[level-membership-for-cardiovascular-category]53
Inheritable Phenotypes Associated With Altered Intracellular Calcium Regulation
Overview of the Function of the Calcium-Handling System
Growing clinical and experimental evidence highlights the relevance of cardiac calcium handling in the pathogenesis of inherited arrhythmias.1 The control of Ca2+ fluxes in myocardial cells requires the timely coordination of several events that ultimately lead to contraction. Any perturbation of this process has the potential to determine an arrhythmogenic substrate.
The core events of Ca2+ ion movements in the myocardial cells are the opening of the L-type Ca2+ channels followed by the release of Ca2+ from the sarcoplasmic reticulum (SR) through the opening of the ryanodine receptors (RyR2).2 This process is called calcium-induced calcium release (CICR).3 The L-type calcium channel (CaV1.2) activation is therefore the first step of CICR. CaV1.2 belongs to the family of the voltage-gated calcium channels that are macromolecular complexes consisting of an ion conducting protein (the α1-subunit) and additional accessory peptides with regulatory function called α2δ, the β1-4, and γ subunits.4 Among the biophysical properties of voltage-dependent calcium current (ICa), the inactivation process is relevant to inherited arrhythmias (Timothy syndrome, see Chapter 95). Two components have been identified: voltage- (VDI) and Ca2+ (CDI)-dependent inactivation. The Ca2+-mediated component of inactivation is modulated by intracellular (cytosolic) concentration of calcium (Ca2+).
CaV1.2 channels tend to cluster in the T-tubules in close proximity with the ryanodine receptors sitting across the membrane of the SR.5 CaV1.2 activation constitutes the signal for the activation of the RyR2. This latter event can be detected as a calcium transient, which is the sum of coordinated local releases that occur at specialized structures: the calcium release units (CRUs; Figure 53-1). One CRU is formed by clusters of RyR2 receptors that are in close proximity to L-type Ca2+ channels in the T-tubules.6 CRUs also include cardiac calsequestrin (CASQ2), triadin (TRDN), and junctin (JTC) that contribute to the control of the calcium release process. These peptides form a macromolecular complex that acts in coordination to control Ca2+ release. The number of CRUs recruited for release at each cardiac cycle is an important modulator of the systolic Ca2+ transient amplitude,7,8 and the loss of integrity of CRU is part of the pathophysiology of CPVT.9
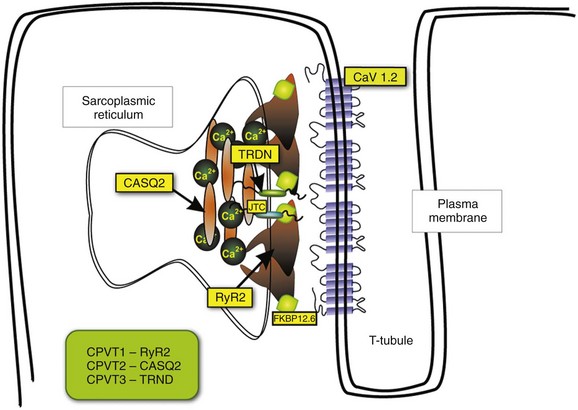
Figure 53-1 Schematic representation of CRU and CPVT gene localization. The figure depicts a schematic representation of a T-tubule with the LTCC sitting across the plasmalemma juxtaposed to the RyR2/CASQ2/TRND/JTC macromolecular complex from the SR side. The SR component of CRU is involved in CPVT pathogenesis. CASQ2, cardiac calsequestrin; CPVT, catecholaminergic polymorphic ventricular tachycardia; CRU, calcium release unit; JTC, junctin; LTCC, L-type calcium channel; RyR2, cardiac ryanodine receptor; SR, sarcoplasmic reticulum; TRDN, triadin.
During the relaxation phase, SR Ca2+ release terminates, and Ca2+ is taken up in the SR by the SR Ca2+-ATPase (SERCA) or extruded from the cell by the Na+/Ca2+ exchanger (NCX).10 NCX extrudes one Ca2+ ion (two positive charges) for every three Na+ ions (three positive charges) that are transferred into the cell. Thus, NCX generates a net inward depolarizing current, the transient inward current—Iti. In physiological conditions, SERCA is responsible for approximately 63% of calcium removal, and NCX mediates the remaining 37%.11 However, NCX becomes important to remove Ca2+ in any condition of calcium overload (e.g., in patients with genetic mutations of the RyR2 gene [CPVT] and during heart failure).11 Excessive activation of NCX can be arrhythmogenic.
Sarcoplasmic Reticulum Ca2+ Release Threshold and the Adrenergic Signaling
Activation of the adrenergic nervous system, mainly through β-adrenergic receptors, has profound effects on calcium handling, and it is often the initiator of calcium-mediated arrhythmogenesis.12 Adrenergic activation has two major effects on calcium handling: the enhancement of the amplitude of the L-type calcium current (ICa) and the increase of SR Ca2+ levels induced through the activation of SERCA13. The latter is responsible for an increase of Ca2+-transient amplitude. The effects of adrenergic activation can take place because of the phosphorylation target proteins14 induced by the activation of two enzymes with kinase activity: protein kinase A14 and Ca2+ calmodulin kinase II (CAMKII).15 Among the several target proteins, the adrenergic-dependent changes of Ca2+ handling are mainly due to the phosphorylation of L-type Ca2+ channel (increased current amplitude) phospholamban (removal of SERCA inhibition and increase of SR Ca reuptake) and RyR2 (increased open probability and increased transients).16 Thus, protein phosphorylation is an important mechanism that enables adrenergic activation to enhance the SR calcium release. In physiological conditions, this response is useful to react to environmental stressors by improving myocardial contractility.
