adulthood. Likewise, nail dystrophy can begin in infancy and is progressive and marked with increasing age. Laryngeal involvement may occur, sometimes resulting in respiratory failure. Although the overall prognosis for JEB-nH gen is generally better than for JEB-H, the fatalities in the neonatal period and infancy are not uncommon. Data from the NEBR reported a cumulative risk of death of 40% and 49% in the first year and 2 years of life, respectively.30
EB with pyloric atresia (EBS-PA or JEB-PA) presents at birth with upper gastrointestinal obstruction, most often affecting the pylorus. The degree of skin involvement is variable and ranges from extensive CLAS to normal skin, with the onset of blistering at several months of age.33 Prenatal signs of an affected fetus include polyhydramnios and abdominal masses appreciated on ultrasound.34 The ocular, respiratory, and urogenital epithelia are often affected. The prognosis for neonates with EB with pyloric atresia is generally poor, but nonlethal cases are reported.35 Infants with extensive erosions often expire quickly due to fluid and electrolyte imbalances as well as sepsis. If surgical correction of the pyloric atresia is successful, infants may still succumb to sepsis, feeding intolerance, failure to thrive, and respiratory or renal disease before 1 year of age.36
Genetics and pathogenesis
JEB-H and a subset of JEB-nH gen are caused by mutations in three genes, LAMA3, LAMB3, and LAMC2, which encode the constitutive subunits of the basement membrane protein laminin-332.37 As a component of the basal lamina, laminin-332 plays a critical role in the adhesion of keratin intermediate filaments to the basement membrane, and absent or abnormal laminin-332 leads to structural instability, manifesting as blistering through the lamina lucida. In the majority of JEB-H cases, mutations that result in premature termination codons and subsequently, absence of laminin-332 expression, are seen.38,39 In the case of JEB-nH, less deleterious mutations result in an abnormal laminin-332 protein that retains some degree of function.39 Mutations in COL17A1 which encodes type XVII collagen, a component of the hemidesmosome, also produce the JEB-nH gen phenotype, as well as JEB-non-Herlitz, localized.40,41 Mutations in three genes, ITGB4 (~80%), ITGA6 (~5%), and PLEC1 (~15%) produce the EB with pyloric atresia phenotype.42
Dystrophic epidermolysis bullosa
DDEB may be more common than reported, as individuals with mild phenotypes may not seek medical attention. The NEBR estimated the prevalence of DDEB to be approximately 1/million,3 while Scottish data suggests a point prevalence of 1/14.6 per million.13 Infants with DDEB may present with CLAS and/or with blistering in the newborn period. As the child ages, the tendency for blistering decreases. Blistering most commonly affects areas predisposed to trauma, such as the hands, feet, elbows, and knees, and heals with atrophy and milia. Oral erosions occur but are often mild. Nail dystrophy is common and may be the only clinical sign of disease.43 Extracutaneous complications such as esophageal strictures are uncommon. Transient bullous dermolysis of the newborn is a distinctive variant of DEB that is benign and self-limited. Blistering begins at birth or in the newborn period and dramatically improves or even remits completely, usually within the first year of life. Most reported cases are sporadic, but familial cases are described.44,45
The incidence of RDEB is approximately 1–2/million live births.3,13 The current classification includes RDEB, severe generalized (RDEB-sev gen, formerly Hallopeau–Siemens), RDEB, generalized other (RDEB-O, formerly non-Hallopeau–Siemens), and several rare subtypes with more limited involvement (Table 11.1).
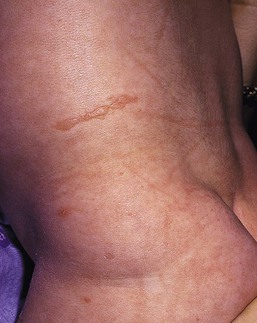
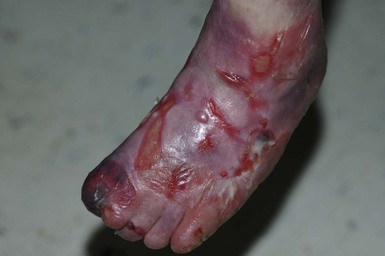
In RDEB, blistering and erosions begin at birth and can be extensive. The compromised skin barrier is a risk for infection, and during the neonatal period sepsis is the most worrisome complication. Although death can occur, infants with RDEB generally do fairly well, especially compared with infants with JEB. Affected areas heal with scarring that can lead to the development of joint contractures over time. Recurrent scarring of the hands and feet leads to loss of the interdigital spaces and eventual contracture of the digits, called pseudosyndactyly or ‘mitten’ deformities (Fig. 11.10).46 Joint contractures and pseudosyndactyly may begin in the first year of life. Nail involvement is common with anonychia due to nail shedding and scarring of the nail bed at an early age.
Extracutaneous involvement is common in RDEB, and generalized RDEB (and JEB) are effectively multisystem disorders. Mucosal involvement includes the gastrointestinal tract, ocular, and genitourinary system. Oral ulcers are painful and make eating difficult, limiting the ability of the child to take in adequate calories. Oral scarring leads to microstomia, ankyloglossia, and loss of the vestibule.47 Esophageal involvement is extremely common; erosions lead to progressive strictures that produce dysphagia and limit adequate caloric intake. Malabsorption is another gastrointestinal complication that is poorly understood.19 Anal erosions make defecation painful, exacerbating constipation. The cornea and conjunctiva are frequent sites of ocular injury. Recurrent abrasions and ulcers lead to scarring that can affect visual acuity.7 Urinary tract involvement may present with dysuria, hematuria, meatal stenosis, or even sepsis. Ureterovesical obstruction and hydronephrosis can occur.48
Failure to thrive is a common complication of generalized RDEB and results from unmet nutritional needs. Affected neonates and infants demonstrate failure to gain weight adequately before height velocity drops off. Chronic wound healing, blood and protein losses from erosions, and infection increase the body’s need for calories. Oral, esophageal and intestinal involvement hinders adequate intake and absorption of nutrients. In addition to causing growth failure, chronic malnutrition contributes to poor wound healing, the development of anemia, deficiencies of essential minerals and trace elements, and increased susceptibility to infection.49,50 Anemia likely results from both iron deficiency and poor iron utilization due to chronic inflammation.51 Additional long-term complications of RDEB that are most often seen in older children, adolescents or adults include renal failure, osteopenia and osteoporosis, cardiomyopathy, and aggressive squamous cell carcinomas.7,46
Genetics and pathogenesis
All forms of DEB are caused by mutations in the gene COL7A1, which encodes type VII collagen, the major component of anchoring fibrils.52 Each collagen VII molecule is made of three polypeptide chains that associate and assemble into a triple helix. Two collagen VII molecules align in an antiparallel fashion, and groups of these dimers form the anchoring fibrils that connect the lamina densa to anchoring plaques in the dermis. As with other forms of EB, genotype–phenotype correlations have emerged. In the case of RDEB-sev gen, homozygous or compound heterozygous mutations resulting in premature termination codons and a subsequent lack of collagen VII are most commonly found.52 Less deleterious mutations, such as missense mutations or the combination of a missense mutation and a premature termination codon, are found in other forms of RDEB. Finally, DDEB is typically caused by heterozygous mutations resulting in glycine substitutions in the triple helical region of COL7A1.52 These mutations allow a full-length protein to be formed, but the substitution leads to conformational instability of the mature collagen VII protein (the so-called ‘dominant negative’ effect).
Kindler syndrome
Kindler syndrome (KS) is a rare, autosomal recessive disorder in which trauma-induced blistering begins at birth or early in infancy. Based on clinical and laboratory features, it is often diagnosed as a form of EB in the neonate. However, patients with KS develop progressive poikiloderma and cutaneous atrophy in childhood.53 Photosensitivity is variable. Gingivitis and periodontitis are common mucosal findings. In addition, esophageal and urogenital stenosis can occur. KS is caused by mutations in FERMT1 which encodes fermitin family homolog 1 (kindlin-1), involved in cell–matrix interactions.54
Differential diagnosis
When faced with a newborn with blisters and erosions, the most critical immediate task is to exclude infection, especially intrauterine herpes simplex infection. Other potential infectious etiologies include bacterial and fungal infections and neonatal varicella. Other genetic disorders can present with bullous or erosive lesions, although these are not the predominant disease features over time. Incontinentia pigmenti presents with vesicles, and neonates with epidermolytic ichthyosis and ankyloblepharon-ectodermal dysplasia-clefting syndrome55 can present with widespread erosions. Infants born to mothers with immune-mediated blistering disorders may also manifest transient blisters and erosions as neonates. In addition, infants may develop immune-mediated blistering disorders de novo (see below). Diffuse cutaneous mastocytosis may also produce extensive blistering and erosion in newborns and infants.
