Inflammatory Disorders of the Stomach
Richard H. Lash
Gregory Y. Lauwers
Robert D. Odze
Robert M. Genta
Historical Perspective
In 1947, at the dawn of gastroscopy, Rudolf Schindler deemed gastritis “one of the most debated diseases of the human body” and predicted that its significance would be discussed “for some time to come.”1 From the mid-1800s, when Cruveiller exposed the inaccuracies of Broussais’s first descriptions of gastritis in autopsy material, to the early 20th century, the concept of gastritis as a disease had been virtually abandoned.2
After gastritis was acknowledged as a distinct entity, the search for its cause began. Since 1870, tiny, curved bacteria within gastric mucosa have been described by human and veterinary pathologists, but the organisms were dismissed as irrelevant contaminants.3 Schindler claimed that the bacteriologic cause of chronic gastritis had not been proved convincingly in a single case.1 Instead, a wide range of etiologic theories were proposed, such as improper mastication, a “coarse” or “miserable” diet, alcohol, caffeine, nicotine, condiments and spices, drugs, heavy metals, thermal injury, chronic infections of tonsils and sinuses, circulatory disturbances, and psychogenic factors. Not surprisingly, researchers were successful in debunking theories put forth by their colleagues but were quite unsuccessful in proving their own.
Subsequently, accurate morphologic data were gathered by pathologic examination of autopsy material4 and from endoscopic biopsy specimens. Distinct types and patterns of gastritis were recognized, which led to the conception, presentation, dismissal, and replacement of many classification systems. Some systems were based on solid morphologic information and proposed valid clinicopathologic associations (e.g., peptic ulcer and gastric cancer), but the lack of therapeutic implications reduced almost all classifications to little more than an academic exercise.
In 1984, when Warren and Marshall proposed that chronic idiopathic gastritis had a bacterial cause (i.e., Helicobacter pylori),5 it was not surprising that their hypothesis was met with skepticism. However, within a few years, the associations of H. pylori gastritis, peptic ulcer, and gastric cancer were recognized and, ultimately, accepted.6 In 1990, guidelines for the classification and grading of gastritis were developed by a group of investigators in Sydney, Australia. The Sydney System correlates topographic, morphologic, and etiologic information with clinically useful diagnoses.7
Four years after its introduction, the Sydney System was updated by an international group of pathologists who established consensus terminology for gastritis, improved the guidelines for histologic grading, and streamlined the diagnostic process.8 Subsequently, modifications were made to improve the criteria for evaluation of atrophy.9,10 In 2006, an international consensus was reached on a diagnostic reporting system designed to use the staging of gastritis as a tool for assessing the risk of gastric cancer.11–15 This chapter provides the terminology and diagnostic approach proposed by the updated Sydney System and its subsequent modifications.
General Pathologic Features of Gastritis
Injury to the gastric mucosa elicits a variety of inflammatory and reactive responses that depend on the type, location, and duration of injury. Various degrees of tissue responses may occur synchronously or metachronously. The correct diagnosis of gastritis rests on the pathologic recognition of the types of tissue responses, their intensity, and their location. To recognize pathologic tissue responses, it is essential to understand the spectrum of the histologic appearance of normal gastric mucosa (Figs. 15.1 and 15.2; Table 15.1). The following sections review the normal histologic variability and common pathologic tissue reactions of gastric mucosa.
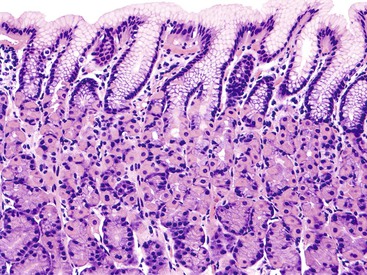
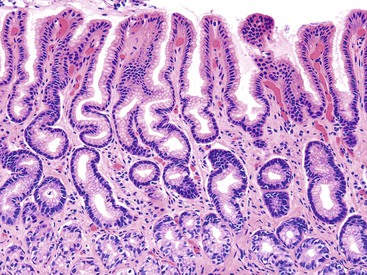
Table 15.1
Normal and Pathologic Histology of the Stomach
| Histopathologic Component | Normal Setting | Pathologic Settings |
| Neutrophils | Rare in lamina propria Not found in normal epithelium |
In active gastritis (Helicobacter pylori) and near erosion or ulcer |
| Mononuclear cells | Scattered isolated lymphocytes and plasma cells in lamina propria (antrum > corpus), increase with age None or rare cells in epithelium |
In chronic gastritis (H. pylori, autoimmune) and lymphocytic gastritis (if intraepithelial) |
| Lymphoid aggregates | Rare aggregates, basally located in oxyntic mucosa, without germinal centers | In H. pylori infection |
| Lymphoid follicles | None | In H. pylori infection |
| Eosinophils | Scattered in lamina propria, increase with age Not present in normal epithelium |
Moderate increase in H. pylori gastritis Severe with clusters or intraepithelial in eosinophilic gastroenteritis |
| Edema of the lamina propria | None | In chemical injury, particularly bile reflux |
| Hyperemia, congestion | None | In any form of active inflammation (H. pylori), chemical injury, or vasculopathies (e.g., GAVE) |
| Surface epithelial degeneration | None Regular, tall cuboidal cells with distinct apical mucin droplet is normal |
In H. pylori infection, chemical injury |
| Erosions | None | In chemical injury (flat, not inflamed); H. pylori infection (elevated, inflamed) |
| Foveolar hyperplasia | None | In chronic chemical injury; mucosa adjacent to ulcer; H. pylori infection |
| Intestinal metaplasia | None | Antrum: H. pylori infection; chemical injury Corpus and fundus: multifocal atrophic gastritis, autoimmune gastritis, H. pylori infection |
| Atrophy | None Antrum: orderly pits separated by little matrix, no fibrosis Corpus and fundus: parallel, tightly packed oxyntic glands reaching muscularis mucosae |
Antrum: H. pylori infection (rare without intestinal metaplasia) Corpus and fundus: multifocal atrophic gastritis, autoimmune gastritis, H. pylori infection |
| Endocrine cell hyperplasia | None; no obvious G cells on H&E staining; no clusters or nests | With chronic PPI therapy; atrophy, particularly autoimmune gastritis |
| Parietal cell alterations | No prominent protruding parietal cells; no lumen or dilations in oxyntic glands | With chronic PPI therapy |
| Interfoveolar smooth muscle hyperplasia | Scattered fibers between antral foveolae; no bundles | In chronic chemical injury, GAVE |
GAVE, Gastric antral vascular ectasia; H&E, hematoxylin and eosin stain; PPI, proton pump inhibitor.
Neutrophil Infiltration
The lamina propria of normal gastric mucosa may contain a few scattered neutrophils. However, infiltration of the epithelium by neutrophils represents a pathologic tissue response and is considered an active component of gastritis. The term active is used to indicate a sustained release of inflammatory mediators and a persistent cause for this process. In the United States, neutrophilic infiltration of the epithelium is most commonly caused by H. pylori gastritis, but other infectious and inflammatory conditions (e.g., syphilis,16 Crohn’s disease17) may cause neutrophil infiltration.
If sufficient numbers of biopsy specimens are examined, neutrophils are detected in almost all patients with H. pylori infection.18–20 Evaluation of the intensity of neutrophil infiltration may help to differentiate the various types of gastritis. For instance, the acute phase of infectious gastritis (i.e., phlegmonous gastritis) may have abundant neutrophils, and the active phase of Helicobacter-induced gastritis may have moderate to severe levels of neutrophils. Acute hemorrhagic gastritis resulting from nonsteroidal antiinflammatory drugs (NSAIDs)– or alcohol-induced chemical injury, often shows only minimal neutrophil infiltration, unless ulceration exists.
Mononuclear Infiltration
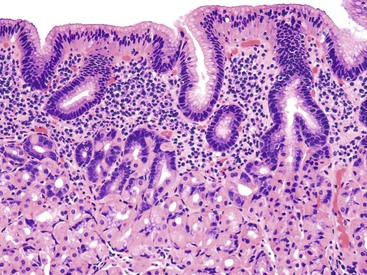
Normal antral mucosa contains few mononuclear inflammatory cells, whereas normal corpus mucosa contains virtually none. However, the density of mononuclear cells (i.e., lymphocytes and plasma cells) in the lamina propria of normal persons varies considerably according to their geographic location and ethnicity.21 To properly gauge the presence of gastritis, pathologists should set a standard for normal that is relevant to the patient population in their geographic region. Scattered lymphocytes and plasma cells in the lamina propria of the antrum are normal in individuals from most countries (see Fig. 15.2). In the absence of established norms for the particular geographic region, it is recommended that gastritis not be diagnosed unless there are at least several clusters of five or more mononuclear cells in the lamina propria or the infiltrate is diffuse. In the corpus and fundus, rare mononuclear cells in the lamina propria are considered a normal finding. Under normal conditions, intraepithelial lymphocytes are not found anywhere in the gastric mucosa.