Phenotypes Associated With Mutation in Calcium Handling Proteins
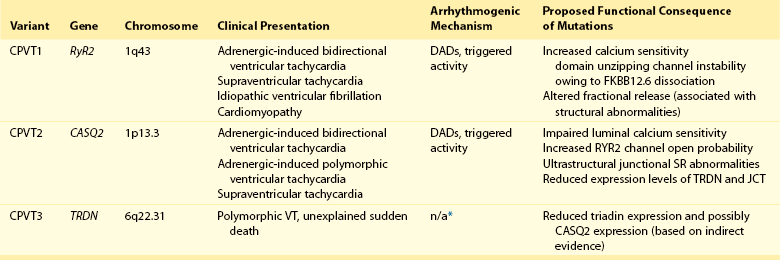
CPVT is the most frequent phenotype associated with mutations involved in intracellular calcium handling. Three genes have been implicated in its pathogenesis (Table 53-1): ryanodine receptor (RyR2 [CPVT1]),17 cardiac calsequestrin (CASQ2 [CPVT2]),18 and cardiac triadin (TRDN [CPVT3]).19 CPVT1 is an autosomal dominant trait, whereas CPVT2 and CPVT3 are rare autosomal recessive disorders. In 2001, Priori et al17 identified RyR2 as the gene responsible for the most frequent variant of CPVT. The prevalence of RyR2 mutations in patients with a clearly diagnostic phenotype is high (≈60% to 70%).20,21 Mutations concentrate in three specific areas of the RyR2 protein (amino acids 77-466, 2246-2534, and 3778-4967); however, 14% of CPVT patients harbor RyR2 mutations located outside these areas.21 Recent data suggest that there is no significant difference of outcome of CPVT according to mutation site, including the outcome of the subgroup with mutations outside the canonical clusters.22 N, RyR2 mutations have been reported also in patients referred for idiopathic ventricular fibrillation and in relatives of subjects who died suddenly.23,24 Autosomal recessive CPVT is rarely identified in the clinical setting. Combined estimated prevalence of the two recessive variants is approximately 5%.
Clinical Manifestations and Management of CPVT
CPVT is a severe inherited arrhythmogenic disease manifesting with adrenergically mediated arrhythmias, often leading to syncope or cardiac arrest.17,23,25–27 Although the resting electrocardiogram is unremarkable, the reproducible inducibility of ventricular tachycardia (VT) during exercise stress test is the hallmark of the disease. Most patients with CPVT show a bidirectional VT pattern characterized by beat-to-beat 180-degree rotation of the QRS axis (Figure 53-2)17,23,26; however, polymorphic tachycardia or ventricular fibrillation may also be part of the picture.23,27 This unstable, catecholamine-sensitive, substrate can lead to sudden death as the first manifestation of the disease in up to 30% of cases.23 Symptoms suggesting the presence of arrhythmias tend to manifest early in life (median age, 12 years), although later onset is possible. The development of palpitations and syncopal events during adrenergic stress is a critical element to suspect the diagnosis of CPVT, which is confirmed by the induction of bidirectional VT on an exercise stress test. In untreated patients, the occurrence of severe arrhythmias is approximately 60% to 70%, and approximately 30% experience a cardiac arrest or sudden death upon first manifestation (see Figure 53-2).23,28 CPVT patients typically present with structurally normal heart. However, some RyR2 mutations have been associated anecdotally with structural cardiomyopathies. One of the first reports on RyR2 gene mutations suggested an association with arrhythmogenic right ventricular cardiomyopathy (ARVC).29 More recently, other authors have reported preliminary data linking RyR2 with hypertrophic cardiomyopathy (HCM).30 Although careful scrutiny of these clinical reports does not establish a causal link, recent experimental findings suggest a mechanistic explanation as to why some RyR2 variants could in principle make the heart more prone to develop structural abnormalities (discussed later).
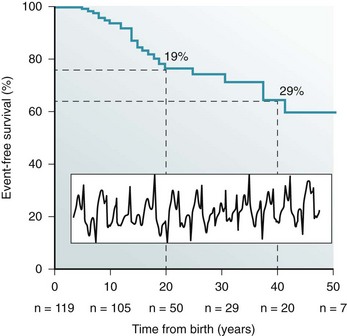
Figure 53-2 Kaplan-Maier curve showing the cumulative occurrence of cardiac arrest, sudden death, or ICD shocks in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) in the absence of therapy. An example of bidirectional ventricular tachycardia, the typical CPVT arrhythmia, is depicted in the inset.
β-Adrenergic receptor block represents the first-choice therapeutic approach. The use of β-blockers is based on the evidence of the direct link between adrenergic activation and cardiac events in CPVT. Clinical data show that β-blockers have an effect on the natural history of CPVT by achieving a significant reduction of cardiac events. In this context, exercise stress testing is important for dosage titration because the threshold for arrhythmias in CPVT is reproducible. β-Blockers can prevent the onset of arrhythmia in 70% to 80% of cases.23,26 In addition, nonselective β-blockers (e.g., nadolol, propranolol) confer the highest degree of protection against the onset of VT and SCD. However, the nontrivial incidence of recurrent cardiac on optimal β-blocker dosage (approximately 30%)22 calls for the identification of additional therapeutic strategies. Implantable cardioverter defibrillator (ICD) implant, left cardiac sympathetic denervation, and other pharmacologic approaches have been proposed, and flecainide appears to be the most promising strategy. Although the mechanisms for its effectiveness are debatable (discussed later), clinical data and personal experience suggest that flecainide affords additional protection when added to β-blockers in CPVT.31,32
Pathophysiology
Abnormalities of Calcium Handling in CPVT
From a mechanistic standpoint, similarities and significant differences exist among CPVT1-3 genetic variants. In all three forms of CPVT, arrhythmogenesis is a consequence of the occurrence of spontaneous calcium release (SCR; Figure 53-3). This term is used to refer to SR Ca2+ release events that are not driven by a stimulated action potential. Whenever an SCR occurs and the levels of Ca2+ in the sarcolemma increase, the NCX activates to extrude the excess of ions. As mentioned earlier, NCX activation generates the Iti current, which is the cause of the development of delayed afterdepolarizations (DADs)33 and transient membrane depolarizations occurring during phase 4 of the action potential (see Figure 53-3). When DAD amplitude reaches the voltage threshold for sodium channel activation, triggered beats occur.
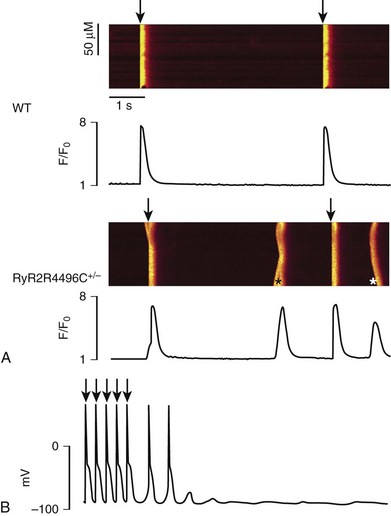
Figure 53-3 A, Representative line-scan images and Ca2+ transients in a wild type myocyte and a RyR2R4496C+/− myocyte at 0.2 Hz pacing in the presence of isoproterenol (100 nM). Asterisk indicates spontaneous Ca2+ release in the RyR2R4496C+/− myocytes. Arrows indicated the field stimulations. B, Action potential recording in a myocyte isolated from a RyR2R4496C+/− during exposure to isoproterenol 30 nmol. Arrows indicate stimulated beats, which are followed by two triggered beats and delayed afterdepolarizations.