Evaluating suspected EB
Because an accurate diagnosis of the type and subtype of EB based on clinical findings alone is often impossible in the neonatal period, laboratory evaluation is crucial. Biopsy of an affected area for routine light microscopy is helpful in differentiating EB from other diagnoses. Additional biopsies for immunofluorescence microscopy (IFM) and, in some cavses, transmission electron microscopy (TEM) are needed to rapidly and accurately diagnose the EB type and subtype. This information improves the care provider’s ability to counsel and educate parents on the baby’s prognosis and to guide future therapy.
While TEM was formerly considered the ‘gold standard’ for EB diagnosis, IFM testing is preferred now due to broader availability, rapid turnaround time, and lower cost.56 The initial and most common application of IFM involves a limited panel of antibodies to BMZ proteins to map the level of the blister, rapidly diagnosing the EB type. A more specific use of IFM is an expanded panel of monoclonal antibodies to proteins affected in EB, permitting the precise determination of the underlying molecular defect, based on the staining intensity (normal, reduced, or absent) of the targeted proteins (see Fig. 11.11 and Table 11.2). Unfortunately, many dermatopathology services do not maintain the antibodies needed for EB-specific IFM studies, and referral to a laboratory experienced in EB diagnosis may be required. The Stanford Dermatopathology Service (http://dermatopathology.stanford.edu) or Beutner Laboratories (http://www.beutnerlabs.com) are two US laboratories that are recommended.
TABLE 11.2
Laboratory evaluation for epidermolysis bullosa – characteristic IFM and TEM findings
| EB subtype | IFM | TEM |
| Lethal acantholytic EB | Absent desmoplakin (C-terminal directed antibodies must be used, limited availability) | Perinuclear retraction of KF |
| Plakophilin deficiency | Absent plakophilin-1 (limited availability) | Perinuclear retraction of KF, small suprabasal desmosomes |
| EBS, localized and other generalized | Normal | Basal cytolysis |
| EBS, Dowling–Meara | Normal | Basal cytolysis, clumped KF |
| EBS with muscular dystrophy | Reduced to absent plectin | Basal cytolysis, KF not attached to HD |
| EBS with pyloric atresia | Reduced to absent plectin or α6β4 integrin | Cleavage in basal layer, KF not attached to HD |
| EBS, autosomal recessive | Absent keratin 14 | Few to absent KF in basal layer |
| JEB, Herlitz | Absent laminin-332 | Cleavage in lamina lucida, reduced to absent HD |
| JEB, non-Herlitz | Reduced laminin-332 or reduced to absent collagen XVII | Cleavage in lamina lucida, rudimentary HD |
| JEB with pyloric atresia | Absent α6β4 integrin | Cleavage in lamina lucida, small HD |
| Bullous dermolysis of the newborn | Intraepidermal, granular collagen VII | Sub-lamina densa cleavage, stellate bodies in basal keratinocytes, reduced AF |
| DDEB, generalized | Normal | Sub-lamina densa cleavage, normal to decreased AF |
| RDEB, severe generalized | Absent collagen VII | Sub-lamina densa cleavage, absent AF |
| RDEB, generalized other | Normal to reduced collagen VII | Sub-lamina densa cleavage, reduced or rudimentary AF |
| Kindler syndrome | Normal, reduced or absent Kindlin-1; reduplication of lamina densa can be demonstrated using anti-type IV and anti-type VII collagen antibodies | Variable cleavage planes, fragmentation and reduplication of lamina densa |
IFM, immunofluorescence microscopy; TEM, transmission electron microscopy; AF, anchoring fibril; HD, hemidesmosome; KF, keratin filament.
TEM is a complementary test to IFM and has advantages in certain situations. Particularly, its ability to directly visualize ultrastructural morphology is useful for diagnosing EBS-DM (keratin clumps) or in supporting the diagnosis of other forms of EB where the IFM findings are inconclusive. For example, in the case of DDEB, the diagnosis may be better supported by showing wispy or reduced anchoring fibrils on TEM than by slightly reduced collagen VII staining on IFM.
Yiasemides and colleagues57 demonstrated the superiority of IFM over TEM in a prospective study of 30 cases of EB, in which IFM using an expanded panel of 13 monoclonal antibodies was compared to TEM, with genetic testing as the gold standard. IFM was 100% sensitive and specific for the diagnosis of either EBS or DEB, compared with 75–80% for TEM, and for the diagnosis of JEB, IFM demonstrated 90% sensitivity and 100% specificity vs 60% and 92% for TEM.
It is imperative that a freshly induced blister is biopsied, and the steps involved in inducing a blister are outlined in Box 11.1. Existing blisters, even if intact, may show signs of re-epithelialization that interferes with proper interpretation. The decision whether to use a punch or shave technique for biopsy depends on the pathologist’s preference, and consultation before the biopsy is taken. At least two biopsies (one each for IFM and TEM studies) should be taken, and each should include a portion of the induced blister and a portion of normal skin. Specimens sent for IFM are placed in Zeus or Michel’s fixative, and TEM specimens should be stored in glutaraldehyde.
DNA mutation analysis can be used to confirm the clinical and pathologic diagnosis. It is most accurate when testing can be directed toward a particular gene. Therefore, it is recommended that the EB subtype be diagnosed by IFM and/or TEM analysis before genetic testing is ordered. Genetic testing may be helpful in cases where the clinical and pathologic findings are indistinguishable, such as distinguishing mild RDEB from DDEB. Knowing a child’s mutation also helps guide genetic counseling and facilitates prenatal diagnosis in future pregnancies. Testing all of the known genetic causes of EB simultaneously is now possible. However, gene testing is time-consuming, and results will probably not be available until several weeks after the test is ordered. Thus, it is not reasonable at this time to rely solely on genetic testing for diagnostic or prognostic counseling in the neonatal period. DNA analysis for EB is available in the USA through the commercial company GeneDx (www.genedx.com).
Prenatal diagnosis of EB was formerly available only by fetal skin biopsy, but is now more commonly performed using DNA mutation analysis from cells obtained via chorionic villus sampling or amniocentesis.58 In these cases, known mutations in affected siblings and/or parents are needed to guide testing and decision-making regarding the fetus. Preimplantation genetic diagnosis for potentially lethal EB phenotypes is also possible.58
Management of EB in neonates and infants
The prospect of caring for a child with EB often induces anxiety in both parents and care providers. Many activities integral to the newborn period, such as diapering, feeding, or simply picking up the infant, can induce more blisters and compound this unease. While EB is not currently curable, treatment is aimed at maintaining the health of the child and supporting the psychosocial well-being of the family. Immediate goals in the newborn period are to: (1) promote wound healing; (2) prevent and treat infection; (3) minimize pain and discomfort; (4) provide optimal nutrition and support adequate growth, and (5) facilitate normal bonding between the infant and family.
Newborns with presumed EB are best cared for in a nursery or neonatal intensive care unit with experience in treating EB. A neonatologist or pediatrician may oversee the care, with the dermatologist providing guidance on the diagnostic work-up, wound care, and other disease-specific aspects of treatment. Other specialists may also be needed, depending on the baby’s particular complications. A skilled nurse, particularly one with experience in wound care, is another critical component of the EB care team.
The goals of EB management in the newborn period are outlined in Table 11.3. In addition, attention should be paid to basic needs such as fluid and electrolyte balance and temperature control. Skin erosions may result in increased fluid loss and electrolyte alterations, such as hypernatremia. If the infant is not feeding well, intravenous fluids may be needed, and electrolytes should be checked as needed until stable. Monitoring the baby will likely be easier from an open bed instead of an enclosed isolette. Temperature control can be aided by the use of a radiant warmer with heat output at a minimal setting. The environment should not be too warm, however, as this may promote blistering.59,60 Temperature probes or other monitoring devices should not be taped to the skin.