Infiltration of the mucosa with lymphocytes, plasma cells, and a number of eosinophils and mast cells is characteristic of chronic H. pylori gastritis. In autoimmune gastritis, the infiltrate is often diffuse, consists mainly of plasma cells and lymphocytes,22 and usually extends to the deep portions of the mucosa, and the corpus and fundus of the stomach are selectively involved.
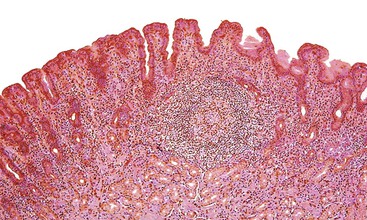
Lymphoid Aggregates and Follicles
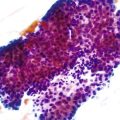
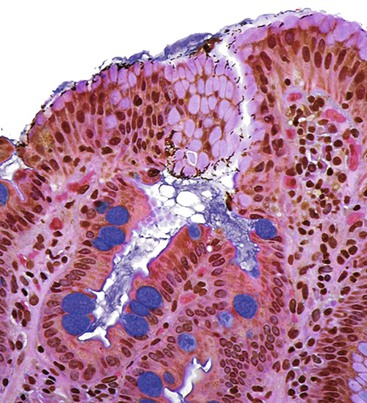
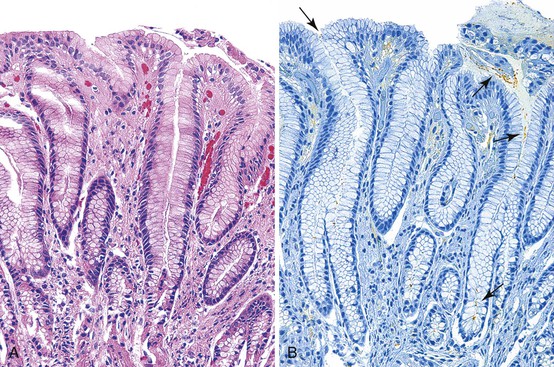
Normal gastric mucosa, particularly the corpus, may contain occasional, small lymphoid aggregates, usually located close to the muscularis mucosae at the base of the lamina propria. In contrast, lymphoid aggregates with germinal centers (i.e., follicles) are rare in gastric mucosa of normal, H. pylori–negative adults.23 In studies that have used an extensive standardized biopsy protocol, lymphoid follicles or aggregates have been detected in almost all individuals with H. pylori gastritis. The finding of lymphoid follicles is highly specific for H. pylori infection (Fig. 15.3). In H. pylori–infected children and young adults, lymphoid follicles may be quite numerous, which may produce an endoscopic appearance of nodularity that is often referred to as follicular gastritis.24,25
Eosinophil Infiltration
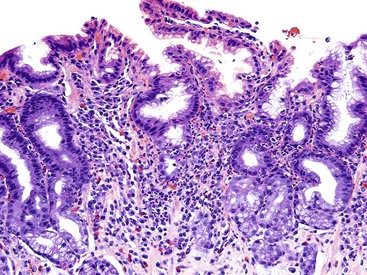
Rare, scattered eosinophils may be seen in the gastric mucosa of normal healthy patients, particularly in individuals who live in a suboptimal public health environment. A prominent eosinophilic infiltration usually represents a pathologic process, such as eosinophilic gastritis or gastroenteritis.26,27 Eosinophil infiltration may occur in a wide variety of disorders, such as gastric anisakiasis and other granulomatous or parasitic infections of the stomach.28
In adults with H. pylori gastritis, a usually mild eosinophil infiltration often composes a portion of the inflammatory infiltrate. A greater eosinophilic component of the inflammatory infiltrate may manifest in children with H. pylori infection (Fig. 15.4).29 After H. pylori eradication, eosinophils may persist for a long time, similar to mononuclear cells.18
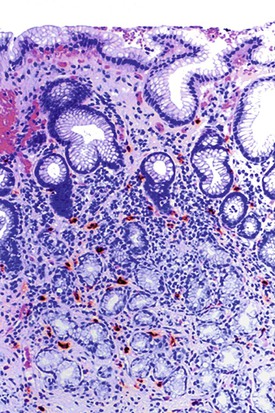
Mucosal Hyperemia and Edema
Mucosal hyperemia (congestion) and edema are often endoscopic or pathologic indicators of chemical injury. In one study, a significant correlation was found between the degree of hyperemia and the concentration of bilirubin within gastric fluid.30 However, congestion and edema may also be features of H. pylori gastritis, which may be caused by an increase in the number of mast cells (Fig. 15.5) that occurs in this condition.31,32
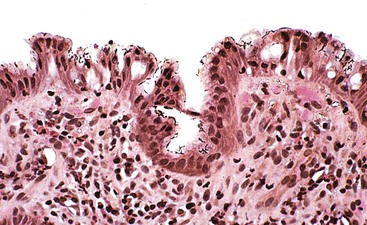
Surface Epithelium Degeneration
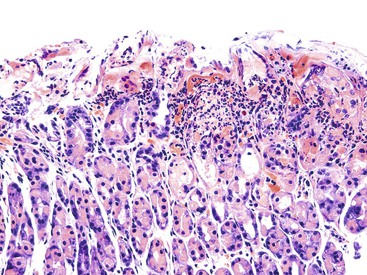
Surface epithelium degeneration is considered a nonspecific response to injury that occurs to some degree in all forms of gastritis. However, degenerative changes are particularly conspicuous in two disorders: chemical gastritis (resulting from bile reflux, ethanol, or NSAIDs) and H. pylori gastritis.33–35 Regardless of the cause, epithelial injury and necrosis lead to surface erosions. Degenerative changes may result in the appearance of cuboidal (rather than columnar) cells and mucin depletion. The former is particularly common in gastritis occurring after Billroth II surgery and thought to result from bile reflux. However, these changes may also be found in H. pylori gastritis, even in areas where bacteria are few in number. Alternatively, epithelial regeneration, a common feature of H. pylori gastritis, may result in the accumulation of buds of cells at the surface of the mucosa (Fig. 15.6).36
Surface Erosion
Surface erosions are the result of severe epithelial injury and necrosis. By definition, erosions do not extend beyond the muscularis mucosae. Endoscopically flat surface erosions often result from the acute effects of drugs, alcohol, bile reflux, or ischemia (Fig. 15.7).37,38 Endoscopically elevated surface erosions are typically associated with H. pylori gastritis. These erosions are usually characterized by a superficial layer of fibrinopurulent debris (i.e., fibrinoid necrosis, neutrophils, and cellular debris) with hyperplastic, regenerative-appearing epithelium at the margins (Fig. 15.8).39
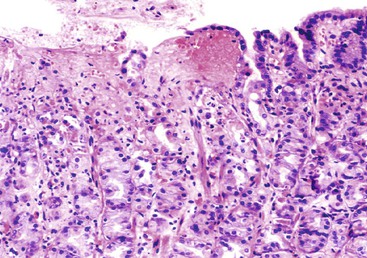
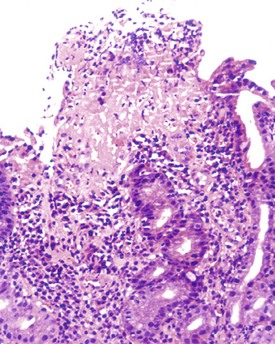
Foveolar Hyperplasia
Foveolar hyperplasia is defined as proliferation, elongation, and tortuosity of gastric pits that result in a corkscrew configuration. It is a compensatory tissue response to increased exfoliation of the surface epithelium and represents a visual surrogate for increased cell proliferation and turnover (Fig. 15.9). Although markedly hyperplastic foveolae are normally recognized microscopically, minimal or mild foveolar hyperplasia can easily be overlooked. True foveolar hyperplasia may be diagnosed reliably when more than four cross-sections of the same gastric pit are visualized in a single, well-oriented gastric biopsy specimen.40,41 Other features of foveolar hyperplasia include hyperchromatic nuclei, cells with a high nucleus-to-cytoplasm ratio, upper pit mitoses, mucin depletion, and cuboidalization of the epithelial cells.