The effects of CPVT mutations have been analyzed in a variety of experimental settings, including lipid bilayer, heterologous expression systems, murine transgenic models, and in myocytes derived from patient-specific induced pluripotent stem cells.34 The following sections outline the key concepts about arrhythmogenesis in each form of CPVT.
Ryanodine Receptor, Mutations, and CPVT
The RyR2 channel is a homotetramer; each subunit is formed by 4967 amino acids with a long (≈4300 amino acids) N-terminal cytoplasmic domain.35 The last 500 amino acids at the C-terminal of RyR2 form the transmembrane segments encircling the channel pore. The regulation of RyR2 opening and closing (gating) is mainly controlled by Ca2+ levels at cytoplasmic and luminal SR sides, and it is facilitated when the Ca2+ concentration at either side increases.36,37 Therefore, RyR2 calcium sensitivity is an important physiological function that controls CICR. The majority of RyR2 mutations identified in CPVT patients cause increased calcium sensitivity (defined as “gain-of-function”).
Marks et al. showed that mutant RyR2 exhibits an increased sensitivity to cytosolic Ca2+ after protein kinase A phosphorylation. They also suggested that such change in sensitivity is due to an abnormal dissociation of FKBP12.6, a putative RyR2 stabilizing protein.38 During adrenergic stimulation, phosphorylation of RyR2 would promote SCRs by further dissociation of FKBP12.6 with a consequent excessive increase of open probability.
An alternative (and strongly supported by the experimental evidence) hypothesis attributes a central role to the concept of SR threshold for calcium release. This mechanism is called store overload-induced Ca2+ release to highlight the idea of the tight interplay between SR calcium content and release threshold. Researchers have demonstrated that several mutations of the RyR2 channel cause a reduction of the threshold for Ca2+ release; on the contrary only few mutations are associated with a different sensitivity to cytosolic Ca2+ (i.e., they reduce the Ca2+ release threshold).9,39,40 In the context of a lowered SR threshold (the effect of the RyR2 mutation), β-adrenergic activation, which physiologically increases SR [Ca2+], greatly enhances the propensity for SCR events because the threshold for release is reached more easily.
Yamamoto et al.41 have demonstrated that some mutations disrupt the three-dimensional conformation of the channel. They initially showed that the closed state of the RyR2 channel is stabilized by tight contacts between the central and N-terminal regions. The presence of a reduced “stickiness” of these regions is defined as “domain unzipping.” The same authors performed follow-up studies demonstrating that RyR2 mutations result in domain unzipping and enhance Ca2+ sensitivity, thus facilitating spontaneous Ca2+ release.42,43 The unzipping mechanism can complement the store overload-induced Ca2+ release hypothesis to provide a structural explanation of the reduced threshold for SR release.
Although there is still debate over the subcellular mechanisms of RyR2 mutations, animal models consistently show that DADs triggering action potentials12,44 is the cause of the onset of bidirectional or polymorphic VTs. Adrenergic activation exacerbates the arrhythmogenic substrate, but the propensity for DAD and triggered beats can be observed even at baseline in isolated cells.12
The use of cardiac myocytes derived from induced pluripotent stem cells (iPS) has been proposed recently as a means to study the consequences of mutations in cells derived directly from the affected patients and to overcome the limitation of preceding experimental models. Fatima et al.45 studied iPS-derived myocytes from a carrier of the F2483I mutation. Calcium imaging studies showed that, in agreement with a CPVT phenotype, iPS-derived myocytes from the patient with CPVT presented abnormal spontaneous calcium transients. Upon administration of isoproterenol, the same cells had a negative chronotropic response similar to that observed in CPVT mice.46 This finding suggests that the myocytes obtained by these authors are indeed similar to nodal or embryonic automatic cells. The onset of DADs and increased Ca2+ sparks frequency during adrenergic stimulation was observed by Jung et al.47 in myocytes from a carrier of the S406L mutation. Overall, the data obtained in iPS-myocytes from CPVT patients confirm the data observed previously in other experimental models, although the limitations of this approach currently prevent significant steps in the understanding of CPVT pathophysiology.34
Additional functional studies have investigated the effects of RyR2 mutation in specific anatomical structures. It is known that Purkinje fibers are more susceptible to Ca2+ overload than ventricular muscle, possibly because of their greater sodium load and longer action potential duration. Recently, two groups reported that Purkinje fibers isolated from R4496C mutant mice display a greater propensity to develop intracellular Ca2+ handling disorder than do ventricular myocytes, suggesting that focally activated arrhythmias might originate in the specialized electrical conducting cells of the His-Purkinje system in CPVT.48,49 Direct mapping of bidirectional VT using voltage sensitive dyes further supports this finding.50
The effect of the R4496C mutation, a typical CPVT mutation, has also been investigated at the level of sinus node.46 The rational for this study is the possible presence of lower than normal heart rate in patients with CPVT.28 In sinus cells isolated from mice harboring the R4496C mutation, the study provided initial evidences for a reduced pacemaker activity and impaired chronotropic response under β-adrenergic stimulation. This decreased automaticity appears to be mediated by a Ca2+-dependent decrease of ICa and sarcoplasmic reticulum Ca2+ depletion during diastole upon adrenergic activation.46
RyR2 Mutations and Cardiomyopathy
The possible link between RyR2 mutations and structural abnormalities has stimulated a recent experimental study.51 The authors compared mutations possibly causing ARVC and HCM and focused the attention not only on the threshold for SR Ca2+ release activation (affected in CPVT mutations), but also on the threshold for Ca2+ release termination. The two thresholds contribute to define the fractional release (i.e., the amount of total SR Ca2+ released at each CICR cycle). This study showed that ARVC mutations cause reduced threshold for both activation and termination, with termination to a greater extent; this increased the fractional SR Ca2+ release, which is unaltered in RyR2-CPVT mutations.51 On the contrary the HCM-associated mutation A1107M induces a significant reduction of fractional release because of increased termination threshold. The authors conclude that mutations affecting termination threshold and fractional release are associated with structural abnormalities, but they could not provide an explanation for why fractional release abnormalities should lead to rearrangements of the contractile machinery.51
Calsequestrin and CPVT
CASQ2 is a 399 amino acid protein expressed at the level of the CRUs, where it acts as an intra-SR Ca2+ buffer and modulates RyR2 activity.52 At low Ca2+ levels (<0.6 mM) CASQ2 is present as a monomer. However, at Ca2+ levels between 0.6 and 3.0 mM, the protein starts to form dimers by N-terminal interactions. Further increase of SR Ca2+ concentration (>3 mM) induces CASQ2 polymerization by promoting additional binding through C-terminal interactions.
