CHAPTER 89 INFLAMMATORY MYOPATHIES
The inflammatory myopathies are a heterogeneous group of acquired diseases of skeletal muscle. They have in common the presence of varying degrees of muscle weakness and inflammation. Based on clinical, histological, and immunopathological criteria, they form three major groups: polymyositis, dermatomyositis, and sporadic inclusion-body myositis.1,2 Dermatomyositis is easily recognized by specific skin changes that occur early in the course of the disease. Sporadic inclusion-body myositis is easily suspected based on its slow progression, unique distribution of weakness and atrophy, characteristic muscle biopsy findings, and resistance to conventional immunotherapies.2,3 Polymyositis, however, remains a diagnostic challenge and is often misdiagnosed as inclusion-body myositis, dystrophy, or toxic or metabolic myopathy.4 The old assumption that dermatomyositis is like polymyositis without a rash, and inclusion-body myositis is like polymyositis without vacuoles, is overly simplistic and incorrect.5,6
CLINICAL PRESENTATION
Dermatomyositis
Dermatomyositis is seen in both children and adults, and more often in women than in men1 (Table 89-1). Juvenile dermatomyositis is the most common form of inflammatory myopathy in children.7,8 Although the most obvious manifestations are due to involvement of skeletal muscle and skin, rarely other organ systems are affected, including the gastrointestinal tract, heart, and lungs. The muscle weakness in dermatomyositis is classically proximal, symmetrical, and frequently progressive. It can vary from mild to severe, occasionally resulting in quadriparesis. The weakness usually develops slowly, over weeks to months, but in rare cases there is an acute onset. Patients usually have problems with physical tasks such as rising up from a chair or climbing steps, stepping onto a curb, lifting objects, or combing their hair. Fine motor movements that depend on the strength of distal muscles, such as manipulating small objects, are spared until late in the course of the disease. Involvement of the neck extensor muscles may lead to head drop. In advanced stages of the disease or during an acute course, patients might have respiratory muscle weakness or dysphagia, causing choking episodes. Facial muscles are spared and extraocular muscles are never affected.1 Sensation remains normal, and tendon reflexes are preserved but may be absent in severely weakened or atrophied muscles. Myalgia is not a common feature and occurs in less than 30% of the patients.1
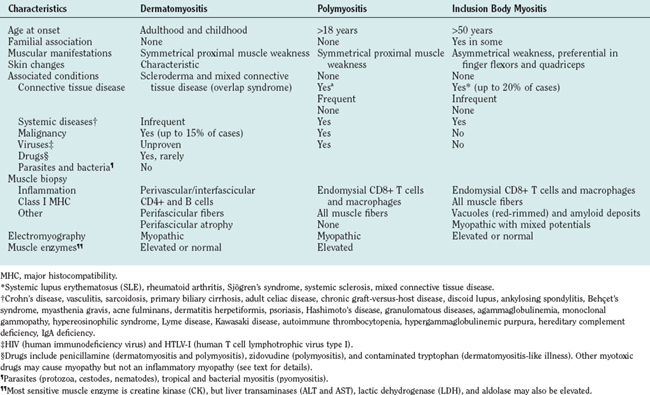
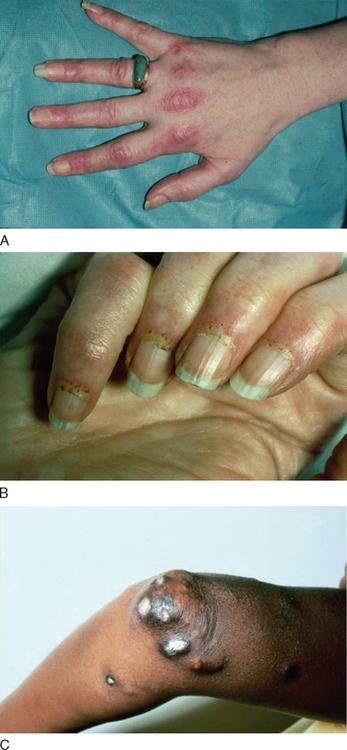
The cutaneous manifestations of dermatomyositis usually precede or accompany the weakness and include the following: erythematous (and later dry and scaly) lesions over the metacarpophalangeal, proximal interphalangeal, or distal interphalangeal joints (Gottron papules); a violaceous hue over the eyelids (heliotrope rash) and periorbital edema; periungual telangiectasia characterized by dilated capillary loops at the base of the fingernails, with irregular, thickened, and distorted cuticle; malar erythema; and erythematous scaly rash over the neck and upper back (shawl sign), anterior chest (V sign), and extensor surfaces of the extremities (Fig. 89-1). The rash can be exacerbated after exposure to the sun and is pruritic in some cases.1 At times, muscle strength appears normal—hence the term dermatomyositis sine myositis, or amyopathic dermatomyositis. When a muscle biopsy is performed in such cases, however, perivascular and perimysial inflammation can be seen. Juvenile dermatomyositis resembles dermatomyositis in adults, except for the presence of more frequent extramuscular manifestations. A child with evolving dermatomyositis is irritable, does not socialize, is uncomfortable, complains of fatigue, and has a red flush on the face with varying degrees of muscle weakness.9 A tiptoe gait due to flexion contracture of the ankles is also common in juvenile dermatomyositis.1 In 3% to 5% of children with juvenile dermatomyositis, the cutaneous manifestations of the disease are present in the absence of clinically evident muscle weakness: these patients are classified as amyopathic.10 Subcutaneous calcinosis is not an uncommon finding in juvenile dermatomyositis, especially when initiation of therapy is delayed, or when applied therapies have not been fully effective, resulting in chronicity of the disease, muscular atrophy, and joint contractures8,11,12 (Fig. 89-1).