Foveolar hyperplasia is a characteristic and prominent feature of bile reflux gastritis30 and NSAID gastropathy, particularly in long-term users.30,40,41 A mild degree of foveolar hyperplasia is common in patients with H. pylori gastritis; however, marked hyperplasia indicates coexistent chemical injury (Fig. 15.10).42
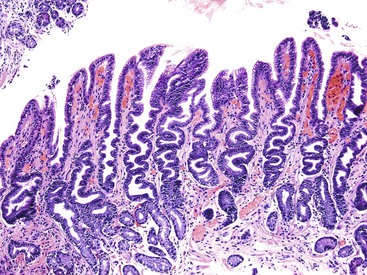
Intestinal Metaplasia
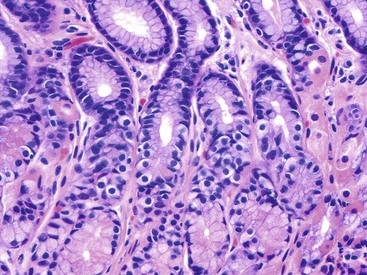
Intestinal metaplasia is defined as the replacement of gastric-type mucinous epithelial cells with small intestinal cells (e.g., goblet cells, enterocytes) (Fig. 15.11).43 Two types of intestinal metaplasia (I and II) have been described but are of limited clinical importance. Metaplastic epithelium that closely resembles normal small intestinal epithelium, containing acid mucin–producing goblet cells and absorptive enterocytes with a brush border, is considered complete metaplasia (type I). Incomplete metaplasia (type II) shows a disorderly mixture of irregularly shaped goblet cells (intestinal and immature intermediate mucous cells) that contain acidic sialomucins and sulfomucins. Incomplete metaplasia is further subdivided into type IIa and type IIb by the finding of sulfomucins within nongoblet (mucinous) cells in the latter.44 Although the incomplete forms of intestinal metaplasia (especially type IIb, also unfortunately referred to as type III) are associated with an increased risk of gastric cancer, the practical application of defining the precise type of metaplasia in any single individual is limited because most patients have a mixture of the incomplete and complete types when extensive areas of mucosa are sampled.43 However, because the degree of incomplete intestinal metaplasia (type IIb) parallels the extent of intestinal metaplasia in general, there is a positive correlation between the degree of any type of intestinal metaplasia and the risk of progression to carcinoma.
Intestinal metaplasia can be identified and its extent evaluated with the use of specific mucin histochemical stains. For instance, the Alcian blue/periodic acid–Schiff (AB/PAS) stain at pH 2.5 is an excellent method for demonstrating the type and extent of intestinal metaplasia, particularly when used in combination with a hematoxylin counterstain (Fig. 15.12). Mucin histochemical stains (e.g., high-iron diamine) that were traditionally used to determine the specific type of intestinal metaplasia by detecting sulfated mucins have been largely replaced by immunohistochemical stains that identify proteins associated with particular mucin-encoding genes. Although more than 20 such mucin genes have been identified, only a few (i.e., MUC1, MUC2, MUC5AC, and MUC6) are used routinely in practice, and even those are used mainly in research settings (Table 15.2).
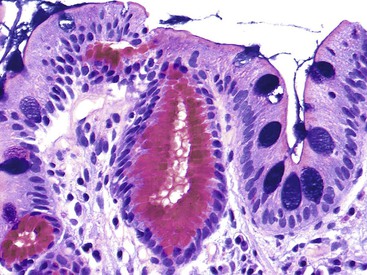
Table 15.2
Mucin Immunohistochemistry
| Mucin Expressed* | Normal Gastric Epithelium | Intestinal Metaplasia Type I | Intestinal Metaplasia Type II | Intestinal Metaplasia Type III | |||
| G | C | G | C | G | C | ||
| MUC1 | ++ (foveolar epithelium; chief and parietal cells) | − | − | ++ | ++ | +++ | +++ |
| MUC2 | − | +++ | − | ++ | + | ++ | ± |
| MUC5AC | +++ (foveolar epithelium; all mucous neck cells) | − | − | ++ | ++ | ++ | ++ |
| MUC6 | + (antral glands; mucopeptic cells of neck zone in corpus) | − | − | − | ± | ± | ± |

* Expression of the most common mucin proteins in the normal stomach and the three types of intestinal metaplasia is detected by immunohistochemical staining. Because the intensity of the staining and the percentages of cells assessed as positive vary and interpretation is highly subjective, the use of these stains to determine the type of intestinal metaplasia and any inference of cancer risk in individual patients is strongly discouraged.
C, Columnar or absorptive cells; G, goblet cells.
Intestinal metaplasia may develop in a variety of pathologic settings, but it usually indicates an underlying chronic atrophic gastritis (chronic atrophic gastritis does not equal autoimmune chronic atrophic gastritis). Several studies have shown that intestinal metaplasia occurs more frequently in patients with H. pylori gastritis.45,46 Because the H. pylori organism does not normally adhere to intestinal-type epithelium (see Fig. 15.11) but disappears in mucosa with extensive intestinal metaplasia and atrophy, one theory posits that intestinal metaplasia represents a host defense against H. pylori infection. Intestinalized epithelium may provide additional defense against H. pylori through changes in the composition of the gastric mucus resulting from metaplasia. Alternatively, metaplasia may represent a type of physiologic adaptation to altered bacterial flora; for instance, bacterial overgrowth may underlie the development of intestinal metaplasia in late-stage autoimmune gastritis, and sulfomucins are more resistant than other mucin types to bacterial enzyme–related degradation.
Intestinal metaplasia is frequently found in the corpus of patients with autoimmune gastritis, and foci of intestinal metaplasia are common in patients with reactive gastropathy after Billroth II surgery.41 Metaplasia is found in individuals with an otherwise completely normal stomach. The biologic significance of intestinal metaplasia remains poorly understood.
Atrophy
Gastric atrophy is defined as a loss of gastric glands.10,47 Atrophy is a histologic finding, not a nosologic entity. When gastric mucosa is damaged, regardless of the cause, it may regenerate to normal (restitutio ad integrum) or undergo an adaptive reparative change that leads to the replacement of native glands with other types of tissue.48 When injured glands fail to regenerate, the stromal space they previously occupied within the lamina propria may be replaced by fibroblasts and extracellular matrix. The result of this process is loss of functional epithelium (i.e., atrophy).
The native glands may be replaced by those with a pyloric phenotype (i.e., pyloric or pseudopyloric metaplasia) (Fig. 15.13) or an intestinal phenotype (i.e., intestinal metaplasia), comprising goblet cells and absorptive cells (with or without a brush border). It is important to differentiate true atrophy from mimics, including normal antrum-body transition mucosa, in which pyloric-type glands may be mixed with oxyntic glands at the base of the mucosa, and normal fundus-cardia transition mucosa, in which mucous-type glands may be associated with oxyntic glands.
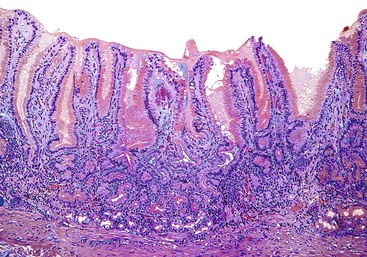
Diffuse fundic atrophy typically occurs in autoimmune gastritis as a consequence of immune-mediated destruction of oxyntic epithelium. Less severe and more focal atrophy, which is usually limited to the antrum, may also occur in reactive gastropathy (Fig. 15.14). The updated Sydney System recognizes multifocal atrophic gastritis (MAG), referred to as environmental atrophic gastritis in the pre–H. pylori era, as an entity distinct from nonatrophic gastritis and autoimmune atrophic gastritis.8,49
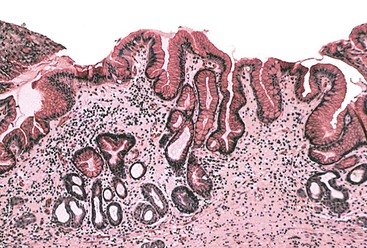
The border between gastritis with focal atrophy and atrophic gastritis has not been well defined (Fig. 15.15). This issue is important because scattered foci of intestinal metaplasia are found in the antrum of most patients with H. pylori gastritis and of a small percentage of noninfected adults. It is not appropriate to classify these individuals with atrophic gastritis, a diagnosis that implies altered gastric function and an increased risk of cancer. In the absence of established guidelines, we suggest that a diagnosis of MAG should be made only when there is evidence of atrophy with or without intestinal metaplasia in at least 50% of a properly biopsied stomach (i.e., minimum of two samples from the antrum and two from the corpus or fundus).