CASQ2 also modulates RyR2 via triadin and junctin.53 A total of fourteen CASQ2 CPVT-related mutations have been described. Functional characterization has shown multiple effects: reduced CASQ expression levels,54,55 impaired polymerization and buffering capacity,56,57 and reduced CASQ2-RyR2 binding with loss of RyR2 modulation.58,59 These mechanisms work in conjunction to generate SCRs and DADs and are further exacerbated upon adrenergic activation that induces triggered arrhythmias.
Although RyR2 Ca2+ sensitivity is directly proportional to SR-free Ca2+ content,3,60,61 the presence of CASQ2 mutations can increase RyR2 sensitivity by impairing the polymerization capacity or by reducing the CASQ2 expression levels. Less clear is the role of calcium buffering properties, because SR calcium content was found to be either reduced or normal in CASQ2-deficient mice.55,62
Another interesting and puzzling effect of CASQ2 mutations is the presence of ultrastructural abnormalities. Loss of calsequestrin causes a spectrum of ultrastructural changes at the SR level, detectable with electron microscopy. The chainlike polymer at the level of junctional SR (representing CASQ2 protein) is absent54,62; couplons are shorter or junctional SR is fragmented (Figure 53-4).
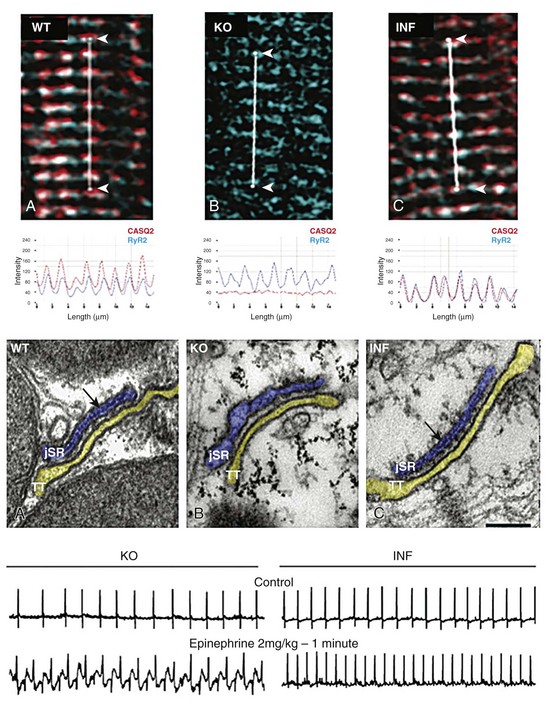
Figure 53-4 High-magnification immunofluorescent image of showing colocalization of CASQ2 (red) and RyR2 (blue) in a wild type cell (A-WT). As expected, no CASQ2 signal is evident in the CASQ2 knockout cell (B-KO). Conversely knockout cells infected with AAV9-CASQ2 (C-inf) show restoration of correct RyR2/CASQ2 colocalization at the z-line level. The middle panel shows electron micrographs of exemplificative cells from wild type (WT), knockout (KO), and KO 20 weeks after AAV9-CASQ2 infection. The electron-dense material inside the junctional sarcoplasmic reticulum (jSR) reappears in the infected cell (INF), whereas the ultrastructural abnormalities (jSR enlargement and fragmentation) are completely reversed. When challenged with epinephrine to mimic adrenergic activation, the infected mice (lower panel, INF) show a clear antiarrhythmic effect in vivo compared with KO animals (lower panel, KO).
Whether these ultrastructural abnormalities have consequences for, or are the cause of, Ca2+ releasing abnormalities is still a matter of debate; however, this peculiar feature makes recessive CPVT a form of ultrastructural cardiomyopathy. It is of considerable interest to note that ultrastructural abnormalities have also been observed in human cardiac myocytes harboring the D307H mutation derived from iPS.63 Electron microscopy has shown that CPVT myocytes present an immature morphology with less-organized myofibrils, enlarged sarcoplasmic reticulum cisternae, and a reduced number of caveolae.
Triadin and CPVT
Only one study reporting two small human TRND families exists in the literature.19 As a consequence, there is only scanty direct evidence of the functional consequences of mutations. Data from a TRDN null mouse have demonstrated the propensity toward adrenergically induced arrhythmias and, interestingly, ultrastructural abnormalities not dissimilar from that of CASQ2 mice.64 Heterologous expression of the human TRDN-T59R mutant in COS-7 resulted in intracellular retention and degradation of the mutant protein.19 No calcium dynamics data are available, but given the tight physiological activity coordination among RyR2, CASQ2, and TRND, it is conceivable that the lack of triadin might produce an abnormal RyR2 open kinetics by altering SR calcium sensitivity and reducing the SR releasing threshold, as observed for CASQ2 mutants. The evidence of the tight interplay between CASQ and TRDN supports this hypothesis. Indeed, it is known that a reduction of CASQ2 expression is associated with a reduction of TRDN levels and vice versa.54,64
Experimental Therapies for Calcium Handling Disorders
Remodulation of Abnormal Calcium Dynamics
Experimental evidence has elucidated the possible role of flecainide65 on the basis of the observation that this drug reduces the durations of channel openings and disrupts the propagation of calcium waves. Thus, a direct effect on the RyR2 channel was postulated. Subsequent work challenged this concept by providing evidence that the most important action of flecainide in counteracting a leaky RyR2 channel occurs through the sodium current inhibition and its negative bathmotropic effect.44 Indeed, flecainide does not inhibit SCRs, but it reduces the propensity for triggered beats.44 Although the mechanism is still debated, the clinical effectiveness of flecainide has been confirmed by independent investigators in small series.66,67 This approach should be considered in addition to β-blockers when insufficient protection is demonstrated. Theoretical studies and anecdotal clinical cases have also proposed a role for calcium channel blockers as a mean to remodulate calcium dynamics in CPVT,68,69 but a clear demonstration of effectiveness is lacking.