Polymyositis
Polymyositis is usually seen after the second decade of life, rarely in children.2,1 It is manifested by muscle weakness of subacute onset, similar to what is seen in dermatomyositis but without any of the cutaneous manifestations. Polymyositis can mimic many other myopathies and remains a diagnosis of exclusion. The most common myopathy misdiagnosed as polymyositis is inclusion-body myositis. This disease is often suspected in retrospect, when a patient with presumed polymyositis has not responded to therapy.2 Other myopathies erroneously diagnosed and treated as polymyositis include acute necrotizing myopathies, toxic and endocrine myopathies, dermatomyositis sine dermatitis, certain dystrophies, and some slowly progressive myopathies starting in late childhood.1 One of the main reasons for misdiagnosing these disorders as polymyositis is that several myopathies, especially certain dystrophies such as Duchenne’s, Becker’s, fascioscapulohumeral, or dysferlinopathies, may also show prominent inflammation in their muscle biopsies.4 Thus, we have to apply new diagnostic criteria that more specifically characterize the type of inflammation seen in polymyositis, as discussed later.
Inclusion Body Myositis
Inclusion-body myositis is the most common form of inflammatory myopathy, especially in patients above the age of 50.2 The weakness and atrophy are usually observed first in the quadriceps femoris muscles of the legs, leading to instability of the knees and increased falls, and in the flexor digitorum profundus muscles in the forearm, resulting in difficulties with fine motor movements such as holding golf clubs, turning keys, or tying knots.13 The weakness and atrophy frequently affect the iliopsoas, triceps, biceps, and foot extensor muscles9 and it may be asymmetrical, resembling lower motor neuron disease or motor neuropathies. The facial muscles are very often affected even early in the disease. Dysphagia is common, occurring in up to 60% of inclusion-body myositis patients, and may lead to episodes of choking. Sensory examination is generally normal, although some patients have mildly diminished vibratory sensation at the ankles that is presumably age related.9 Disease progression is slow but steady, and most patients require assistive devices such as a cane, walker, or a wheelchair after several years.
Associated Clinical Manifestations
Inflammatory myopathies, in particular dermatomyositis and polymyositis, can have non–skeletal muscle manifestations. Constitutional symptoms such as fever, malaise, weight loss, arthralgia, or Raynaud’s phenomenon can occur in the subacute stages of dermatomyositis and polymyositis, especially when the disease is associated with another connective-tissue disorder.14 Cardiovascular abnormalities, including conduction defects, tachyarrhythmias, or myocarditis, are rarely seen during the active phase of the disease. Hypertension and heart failure are more frequently seen late in the course of the disease, probably as a result of longterm steroid therapy or lung disease.1 Pulmonary involvement may occur, either due to thoracic muscle weakness or interstitial lung disease. The latter is most commonly seen in patients with autoantibodies against t-RNA synthetases (Jo-1), signal recognition particle (SRP),15 or a mucin-like glycoprotein (KL-6).16,17 Antisynthetase syndrome refers to the presence of inflammatory myopathy, anti-Jo-1 antibodies, and interstitial lung disease, often in association with low-grade fever, subluxation of the phalangeal joints, Raynaud’s phenomenon, and thick cracked skin on the fingers (mechanic’s hands).18
Malignancy
Among the inflammatory myopathies, dermatomyositis is associated with increased incidence of malignant diseases, in up to 15% of the adult cases.19 The most common cancers are those of the ovaries, gastrointestinal tract, lung, breast, and non-Hodgkin lymphomas20 or nasopharyngeal cancer in Asian populations. Increased vigilance, with careful periodic clinical examination and the prudent use of appropriate laboratory studies, are required for early detection of cancer, especially in older patients, and during the first 3 years after disease onset.2,21
DIAGNOSIS
The criteria of Bohan and Peter22 used for several years, have now become obsolete because they do not distinguish polymyositis from inclusion-body myositis or certain muscular dystrophies (Table 89-2). The need for new criteria was recognized in 1991.2 The inclusion of simple immunopathology in processing the muscle biopsy specimens was recently emphasized as the best means of differentiating inflammatory from noninflammatory myopathies.1 The diagnosis is based on the characteristic clinical features of polymyositis, dermatomyositis, or inclusion-body myositis outlined earlier, combined with the triad of serum muscle enzyme levels, electromyography, and muscle biopsy. Imaging studies have a limited role in the diagnosis of inflammatory myopathies. Subcutaneous calcinosis, an uncommon finding in adult but more common in juvenile dermatomyositis, can be detected on plain radiographs.11 Magnetic resonance imaging (MRI) may reveal the involved muscles in inclusion-body myositis23,24 but is not needed for diagnostic purposes. The only usefulness of MRI is to help us select the appropriate site for biopsy, when the clinical pattern of weakness is not uniform or typical. We do not recommend routine MRI for our patients; we select the biopsy site on the basis of clinical assessment. In the chronic stages of these diseases, when muscle atrophy and fatty infiltration dominate the picture, MRI is of no diagnostic value.