TABLE 11.3
Objectives of EB management in the neonatal period
| Objective | Plan |
| Accurately diagnose the EB type and subtype | Use clinical history, cultures, histology to rule out etiologies other than EB |
| Biopsy an induced blister for immunofluorescence microscopy and transmission electron microscopy studies | |
| Consider genetics consultation and genetic testing | |
| Minimize new blisters and erosions | Mandate gentle handling and reduce friction on the skin |
| Forbid tape or adhesives on the skin | |
| Promote wound healing | Inspect the skin and bandages daily |
| Plan for daily to every other day cleansing of the skin and bandage changes, depending on the degree of exudate from the wounds | |
| Prevent infection | Thoroughly wash hands or use alcohol-based hand sanitizers before and after patient care |
| Place the baby in protective contact isolation (i.e., use gowns and gloves when handling the baby) | |
| Culture areas suspicious for infection and treat infection accordingly | |
| Promote comfort and control pain | Use oral sucrose with dressing changes |
| Consider acetaminophen or short-acting narcotics | |
| Use sucralfate for oral ulcers | |
| Treat gastroesophageal reflux | |
| Optimize nutrition | Follow weight daily |
| Fortify breast-milk or formula | |
| Use soft nipples or cleft lip and palate feeder | |
| Begin a daily multivitamin | |
| Support the family | Promote normal bonding between the baby and family |
| Referral to Dystrophic Epidermolysis Bullosa Research Association (DEBRA) |
Given the nature of EB, it is impossible to completely prevent new blisters from forming, but by minimizing trauma to the skin and mucosa, it is possible to limit their frequency and severity. As even minimal trauma can produce blisters, careful handling of the baby is paramount.59,60 Tape or adhesives should not be used on the skin as their removal produces blistering. If medical devices need to be secured, they may be fastened to dressings or a self-adhesive compression wrap (e.g., Coban™, 3M) can be used. Mepitac® (Mölnlycke Health Care), a silicone ‘tape’ that does not contain adhesives, can be used on fragile skin, but should be used with care in those EB subtypes with excessive skin fragility. Friction often occurs where clothes or diapers rub the skin. Cloth diapers may be less traumatic around the waistband and thigh areas. Alternatively, the elastic cuffs can be cut out of disposable diapers, minimizing blistering on the thighs. Seams from clothing may also produce blisters. Clothing should be soft and loose fitting, and if flat seams are not available the clothing should be turned inside out. The baby should not be lifted under the arms; instead, the head and buttocks should be supported at all times. To minimize blistering on the back, soft padding such as a sheepskin should be used to line the bed (which should remain flat), and a disposable underpad covered with a thin layer of petroleum jelly or a similar ointment will help minimize friction.
For existing blisters and erosions, wound care is essential to promote healing and prevent infection. Bandaging also provides protection to the skin and helps limit future blistering.61,62 Wound care should be performed daily to every other day, depending on the nature of the wounds. The complete task of bathing and redressing the baby can take up to 2 h to complete and needs two people working together to run smoothly. Having all the necessary materials prepared ahead of time greatly speeds up the task (Table 11.4 ).
).
TABLE 11.4
Typical items needed for epidermolysis bullosa dressing changes
| Item | Purpose |
| Warm saline or water and mild cleanser | To clean the skin and wounds |
| Sterile needles and sterile gauze | To lance and drain intact blisters |
| Sterile sharp scissors | To trim excess or hanging skin |
| Preservative-free, petrolatum-based ointment | To emolliate the skin and provide moisture to the wounds; to prevent bandages from adhering to wounds |
| Petrolatum-impregnated gauze, Mepitel®, or similar contact layer | Contact layer, applied directly over wounds |
| Foam or absorbent dressing | Absorbent layer, soaks up exudate and protects noninjured skin |
| Retention dressing (rolled gauze, elastic tubular dressing) | Holds dressings in place |
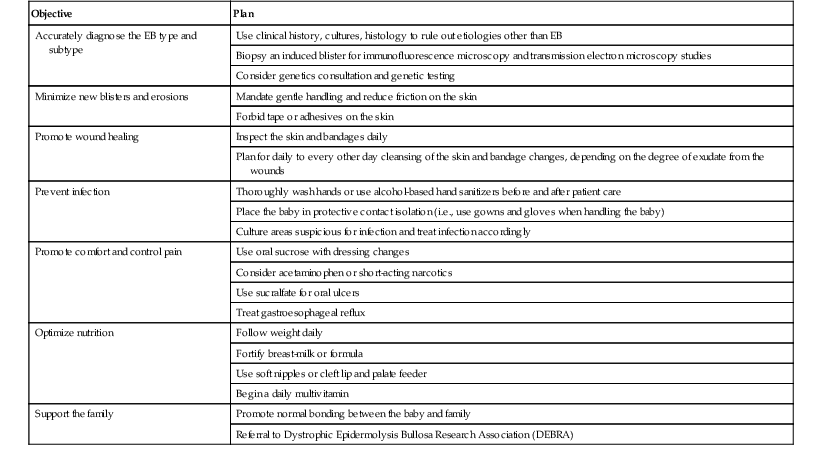
To minimize trauma to exposed skin, it is best to clean and bandage the baby one part at a time. For instance, wound care can be performed on each extremity, followed by the torso and then the face and scalp. Bandages should be removed gently. If they are adherent to the wound, they should be soaked off with saline or dabbed with a petrolatum-based ointment until soft. Bandages should never be forcibly removed – this is both painful and disrupts the healing process.62 The skin should then be cleaned with a saline or mild soap or cleanser and water, and gently patted dry with a soft towel. The skin should not be rubbed. Existing erosions should be assessed for signs of infection and for evidence of healing. In order to prevent extension of existing lesions, intact blisters should be drained by puncturing the dependent side using a sterile needle or lancet (Fig. 11.12) and using sterile gauze to wick away blister fluid.59–61 The blister roof should be left in place unless it hangs freely. Redundant skin or crust can be gently trimmed away with clean, sharp scissors, but aggressive debridement is not indicated.
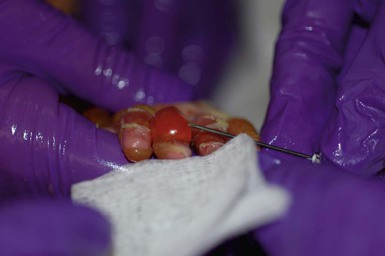
The skin should then be covered with dressings. Ideal dressings for EB patients should be nonadherent and promote a moist, healthy wound bed.61,63,64 Many products fulfill these criteria, and the ultimate decision on which ones are used depends on their availability, cost, and the physician (and later parent) preference. The contact layer, the first bandage to cover the wound, should be nonadherent. Silicone dressings are generally preferred as they release easily from the wound and intact skin without causing additional trauma. Mepitel® (Mölnlycke Health Care) is a fenestrated silicone contact layer that clings gently to noninjured skin and is easily removed by moistening it with water. Another common nonadherent dressing is petrolatum-impregnated gauze. If such a product is used, it should be ‘buttered’ with a liberal amount of an emollient such as petroleum jelly or Aquaphor® healing ointment (Beiersdorf Inc.) first. This prevents the wound from drying out and reduces the likelihood of the bandage sticking to the wound.
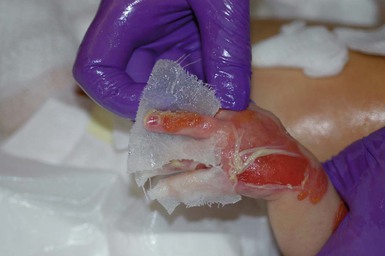
Absorptive dressings are then applied over the contact layer. These soak up exudate and provide padding and protection for the child. Foam dressings such as Mepilex®, Mepilex Lite®, and Mepilex Transfer® (Mölnlycke Health Care) and Allevyn® (Smith & Nephew) combine a contact layer with absorptive material and can be used directly on the skin. Dressings should then be secured with layers of conformable rolled gauze and stretchable burn netting or a tubular retention dressing. For infants with RDEB and digital erosions, separating the digits with strips of petrolatum-impregnated gauze and wrapping the fingers individually may help delay the onset of webbing and pseudosyndactyly (Fig. 11.13). With good wound care, even extensive erosions will show excellent healing within 1–2 weeks (Fig. 11.14).
The prophylactic use of topical antibiotics is not recommended for wounds that appear clean. Signs of wound infection include increased erythema, drainage, odor, crusting, or tenderness. Although wound infection is a clinical diagnosis, wound cultures can be used to guide antibiotic therapy. Minor wound infections often improve with the use of topical antibiotics and dressings that help to wick away excess exudate. Common topical antibiotics include bacitracin, mupirocin, and gentamicin ointments. It is best to limit the duration of use of topical antibiotics to periods of suspected infection. They should be discontinued when the wound appears clean, or rotated every 1–2 months to prevent resistance.63,65 Bathing with dilute bleach or vinegar in the bath may also be useful to reduce the bacterial load on the skin and may help minimize reliance on topical antibiotics. Two teaspoons of undiluted bleach per gallon of water is one recommended concentration. Systemic antibiotics are indicated for extensive and/or invasive infection. Open erosions act as portals of entry for bacteria, and sepsis is a complication of EB in the neonatal period. Neonates with presumed cellulitis or sepsis should be treated with broad-spectrum systemic antibiotics, which should be subsequently narrowed based on wound and blood culture results.
In addition to the calories needed for normal growth, neonates and infants with extensive erosions have added metabolic demands due to wound healing. Failing to account for this contributes to failure to thrive in children with JEB and RDEB. However, obtaining adequate nutrition in EB can be a management challenge, especially when oral erosions and ulcers make feeding painful and laborious. Mothers who choose to breast-feed should be supported, but suckling can aggravate oral erosions, and breast milk alone may not meet the nutritional needs of an infant with EB. For those infants at high risk for failure to thrive, it may be preferable to express and then fortify the breast-milk. Likewise, high-calorie formulas should be started for infants with JEB and RDEB that are formula fed, even before signs of poor weight gain are seen.49 The simple act of using growth charts to follow weight and height cannot be forgotten. Declines in the velocity of weight gain signify the patient is not getting adequate calories and need to be addressed promptly. The early input of the gastroenterology and nutrition team can be very helpful. Nipples should be soft and preferably high flow, so that the infant does not need to suck vigorously. A cleft lip and palate nurser, such as the Haberman® feeder (Medela) may also be helpful. In rare instances, a soft nasogastric (NG) tube may be used as a temporary intervention to aid in feeding. However, NG tubes should be placed with caution as they may cause trauma to the oral mucosa and esophagus and promote the development of strictures.