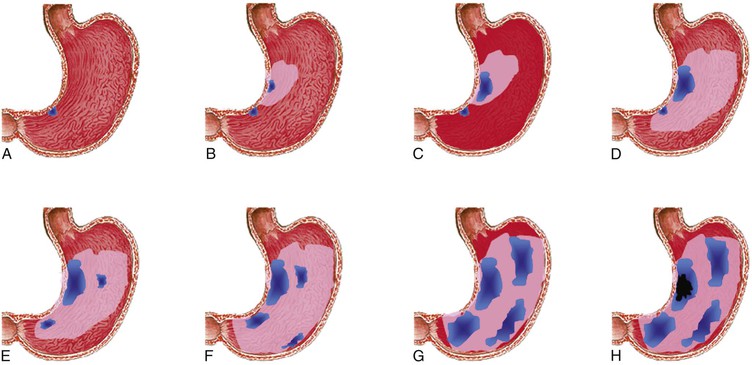
Several pathology workshops have been devoted to developing a reproducible method for grading atrophy in mucosal biopies.9,10 Pathologists have recommended that atrophy be evaluated according to its two subtypes: nonmetaplastic or metaplastic. The subtype known as nonmetaplastic atrophy is an area of mucosa with true glandular depletion (relative to the normal complement for that site) with or without fibrosis. The subtype known as metaplastic atrophy is replacement of native glands by intestinal-type glands anywhere in the stomach or pyloric (pseudopyloric) glands in the corpus (i.e., metaplasia equals atrophy). In both types of atrophy, the degree of gland loss may be graded as mild, moderate, or severe, which corresponds to a scale of 1 to 3 (Fig. 15.16).
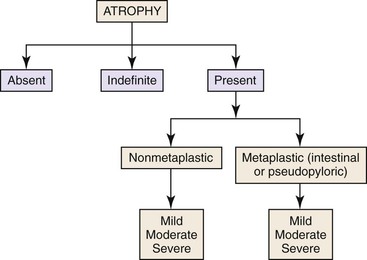
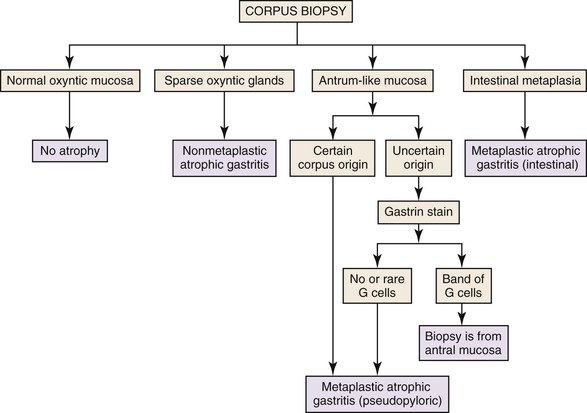
Problems may arise when gastric biopsies are labeled as “stomach” or “antrum-body,” in which case it may be difficult to determine whether there is true pyloric metaplasia or the biopsy is simply representative of the antrum or antral-fundic transition. In this instance, immunohistochemical staining for gastrin-positive endocrine cells can help to identify mucosa derived from the antrum. The algorithm depicted in Figure 15.17 summarizes an approach to the evaluation of atrophy in gastric biopsies.
Endocrine Cell Proliferation
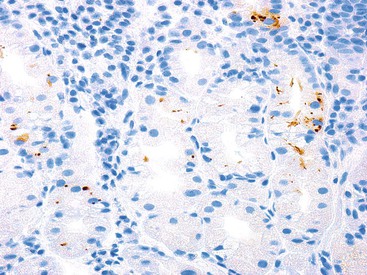
Endocrine cell hyperplasia develops as a consequence of functional changes in the stomach and is most prominent in autoimmune atrophic gastritis. In this condition, hypochlorhydria or achlorhydria may lead to antral G cell hyperplasia and a secondary elevation of the serum gastrin level.50,51 Hypergastrinemia causes histamine-producing enterochromaffin-like (ECL) cells within oxyntic mucosa to proliferate. Once thought to be rare and limited to patients with advanced atrophic gastritis, neuroendocrine proliferation is now frequently seen in routine gastric biopsies as a result of widespread use of proton pump inhibitors (PPIs).52 Although endocrine hyperplasia can be detected easily on hematoxylin and eosin (H&E)–stained tissue sections of the antrum (Fig. 15.18), hyperplasia of ECL cells in oxyntic mucosa is best visualized and quantified with specific immunostains (Fig. 15.19). Immunostains have largely replaced the use of argentaffin and argyrophil-based histochemical stains. In routine practice, staining of endocrine cells helps to identify cases of autoimmune atrophic gastritis of the corpus. A mild to moderate degree of ECL cell hyperplasia may occur in a subset of patients who use PPIs for extended periods. However, in these patients, the oxyntic mucosa is not atrophic and may display parietal cell hypertrophy and oxyntic gland dilations, which are characteristically seen in chronic PPI users.
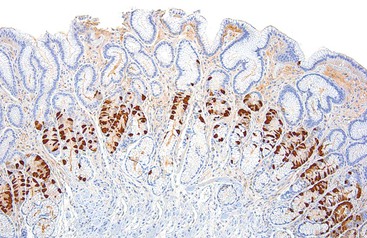
The most widely used criteria for the diagnosis and classification of gastric endocrine cell proliferation are those proposed by Solcia and colleagues (see Chapter 29).53 The classification system distinguishes simple or diffuse, linear, micronodular and adenomatoid hyperplasia, dysplasia, and neoplasia (i.e., carcinoid tumors) and is reviewed in the section on Autoimmune Gastritis and in Table 15.3.
Table 15.3
Classification of Enterochromaffin-Like Cell Proliferations
| Diagnosis | Criteria for Increased Endocrine Cells | Common Disorders |
| Simple or diffuse hyperplasia | >2 × standard deviations (age and gender matched) | ZES, primary gastrin cell hyperplasia |
| Linear hyperplasia | Linear groups of 5 or more cells inside the glandular BM | ZES, pernicious anemia |
| Micronodular hyperplasia | Clusters of 5 or more cells within epithelium measuring <150 µm in diameter | Autoimmune atrophic gastritis |
| Adenomatoid hyperplasia | Aggregates of 5 or more micronodules in lamina propria | Autoimmune atrophic gastritis, MEN-ZES |
| Dysplasias | Autoimmune atrophic gastritis, MEN-ZES | |
| Enlarged micronodules | >150 µm | |
| Adenomatous micronodules | Collections of at least 5 closely adherent micronodules, intervening BM only | |
| Fused micronodules | Adenomatous micronodules with no intervening BM | |
| Microinfiltrative lesions | Infiltration of the lamina propria | |
| Carcinoids | Autoimmune atrophic gastritis, MEN-ZES | |
| Intramucosal | Expansile or infiltrative nodules > 0.5 mm | |
| Invasive | Any size tumor within submucosa |
BM, Basement membrane; LP, lamina propria; MEN, multiple endocrine neoplasia; ZES, Zollinger-Ellison syndrome.
Data from Solcia E, Fiocca R, Villani L, et al. Hyperplastic, dysplastic, and neoplastic enterochromaffin-like cell proliferations of the gastric mucosa: classification and histogenesis. Am J Surg Pathol. 1995;19(suppl 1):S1-S7.