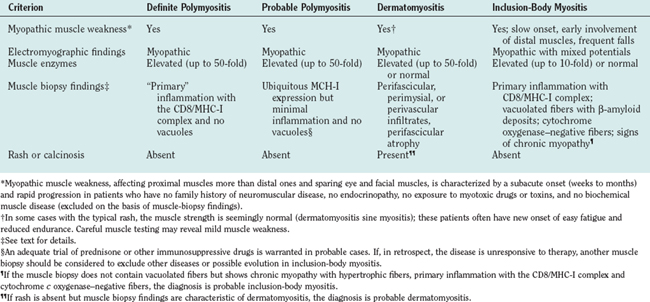
Muscle Enzymes
Creatine kinase, aspartate and alanine aminotransferases, lactate dehydrogenase, and aldolase levels are usually increased in inflammatory myopathies, particularly in the active phases of the disease, and parallel the disease activity.1 Elevation of serum creatine kinase, although not specific to inflammatory myopathies, is a useful laboratory marker.9 Creatine kinase, however, may remain within the normal range in some patients, especially with dermatomyositis or inclusion-body myositis, and should not be used as the sole marker for determining disease activity.
Electrodiagnostic Studies
Needle electromyography shows myopathic potentials, characterized by increased spontaneous activity, fibrillations, complex repetitive discharges, and positive sharp waves. The voluntary motor units have low amplitude and short duration and are polyphasic. Mixed potentials (polyphasic units of short and long duration) indicating a chronic process or muscle fiber regeneration are often present in inclusion-body myositis.9 These findings, however, are not disease specific, as they can occur in any other active myopathy.9 At times, the presence of active myopathic potentials can distinguish exacerbation of the primary disease from steroid-induced myopathy.1
Muscle Biopsy
Muscle biopsy is the most important test for establishing the diagnosis, but it can also lead to misdiagnosis due to erroneous interpretation.1
Occasionally, due to the spotty nature of the inflammation, a repeat muscle biopsy may become necessary. This needs to be especially considered in the patients who meet the clinical criteria described earlier but whose initial muscle biopsies are nondiagnostic.1 We also recommend a repeat muscle biopsy when a patient carries the diagnosis of polymyositis but remains unresponsive to therapies.
The histopathology of dermatomyositis is characterized by inflammation, which is predominantly perivascular and in the interfascicular septa rather than in the fascicles.3,9,25 Necrosis and phagocytosis of muscle fibers commonly occur in groups (microinfarcts) and involves the peripheral portion of the muscle fascicle. The resultant perifascicular atrophy (Fig. 89-2) is characterized by 2 to 10 layers of atrophic fibers and is diagnostic of dermatomyositis irrespective of the presence of inflammation. In the absence of typical features of inflammatory infiltrates or perifascicular atrophy, increased perifascicular class I major histocompatibility (MHC) expression, may be of diagnostic value.26 Biopsy of skin lesions in dermatomyositis shows perivascular inflammation in the dermis with CD4+ cells and dilatation of superficial capillaries in more chronic stages.1
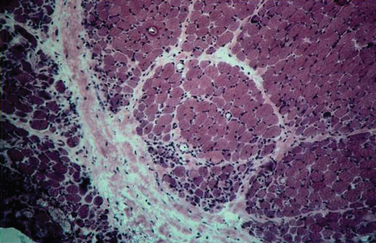
Figure 89-2 Perifascicular atrophy in a cross section of a muscle biopsy in a patient with dermatomyositis.
Muscle biopsies of patients with polymyositis show varying degrees of endomysial inflammation (Fig. 89-3). In polymyositis and inclusion-body myositis, the inflammation is characterized as primary to denote the partial invasion of still intact and nonnecrotic muscle fibers by activated CD8+ T lymphocytes. B lymphocytes form a minor fraction of perivascular infiltrates, but they are mostly absent from the endomysium.27,28 In chronic stages of the disease, there is an increase in connective tissue, which may react with alkaline phosphatase. Both invaded and noninvaded muscle fibers express class I MHC antigen on their surface29 (Fig. 89-4). The ubiquitous sarcolemmal expression of class I MHC antigen has now become essential in diagnosing polymyositis, not only because it is part of the specific lesion but also because it is not present in other non-immune myopathies and it is not affected by the use of immunosuppressive agents. The “CD8+/MHC-I complex” (MHC-I along with CD8+ cells) is specific for the inflammation in polymyositis and inclusion-body myositis and needs to be incorporated into the histological evaluation of polymyositis patients, in an effort to rule out the secondary inflammation seen in various dystrophies or toxic myopathies.1
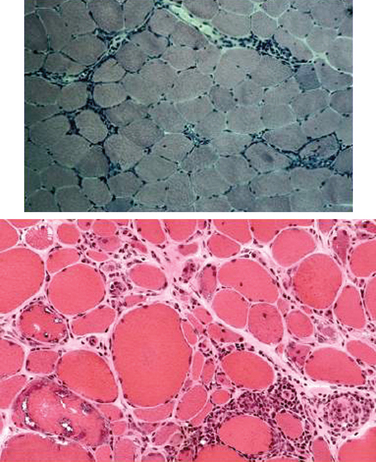
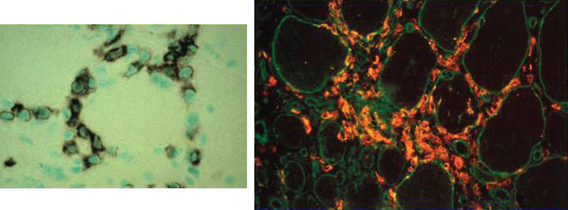
Muscle biopsies from patients with inclusion-body myositis show the same pattern of CD8/MHC-I expression as in polymyositis, even in the late stages of the disease. In inclusion-body myositis, however, in addition to inflammation, a number of muscle fibers not invaded by T cells contain single or multiple vacuoles, which seem to increase as the disease progresses4 (see Fig. 89-3). The vacuoles contain small, basophilic granules in their centers or against their walls and appear as “red-rimmed vacuoles” on the trichrome stain. Frozen sections and enzyme histochemistry are required for optimal visualization of vacuoles, because the granules dissolve on paraffin sections and might be easily overlooked if the biopsy is processed only with paraffin.4 Deposits of amyloid can be seen in vacuolated fibers as green birefringence on