Introduction of soft solid foods should begin at 4–6 months of age and advanced slowly as tolerated. Patients with severe EB may not tolerate solid foods well due to trauma and scarring of the oral mucosa, and the development of feeding coordination can be delayed. The input of an experienced feeding therapist can be invaluable. Many patients remain dependent on calorically-dense liquid supplements to meet their caloric needs throughout their life. Infants with feeding difficulties or failure to thrive may benefit from the placement of a gastrostomy tube to deliver nutrition and medications. Gastrostomy tubes can be used to meet some, most, or all of a patient’s nutritional needs, depending on the ability to take foods by mouth. Daily requirements for vitamins and minerals are likely increased in patients with EB, possibly due to increased utilization for metabolism and wound healing, along with inadequate intake and absorption. It is not uncommon to find patients with low levels of iron, zinc, and vitamin D.66 All infants with EB should be started on a daily multivitamin, ideally with iron.
The blisters and erosions of EB, both on the skin and internally, are painful, and most patients report living with chronic pain.67,68 In addition, pain is often exacerbated by procedures such as dressing changes and bathing. Pain management in EB is a challenge, owing to the chronic nature of the disease. Proper wound care is a ‘topical’ form of pain control.61 The discomfort of bathing may be alleviated by adding pool salts to the water to approximate normal saline and allow the water to be isotonic with open wounds.69 In infants with extensive erosions, systemic pain medications may be needed, especially with dressing changes. Medications should be given 20–30 min before beginning bandaging. Acetaminophen may be tried first, and if this is not sufficient, an opiate agonist can be added.68 Oral sucrose is effective to control procedural pain in neonates and may also be used,70 although it has not been specifically evaluated for EB. Oral discomfort may be controlled with topical analgesics before feeding. The use of sucralfate to coat ulcers is also effective.71 The use of eye ointments to prevent and treat corneal erosions reduces eye pain. Gastroesophageal reflux may also be painful, especially if the child has esophageal erosions. The use of an H2 blocker or proton-pump inhibitor in these cases reduces pain.19
It should not be forgotten that a baby with EB is a baby, and normal bonding with the parents should be encouraged and a caregiver’s voice and touch is important to the child. However, having a newborn with EB is a life-altering experience, and the parents may need time to adjust. The medical team must be able to provide psychosocial support for the family. An excellent resource for families affected by EB is the Dystrophic Epidermolysis Bullosa Research Association (DEBRA, www.debra.org, www.debra-international.org). In addition to providing excellent information geared toward a lay audience, DEBRA offers consultation with an EB nurse specialist.
Additional considerations
In those cases where neonates present with extracutaneous manifestations requiring surgical intervention, such as pyloric stenosis or respiratory distress in the case of suspected JEB-H, the risks and benefits of surgical intervention must be weighed against the child’s overall prognosis. With careful attention, patients with EB can tolerate intubation (either nasopharyngeal or endotracheal) and anesthesia for procedures.72,73 Repair of pyloric atresia36 and early placement of tracheostomy tubes29 have been performed successfully in EB patients, but these infants often succumb to other complications of their disease. Thus, comfort care alone is a valid treatment option for infants with EB subtypes that carry a poor prognosis, such as JEB-PA and JEB-H.31,74
In order to be discharged after birth, infants with EB need to be medically stable. From a dermatologic standpoint, a reasonable goal is no more than 10% skin involvement, minimal development of new blisters, and evidence of adequate feeding and weight gain. In addition, parents or care providers should demonstrate comfort with and competence at changing the baby’s dressings, and support services such as a home health nurse and sufficient medical supplies need to be in place. Infants should have follow-up with care providers who are experienced in managing EB. A multidisciplinary approach is needed to monitor for and treat the many complications that may develop as the child ages.
Protocols for optimal follow-up after discharge are not standardized, but clinic visits at least every 3 months in the first year of life are a good rule of thumb, with more frequent follow-up being needed for infants with severe disease. In addition to monitoring skin involvement, growth and development should be followed closely with the pediatrician. Many infants with EB will lag in motor development, and having a physical therapist and occupational therapist involved in the treatment plan to address gross and fine motor development is important. Due to a high risk for dental complications, visits with a pediatric dentist should also begin in the first year of life.75 Any infant with EB and signs and symptoms of anemia, such as fatigue, pallor, and tachycardia, should have blood work performed. However, as anemia is common in severe forms of EB and as the signs and symptoms may be difficult to appreciate, all infants with JEB and RDEB should have a screening complete blood count and iron studies performed by 1 year of age, and repeated every 6–12 months thereafter.
Finally, families of infants with EB should be encouraged to maintain hope as disease-specific treatments that show promise are becoming available. A landmark study demonstrated that keratinocyte stem cells from an adult patient with JEB-nH were genetically corrected and successfully transplanted back to affected areas of the legs, resulting in normal, robust skin during the 1-year follow-up period.76 This technique is being investigated as a treatment for RDEB.77 Bone marrow transplant has been shown to increase expression of collagen VII at the basement membrane zone and reduces the severity of disease, although there is significant risk with this approach.78 Replacement of collagen VII,79,80 either locally or systemically, is another therapy that is hoped will be available for RDEB on an experimental basis soon.
Acquired blistering diseases
Autoimmune bullous disorders
Autoimmune bullous disorders (AIBD) are a heterogeneous group of acquired blistering skin diseases due to pathogenic circulating antibodies targeted against structural adhesion molecules of the skin and mucous membranes. Transient bullous disorders (see Chapter 10) can occur in neonates born to mothers with active or quiescent AIBD, such as pemphigus vulgaris,81–83 pemphigus foliaceus,84,85 epidermolysis bullosa acquisita,86 and pemphigoid gestationis.87,88 In these cases, maternal IgG antibodies cross the placenta and cause symptoms in the neonate. The estimated plasma elimination half-life of IgG is 15 days, and disease resolution typically parallels clearance of the antibodies. Supportive therapy is usually sufficient for this duration, although topical corticosteroids may be beneficial for patients with a significant inflammatory presentation.89
AIBD also arise in neonates and children due to the de novo generation of autoantibodies. These disorders are quite rare, and presentation in the first 2 years is uncommon. There is significant morphologic overlap between the variants, making clinical diagnosis imperfect. Histopathology and immunofluorescence (IF) are required to identify the target antigen(s) and establish a definitive diagnosis. In addition to the disorders discussed below, pemphigus vulgaris, pemphigus foliaceus, paraneoplastic pemphigus, and dermatitis herpetiformis are AIBD to consider in the differential diagnosis of bullous and erosive lesions,90 but their presentation in infants is unlikely.
Linear IgA disease
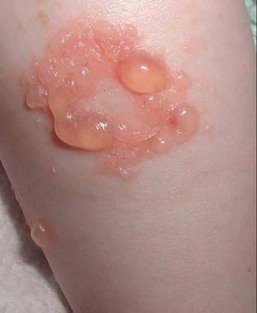
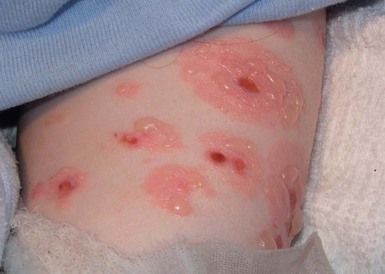
Linear IgA disease (LAD, also known as linear IgA bullous dermatosis, chronic bullous disease of childhood) is the most common AIBD of childhood, typically occurring between 6 months and 5 years of age. Disease onset in the neonatal period has been reported, albeit infrequently, and case reports suggest that neonatal onset can be associated with significant morbidity due to mucosal involvement.91–93 As the name suggests, the diagnosis relies on immunopathological findings: the exclusive or predominant presence of linear deposits of IgA along the BMZ.94 Disease onset is typically abrupt, and may be associated with pyrexia, malaise or other prodromal symptoms.95 The mean disease duration is 3–5 years, after which the majority of patients achieve complete remission, and disease rarely persists or recurs in adulthood.96,97
Clinically, LAD is characterized by the presence of clear or hemorrhagic vesicles, bullae or erosions that vary in size, arising from normal skin or urticarial plaques (Fig. 11.15). New lesions tend to occur at the periphery of older ones forming rosettes or clusters.98 Bullae are usually tense, and may mimic lesions of bullous pemphigoid (Fig. 11.16). Lesions are predominantly localized to the lower abdomen, neck and perineal areas, with prominent anogenital involvement. Mucosal involvement is more common in the pediatric setting, affecting up to 76% of children, and may be associated with significant morbidity.99,100 Oral lesions usually present as erosions and ulcerations, and predominantly affect the hard and soft palate, buccal mucosa and gingiva, with relative sparing of the lips. Ocular involvement is also common in the pediatric setting and occurs in up to 65% of patients, with scarring occurring in 43% of patients who report symptoms.101 It may appear as isolated conjunctival injection and can result in scarring with subconjunctival fibrosis and symblepharon formation.101,102 Other mucosal membranes may be involved, and are also at risk of scarring, including the larynx, pharynx, trachea, esophagus, bronchial tree, and vaginal mucosae.