Calmodulin Kinase Pathway
Another interesting approach is that of inhibiting the effects of β-adrenergic stimulation by acting on the downstream targets of RyR2 phosphorylation. Calmodulin kinase (CAMKII) phosphorylates the RyR2 at different sites. Moreover, CAMKII inhibition reduces ICa, diastolic Ca2+ leakage, and the Iti current.70 There is also evidence that a specific CAMKII inhibitor, KN93, could prevent arrhythmias both in vitro and in vivo.71
RyR2 Channel Stabilization
An increased FKBP12.6 dissociation could be one of the mechanisms for arrhythmogenesis in the context of a mutant RyR2 channel. Accordingly, pharmacologic enhancement or restoration of FKBB12.6 binding might represent a way to revert CPVT arrhythmias. It has been shown that the FKBP12.6 knockout mouse model develops stress-induced arrhythmias mainly because of increased RyR2 open probability.38 In this model, the treatment with the benzothiazepine compound K201 was able to restore FKBP12.6-RyR2 binding and to prevent arrhythmias.38 Similar results were also observed directly in a CPVT knock-in mouse model harboring the R2474S mutation.72 More recently, S107 (a drug similar to K201) was found to inhibit the inducibility of atrial fibrillation in other CPVT mouse models73; however, conflicting evidence has challenged these results. George et al.74 and Jiang et al.39 showed that mutant RyR2 could interact normally with FKBP12.6, and Liu et al.12 observed normal FKBP12.6 binding and no significant antiarrhythmic effect of K201 both in vivo and in vitro.
Gene Therapy
Restoration of a functionally normal gene function is clearly an attractive goal for genetic diseases. This approach has been attempted for autosomal recessive CPVT, and it was demonstrated that infection of an adeno-associated viral (AAV) vector harboring wild type calsequestrin achieves long-term (at least 3 months) reexpression of properly localized CASQ2 peptides.75 In parallel, there was complete protection from adrenergically induced arrhythmias and a regression of ultrastructural abnormalities (see Figure 53-4). Because AAV vectors have been used in the clinical setting for gene therapy of cardiac disease,76 this approach could possibly be transferred to clinical practice.
Dantrolene
It has long been known that dantrolene suppresses spontaneous SR Ca2+ release in skeletal muscle. Dantrolene does not seem to inhibit the function of normal RyR2 channels; it can bind to and inhibit abnormal RyR2 channels with defective domain interactions. Although the action of dantrolene on spontaneous SR Ca2+ release in cardiac muscle is unclear, it has been shown to suppress ventricular arrhythmias in the R2474S+/− mouse model of CPVT.77 Interestingly these data have been confirmed in cardiomyocytes derived from iPS of a patient with CPVT.47
References
1. Napolitano, C, Bloise, R, Monteforte, N, et al. Sudden cardiac death and genetic ion channelopathies: long QT, Brugada, short QT, catecholaminergic polymorphic ventricular tachycardia, and idiopathic ventricular fibrillation. Circulation. 2012; 125:2027–2034.
2. Shannon, TR, Guo, T, Bers, DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003; 93:40–45.
3. Fabiato, A, Fabiato, F. Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, and frog hearts and from fetal and new-born rat ventricles. Ann N Y Acad. Sci. 1978; 307:491–522.
4. Napolitano, C, Antzelevitch, C. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac voltage-dependent L-type calcium channel. Circ Res. 2011; 108:607–618.
5. Franzini-Armstrong, C, Protasi, F, Ramesh, V. Shape, size, and distribution of Ca(2+) release units and couplons in skeletal and cardiac muscles. Biophys J. 1999; 77:1528–1539.
6. Franzini-Armstrong, C, Protasi, F, Tijskens, P. The assembly of calcium release units in cardiac muscle. Ann N Y Acad Sci. 2005; 1047:76–85.
7. Cannell, MB, Cheng, H, Lederer, WJ. The control of calcium release in heart muscle. Science. 1995; 268:1045–1049.
8. Satoh, H, Blatter, LA, Bers, DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol. 1997; 272:H657–H668.
9. Priori, SG, Chen, SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011; 108:871–883.
10. Bers, DM. Cardiac excitation-contraction coupling. Nature. 2002; 415:198–205.
11. Pieske, B, Maier, LS, Bers, DM, et al. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ Res. 1999; 85:38–46.
12. Liu, N, Colombi, B, Memmi, M, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia. Insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006; 99:292–298.
13. Ginsburg, KS, Bers, DM. Modulation of excitation-contraction coupling by isoproterenol in cardiomyocytes with controlled SR Ca2+ load and Ca2+ current trigger. J Physiol. 2004; 556:463–480.
14. Hussain, M, Orchard, CH. Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during beta-adrenergic stimulation. J Physiol. 1997; 505(Pt 2):385–402.
15. Grimm, M, Brown, JH. Beta-adrenergic receptor signaling in the heart: role of CaMKII. J Mol Cell Cardiol. 2010; 48:322–330.
16. Wehrens, XH, Lehnart, SE, Reiken, SR, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004; 304:292–296.
17. Priori, SG, Napolitano, C, Tiso, N, et al. Mutations in the cardiac ryanodine receptor gene (hryr2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001; 103:196–200.
18. Lahat, H, Pras, E, Olender, T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001; 69:1378–1384.
19. Roux-Buisson, N, Cacheux, M, Fourest-Lieuvin, A, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012; 21:2759–2767.
20. Ackerman, MJ, Priori, SG, Willems, S, et al. HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011; 13:1077–1109.
21. Cerrone, M, Priori, SG. Genetics of sudden death: focus on inherited channelopathies. Eur Heart J. 2011.
22. Cerrone, M, De Giuli, L, Bloise, R, et al. Value of entire open reading frame screening of the cardiac ryanodine receptor gene RyR2. Evidence from the Italian CPVT registry. Heart Rhythm. 2011; 6:S462.
23. Priori, SG, Napolitano, C, Memmi, M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002; 106:69–74.
24. Tester, DJ, Kopplin, LJ, Will, ML, et al. Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm. 2005; 2:1099–1105.
25. Coumel, P, Fidelle, J, Lucet, V, et al. Catecholaminergic-induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: report of four cases. Br Heart J. 1978; 40:28–37.
26. Leenhardt, A, Lucet, V, Denjoy, I, et al. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation. 1995; 91:1512–1519.