Congo red staining30 or as red dichroism when visualized with Texas red filters.31 Characteristically, the vacuolated fibers in inclusion-body myositis are not invaded by T cells, and those fibers that are partially invaded by inflammatory cells are never vacuolated.4 Vacuolated fibers, even the ones containing amyloid deposits, are not specific for inclusion-body myositis but can be seen in other chronic distal myopathies (e.g., myofibrillar, facioscapulohumeral, and dysferlin myopathies), and even in chronic neurogenic disorders such as old paralytic poliomyelitis.32 Other findings in inclusion-body myositis include ragged red fibers or cytochrome c oxidase–negative fibers, which contain abnormal mitochondria harboring multiple mitochondrial DNA deletions.33 These fibers are common in inclusion-body myositis and more frequent than expected for the patient’s age. On electron microscopy, the basophilic granules seen in cryostat sections represent membranous whorls of various sizes. In their vicinity, abnormal filaments 12 to 18 nm in diameter can be found. They have a tubular structure with a central lumen approximately 3.5 nm in diameter. On longitudinal view, these filaments often have a periodic cross-hatched appearance, which seems to be outside the axial core of the filament. The nature of the inclusion-body myositis filaments remains obscure. They immunoreact with various amyloid-related proteins, identical to those seen in Alzheimer’s disease, such as ubiquitin, phosphorylated tau, presenilin-1, apolipoprotein E (apoE), and others.34
Immunopathogenesis
Polymyositis, dermatomyositis, and inclusion body myositis, although immunopathologically distinct, share common histopathological features of inflammation, fibrosis, and loss of muscle fibers.9 In all three, the transmigration of activated T cells, and their adhesion to the muscle fibers, are facilitated by cytokines, chemokines, and adhesion molecules.5 Rare familial occurrences and association with certain HLA genes, such as DRB1*0301 alleles for polymyositis and inclusion-body myositis and HLA DQA1*0501 for juvenile dermatomyositis,35 suggest that genetic factors may also play a role in their pathogenesis.
Dermatomyositis
Dermatomyositis is a microangiopathy, involving arterioles and endomysial capillaries. The primary antigenic target in dermatomyositis is unknown but is believed to be the endothelium of the endomysial capillaries.1 The disease begins when putative antibodies directed against endothelial cells activate complement, leading to the formation and deposition of the membrane attack complex (MAC) on the endomysial microvasculature.1,36,37 This leads to lysis of endothelial cells, capillary necrosis, ischemia and microinfarcts, inflammation, endofascicular hypoperfusion, and eventual perifascicular atrophy1,4 (Fig. 89-5). The predominant cells infiltrating the muscle are B lymphocytes and CD4+ T cells, consistent with a humorally mediated process.1,28 Recently, a large number of plasmacytoid dendritic cells have been observed.38 Cytokines, including signal transducer and activator of transcription (STAT) and chemokines, are activated and overexpressed in the muscle of patients with dermatomyositis, facilitating the transmigration of T cells and further enhancing the inflammatory cascade and eventual tissue necrosis. Fibrogenic cytokines, such as transforming growth factor-β, and chemokines are also overexpressed in the perimysium and may have a role in facilitating the formation of fibrosis, as seen in later stages of the disease.39,40 Genes induced by interferon-α/β are also overexpressed in the patient’s muscle; one of them, the interferon-α/β inducible myxovirus resistance-A protein, is upregulated in the perifascicular region and in the capillaries and to a lesser degree in the myofibers.38
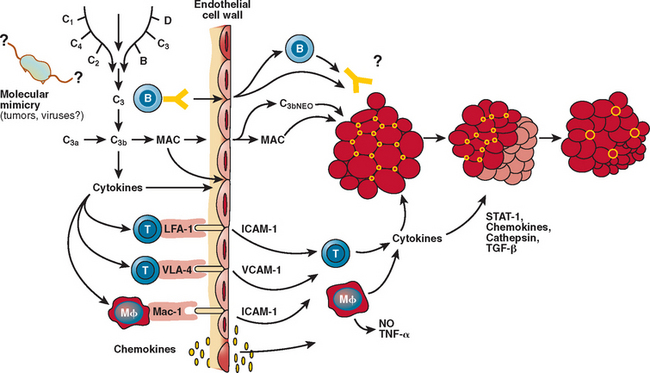
Polymyositis and Inclusion-Body Myositis
The immunopathogenesis in polymyositis is the same as in inclusion-body myositis. In these disorders, there is evidence for a MHC class I antigen-restricted process mediated by cytotoxic CD8+ T cells directed against muscle fibers.5 This concept is supported by several observations. First, endomysial T cells are cytotoxic to autologous myotubes in vitro and perforin granules are expressed in vivo and are released toward the muscle fibers they invade, leading to muscle fiber necrosis.41–43 Second, there is clonal expansion of the autoinvasive T cells, with restricted use of T cell receptor gene families.44–47 Third, costimulatory molecules are upregulated.43,48,49 The increased expression of costimulatory molecules on T cells, such as inducible costimulatory molecule and CD28, along with upregulation of their respective counterreceptors (inducible costimulatory ligand and BB1) on muscle fibers, suggests that muscle fibers in polymyositis and inclusion-body myositis can behave as antigen-presenting cells5,46 (Fig. 89-6). Although in both polymyositis and inclusion-body myositis the exact antigens that trigger the inflammatory process remain unknown, the presence of similar T cell clones in different muscles and their persistence over time in each patient suggest that the same antigenic stimuli may continue to drive the inflammatory response.5,44,45,47