Fresh blisters on routine histopathology may be suggestive, but not diagnostic, showing subepidermal blistering with an inflammatory infiltrate consisting of neutrophils and eosinophils. Skin biopsy of perilesional unaffected skin for direct IF is mandatory to establish the diagnosis. IF shows a linear deposit of IgA antibodies at the BMZ. C3, IgG or IgM may be seen as well, but the predominance of IgA is needed to establish the diagnosis.98 The multiplicity and complexity of target antigens is likely due to epitope spreading. Circulating IgA antibodies, although usually present in low titers, are detectable in 80% of children, a higher proportion than occurs in adults.100,103
Therapeutic options include potent topical steroids for mild cases or mucosal lesions which are often resistant to systemic therapies. Systemic therapy is required until patients enter complete clinical remission, followed by maintenance therapy until the disease remits. Prednisolone (0.5–2 mg/kg per day) is often used first to attain disease control, but should be combined with other therapies and tapered as tolerated in order to minimize adverse effects. Dapsone (0.5–3 mg/kg per day) is considered the treatment of choice beyond the neonatal period. Other options include erythromycin (50 mg/kg per day), IVIG (400–2000 mg/kg per cycle), and tacrolimus 0.03% topically BD. Mycophenolate mofetil (300–600 mg/m2), and cyclosporine (2.5–5 mg/kg per day) have shown good efficacy in older children refractory to other therapies.104,105
Bullous pemphigoid
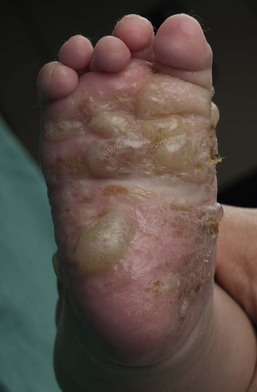
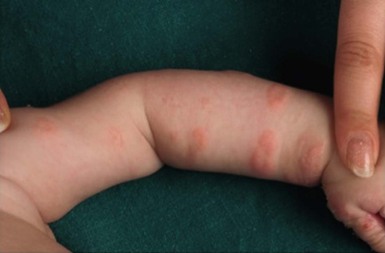
Childhood bullous pemphigoid (BP) is an IgG-mediated subepidermal bullous disorder. It is rare in childhood, with peak prevalence in the 1st and 8th years of life.106–109 Some 50% of cases present under 12 months of age, with the earliest reported cases occurring at 2 months of age.108,110 Mucous membrane involvement, acral distribution, and facial involvement appear to occur more frequently in childhood BP, and lesions tend to involve the palms and soles of infants of less than 1 year (Fig. 11.17).108 Lesions of BP may begin as urticarial, irregularly bordered plaques reminiscent of erythema multiforme, evolving into widespread, tense blisters on erythematous or clinically unaffected skin, frequently located on the trunk and flexural areas (Fig. 11.18).109 Pruritus is often prominent. A limited vulvar variant is recognized and can be mistaken for sexual abuse.111 The course of childhood BP is usually indolent, and once appropriate treatment is instigated, remissions typically occur within 1 year.112,113
The target autoantibodies in BP are the 180 kD or 230 kD bullous pemphigoid antigens (BP180 or BP 230).106 Lesional histology shows subepidermal blister formation with an intact epidermis and a predominantly eosinophilic infiltrate. Direct IF is characterized by linear deposition of IgG and C3, however IgM and IgA may be found.114,115 Indirect IF can show circulating autoantibodies directed against the epidermal side of 1.0M sodium chloride (NaCl) split skin.
Oral prednisolone (0.5–2 mg/kg per day) is the mainstay of therapy, but erythromycin, IVIG, and dapsone may be useful alternative or adjunctive therapies.108,110,116–119
Epidermolysis bullosa acquisita
Epidermolysis bullosa acquisita (EBA) is a rare AIBD characterized by autoantibodies to type VII collagen, the major component of anchoring fibrils. EBA has been reported as early as 3 months of age. It is exceptionally rare; less than 50 cases have been reported in the pediatric literature.120,121 Two phenotypes of EBA are recognized: the classic presentation with a noninflammatory mechanobullous phenotype, resembling the dystrophic form of inherited EB; and less frequently an inflammatory presentation that mimics other AIBD. Children less than 5 years of age are more likely to present with the inflammatory type, and mucosal involvement is also thought to occur more frequently.121 Ocular involvement, which may be associated with visual loss, has been reported to occur more commonly in IgA-mediated EBA. The IgA variant, however, accounts for a minority of childhood EBA; the majority are mediated by IgG autoantibodies.102,122–124 In contrast to adult EBA, which is often refractory to therapy, children with EBA typically respond well to therapies such as low-dose corticosteroids and dapsone.125 Other therapeutic options include colchicine, dapsone, infliximab, and IVIG.126,127
Bullous systemic lupus erythematosus
The bullous variant of systemic lupus erythematosus (SLE) is rarely seen in children, although an isolated case of neonatal lupus erythematosus presenting with overt blistering has been reported.128 Autoantibodies to type VII collagen have been reported as pathogenic in bullous SLE, however autoantibodies to a variety of other molecules involved in dermal–epidermal adhesion have been found, including BP antigen 1, laminin-332, and laminin-331.129,130 Bullous SLE may be difficult to differentiate from EBA, as the direct IF pattern is similar; patients should fulfill diagnostic criteria for SLE to establish the diagnosis.131
Non-autoimmune bullous disorders
A range of other disorders may present with blisters and erosions in addition to other changes and must be considered in the differential diagnosis. Toxin-producing strains of Staphylococcus aureus (see Chapter 12) can produce localized flaccid bullae that quickly progress to erosions (bullous impetigo) or the generalized eruption of staphylococcal scalded skin syndrome. Viral infections with herpes simplex virus and varicella zoster virus (see Chapter 13) produce grouped or isolated vesicles on a base of erythematous skin. In any ill-appearing child with the abrupt onset of blisters, toxic epidermal necrosis (TEN) and Stevens–Johnson syndrome (SJS) (see Chapter 20) must be excluded. Wells syndrome, a rare disorder characterized by pruritic, erythematous plaques mimicking cellulitis, can also sometimes be bullous (see Chapter 20).
Mastocytosis (see Chapter 28) is a disorder of clonal mast cell proliferation that commonly affects the skin. Three cutaneous variants are recognized: mastocytomas (discrete plaques or nodules), maculopapular (often called urticaria pigmentosa) and diffuse cutaneous mastocytosis (DCM). All cutaneous variants can manifest with cutaneous blistering,132–136 although DCM is most likely to be problematic. In DCM, there is widespread erythroderma and blistering which usually presents in the first year of life, with a congenital presentation in 25% of cases.137 The clinical presentation is broadly divided into two phenotypes; patients presenting with blistering on a background of erythema and urticarial plaques, or presentation with indurated leathery skin and minimal blistering. Sudden release of mast cell mediators results in pruritus, flushing, urticaria, blistering, tachycardia, hypotension, wheeze, diarrhea, abdominal pain, melena, and shock.133 Mortality in the neonatal period has been reported as high as 23% of cases, due to overwhelming mast cell degranulation, sepsis or mast cell leukemia.138
Blistering in mastocytosis usually resolves by 1–3 years; skin lesions improved in 71% in one series by 1 year of life, and 90% are symptom-free by puberty.139 Therapeutic strategies aim to control symptoms of mast cell degranulation and to block their proliferation. The key to management is avoidance of agents that trigger histamine release, including heat, friction, polymyxin B sulfate, narcotics and NSAIDs. Symptomatic approaches include non-sedating H1 and H2 antihistamines, topical and systemic corticosteroids, oral sodium cromoglycate for intestinal complaints, ketotifen, PUVA, and antiseptic measures because of high risk of sepsis.