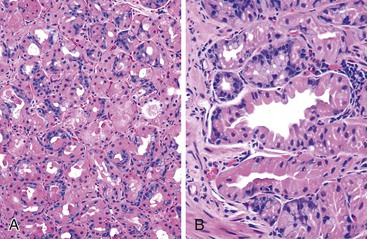
Parietal Cell Alterations
Apocrine-like protrusion and pseudohypertrophy of oxyntic cells are frequently attributed to chronic use of PPIs (Fig. 15.20).54,55 However, identical histologic changes have been reported in other clinical settings, and the findings are therefore not pathognomonic.56 Use of PPIs may also lead to dilation of oxyntic glands, which in extreme cases may produce multiple fundic gland–type polyps that impart an appearance of the gastric mucosa that inspired the picturesque description of gastric acne.57,58 Because most chronic users of NSAIDs also take PPIs to reduce the risk of NSAID-induced ulceration, oxyntic gland alterations are frequently seen in conjunction with reactive (chemical) gastropathy.41,59,60
Interfoveolar Smooth Muscle Hyperplasia
In the normal stomach, smooth muscle fibers are confined to the muscularis mucosae, where they run parallel to the mucosal surface. Several studies have shown that chemical injury is often associated with the smooth muscle fibers that run perpendicular to the muscularis mucosae within the interfoveolar lamina propria (see Fig. 15.9). This proliferation may be caused by the pulling effect of prolapsing mucosa, similar to that seen in the so-called solitary rectal ulcer syndrome. It is unclear whether peristalsis may exert analogous forces on injured gastric mucosa. Another proposed mechanism is the release of platelet-derived growth factor (PDGF), a known smooth muscle stimulant, as a result of epithelial damage. However, smooth muscle hyperplasia is not specific for NSAID-induced gastritis and may be seen in a variety of other conditions, such as gastric antral vascular ectasia (GAVE), bile reflux gastritis, and reactive mucosa adjacent to an ulcer.41,60
Updated Sydney System
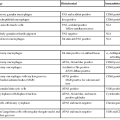
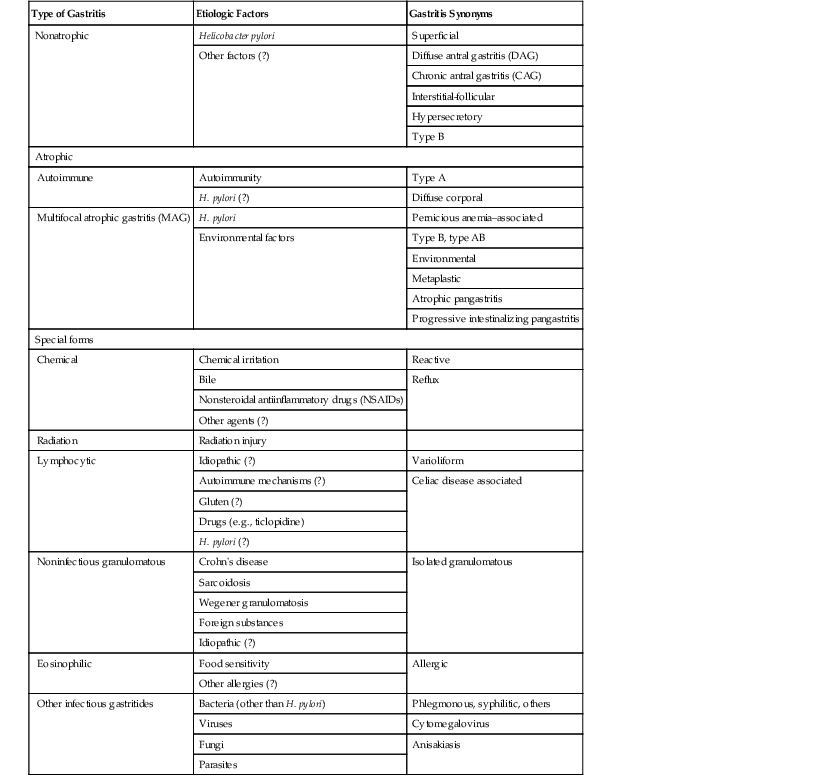
The updated Sydney System provides pathologists with guidelines for generating systematic, uniform diagnostic reports. The goal of the Sydney System is to make gastric biopsy pathology reporting consistent so that pathologists create clinically relevant and precise diagnoses known to all clinicians and to allow clinical studies to be performed and evaluated in an unambiguous manner. To create a pathology report suggested by the updated Sydney System, at least five biopsy specimens should be evaluated and the findings synthesized (Fig. 15.21). The system (Table 15.4) classifies chronic gastritis into three broad categories on the basis of topography, morphology, and when possible, on the basis of cause as acute, chronic, or special (distinctive).8 The latter category includes entities of uncertain pathogenesis and gastropathies. This system also separates chronic gastritis into atrophic and nonatrophic forms (Fig. 15.22).
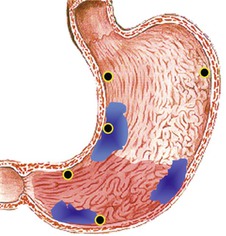
Table 15.4
Sydney System Classification of Gastritis
| Type of Gastritis | Etiologic Factors | Gastritis Synonyms |
| Nonatrophic | Helicobacter pylori | Superficial |
| Other factors (?) | Diffuse antral gastritis (DAG) | |
| Chronic antral gastritis (CAG) | ||
| Interstitial-follicular | ||
| Hypersecretory | ||
| Type B | ||
| Atrophic | ||
| Autoimmune | Autoimmunity | Type A |
| H. pylori (?) | Diffuse corporal | |
| Multifocal atrophic gastritis (MAG) | H. pylori | Pernicious anemia–associated |
| Environmental factors | Type B, type AB | |
| Environmental | ||
| Metaplastic | ||
| Atrophic pangastritis | ||
| Progressive intestinalizing pangastritis | ||
| Special forms | ||
| Chemical | Chemical irritation | Reactive |
| Bile | Reflux | |
| Nonsteroidal antiinflammatory drugs (NSAIDs) | ||
| Other agents (?) | ||
| Radiation | Radiation injury | |
| Lymphocytic | Idiopathic (?) | Varioliform |
| Autoimmune mechanisms (?) | Celiac disease associated | |
| Gluten (?) | ||
| Drugs (e.g., ticlopidine) | ||
| H. pylori (?) | ||
| Noninfectious granulomatous | Crohn’s disease | Isolated granulomatous |
| Sarcoidosis | ||
| Wegener granulomatosis | ||
| Foreign substances | ||
| Idiopathic (?) | ||
| Eosinophilic | Food sensitivity | Allergic |
| Other allergies (?) | ||
| Other infectious gastritides | Bacteria (other than H. pylori) | Phlegmonous, syphilitic, others |
| Viruses | Cytomegalovirus | |
| Fungi | Anisakiasis | |
| Parasites | ||
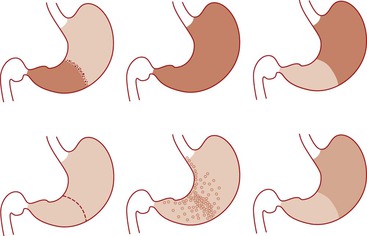
Biopsy Protocol
The biopsy protocol depicted in Figure 15.21 is recommended to obtain satisfactory mucosal sampling. Specimens from three compartments (i.e., antrum, incisura angularis, and corpus) should be separately designated when submitted to the pathology laboratory. Proper specimen orientation is critical for optimal histologic evaluation and is best accomplished in the pathology laboratory at the time of tissue embedding. Unfortunately, in routine clinical practice, gastroenterologists do not often adhere to such a rigorous sampling and labeling protocol (see Reporting Gastritis in the Absence of a Complete Biopsy Set), and this limitation is not addressed by the Sydney classification.
Evaluation of Histologic Variables
Each mucosal biopsy specimen should be assessed for its suitability for pathologic examination. An acceptable slide is one that shows several well-oriented sections with the mucosal surface and the muscularis mucosae visible. Each relevant pathologic feature (e.g., density of H. pylori, intensity of neutrophilic and mononuclear inflammation, atrophy of the antrum and corpus, intestinal metaplasia) should be graded on a standardized visual analogue scale (Fig. 15.23). Each feature is assigned a numeric or descriptive value: 0 for absent, 1 for mild, 2 for moderate, and 3 for marked or severe. The values of each specimen are determined separately for each anatomic compartment (i.e., antrum and corpus). A minimum of two specimens from the antrum, one from the incisura angularis, and two from the corpus should be evaluated.