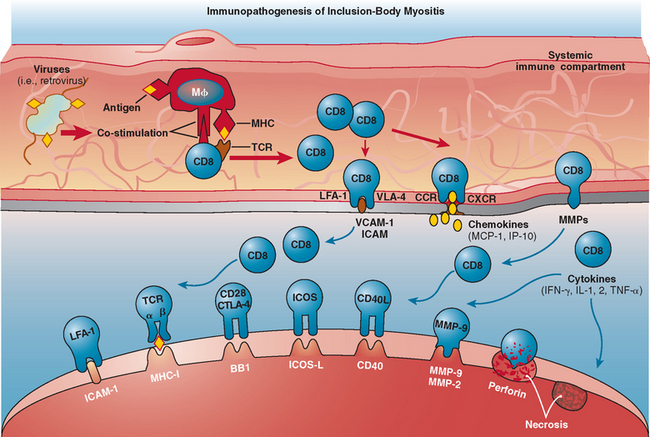
In inclusion-body myositis, in addition to the inflammatory process, a degenerative mechanism is taking place. This conclusion is based on the presence of vacuolated muscle fibers away from the inflammation and on the intracellular accumulation of various degeneration-associated molecules, such as amyloid, β-amyloid precursor protein, phosphorylated tau, ubiquitin, α1-antichymotrypsin, presenelin-1, prion proteins, and others.50 Although these features are characteristics of inclusion-body myositis, they are not specific to this disease. Similar vacuoles are observed in several other myopathies, such as hereditary inclusion-body myopathy, Emery-Dreifuss muscular dystrophy, rigid spine syndrome, and distal myopathies and even in chronic neurogenic conditions such as postpolio syndrome.9,32,51,52 In sporadic inclusion-body myositis, there may be a relationship between cytokines, amyloid, and chronic inflammation. The increase in cytokines such as interleukin 1β, their co-localization with β-amyloid precursor protein βAPP, and the ability of interleukin 1β and amyloid to upregulate the production of one another suggests an interaction between amyloid and inflammatory mediators.53 Preliminary observations at the mRNA level have shown a linear relationship between cytokines and amyloid-related molecules, such as tau and βAPP.54 In Alzheimer’s disease, the presence of strong β-amyloid–reactive and HLA-restricted T cell responses against β-amyloid 1–4255 suggest an immunogenic role for amyloid and its presentation to T cells by antigen-presenting cells.56 Whether in inclusion-body myositis the antigen-presenting cell–functioning muscle fibers are able to present amyloid and lead to an antigenspecific T cell response remains unclear.5 Recent findings suggest that thrombospondin-1 and its binding partners, CD36 and CD47, may also play a role in the inflammatory process of inclusion-body myositis and might provide a link between inflammation and degeneration.57 CD36 has been shown to be important in the uptake of antigen by immature dendritic cells,58 and it is a key player in the inflammatory response to β-amyloid in Alzheimer’s disease.59
Autoantibodies
Antibodies to nuclear or cytoplasmic constituents can be seen in patients with polymyositis, dermatomyositis, and inclusion-body myositis. Although some are classified as myositis specific, their specificity for these disorders has not been proved. Some of the myositis-associated autoantibodies seen in polymyositis, dermatomyositis, and, rarely, inclusion-body myositis include antisynthetase antibodies, such as anti–Jo-1, directed against various aminoacyl tRNA synthetases. With the possible exception of anti–Jo-1, which has a high specificity for the antisynthetase syndrome, none of the other antibodies are of sufficient specificity or sensitivity to be of diagnostic or prognostic value.60
TREATMENT
The goal of therapy in inflammatory myopathies is to improve the patient’s ability to carry out activities of daily living by increasing muscle strength and ameliorating the extramuscular manifestations of rash, dysphagia, dyspnea, arthralgia, and fever. Overall, dermatomyositis shows the best response to treatment, whereas inclusion-body myositis is the most resistant.1 Although immunosuppressive therapy may decrease the level of serum creatine kinase and increase strength, the reverse is not always true. Treatment decisions, therefore, should be based on clinical response and not on chasing the creatine kinase levels. It is also prudent to discontinue a particular drug if an adequate trial fails to cause an objective improvement in strength, regardless of the change in creatine kinase levels.9
Dermatomyositis and Polymyositis
Oral prednisone is the initial treatment of choice, at dosages of at least 1 mg/kg per day. Steroid treatment, as most other therapies, remains empirical because their efficacy has not been tested in a controlled study. After 3 to 4 weeks, prednisone is tapered slowly over a period of 10 weeks to 1 mg/kg every other day. It is further decreased, by 5 or 10 mg every 3 to 4 weeks, until the lowest possible dose sufficient to control the disease is attained.9 Most patients with dermatomyositis and polymyositis show a positive response to prednisone, as evidenced by an objective increase in muscle strength and in the activities of daily living.9 The failure of prednisone to provide such a benefit after 3 months of high-dose therapy should be taken as a sign of unresponsiveness to the drug. In these circumstances, tapering should be accelerated and consideration should be given to initiating therapy with another immunosuppressive agent or with intravenous immunoglobulin (IVIg).9 In acute cases, intravenous steroid, at a dose of 1 g/day, has been used for a faster response.