27. Hayashi, M, Denjoy, I, Extramiana, F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009; 119:2426–2434.
28. Postma, AV, Denjoy, I, Kamblock, J, et al. Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet. 2005; 42:863–870.
29. Tiso, N, Stephan, DA, Nava, A, et al. Identification on mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet. 2001; 10:189–194.
30. Fujino, N, Ino, H, Hayashi, K, et al. A novel missense mutation in cardiac ryanodine receptor gene as a possible cause of hypertrophic cardiomyopathy: evidence from familial analysis. Circulation. 2006; 114(Suppl II):164.
31. van der Werf, C, Hofman, N, Tan, HL, et al. Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: the experience of a tertiary referral center in The Netherlands. Heart Rhythm. 2010; 7:1383–1389.
32. Hong, RA, Rivera, KK, Jittirat, A, et al. Flecainide suppresses defibrillator-induced storming in catecholaminergic polymorphic ventricular tachycardia. Pacing Clin Electrophysiol. 2012; 35:794–797.
33. Lederer, WJ, Tsien, RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibres. J Physiol. 1976; 263:73–100.
34. Priori, SG, Napolitano, C, Di Pasquale, E, et al. Induced pluripotent stem cell-derived cardiomyocytes in studies of inherited arrhythmias. J Clin Invest. 2012.
35. George, CH, Jundi, H, Thomas, NL, et al. Ryanodine receptors and ventricular arrhythmias: emerging trends in mutations, mechanisms and therapies. J Mol Cell Cardiol. 2007; 42:34–50.
36. Sitsapesan, R, Williams, AJ. Gating of the native and purified cardiac SR Ca(2+)-release channel with monovalent cations as permeant species. Biophys. J. 1994; 67:1484–1494.
37. Sitsapesan, R, Williams, AJ. Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2+)-release channel by luminal Ca2+. J Membr Biol. 1994; 137:215–226.
38. Wehrens, XH, Lehnart, SE, Huang, F, et al. FKBP12. 6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003; 113:829–840.
39. Jiang, D, Wang, R, Xiao, B, et al. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005; 97:1173–1181.
40. Jiang, D, Xiao, B, Yang, D, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A. 2004; 101:13062–13067.
41. Yamamoto, T, Yano, M, Xu, X, et al. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation. 2008; 117:762–772.
42. Tateishi, H, Yano, M, Mochizuki, M, et al. Defective domain-domain interactions within the ryanodine receptor as a critical cause of diastolic Ca2+ leak in failing hearts. Cardiovasc Res. 2009; 81:536–545.
43. Uchinoumi, H, Yano, M, Suetomi, T, et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010; 106:1413–1424.
44. Liu, N, Denegri, M, Ruan, Y, et al. Short communication: flecainide exerts an antiarrhythmic effect in a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the Threshold for triggered activity. Circ Res. 2011; 109:291–295.
45. Fatima, A, Xu, G, Shao, K, et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem. 2011; 28:579–592.
46. Neco, P, Torrente, AG, Mesirca, P, et al. Paradoxical effect of increased diastolic Ca2+ release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation. 2012; 126:392–401.
47. Jung, CB, Moretti, A, Mederos y Schnitzler, M, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2011; 4:180–191.
48. Hilliard, FA, Steele, DS, Laver, D, et al. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2009; 48:293–301.
49. Herron, TJ, Milstein, ML, Anumonwo, J, et al. Purkinje cell calcium dysregulation is the cellular mechanism that underlies catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2010; 7:1122–1128.
50. Cerrone, M, Noujaim, SF, Tolkacheva, EG, et al. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007; 11:1039–1048.
51. Tang, Y, Tian, X, Wang, R, et al. Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ Res. 2012; 110:968–977.
52. Gyorke, S, Terentyev, D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008; 77:245–255.
53. Gyorke, I, Hester, N, Jones, LR, et al. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004; 86:2121–2128.
54. Rizzi, N, Liu, N, Napolitano, C, et al. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ Res. 2008; 103:298–306.
55. Song, L, Alcalai, R, Arad, M, et al. Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2007; 117:1814–1823.
56. Valle, G, Galla, D, Nori, A, et al. Catecholaminergic polymorphic ventricular tachycardia-related mutations R33Q and L167H alter calcium sensitivity of human cardiac calsequestrin. Biochem J. 2008; 413:291–303.
57. Kalyanasundaram, A, Bal, NC, Franzini-Armstrong, C, et al. The calsequestrin mutation CASQ2D307H does not affect protein stability and targeting to the junctional sarcoplasmic reticulum but compromises its dynamic regulation of calcium buffering. J Biol Chem. 2009; 285:3076–3083.
58. Terentyev, D, Nori, A, Santoro, M, et al. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ Res. 2006; 98:1151–1158.
59. Houle, TD, Ram, ML, Cala, SE. Calsequestrin mutant D307H exhibits depressed binding to its protein targets and a depressed response to calcium. Cardiovasc Res. 2004; 64:227–233.
60. Qin, J, Valle, G, Nani, A, et al. Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J Gen Physiol. 2008; 131:325–334.
61. Gyorke, I, Gyorke, S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998; 75:2801–2810.
62. Knollmann, BC, Chopra, N, Hlaing, T, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006; 116:2510–2520.
63. Novak, A, Barad, L, Zeevi-Levin, N, et al. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta-adrenergic stimulation. J Cell Mol Med. 2012; 16:468–482.
64. Chopra, N, Yang, T, Asghari, P, et al. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009; 106:7636–7641.
65. Watanabe, H, Chopra, N, Laver, D, et al. Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 2009; 15:380–383.
66. Pott, C, Dechering, DG, Reinke, F, et al. Successful treatment of catecholaminergic polymorphic ventricular tachycardia with flecainide: a case report and review of the current literature. Europace. 2010.
67. van der Werf, C, Kannankeril, PJ, Sacher, F, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol. 2011; 57:2244–2254.
68. Rosso, R, Kalman, JM, Rogowski, O, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007; 4:1149–1154.
69. Sung, RJ, Lo, CP, Hsiao, PY, et al. Targeting intracellular calcium cycling in catecholaminergic polymorphic ventricular tachycardia: a theoretical investigation. Am J Physiol Heart Circ Physiol. 2011; 301:H1625–H1638.
70. Wu, Y, Temple, J, Zhang, R, et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002; 106:1288–1293.
71. Liu, N, Ruan, Y, Denegri, M, et al. Calmodulin kinase II inhibition prevents arrhythmias in RyR2R4496C+/− mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol. 2011; 50:214–222.