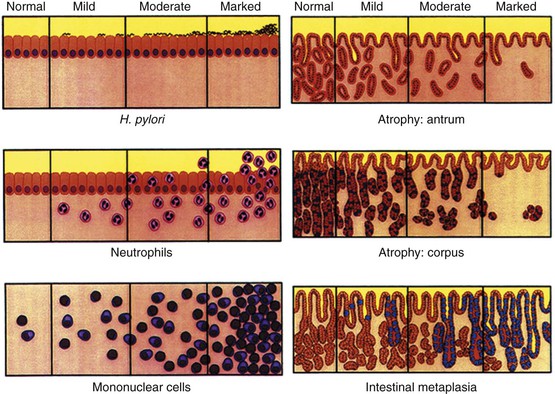
Determination of Gradient of Inflammation
After evaluation of the histologic variables, the next step in the Sydney System is to document the degree of inflammation in the two main gastric compartments to determine whether the inflammation is similar in intensity (i.e., pangastritis) or more severe in the antrum (i.e., antrum-predominant gastritis) or the corpus (i.e., corpus-predominant gastritis). To conclude that inflammation in one compartment is predominant, the difference in the inflammatory variables should be at least two grades. This helps to minimize the effect of interobserver variability. The degree of atrophy and metaplasia can also be assessed according to the Atrophy 2000 guidelines.10 The last step in the Sydney classification is to decide whether focal atrophy or diffuse atrophy (metaplastic or nonmetaplastic) is present.
Diagnosis
The final diagnosis according to the Sydney system represents a synthesis of the observations outlined previously, including information regarding possible cause. Examples of diagnostic reports are “H. pylori antrum-predominant gastritis” and “corpus-restricted atrophic gastritis without H. pylori infection, suggestive of autoimmune gastritis” (Fig. 15.24).
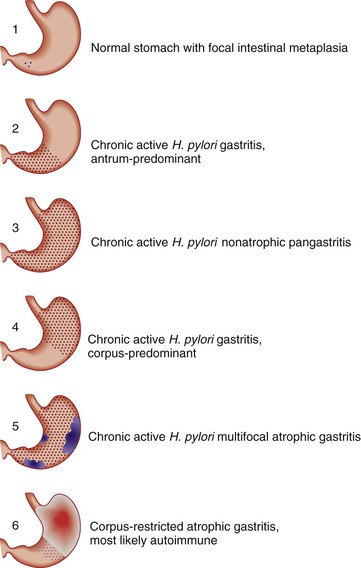
Reporting Gastritis in the Absence of a Complete Biopsy Set
The Sydney System guidelines can be applied only when a full set of biopsy specimens is available (see Fig. 15.21). In routine practice, pathologists are usually asked to make a diagnosis based on only one or two gastric biopsy specimens and often from unspecified sites. In these cases, no attempt should be made to use the Sydney System. Instead, an empiric approach is recommended.
Most types of gastritis, including those caused by H. pylori, can be diagnosed without extensive tissue sampling. In contrast, assessment of the degree of atrophy requires adequate sampling and knowledge of the site of origin of each specimen to distinguish MAG from autoimmune metaplastic atrophic gastritis. A diagnosis of gastritis based on only a few nonrepresentative or inadequately identified biopsy specimens should not include any statements regarding topographic distribution of disease. If a specimen labeled as “corpus” contains pyloric-appearing glands and lacks parietal and chief cells, suggesting that it may be derived from the antrum, an immunostain for gastrin can help to resolve the issue. The finding of numerous gastrin-positive cells, typically in the midgland region, indicates that the biopsy was derived from the antrum, whereas atrophic pyloric-type glands from metaplastic corpus contain few or no gastrin-positive cells. If uncertainty persists, a less specific diagnosis (e.g., chronic inactive gastritis with atrophy and intestinal metaplasia) may be used, along with a comment regarding the differential diagnosis, including a suggestion that broader and more extensive sampling would be helpful to determine an exact cause. Other information, such as clinical evidence of pernicious anemia, hypergastrinemia, achlorhydria, and the presence or absence of circulating anti–parietal cell antibodies, may help to establish a precise diagnosis of autoimmune metaplastic atrophic gastritis.
OLGA System
Building on the current knowledge of the natural history of gastritis and its associated cancer risk, an international group of gastroenterologists and pathologists proposed a system of gastritis reporting in terms of stage: the Operative Link for Gastritis Assessment (OLGA) staging system.11,61 The system places the histologic phenotypes of gastritis on a scale of progressively increasing gastric cancer risk, from the lowest (stage 0) to the highest (stage IV).
Use of a well-defined biopsy sampling protocol (Sydney System) is considered the minimum required for reliable staging of chronic gastritis. The stage of gastritis is determined by the combination of the extent of atrophy (scored histologically) with its topographic location (resulting from the mapping protocol). Consistent with the Sydney recommendations, OLGA staging system reporting includes information about the likely cause of gastritis (e.g., H. pylori, autoimmune).
The OLGA system is based on the atrophy score. Atrophy is scored as a percentage of the atrophic glands. Nonmetaplastic and metaplastic subtypes are considered together. At each biopsy sample level (irrespective of the area from which it originates), atrophy is scored by using a four-tiered scale: atrophy absent (0%) = 0; mild (1% to 30%) = 1; moderate (31% to 60%) = 2; and severe (>60%) = 3. The OLGA gastritis stage results by combining the overall antrum score with an overall corpus score for atrophy (Table 15.5). Details on how to apply the OLGA scores in clinical practice are provided in a step-by-step tutorial by Rugge and colleagues.13,14 Several retrospective studies have shown that gastric adenocarcinoma rarely or never develops in patients with OLGA scores of 0 to 2; the disease tends to occur in patients with scores of 3 or 4.13,15,62
Table 15.5
Operative Link for Gastritis Assessment (OLGA) Staging System
| Antrum | Corpus | |||
| No Atrophy (score 0) | Mild Atrophy (score 1) | Moderate Atrophy (score 2) | Severe Atrophy (score 3) | |
| No atrophy (score 0), including incisura angularis | Stage 0 | Stage I | Stage II | Stage III |
| Mild atrophy (score 1), including incisura angularis | Stage I | Stage I | Stage II | Stage III |
| Moderate atrophy (score 2), including incisura angularis | Stage II | Stage II | Stage III | Stage IV |
| Severe atrophy (score 3), including incisura angularis | Stage III | Stage III | Stage IV | Stage IV |
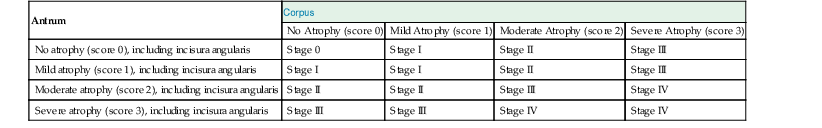
A Dutch group modified the OLGA staging system using intestinal metaplasia rather than atrophy as the main parameter for the assessment of gastric cancer risk. The investigators suggested that their system (i.e., Operative Link for Gastric Intestinal Metaplasia [OLGIM]) may be less vulnerable to interobserver variability because intestinal metaplasia can be recognized and quantified more reliably than atrophy.
Acute Gastritis
In the Sydney System, the term acute gastritis refers to the initial phase of H. pylori infection (discussed later) or acute bacterial suppurative gastritis (i.e., phlegmonous gastritis), which is rare.63–66 However, acute hemorrhagic gastritis is characterized by sudden onset and rapid evolution, and it therefore meets the clinical criteria for an acute illness.
Acute Hemorrhagic Gastritis
Clinical Features
Acute hemorrhagic gastritis is characterized by diffuse mucosal hyperemia associated with bleeding, erosions, and ulcers and precipitated by a sudden stress-induced imbalance between injurious and protective factors involved in the maintenance of mucosal integrity. This disorder has been a recognized nosologic entity for at least a century. Thomas Blizard Curling, a British surgeon, documented an association between severe burns and duodenal ulceration (i.e., Curling ulcer) in 1842.67,68 The prevalence and clinical importance of stress-induced gastric ulcers have been appreciated recently. Most patients in intensive care units have mucosal ulcers, approximately 20% develop overt bleeding, and as many as 5% succumb to life-threatening hemorrhage.69
Ingestion of large doses of aspirin or other types of NSAIDs may cause acute mucosal injury, ranging from edema and hyperemia to erosion and ulceration. These lesions may occur suddenly, without prior pain or discomfort, in first-time and chronic NSAID users.59 Similar but usually less severe changes may be caused by ingestion of large quantities of alcohol.70 Because alcohol and aspirin act synergistically to alter mucosal defenses, many cases of hemorrhagic gastritis are caused by alcohol users who consume aspirin to prevent hangover sickness.71 The pathogenesis of stress-induced hemorrhagic gastritis is unknown, but luminal acid is essential. Acid exerts its noxious effect by inhibiting and causing loss of integrity of mucosal defense mechanisms (e.g., mucus-bicarbonate barrier). Vascular compromise associated with stasis, vasoconstriction, and increased vascular permeability also contributes to loss of mucosal integrity.