In most patients, immunosuppressive agents other than prednisone may also be required. The decision to initiate such therapy is based on the need for a steroid-sparing effect when, despite steroid responsiveness, the patients develop complications or show any of the following: resistance to steroid therapy, relapse of the disease with attempts at lowering the prednisone dose, or a rapidly progressive disease course with severe weakness and respiratory failure.1,9 The drugs most commonly used, but never tested in randomized trials, include the following:1,9
 Azathioprine, which is well tolerated and has few side effects. The maximum dose is 3 mg/kg daily, but requires 4-6 months to have an effect.
Azathioprine, which is well tolerated and has few side effects. The maximum dose is 3 mg/kg daily, but requires 4-6 months to have an effect.
 Mycophenolate mofetil is used at a dosage of 2 g/day and is emerging as a promising and well-tolerated drug.61
Mycophenolate mofetil is used at a dosage of 2 g/day and is emerging as a promising and well-tolerated drug.61

In our experience, the benefit of these drugs is mostly to maintain, rather than induce, a response. For this reason, if prednisone is not sufficient to control the disease, we recommend high-dose IVIg, which has been shown to improve strength and diminish the rash in controlled trials of patients with refractory dermatomyositis. Repeated infusions, every 6 to 8 weeks, are often needed to maintain improvement. A dose of 2 g/kg, divided over 2 to 5 days, is recommended per course. Uncontrolled observations suggest that IVIg is also beneficial for the management of patients with polymyositis.9 Plasmapheresis is not effective in polymyositis and dermatomyositis.62 Accordingly, we apply a following step-by-step approach to the treatment of polymyositis and dermatomyositis (Table 89-3). For difficult cases, newer agents such as tacrolimus or sirolimus may be used; for monoclonal antibody against CD20, rituximab may be considered.
Inclusion-Body Myositis
Inclusion-body myositis is generally resistant to immunosuppressive therapies. Prednisone, azathioprine, and methotrexate have been often tried for a few months in newly diagnosed patients, but the results have been generally disappointing. Occasionally, patients feel subjectively weaker after discontinuing these drugs, thus forcing the clinicians to maintain low-dose every-other-day prednisone or weekly methotrexate regimens even in the absence of objective evidence supporting this practice.9 Controlled trials have shown that IVIg does not offer a statistically significant improvement in inclusion-body myositis,63,64 although one third of the patients experience transient benefit.14 However, in our controlled study as well as in other uncontrolled observations, IVIg has shown to be effective in transiently improving swallowing function.9,65 Therefore, IVIg can be tried for 2 or 3 months in selected patients who exhibit significant dysphagia.
Associated Clinical Manifestations
Various immunosuppressive medications have been suggested for the treatment of interstitial lung disease, although a standard regimen has not been established. Oral prednisone can result in improved pulmonary function, particularly when used at an early inflammatory stage. High-dose intravenous pulse methylprednisolone, given as 1 g of methylprednisolone daily for 3 consecutive days, has been used in patients who do not respond to oral prednisone or during acute relapses. In corticosteroid-resistant patients with interstitial lung disease, other immunosuppressive agents have been used, including cyclophosphamide, azathioprine, cyclosporine, and tacrolimus.17
DIFFERENTIAL DIAGNOSIS
The typical skin rash of dermatomyositis, along with the presence of subacute onset of proximal muscle weakness, very rarely raises confusion about the correct diagnosis. The skin rash of lupus is different from that of dermatomyositis, and if a myopathy occurs in patients with lupus, it is usually multifactorial and occurs in patients with a known disease. The skin changes of scleroderma may overlap with those of dermatomyositis, and scleroderma is the main disease from which dermatomyositis needs to be distinguished.9
PROGNOSIS
Although the outcome of polymyositis and dermatomyositis has improved, at least one third of patients are left with mild to severe disability. Dermatomyositis responds more favorably to therapy than does polymyositis and has a better prognosis. Most patients improve with therapy, and many make a full functional recovery, which is often sustained with maintenance therapy, although relapses may occur at any time.9 A small cohort study showed a 5-year survival of 95% and a 10-year survival of 84%.66,67 Death is usually due to pulmonary, cardiac, or other systemic complications.9 Older patients, those with associated cancer, severely affected patients, or those not treated early and effectively carry a poorer prognosis.1,9 Pulmonary fibrosis, frequent aspiration pneumonias due to esophageal dysfunction, and extensive calcinosis in dermatomyositis are associated with increased morbidity.66,67
Of children with juvenile dermatomyositis, 34% to 40% have a monocyclic disease, and after an acute course that resolves within a 2-year period, they enter into indefinite remission.68 The remaining patients have a chronic disease that can be progressive or characterized by remissions and exacerbations (polycyclic course), and may require chronic immunosuppressive therapy.69
Inclusion-body myositis has the least favorable prognosis, and most patients will require assistive devices such as canes, walkers, or wheelchairs within 5 to 10 years of onset. In general, the older the age of onset in inclusion-body myositis, the more rapidly progressive the course of the disease.9
Amato AA, Griggs RC. Treatment of idiopathic inflammatory myopathies. Curr Opin Neurol. 2003;16:569-575.
Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982.
Dalakas MC. Inflammatory disorders of muscle: progress in polymyositis, dermatomyositis and inclusion body myositis. Curr Opin Neurol. 2004;17:561-567.
Kissel JT. Misunderstandings, misperceptions, and mistakes in the management of the inflammatory myopathies. Semin Neurol. 2002;22:41-51.
Mastaglia FL, Garlepp MJ, Phillips BA, Zilko PJ. Inflammatory myopathies: clinical, diagnostic and therapeutic aspects. Muscle Nerve. 2003;27:407-425.