72. Lehnart, SE, Mongillo, M, Bellinger, A, et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest. 2008; 118:2230–2245.
73. Shan, J, Xie, W, Betzenhauser, M, et al. Calcium leak through ryanodine receptors leads to atrial fibrillation in three mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2012.
74. George, CH, Higgs, GV, Lai, FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003; 93:531–540.
75. Denegri, M, Avelino-Cruz, JE, Boncompagni, S, et al. Viral gene transfer rescues arrhythmogenic phenotype and ultrastructural abnormalities in adult calsequestrin-null mice with inherited arrhythmias. Circ Res. 2012.
76. Jessup, M, Greenberg, B, Mancini, D, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011; 124:304–313.
77. Kobayashi, S, Yano, M, Uchinoumi, H, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ J. 2010; 74:2579–2584.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]53
Inheritable Phenotypes Associated With Altered Intracellular Calcium Regulation
Overview of the Function of the Calcium-Handling System
Growing clinical and experimental evidence highlights the relevance of cardiac calcium handling in the pathogenesis of inherited arrhythmias.1 The control of Ca2+ fluxes in myocardial cells requires the timely coordination of several events that ultimately lead to contraction. Any perturbation of this process has the potential to determine an arrhythmogenic substrate.
The core events of Ca2+ ion movements in the myocardial cells are the opening of the L-type Ca2+ channels followed by the release of Ca2+ from the sarcoplasmic reticulum (SR) through the opening of the ryanodine receptors (RyR2).2 This process is called calcium-induced calcium release (CICR).3 The L-type calcium channel (CaV1.2) activation is therefore the first step of CICR. CaV1.2 belongs to the family of the voltage-gated calcium channels that are macromolecular complexes consisting of an ion conducting protein (the α1-subunit) and additional accessory peptides with regulatory function called α2δ, the β1-4, and γ subunits.4 Among the biophysical properties of voltage-dependent calcium current (ICa), the inactivation process is relevant to inherited arrhythmias (Timothy syndrome, see Chapter 95). Two components have been identified: voltage- (VDI) and Ca2+ (CDI)-dependent inactivation. The Ca2+-mediated component of inactivation is modulated by intracellular (cytosolic) concentration of calcium (Ca2+).
CaV1.2 channels tend to cluster in the T-tubules in close proximity with the ryanodine receptors sitting across the membrane of the SR.5 CaV1.2 activation constitutes the signal for the activation of the RyR2. This latter event can be detected as a calcium transient, which is the sum of coordinated local releases that occur at specialized structures: the calcium release units (CRUs; Figure 53-1). One CRU is formed by clusters of RyR2 receptors that are in close proximity to L-type Ca2+ channels in the T-tubules.6 CRUs also include cardiac calsequestrin (CASQ2), triadin (TRDN), and junctin (JTC) that contribute to the control of the calcium release process. These peptides form a macromolecular complex that acts in coordination to control Ca2+ release. The number of CRUs recruited for release at each cardiac cycle is an important modulator of the systolic Ca2+ transient amplitude,7,8 and the loss of integrity of CRU is part of the pathophysiology of CPVT.9
Figure 53-1 Schematic representation of CRU and CPVT gene localization. The figure depicts a schematic representation of a T-tubule with the LTCC sitting across the plasmalemma juxtaposed to the RyR2/CASQ2/TRND/JTC macromolecular complex from the SR side. The SR component of CRU is involved in CPVT pathogenesis. CASQ2, cardiac calsequestrin; CPVT, catecholaminergic polymorphic ventricular tachycardia; CRU, calcium release unit; JTC, junctin; LTCC, L-type calcium channel; RyR2, cardiac ryanodine receptor; SR, sarcoplasmic reticulum; TRDN, triadin.
During the relaxation phase, SR Ca2+ release terminates, and Ca2+ is taken up in the SR by the SR Ca2+-ATPase (SERCA) or extruded from the cell by the Na+/Ca2+ exchanger (NCX).10 NCX extrudes one Ca2+ ion (two positive charges) for every three Na+ ions (three positive charges) that are transferred into the cell. Thus, NCX generates a net inward depolarizing current, the transient inward current—Iti. In physiological conditions, SERCA is responsible for approximately 63% of calcium removal, and NCX mediates the remaining 37%.11 However, NCX becomes important to remove Ca2+ in any condition of calcium overload (e.g., in patients with genetic mutations of the RyR2 gene [CPVT] and during heart failure).11 Excessive activation of NCX can be arrhythmogenic.
Sarcoplasmic Reticulum Ca2+ Release Threshold and the Adrenergic Signaling
Activation of the adrenergic nervous system, mainly through β-adrenergic receptors, has profound effects on calcium handling, and it is often the initiator of calcium-mediated arrhythmogenesis.12 Adrenergic activation has two major effects on calcium handling: the enhancement of the amplitude of the L-type calcium current (ICa) and the increase of SR Ca2+ levels induced through the activation of SERCA13. The latter is responsible for an increase of Ca2+-transient amplitude. The effects of adrenergic activation can take place because of the phosphorylation target proteins14 induced by the activation of two enzymes with kinase activity: protein kinase A14 and Ca2+ calmodulin kinase II (CAMKII).15 Among the several target proteins, the adrenergic-dependent changes of Ca2+ handling are mainly due to the phosphorylation of L-type Ca2+ channel (increased current amplitude) phospholamban (removal of SERCA inhibition and increase of SR Ca reuptake) and RyR2 (increased open probability and increased transients).16 Thus, protein phosphorylation is an important mechanism that enables adrenergic activation to enhance the SR calcium release. In physiological conditions, this response is useful to react to environmental stressors by improving myocardial contractility.
Phenotypes Associated With Mutation in Calcium Handling Proteins
CPVT is the most frequent phenotype associated with mutations involved in intracellular calcium handling. Three genes have been implicated in its pathogenesis (Table 53-1): ryanodine receptor (RyR2 [CPVT1]),17 cardiac calsequestrin (CASQ2 [CPVT2]),18 and cardiac triadin (TRDN [CPVT3]).19 CPVT1 is an autosomal dominant trait, whereas CPVT2 and CPVT3 are rare autosomal recessive disorders. In 2001, Priori et al17 identified RyR2 as the gene responsible for the most frequent variant of CPVT. The prevalence of RyR2 mutations in patients with a clearly diagnostic phenotype is high (≈60% to 70%).20,21 Mutations concentrate in three specific areas of the RyR2 protein (amino acids 77-466, 2246-2534, and 3778-4967); however, 14% of CPVT patients harbor RyR2 mutations located outside these areas.21 Recent data suggest that there is no significant difference of outcome of CPVT according to mutation site, including the outcome of the subgroup with mutations outside the canonical clusters.22 N, RyR2 mutations have been reported also in patients referred for idiopathic ventricular fibrillation and in relatives of subjects who died suddenly.23,24 Autosomal recessive CPVT is rarely identified in the clinical setting. Combined estimated prevalence of the two recessive variants is approximately 5%.