NSAIDs act by interfering with prostaglandin synthesis, whereas alcohol causes direct damage to the gastric mucosa. At a concentration of 12.5%, alcohol induces hyperemia and petechiae. At concentrations greater than 40%, alcohol causes necrosis of the surface epithelium and capillaries, and causes interstitial hemorrhage.37,70 Suppression of acid secretion with PPIs helps to reduce the severity of mucosal damage and facilitates mucosal healing.
Pathologic Features
Acute hemorrhagic gastritis is grossly characterized by hyperemic edematous mucosa, surface erosions, and active bleeding (Fig. 15.25). The clinical history (e.g., shock, burns, ingestion of large doses of NSAIDs) rather than the pathologic features helps to determine the precise cause. In extreme cases, erosion of an underlying large blood vessel may cause catastrophic hemorrhage and death.
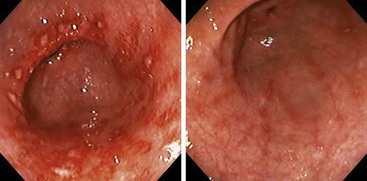
Microscopically, acute hemorrhagic gastritis, regardless of its cause, is characterized by dilation and congestion of mucosal capillaries, edema, and interstitial hemorrhage within the lamina propria. Surface erosions are usually small and appear ischemic. Aggregates of fibrin and neutrophils replace the eroded epithelium and often project above the mucosal surface to form small, elevated clumps of necrotic debris (Fig. 15.26). The foveolar epithelium underneath and adjacent to erosions and ulcers show regenerative changes, such as elongation, increased tortuosity, mucin depletion, enlarged and hyperchromatic nuclei, and increased mitoses; the latter two features should not be misinterpreted as dysplasia. Recalling the location of the normal regenerative zones in the antrum (i.e., gland base) and corpus (i.e., mucous cell neck region) can help to mitigate an overdiagnosis of dysplasia. In the absence of concurrent H. pylori infection, unaffected areas of the stomach do not usually show increased chronic inflammation. In the setting of H. pylori infection, the pathologist may see chronic active inflammation in the mucosa, which may obscure or worsen the changes caused by NSAIDs or alcohol.
Chronic Gastritis
Helicobacter pylori Gastritis
Clinical Features
Chronic H. pylori gastritis affects two thirds of the world’s population and is one of the most common chronic inflammatory disorders of humans.72,73 It is causally related to most non–NSAID-induced duodenal and gastric ulcers, and most gastric mucosa-associated lymphoid tissue (MALT) lymphomas. In certain regions of the world, atrophic gastritis develops in a considerable proportion of infected persons, and this condition is a precursor to gastric carcinoma. H. pylori gastritis is a strong risk factor for development of gastric cancer, particularly the intestinal type.
The early phase of H. pylori infection elicits an acute inflammatory response that is asymptomatic or has short-lived clinical manifestations, such as nausea and vomiting. Because most patients with early H. pylori gastritis do not undergo endoscopy, information regarding the clinical, endoscopic, and pathologic aspects of acute H. pylori infection is limited to a few well-documented case reports, human ingestion studies,72 and analyses of infections acquired from inadequately disinfected endoscopic instruments.74–76
Endoscopically, manifestations of acute H. pylori gastritis are typically found in the antrum and characterized by hemorrhagic lesions and multiple erosions or ulcers. Virtually identical findings were previously reported in patients with epidemic gastritis and achlorhydria, a syndrome reported before the identification of H. pylori as the principal cause of most cases of chronic gastritis.77
Although chronic H. pylori gastritis is asymptomatic in most infected individuals, its impact on human health is profound. H. pylori gastritis confers a 15% to 20% lifetime risk for peptic ulcer disease, and 70% of gastric cancers and most primary gastric MALT lymphomas are directly linked to chronic H. pylori infection. In one British community, H. pylori was estimated to be responsible for approximately 5% of all gastrointestinal (GI) ailments.78
When H. pylori was initially identified, combination treatment with antibiotics and bismuth-containing compounds was recommended only for patients with gastric ulcers and known MALT lymphomas. In time, guidelines were developed to include treatment for patients with dyspepsia and, essentially, all patients in whom infection could be identified.79,80
Although programs for global eradication of H. pylori have been advocated with the goal of preventing gastric carcinoma, insufficient supportive evidence has prevented widespread acceptance of the proposals.81,82 Critics of these programs cite the increasing resistance of H. pylori to the antibiotics used in current protocols. There is also much attention to the development of an effective vaccine to prevent H. pylori infection.83
Epidemiology
The prevalence of H. pylori infection among adults approaches 90% in many developing countries, particularly those in the tropics. Cross-sectional studies have revealed a high prevalence of infection among children, indicating that exposure to the bacterium probably occurs relatively early in life. In industrialized parts of the world (e.g., Western Europe, United States, Canada, Australia), exposure tends to occur later in life, which results in a lower percentage of infected adults. An average of 20% to 30% of adults are infected by 50 years of age.84,85
In eastern Asia (e.g., Japan, South Korea), where improved sanitation methods have been recently introduced, there is a clear trend toward a lower rate of H. pylori infection. The prevalence of H. pylori infection has been steadily declining in industrialized and emerging countries, which probably reflects the improved sanitary conditions and widespread use of antibiotics. Prevalence rates as low as 5% have been reported in Norwegian children between 1 month and 3 years of age,86 and even in China (Guangzhou province), the infection rate for children between 1 and 5 years was only 19% in 2003.87
Despite declining rates of H. pylori infection overall, the prevalence of H. pylori among patients who undergo endoscopy remains significant. H. pylori should be considered in all gastric biopsy specimens examined, regardless of the patient’s age or history.
Pathologic Features
Grossly, there are no distinct endoscopic patterns of chronic H. pylori gastritis. Depending on the stage and type of gastritis, hyperemia, erosions, hypertrophy, and atrophy may coexist in various combinations. Unfortunately, none of these endoscopic features has proved useful for predicting the presence or absence of H. pylori gastritis. Endoscopists should not attempt to diagnose H. pylori gastritis based solely on the gross appearance of the gastric mucosa. The diagnosis of H. pylori gastritis rests on pathologic evaluation of gastric mucosal biopsies or detection of urease in mucosal specimens by the Campylobacter-like organism (CLO) test or the urea breath test.88 Polymerase chain reaction (PCR) testing is discussed later.
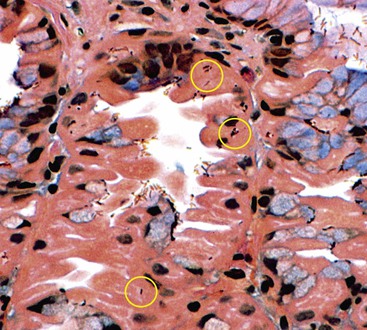
Microscopically, the gastric mucosa is covered by a thick layer of mucus that plays a protective role. This mucous layer, which is best visualized in Carnoy-fixed tissues, is the primary site of H. pylori colonization and often contains large numbers of organisms.89 When in contact with epithelium, H. pylori organisms characteristically attach to but do not penetrate the surface mucous cells. Intracellular invasion has been observed in surface mucous cells, chief cells, and parietal cells (rare), particularly in patients who have received PPIs (Fig. 15.27).90–93 Although some authorities have suggested that penetration and intracellular invasion may be related to the organism’s ability to promote carcinogenesis,92,94 the significance remains unclear.