1 Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982.
2 Dalakas MC. Polymyositis, dermatomyositis and inclusion-body myositis. N Engl J Med. 1991;325:1487-1498.
3 Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am. 2002;28:723-741.
4 Dalakas MC. Muscle biopsy findings in inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:779-798. vi.
5 Dalakas MC. Inflammatory disorders of muscle: progress in polymyositis, dermatomyositis and inclusion body myositis. Curr Opin Neurol. 2004;17:561-567.
6 Amato AA, Griggs RC. Unicorns, dragons, polymyositis, and other mythological beasts. Neurology. 2003;61:288-289.
7 Ramanan AV, Feldman BM. Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin North Am. 2002;28:833-857.
8 Pachman LM. Juvenile dermatomyositis: immunogenetics, pathophysiology, and disease expression. Rheum Dis Clin North Am. 2002;28:579-602. vii.
9 Dalakas MC. Polymyositis, dermatomyositis and inclusion body myositis. In Braunwald E, Fauci AS, Kasper DL, editors: Harrison’s Principles of Internal Medicine, 16th ed., New York: McGraw-Hill, 2004.
10 Sontheimer RD. Dermatomyositis: an overview of recent progress with emphasis on dermatologic aspects. Dermatol Clin. 2002;20:387-408.
11 Dalakas MC. Images in clinical medicine. Calcifications in dermatomyositis. N Engl J Med. 1995;333:978.
12 Fisler RE, Liang MG, Fuhlbrigge RC, et al. Aggressive management of juvenile dermatomyositis results in improved outcome and decreased incidence of calcinosis. J Am Acad Dermatol. 2002;47:505-511.
13 Sekul EA, Dalakas MC. Inclusion body myositis: new concepts. Semin Neurol. 1993;13:256-263.
14 Dalakas MC. Progress in inflammatory myopathies: good but not good enough. J Neurol Neurosurg Psychiatry. 2001;70:569-573.
15 Kao AH, Lacomis D, Lucas M, et al. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004;50:209-215.
16 Douglas WW, Tazelaar HD, Hartman TE, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med. 2001;164:1182-1185.
17 Hirakata M, Nagai S. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol. 2000;12:501-508.
18 Imbert-Masseau A, Hamidou M, Agard C, et al. Antisynthetase syndrome. Joint Bone Spine. 2003;70:161-168.
19 Sigurgeirsson B, Lindelof B, Edhag O, et al. Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med. 1992;326:363-367.
20 Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
21 Callen JP. When and how should the patient with dermatomyositis or amyopathic dermatomyositis be assessed for possible cancer? Arch Dermatol. 2002;138:969-971.
22 Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292:344-347.
23 Sekul EA, Chow C, Dalakas MC. Magnetic resonance imaging of the forearm as a diagnostic aid in patients with sporadic inclusion body myositis. Neurology. 1997;48:863-866.
24 Kane D, Grassi W, Sturrock R, et al. Musculoskeletal ultra-sound—a state of the art review in rheumatology. Part 2: clinical indications for musculoskeletal ultrasound in rheumatology. Rheumatology (Oxford). 2004;43:829-838.
25 Hilton-Jones D. Inflammatory muscle diseases. Curr Opin Neurol. 2001;14:591-596.
26 Civatte M, Schleinitz N, Krammer P, et al. Class I MHC detection as a diagnostic tool in noninformative muscle biopsies of patients suffering from dermatomyositis (dermatomyositis). Neuropathol Appl Neurobiol. 2003;29:546-552.
27 Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. I: quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol. 1984;16:193-208.
28 Engel AG, Arahata K. Mononuclear cells in myopathies: quantitation of functionally distinct subsets, recognition of antigenspecific cell-mediated cytotoxicity in some diseases, and implications for the pathogenesis of the different inflammatory myopathies. Hum Pathol. 1986;17:704-721.
29 Karpati G, Pouliot Y, Carpenter S. Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol. 1988;23:64-72.
30 Mendell JR, Sahenk Z, Gales T, et al. Amyloid filaments in inclusion body myositis. Novel findings provide insight into nature of filaments. Arch Neurol. 1991;48:1229-1234.
31 Askanas V, Engel WK, Alvarez RB. Enhanced detection of Congo-red-positive amyloid deposits in muscle fibers of inclusion body myositis and brain of Alzheimer’s disease using fluorescence technique. Neurology. 1993;43:1265-1267.
32 Semino-Mora C, Dalakas MC. Rimmed vacuoles with beta-amyloid and ubiquitinated filamentous deposits in the muscles of patients with long-standing denervation (postpoliomyelitis muscular atrophy): similarities with inclusion body myositis. Hum Pathol. 1998;29:1128-1133.
33 Santorelli FM, Sciacco M, Tanji K, et al. Multiple mitochondrial DNA deletions in sporadic inclusion body myositis: a study of 56 patients. Ann Neurol. 1996;39:789-795.
34 Askanas V, Engel WK. Inclusion-body myositis: newest concepts of pathogenesis and relation to aging and Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:1-14.
35 Shamim EA, Rider LG, Miller FW. Update on the genetics of the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 2000;12:482-491.
36 Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314:329-334.
37 Kissel JT. Misunderstandings, misperceptions, and mistakes in the management of the inflammatory myopathies. Semin Neurol. 2002;22:41-51.
38 Greenberg SA, Pinkus JL, Pinkus GS, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664-678.
39 Amemiya K, Semino-Mora C, Granger RP, et al. Downregulation of TGF-beta1 mRNA and protein in the muscles of patients with inflammatory myopathies after treatment with high-dose intravenous immunoglobulin. Clin Immunol. 2000;94:99-104.