Access the full reference list at ExpertConsult.com 
Table 4 is available online at ExpertConsult.com 
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Inherited and Acquired Blistering Diseases
Anna L. Bruckner, Grainne M. O’Regan
Inherited blistering diseases
Epidermolysis bullosa
Epidermolysis bullosa (EB) is a family of rare, inherited disorders characterized by fragility of the skin and sometimes the mucosa in response to minor mechanical trauma. EB is caused by mutations in at least 14 genes1,2 that encode proteins of the basement membrane zone (BMZ) of the skin (Fig. 11.1). BMZ proteins are structural molecules involved in the adhesion of the epidermis and dermis. When an affected protein is absent or abnormal, adhesion strength is diminished and blistering occurs as a response to frictional stress. Data from the National Epidermolysis Bullosa Registry (NEBR) estimates the incidence of all forms of EB in the USA to be 20 cases/million live births.3
The classification of EB was revised in 2008 and utilizes the level of ultrastructural blistering as the primary categorical designation.1 The four major types of EB are: EB simplex (EBS) where cleavage occurs in the epidermis; junctional EB (JEB) with blistering through the BMZ; dystrophic EB (DEB) with cleavage arising in the superficial dermis, and Kindler syndrome where multiple splits can be seen. Each type is further divided into major and minor subtypes based on inheritance pattern, clinical findings, and the implicated molecular and genetic defects (Table 11.1).
TABLE 11.1
The classification of epidermolysis bullosa
| Major EB type | Major subtype | Minor subtype | Affected gene(s) (protein) | Typical inheritance |
| Simplex | Suprabasal | Lethal acantholytic EB | DSP (desmoplakin) | AR |
| Plakophilin deficiency (ectodermal dysplasia with skin fragility) | PKP1 (plakophilin) | AR | ||
| EBS superficialis | ? | ? | ||
| Basal | Localized (formerly Weber–Cockayne) | KRT5 (keratin 5), KRT14 (keratin 14) | AD | |
| Dowling–Meara | KRT5 (keratin 5), KRT14 (keratin 14) | AD | ||
| Other generalized (formerly Koebner) | KRT5 (keratin 5), KRT14 (keratin 14) | AD | ||
| With mottled pigmentation | KRT5 (keratin 5) | AD | ||
| With muscular dystrophy | PLEC1 (plectin) | AR | ||
| With pyloric atresia | PLEC1 (plectin), ITGA6, ITGB4 (α6β4 integrin) | AR | ||
| Autosomal recessive | KRT14 (keratin 14) | AR | ||
| Ogna | PLEC1 (plectin) | AD | ||
| Migratory circinate | KRT5 (keratin 5) | AD | ||
| Junctional | Herlitz | – | LAMA3, LAMB3, LAMC2 (laminin-332*) | AR |
| Other | Non-Herlitz, generalized | LAMA3, LAMB3, LAMC2 (laminin-332*), COL17A1 (type XVII collagen) | AR | |
| Non-Herlitz, localized | COL17A1 (type XVII collagen) | AR | ||
| With pyloric atresia | ITGA6, ITGB4 (α6β4 integrin) | AR | ||
| Inversa | LAMA3, LAMB3, LAMC2 (laminin-332*) | AR | ||
| Late onset | COL17A1 (type XVII collagen) | AR | ||
| LOC syndrome (Shabbir syndrome) | LAMA3 (laminin-332* α chain) | AR | ||
| Dystrophic | Dominant | Generalized | COL7A1 (type VII collagen) | AD |
| Acral | ||||
| Pretibial | ||||
| Pruriginosa | ||||
| Nails only | ||||
| Bullous dermolysis of the newborn | ||||
| Recessive | Severe generalized (former Hallopeau–Siemens) | COL7A1 (type VII collagen) | AR | |
| Generalized other (formerly non-Hallopeau–Siemens) | ||||
| Inversa | ||||
| Pretibial | ||||
| Pruriginosa | ||||
| Centripetalis | ||||
| Bullous dermolysis of the newborn | ||||
| Kindler syndrome | – | – | FERMT1 (kindlin-1) | AR |
AD, autosomal dominant; AR, autosomal recessive; LOC, laryngo-onycho-cutaneous; *Laminin-332 was formerly called laminin-5.
Clinical features of epidermolysis bullosa
Friction-induced blisters and erosions are the cardinal cutaneous features of EB. The distribution and extent of blistering varies depending on the disease subtype. Some forms of EB, such as EBS and dominant DEB (DDEB), are stereotyped as mild and often localized, whereas blistering in recessive DEB (RDEB) and JEB is often severe and generalized. It is important to remember that these generalizations best apply to older infants and children in whom a ‘mature’ EB phenotype has developed. In contrast, neonates with any type of EB may present with widespread cutaneous blistering and erosions. Mucosal erosions and absent or dystrophic nails can also be seen in neonates with all forms of EB. Thus, diagnosing the specific type and subtype based on clinical findings alone can be difficult in the first weeks of life, and biopsies are indicated (see below). Certain EB subtypes are associated with extracutaneous manifestations and complications that can begin to present in infancy and early childhood.
Neonatal features
Neonates with EB may present at birth with large ulcers, usually on the lower extremities, called congenital localized absence of skin (CLAS) (Fig. 11.2). The edges of such ulcers are well-demarcated and the base is red and shiny. Bart and colleagues4 originally described the association of CLAS, mucosal blistering, and nail dystrophy and proposed that this triad represented a distinct syndrome, later termed Bart syndrome. Since that description, however, CLAS has been reported as a presenting sign in all types of EB, and Bart’s original kindred was further examined and found to have DDEB.5 CLAS likely results from intrauterine friction, such as the leg rubbing against the uterine wall, and is not specific for any one type of EB. The use of the term Bart syndrome is now discouraged.
With or without CLAS, neonates with EB develop friction-induced blistering and erosions after birth. Skin changes may initially correlate with areas traumatized during birthing, such as the scalp and face in cases of vaginal birth. In many instances, blistering may be generalized. After birth, areas that are most subject to friction, such as the hands, diaper area, extensor extremities, and back, are most likely to blister (Fig. 11.3). Intact blisters are filled with serous or hemorrhagic fluid. In JEB and RDEB, the bullae can be quite large, and the pressure of fluid within the blister cavity can cause the lesion to extend (Fig. 11.4). More superficial blisters may rupture easily, leaving open erosions.
The blisters in EBS and JEB often heal without scarring, but macular hypo- or hyperpigmentation may be a transient change after blisters heal. DEB blisters heal with scarring that is often atrophic. Milia are most suggestive of DEB but can be seen in all forms of EB (Fig. 11.5). Granulation tissue, which is often described as ‘exuberant’ or ‘hyper-’ in JEB, can be seen in any EB erosion that has been slow to heal but is not often seen in the neonatal period.
Oral involvement is most often seen in neonates with JEB or DEB but can be seen in EBS as well. Open erosions or intact vesicles can occur on the lips, gums, and palate. These lesions probably result from the trauma of sucking and may result in pain with feeding. In any form of EB, trauma to the periungual skin can result in nails that are absent, dystrophic, or may be shed.
Blisters and erosions are at risk of becoming infected, signaled by the formation of crusts and foul-smelling or purulent drainage. Neonates with EB and extensive erosions are at risk for developing fluid and electrolyte abnormalities as well as sepsis. Neonates with EB and pyloric atresia may present with gastrointestinal obstruction at birth.6 In these cases, polyhydramnios and gastric distension may have been noted on prenatal ultrasound, and genitourinary strictures and obstruction may be present as well. Airway involvement is uncommon in neonates with EB, but can occur. Although it is most often associated with JEB-Herlitz (JEB-H), laryngeal involvement can occur in infants with certain forms of EBS and with RDEB.7 An early sign of blisters and erosions of the larynx is hoarseness, which can progress to stridor as airway obstruction worsens.
Specific epidermolysis bullosa subtypes
Suprabasal epidermolysis bullosa simplex
Lethal acantholytic EB (LAEB) is an extremely rare subtype of EB.8–10 Affected infants present at birth with rapidly progressive erosions with a positive Nikolsky’s sign. Intact vesicles and erosions are not seen. Other common features are complete alopecia, anonychia, and oral erosions. All reported cases have been fatal in the neonatal period. Histology shows suprabasal acantholysis, but immunofluorescence studies will be negative for pemphigus and the typical causes of EB (see below). Ultrastructural studies show that keratin filaments fail to connect properly to desmosomes. Loss of function mutations in DSP that lead to truncation of the desmoplakin protein are causative.
Plakophilin deficiency was initially described in 1997 as ectodermal dysplasia/skin fragility syndrome, by McGrath and colleagues.11 Children with this disorder develop spontaneous erosions and fissures, and the perioral area is commonly affected. Painful, fissured keratoderma, absent, sparse or wooly hair, nail dystrophy, and growth failure are other universal features.12 Biopsies of affected skin show acanthosis and hyperkeratosis and widened spaces between keratinocytes in the spinous layer. Desmosomes are small and poorly formed on electron microscopy. This is an autosomal recessive disorder caused by mutations in the PKP1 gene that encodes plakophilin.