Clinical Manifestations and Management of CPVT
CPVT is a severe inherited arrhythmogenic disease manifesting with adrenergically mediated arrhythmias, often leading to syncope or cardiac arrest.17,23,25–27 Although the resting electrocardiogram is unremarkable, the reproducible inducibility of ventricular tachycardia (VT) during exercise stress test is the hallmark of the disease. Most patients with CPVT show a bidirectional VT pattern characterized by beat-to-beat 180-degree rotation of the QRS axis (Figure 53-2)17,23,26; however, polymorphic tachycardia or ventricular fibrillation may also be part of the picture.23,27 This unstable, catecholamine-sensitive, substrate can lead to sudden death as the first manifestation of the disease in up to 30% of cases.23 Symptoms suggesting the presence of arrhythmias tend to manifest early in life (median age, 12 years), although later onset is possible. The development of palpitations and syncopal events during adrenergic stress is a critical element to suspect the diagnosis of CPVT, which is confirmed by the induction of bidirectional VT on an exercise stress test. In untreated patients, the occurrence of severe arrhythmias is approximately 60% to 70%, and approximately 30% experience a cardiac arrest or sudden death upon first manifestation (see Figure 53-2).23,28 CPVT patients typically present with structurally normal heart. However, some RyR2 mutations have been associated anecdotally with structural cardiomyopathies. One of the first reports on RyR2 gene mutations suggested an association with arrhythmogenic right ventricular cardiomyopathy (ARVC).29 More recently, other authors have reported preliminary data linking RyR2 with hypertrophic cardiomyopathy (HCM).30 Although careful scrutiny of these clinical reports does not establish a causal link, recent experimental findings suggest a mechanistic explanation as to why some RyR2 variants could in principle make the heart more prone to develop structural abnormalities (discussed later).
Figure 53-2 Kaplan-Maier curve showing the cumulative occurrence of cardiac arrest, sudden death, or ICD shocks in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) in the absence of therapy. An example of bidirectional ventricular tachycardia, the typical CPVT arrhythmia, is depicted in the inset.
β-Adrenergic receptor block represents the first-choice therapeutic approach. The use of β-blockers is based on the evidence of the direct link between adrenergic activation and cardiac events in CPVT. Clinical data show that β-blockers have an effect on the natural history of CPVT by achieving a significant reduction of cardiac events. In this context, exercise stress testing is important for dosage titration because the threshold for arrhythmias in CPVT is reproducible. β-Blockers can prevent the onset of arrhythmia in 70% to 80% of cases.23,26 In addition, nonselective β-blockers (e.g., nadolol, propranolol) confer the highest degree of protection against the onset of VT and SCD. However, the nontrivial incidence of recurrent cardiac on optimal β-blocker dosage (approximately 30%)22 calls for the identification of additional therapeutic strategies. Implantable cardioverter defibrillator (ICD) implant, left cardiac sympathetic denervation, and other pharmacologic approaches have been proposed, and flecainide appears to be the most promising strategy. Although the mechanisms for its effectiveness are debatable (discussed later), clinical data and personal experience suggest that flecainide affords additional protection when added to β-blockers in CPVT.31,32
Pathophysiology
Abnormalities of Calcium Handling in CPVT
From a mechanistic standpoint, similarities and significant differences exist among CPVT1-3 genetic variants. In all three forms of CPVT, arrhythmogenesis is a consequence of the occurrence of spontaneous calcium release (SCR; Figure 53-3). This term is used to refer to SR Ca2+ release events that are not driven by a stimulated action potential. Whenever an SCR occurs and the levels of Ca2+ in the sarcolemma increase, the NCX activates to extrude the excess of ions. As mentioned earlier, NCX activation generates the Iti current, which is the cause of the development of delayed afterdepolarizations (DADs)33 and transient membrane depolarizations occurring during phase 4 of the action potential (see Figure 53-3). When DAD amplitude reaches the voltage threshold for sodium channel activation, triggered beats occur.
Figure 53-3 A, Representative line-scan images and Ca2+ transients in a wild type myocyte and a RyR2R4496C+/− myocyte at 0.2 Hz pacing in the presence of isoproterenol (100 nM). Asterisk indicates spontaneous Ca2+ release in the RyR2R4496C+/− myocytes. Arrows indicated the field stimulations. B, Action potential recording in a myocyte isolated from a RyR2R4496C+/− during exposure to isoproterenol 30 nmol. Arrows indicate stimulated beats, which are followed by two triggered beats and delayed afterdepolarizations.
The effects of CPVT mutations have been analyzed in a variety of experimental settings, including lipid bilayer, heterologous expression systems, murine transgenic models, and in myocytes derived from patient-specific induced pluripotent stem cells.34 The following sections outline the key concepts about arrhythmogenesis in each form of CPVT.
Ryanodine Receptor, Mutations, and CPVT
The RyR2 channel is a homotetramer; each subunit is formed by 4967 amino acids with a long (≈4300 amino acids) N-terminal cytoplasmic domain.35 The last 500 amino acids at the C-terminal of RyR2 form the transmembrane segments encircling the channel pore. The regulation of RyR2 opening and closing (gating) is mainly controlled by Ca2+ levels at cytoplasmic and luminal SR sides, and it is facilitated when the Ca2+ concentration at either side increases.36,37 Therefore, RyR2 calcium sensitivity is an important physiological function that controls CICR. The majority of RyR2 mutations identified in CPVT patients cause increased calcium sensitivity (defined as “gain-of-function”).
Marks et al. showed that mutant RyR2 exhibits an increased sensitivity to cytosolic Ca2+ after protein kinase A phosphorylation. They also suggested that such change in sensitivity is due to an abnormal dissociation of FKBP12.6, a putative RyR2 stabilizing protein.38 During adrenergic stimulation, phosphorylation of RyR2 would promote SCRs by further dissociation of FKBP12.6 with a consequent excessive increase of open probability.
[/not-level-membership-for-cardiovascular-category]