40 Figarella-Branger D, Civatte M, et al. Cytokines, chemokines, and cell adhesion molecules in inflammatory myopathies. Muscle Nerve. 2003;28:659-682.
41 Hohlfeld R, Engel AG. The immunobiology of muscle. Immunol Today. 1994;15:269-274.
42 Goebels N, Michaelis D, Engelhardt M, et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest. 1996;97:2905-2910.
43 Schmidt J, Rakocevic G, Raju R, et al. Upregulated inducible co-stimulator (ICOS) and ICOS-ligand in inclusion body myositis muscle: significance for CD8+ T cell cytotoxicity. Brain. 2004;127:1182-1190.
44 Hofbauer M, Wiesener S, Babbe H, et al. Clonal tracking of autoaggressive T cells in polymyositis by combining laser microdissection, single-cell PCR, and CDR3-spectratype analysis. Proc Natl Acad Sci U S A. 2003;100:4090-4095.
45 Amemiya K, Granger RP, Dalakas MC. Clonal restriction of T-cell receptor expression by infiltrating lymphocytes in inclusion body myositis persists over time. Studies in repeated muscle biopsies. Brain. 2000;123(Pt 10):2030-2039.
46 Benveniste O, Cherin P, Maisonobe T, et al. Severe perturbations of the blood T cell repertoire in polymyositis, but not dermatomyositis patients. J Immunol. 2001;167:3521-3529.
47 Benveniste O, Herson S, Salomon B, et al. Longterm persistence of clonally expanded T cells in patients with polymyositis. Ann Neurol. 2004;56:867-872.
48 Murata K, Dalakas MC. Expression of the costimulatory molecule BB-1, the ligands CTLA-4 and CD28, and their mRNA in inflammatory myopathies. Am J Pathol. 1999;155:453-460.
49 Wiendl H, Mitsdoerffer M, Schneider D, et al. Muscle fibres and cultured muscle cells express the B7.1/2-related inducible costimulatory molecule, ICOSL: implications for the pathogenesis of inflammatory myopathies. Brain. 2003;126:1026-1035.
50 Askanas V, Engel WK. Proposed pathogenetic cascade of inclusion-body myositis: importance of amyloid-beta, misfolded proteins, predisposing genes, and aging. Curr Opin Rheumatol. 2003;15:737-744.
51 Fidzianska A, Kaminska A. Congenital myopathy with abundant ring fibres, rimmed vacuoles and inclusion body myositis-type inclusions. Neuropediatrics. 2003;34:40-44.
52 Fidzianska A, Rowinska-Marcinska K, Hausmanowa-Petrusewicz I. Coexistence of X-linked recessive Emery-Dreifuss muscular dystrophy with inclusion body myositis-like morphology. Acta Neuropathol (Berl). 2004;107:197-203.
53 Dalakas MC. Molecular immunology and genetics of inflammatory muscle diseases. Arch Neurol. 1998;55:1509-1512.
54 Schmidt J, Raju R, Salajegheh M, Rakocevic G, et al. Distinct interplay between inflammatory and degeneration-associated molecules in sporadic inclusion body myositis (sIBM). Neurology (suppl 1). 2005;64:A337-A338.
55 Monsonego A, Zota V, Karni A, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112:415-422.
56 Monsonego A, Weiner HL. Immunotherapeutic approaches to Alzheimer’s disease. Science. 2003;302:834-838.
57 Salajegheh M, Raju R, Schmidt J, et al. Thrombospondin-1 (TSP1) and its binding partners CD36 and CD47, as mediators of inflammation in sporadic inclusion body myositis (sIBM). Neurology. 2005;Suppl 1:64. A158.
58 Albert ML, Pearce SF, Francisco LM, et al. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998;188:1359-1368.
59 El Khoury JB, Moore KJ, Means TK, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657-1666.
60 Mastaglia FL, Garlepp MJ, Phillips BA, et al. Inflammatory myopathies: clinical, diagnostic and therapeutic aspects. Muscle Nerve. 2003;27:407-425.
61 Chaudhry V, Cornblath DR, Griffin JW, et al. Mycophenolate mofetil: a safe and promising immunosuppressant in neuromuscular diseases. Neurology. 2001;56:94-96.
62 Miller FW, Leitman SF, Cronin ME, et al. Controlled trial of plasma exchange and leukapheresis in polymyositis and dermatomyositis. N Engl J Med. 1992;326:1380-1384.
63 Dalakas MC. The use of intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: evidence-based indications and safety profile. Pharmacol Ther. 2004;102:177-193.
64 Walter MC, Lochmuller H, Toepfer M, et al. High-dose immunoglobulin therapy in sporadic inclusion body myositis: a double-blind, placebo-controlled study. J Neurol. 2000;247:22-28.
65 Cherin P, Pelletier S, Teixeira A, et al. Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology. 2002;58:326.
66 Marie I, Hachulla E, Hatron PY, et al. Polymyositis and dermatomyositis: short term and longterm outcome, and predictive factors of prognosis. J Rheumatol. 2001;28:2230-2237.
67 Sultan SM, Ioannou Y, Moss K, et al. Outcome in patients with idiopathic inflammatory myositis: morbidity and mortality. Rheumatology (Oxf). 2002;41:22-26.
68 Rennebohm R. Juvenile dermatomyositis. Pediatr Ann. 2002;31:426-433.
69 Wargula JC. Update on juvenile dermatomyositis: new advances in understanding its etiopathogenesis. Curr Opin Rheumatol. 2003;15:595-601.