Basal epidermolysis bullosa simplex
Basal EBS is the most common and often mildest form of EB. Most forms are transmitted in an autosomal dominant manner. Individuals with localized forms of EBS may not present for medical evaluation, thus precise estimates of the true prevalence of EBS are lacking. For example, the NEBR calculated the prevalence of EBS to be 4.60 cases/million, but also estimated that the registry probably captured only 10% of all individuals affected with EBS.3 Population-based data from Scotland show a prevalence of 28.6 cases/million in 1992.13
EBS, localized (EBS-loc, formerly Weber–Cockayne) is characterized by blisters of the hands and feet. Other than mild oral disease, extracutaneous involvement does not occur. Patients may first develop blisters at any age, including birth, but it is common for the first signs to present in the toddler years, or sometimes as late as adolescence, after a period of marked frictional stress.
EBS, other generalized (EBS, gen-nonDM, formerly Koebner) presents at birth and is characterized by intraepidermal blistering in a generalized distribution. Oral involvement can be seen in infancy but improves with age. Atrophic scarring, dyspigmentation, and nail dystrophy may be seen. Extracutaneous involvement in this type of EB is uncommon. For both EBS-loc and EBS, gen-nonDM, life expectancy is normal and the overall prognosis is good. However, affected adults report that acral blistering can be painful and limits daily activities such as walking, thereby affecting quality of life.14,15
EBS Dowling–Meara (EBS-DM), on the other hand, is more severe and can even be fatal in the newborn period.14,16,17 Blistering presents at birth or within the first days of life. Skin involvement ranges from generalized to localization of blisters and erosions to areas of friction. A clinical clue to the diagnosis of EBS-DM is the occurrence of blisters in grouped or annular configurations (Fig. 11.6). This feature, however, is not always reliable and may not be present until after 1 year of age. Milia and atrophic scarring may occur, and nail dystrophy in which nails may be thickened, ridged, or shed, is fairly common. In childhood, a confluent palmoplantar keratoderma develops and becomes more prominent with age, persisting into adulthood. This hyperkeratosis can interfere with ambulation, and joint contractures may be a subsequent complication.14,17 Oral blistering is often present and varies in severity. Laryngeal involvement, presenting as hoarseness, has been reported, but unlike in JEB, does not signal a poor prognosis.18 Gastroesophageal reflux and constipation may also be seen as complications.19 The severity of EBS-DM generally improves over time, with less blistering in adolescence, and rare blistering in adulthood in many cases. Patients may also report reduced blistering during febrile illnesses.
EBS with mottled pigmentation (EBS-MP) is a rare subtype.20 It is characterized by acral, nonscarring blistering that presents in infancy. In addition, 2–5 mm hypo- and hyperpigmented macules occur in a reticulate pattern around the neck, axillary, and groin areas. This mottled pigmentation may be congenital or can develop later in infancy.
EBS with muscular dystrophy (EBS-MD), EBS with pyloric atresia (EBS-PA), and EBS, Ogna (EBS-Og) are all due to mutations in PLEC1, encoding plectin, although EBS-MD and EBS-PA are inherited recessively and EBS-Og is autosomal dominant.21 EBS-MD is a rare disorder that begins at or shortly after birth with mild generalized blistering and mucous membrane involvement. Other cutaneous findings include milia, atrophic scarring, and nail dystrophy.22 Nonmucosal findings can include respiratory involvement and dental enamel hypoplasia, leading to prominent caries. In most cases, progressive muscle weakness begins in adolescence or adulthood, although onset in infancy has been reported.23 The presentation of neonates with EB with pyloric atresia is discussed below.
Genetics and pathogenesis
EBS-DM, EBS-loc, and EBS, gen-nonDM are dominantly inherited, and mutations in KRT5 and KRT14 can be identified in about 75% of cases.24 Keratin-5 and -14 are complementary intermediate filaments expressed in basal keratinocytes. They are crucial components of the cytoskeleton involved in maintaining structural integrity. In addition, they function in the adhesion of these cells to the BMZ by attaching to the hemidesmosome via plectin (see Fig. 11.1).25 Mutations in these keratin genes lead to abnormal keratin intermediate filaments with reduced ability to withstand frictional stress, resulting in basal cell cytolysis histologically and in blistering clinically. Genotype–phenotype correlations for mutations KRT5 and KRT14 suggest that alterations in the highly conserved boundary domains of the α-helical rod domain lead to the Dowling–Meara phenotype, whereas mutations in the less conserved regions produce milder phenotypes.24
Most cases of EBS-MP are due to a specific point mutation in the nonhelical amino-terminal head domain of keratin-5,26 although a case with a mutation in KRT14 has been reported.27 Exactly how these mutations lead to disruption of keratin intermediate filaments or pigmentation is unclear.
Plectin, the affected protein in EBS-MD, EBS-PA, and EBS-Og anchors basal keratins to the hemidesmosome and is also expressed in the sarcolemma of muscle. The EBS-MD phenotype tends to correlate with mutations affecting the central rod domain of plectin, while EBS-PA is due to mutations outside this domain.28 The Ogna phenotype is due to a heterozygous missense mutation exerting a dominant negative effect.
Junctional epidermolysis bullosa
Junctional epidermolysis bullosa (JEB) is the least common type of EB. Data from the NEBR suggests an incidence of two cases/million live births.3 Inheritance of all forms of JEB is autosomal recessive, and a spectrum of clinical phenotypes can be seen. The JEB–Herlitz (JEB-H) and JEB with pyloric atresia (JEB-PA) subtypes, however, are often incompatible with survival beyond infancy.
JEB-H is characterized by generalized skin and mucosal erosions with a propensity for wounds to form exuberant granulation tissue. Other associated findings include nail dystrophy (Fig. 11.7), dental enamel hypoplasia, and involvement of the respiratory epithelium. Blistering and erosions are seen at, or shortly after, birth (Fig. 11.8A), although the amount of blistering is not predictive of outcome, as infants with little skin involvement can do poorly. Severe involvement of the back and buttocks is common but not pathognomonic (Fig. 11.8B). In neonates who survive into infancy, the development of exuberant or hypergranulation tissue within erosions is a finding with high specificity for JEB-H. The central face, especially the periorificial areas (Fig. 11.9), periungual skin (paronychial inflammation), and nape of the neck are commonly affected. Absent or dystrophic nails or nail shedding are often present. Ocular erosions can also occur.7 Oral blistering and erosions are often present in the neonatal period and can make feeding difficult. The laryngeal and respiratory epithelia may also be affected, typically first presenting as a weak or hoarse cry.29 Stridor suggests worsening airway obstruction, which can be fatal. Involvement of the gastrointestinal and urinary epithelia can also occur. Anemia, probably due to a combination of iron deficiency and chronic inflammation, is common. Patients with JEB-H typically die in the first few years of life due to failure to thrive, sepsis, pneumonia or respiratory failure. Data from the NEBR reported a cumulative risk of death of ~45% in the first year of life,30 while the Dutch EB registry found that over 23 years, all 22 subjects with JEB-H passed away in the first 3 years, with the average age of death being 5.8 months.31
JEB-non-Herlitz, generalized (JEB-nH gen) has features similar to JEB-H, although it is generally less severe and its overall prognosis tends to be better.32 In the neonatal period, however, clinical signs and pathologic analysis may not fully distinguish between JEB-H and JEB-nH gen, and a final diagnosis may be based on long-term outcome and/or results of genetic mutation analysis. Generalized blistering and mucous membrane involvement are seen in the neonatal period, and both improve as the child ages. Healing with granulation tissue is less common and less pronounced than in JEB-H, but can occur. Cutaneous lesions may heal with atrophy and pigmentary alterations. In hair-bearing areas, progressive alopecia may result, although this finding is most pronounced in adulthood. Likewise, nail dystrophy can begin in infancy and is progressive and marked with increasing age. Laryngeal involvement may occur, sometimes resulting in respiratory failure. Although the overall prognosis for JEB-nH gen is generally better than for JEB-H, the fatalities in the neonatal period and infancy are not uncommon. Data from the NEBR reported a cumulative risk of death of 40% and 49% in the first year and 2 years of life, respectively.30
EB with pyloric atresia (EBS-PA or JEB-PA) presents at birth with upper gastrointestinal obstruction, most often affecting the pylorus. The degree of skin involvement is variable and ranges from extensive CLAS to normal skin, with the onset of blistering at several months of age.33 Prenatal signs of an affected fetus include polyhydramnios and abdominal masses appreciated on ultrasound.34 The ocular, respiratory, and urogenital epithelia are often affected. The prognosis for neonates with EB with pyloric atresia is generally poor, but nonlethal cases are reported.35 Infants with extensive erosions often expire quickly due to fluid and electrolyte imbalances as well as sepsis. If surgical correction of the pyloric atresia is successful, infants may still succumb to sepsis, feeding intolerance, failure to thrive, and respiratory or renal disease before 1 year of age.36
Genetics and pathogenesis
JEB-H and a subset of JEB-nH gen are caused by mutations in three genes, LAMA3, LAMB3, and LAMC2, which encode the constitutive subunits of the basement membrane protein laminin-332.37 As a component of the basal lamina, laminin-332 plays a critical role in the adhesion of keratin intermediate filaments to the basement membrane, and absent or abnormal laminin-332 leads to structural instability, manifesting as blistering through the lamina lucida. In the majority of JEB-H cases, mutations that result in premature termination codons and subsequently, absence of laminin-332 expression, are seen.38,39 In the case of JEB-nH, less deleterious mutations result in an abnormal laminin-332 protein that retains some degree of function.39 Mutations in COL17A1 which encodes type XVII collagen, a component of the hemidesmosome, also produce the JEB-nH gen phenotype, as well as JEB-non-Herlitz, localized.40,41 Mutations in three genes, ITGB4 (~80%), ITGA6 (~5%), and PLEC1 (~15%) produce the EB with pyloric atresia phenotype.42
Dystrophic epidermolysis bullosa
DDEB may be more common than reported, as individuals with mild phenotypes may not seek medical attention. The NEBR estimated the prevalence of DDEB to be approximately 1/million,3 while Scottish data suggests a point prevalence of 1/14.6 per million.13 Infants with DDEB may present with CLAS and/or with blistering in the newborn period. As the child ages, the tendency for blistering decreases. Blistering most commonly affects areas predisposed to trauma, such as the hands, feet, elbows, and knees, and heals with atrophy and milia. Oral erosions occur but are often mild. Nail dystrophy is common and may be the only clinical sign of disease.43 Extracutaneous complications such as esophageal strictures are uncommon. Transient bullous dermolysis of the newborn is a distinctive variant of DEB that is benign and self-limited. Blistering begins at birth or in the newborn period and dramatically improves or even remits completely, usually within the first year of life. Most reported cases are sporadic, but familial cases are described.44,45
The incidence of RDEB is approximately 1–2/million live births.3,13 The current classification includes RDEB, severe generalized (RDEB-sev gen, formerly Hallopeau–Siemens), RDEB, generalized other (RDEB-O, formerly non-Hallopeau–Siemens), and several rare subtypes with more limited involvement (Table 11.1).
In RDEB, blistering and erosions begin at birth and can be extensive. The compromised skin barrier is a risk for infection, and during the neonatal period sepsis is the most worrisome complication. Although death can occur, infants with RDEB generally do fairly well, especially compared with infants with JEB. Affected areas heal with scarring that can lead to the development of joint contractures over time. Recurrent scarring of the hands and feet leads to loss of the interdigital spaces and eventual contracture of the digits, called pseudosyndactyly or ‘mitten’ deformities (Fig. 11.10).46 Joint contractures and pseudosyndactyly may begin in the first year of life. Nail involvement is common with anonychia due to nail shedding and scarring of the nail bed at an early age.
Extracutaneous involvement is common in RDEB, and generalized RDEB (and JEB) are effectively multisystem disorders. Mucosal involvement includes the gastrointestinal tract, ocular, and genitourinary system. Oral ulcers are painful and make eating difficult, limiting the ability of the child to take in adequate calories. Oral scarring leads to microstomia, ankyloglossia, and loss of the vestibule.47 Esophageal involvement is extremely common; erosions lead to progressive strictures that produce dysphagia and limit adequate caloric intake. Malabsorption is another gastrointestinal complication that is poorly understood.19 Anal erosions make defecation painful, exacerbating constipation. The cornea and conjunctiva are frequent sites of ocular injury. Recurrent abrasions and ulcers lead to scarring that can affect visual acuity.7 Urinary tract involvement may present with dysuria, hematuria, meatal stenosis, or even sepsis. Ureterovesical obstruction and hydronephrosis can occur.48
Failure to thrive is a common complication of generalized RDEB and results from unmet nutritional needs. Affected neonates and infants demonstrate failure to gain weight adequately before height velocity drops off. Chronic wound healing, blood and protein losses from erosions, and infection increase the body’s need for calories. Oral, esophageal and intestinal involvement hinders adequate intake and absorption of nutrients. In addition to causing growth failure, chronic malnutrition contributes to poor wound healing, the development of anemia, deficiencies of essential minerals and trace elements, and increased susceptibility to infection.49,50 Anemia likely results from both iron deficiency and poor iron utilization due to chronic inflammation.51 Additional long-term complications of RDEB that are most often seen in older children, adolescents or adults include renal failure, osteopenia and osteoporosis, cardiomyopathy, and aggressive squamous cell carcinomas.7,46
Genetics and pathogenesis
All forms of DEB are caused by mutations in the gene COL7A1, which encodes type VII collagen, the major component of anchoring fibrils.52 Each collagen VII molecule is made of three polypeptide chains that associate and assemble into a triple helix. Two collagen VII molecules align in an antiparallel fashion, and groups of these dimers form the anchoring fibrils that connect the lamina densa to anchoring plaques in the dermis. As with other forms of EB, genotype–phenotype correlations have emerged. In the case of RDEB-sev gen, homozygous or compound heterozygous mutations resulting in premature termination codons and a subsequent lack of collagen VII are most commonly found.52 Less deleterious mutations, such as missense mutations or the combination of a missense mutation and a premature termination codon, are found in other forms of RDEB. Finally, DDEB is typically caused by heterozygous mutations resulting in glycine substitutions in the triple helical region of COL7A1.52 These mutations allow a full-length protein to be formed, but the substitution leads to conformational instability of the mature collagen VII protein (the so-called ‘dominant negative’ effect).
Kindler syndrome
Kindler syndrome (KS) is a rare, autosomal recessive disorder in which trauma-induced blistering begins at birth or early in infancy. Based on clinical and laboratory features, it is often diagnosed as a form of EB in the neonate. However, patients with KS develop progressive poikiloderma and cutaneous atrophy in childhood.53 Photosensitivity is variable. Gingivitis and periodontitis are common mucosal findings. In addition, esophageal and urogenital stenosis can occur. KS is caused by mutations in FERMT1 which encodes fermitin family homolog 1 (kindlin-1), involved in cell–matrix interactions.54
Differential diagnosis
When faced with a newborn with blisters and erosions, the most critical immediate task is to exclude infection, especially intrauterine herpes simplex infection. Other potential infectious etiologies include bacterial and fungal infections and neonatal varicella. Other genetic disorders can present with bullous or erosive lesions, although these are not the predominant disease features over time. Incontinentia pigmenti presents with vesicles, and neonates with epidermolytic ichthyosis and ankyloblepharon-ectodermal dysplasia-clefting syndrome55 can present with widespread erosions. Infants born to mothers with immune-mediated blistering disorders may also manifest transient blisters and erosions as neonates. In addition, infants may develop immune-mediated blistering disorders de novo (see below). Diffuse cutaneous mastocytosis may also produce extensive blistering and erosion in newborns and infants.
Evaluating suspected EB
Because an accurate diagnosis of the type and subtype of EB based on clinical findings alone is often impossible in the neonatal period, laboratory evaluation is crucial. Biopsy of an affected area for routine light microscopy is helpful in differentiating EB from other diagnoses. Additional biopsies for immunofluorescence microscopy (IFM) and, in some cavses, transmission electron microscopy (TEM) are needed to rapidly and accurately diagnose the EB type and subtype. This information improves the care provider’s ability to counsel and educate parents on the baby’s prognosis and to guide future therapy.
While TEM was formerly considered the ‘gold standard’ for EB diagnosis, IFM testing is preferred now due to broader availability, rapid turnaround time, and lower cost.56 The initial and most common application of IFM involves a limited panel of antibodies to BMZ proteins to map the level of the blister, rapidly diagnosing the EB type. A more specific use of IFM is an expanded panel of monoclonal antibodies to proteins affected in EB, permitting the precise determination of the underlying molecular defect, based on the staining intensity (normal, reduced, or absent) of the targeted proteins (see Fig. 11.11 and Table 11.2). Unfortunately, many dermatopathology services do not maintain the antibodies needed for EB-specific IFM studies, and referral to a laboratory experienced in EB diagnosis may be required. The Stanford Dermatopathology Service (http://dermatopathology.stanford.edu) or Beutner Laboratories (http://www.beutnerlabs.com) are two US laboratories that are recommended.
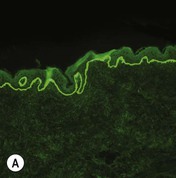
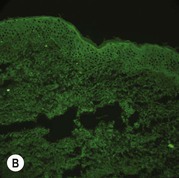
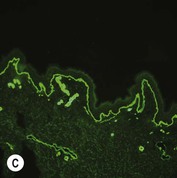
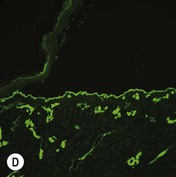
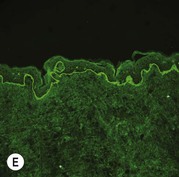
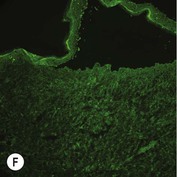
[/not-level-membership-for-dermatology-category]