In severe acute infection, grossly visible pseudomembranes and pus adherent to inflamed mucosa have been described (Fig. 15.28).95 In chronic H. pylori gastritis, epithelial degeneration is particularly prominent because of intimate contact of the H. pylori organisms with the surface cell membrane (see Fig. 15.6).96
The epithelial cells often become irregular and cuboidal, have a decreased apical mucin content, and occasionally drop out, leaving small gaps in the epithelium that contribute to a ragged, disorderly appearance. The characteristic alterations in H. pylori infection are caused by bacterial toxins such as VacA and CagA, urease, ammonia, acetaldehyde, and phospholipases, which have a direct effect on epithelial cells.97–99
H. pylori causes mast cells to release platelet-activating factors, which leads to thrombosis.100 The disturbance of the local microcirculation results in ischemic loss of epithelial integrity and ultimately in surface erosions and ulcers.99
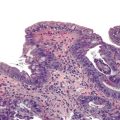
Mucosal neutrophils are a distinctive histologic feature of H. pylori infection. Neutrophils are more abundant in the antrum and cardia than in the corpus, where they may be rare or completely absent despite the presence of organisms. Neutrophils may be seen in the lamina propria (where they are typically mixed with mononuclear cells and eosinophils) and within the surface and foveolar epithelium (Fig. 15.29). Neutrophils may fill the lumen of the gastric pits, forming pit microabscesses and a surface exudate. In patients with atrophy, active neutrophilic inflammation only rarely occurs in areas of metaplasia. After successful eradication therapy, neutrophils disappear rapidly, and their continued presence is considered a valuable indicator of therapeutic failure.18
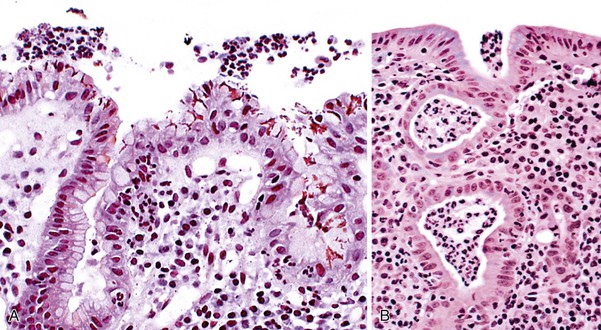
Gastric mucosa chronically infected by H. pylori typically shows a prominent mononuclear (lymphoplasmacytic) infiltrate. In the corpus, the mononuclear infiltrate is usually most intense in the superficial neck region of the lamina propria (Fig. 15.30), which prompted the use of the term chronic superficial gastritis as a synonym for nonatrophic gastritis.49 In exceptional cases, the pathologist may also find a significant lymphocytic infiltrate within the glandular portion of the mucosa. In some cases, a diffuse intraepithelial infiltrate may be prominent, which can be mistaken for lymphocytic gastritis.101–103 In the setting of a focal and destructive lymphocytic infiltrate (i.e., lymphoepithelial lesion), additional evidence of extranodal marginal zone lymphoma should be sought. The intensity of the mononuclear cell infiltrate typically declines quite slowly after successful eradication of infection. As many as 30% of patients have chronic inactive gastritis that persists for several years after eradication of the organism.
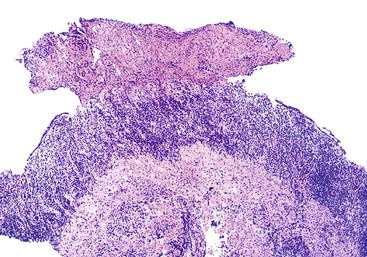
Lymphoid follicles with germinal centers are found in almost all H. pylori–infected patients, and their presence in the gastric mucosa is highly specific for H. pylori–associated gastritis (see Fig. 15.3).23,104,105 The density of lymphoid follicles is usually greatest in the region of the angulus and lowest in the proximal greater curve of the stomach. These anatomic sites correspond, respectively, to the most and least common sites of gastric MALT lymphoma.23 H. pylori infection is a major cause of acquired MALT in the stomach and is a critical factor in the development of primary gastric B-cell lymphoma (MALT lymphoma, which is now classified as extranodal marginal-zone B-cell lymphoma).106–108 Occasionally, lymphoid infiltrates may be large and irregular, with lymphoepithelial lesions at the periphery. In this circumstance, gastric lymphoma should be considered. Pathologic features that warrant further investigation with immunocytochemical and molecular techniques include: an expansile monotypic infiltrate, a monocytoid appearance of the lymphocytes, lymphoepithelial lesions associated with glandular destruction, and endoscopic evidence of a mass or nodularity. Immunohistochemical and molecular tests often yield equivocal results, and a diagnosis of atypical lymphoid infiltrate may be provided with elaboration of the inconclusive findings.
If H. pylori infection is detected by direct or indirect methods, including serology, eradication therapy with follow-up biopsies of the affected area is warranted.109–111 Lymphoid follicles normally decline in number and size, although very slowly, after successful treatment. In some patients, lymphoid follicles may persist indefinitely.
Subtypes of Helicobacter pylori Gastritis
H. pylori infection may show different patterns of gastritis and may ultimately be associated with the development of glandular atrophy. Different patterns of H. pylori gastritis are associated with various outcomes.
Nonatrophic Antrum-Predominant Gastritis
Nonatrophic antrum-predominant gastritis is the most common pattern of H. pylori gastritis (i.e., hypersecretory, diffuse antral, or superficial antral gastritis) in the Western world. It is characterized by moderate to severe inflammation of the antrum, normal or mild inflammation of the corpus, and absence of atrophy. This clinical presentation is associated with normal or increased acid secretion and a 20% estimated lifetime risk of duodenal ulceration.112–114
Nonatrophic Corpus-Predominant Gastritis
Nonatrophic corpus-predominant gastritis is restricted mainly to patients who use PPIs chronically. The density of H. pylori organisms and the intensity of inflammation are low in the antrum and high in the corpus. Several studies have shown that corpus atrophy proceeds at an accelerated rate in these patients.115,116
Nonatrophic Pangastritis
In some individuals chronically infected with H. pylori, marked inflammation is evenly distributed throughout the stomach, with little difference between the antrum and corpus. This pattern of gastritis is particularly common in poorly sanitized areas, where H. pylori infection is highly endemic. Pangastritis is widely thought to be the background condition in which atrophy develops.117–119
Antrum-Restricted Atrophic Gastritis
In Western populations, atrophy and intestinal metaplasia are usually minimal, focal, and detected only in the antrum. These findings may be the consequence of current or previous H. pylori infection or chemical gastropathy. These changes do not represent antrum-restricted atrophic gastritis, a term reserved for conditions in which atrophic metaplastic changes are extensive.
Biopsies in this condition show patchy intestinal metaplasia and atrophy restricted to the antral mucosa (including the incisura angularis) and usually associated with moderate to severe inflammation and show a normal or mildly inflamed corpus without atrophy. The relationship between antrum-restricted atrophy and MAG is unknown. Although it is possible that these entities are biologically different, it is also possible that they represent different stages of the same disease.
Multifocal Atrophic Gastritis
MAG was formerly referred to as environmental chronic atrophic gastritis and, in its most advanced stage, as atrophic pangastritis.7,120 This pattern of gastritis is most prevalent among populations who live or have recently lived in suboptimal sanitary conditions, such as parts of southern and eastern Asia, Latin America, and Europe.121–124 Exceptions to this epidemiologic association include Japan, where despite high levels of sanitation and personal hygiene, there is one of the world’s highest prevalence rates of atrophic gastritis and gastric adenocarcinoma.125–127 In contrast, in Equatorial Africa which has a precarious socioeconomic environment, inadequate sanitary conditions, and an H. pylori prevalence of almost 90%, there is a surprisingly low prevalence of atrophic gastritis and gastric adenocarcinoma.128–130
The determination of H. pylori genotype has not proved reliable in predicting the phenotype of gastritis, including atrophy.131 As a result, other than consideration of the country of origin, it is difficult to predict the risk for atrophic gastritis in an individual patient in industrialized countries.
Atrophic gastritis is a risk factor for gastric ulceration and, more importantly, noninvasive neoplasia (i.e., dysplasia) and intestinal-type adenocarcinoma.132–135 In biopsy specimens with MAG, foci of atrophy and intestinal metaplasia are found in antral and corpus mucosa. In contrast to antrum-restricted atrophic gastritis, MAG often displays severe inflammation in the corpus mucosa, and acid secretion may be reduced, suggesting a more advanced state of disease.
Differential Diagnosis
Methods of Detecting Helicobacter pylori
H. pylori normally infects antral and corpus mucosa, but in the cardia, organisms may be difficult to detect. H. pylori organisms rarely colonize intestinal epithelium, and in patients with extensive areas of metaplastic atrophy, organisms are usually confined to the nonmetaplastic, nonatrophic areas of mucosa. In patients who use PPIs, H. pylori organisms tend to be rare or absent in the antrum, and they may be difficult to detect in the corpus, even in the setting of chronic active inflammation.136 Similarly, patients treated with antibiotics before gastric biopsy may demonstrate a markedly reduced number of organisms and often have atypical (coccoid) forms (Fig. 15.31).137
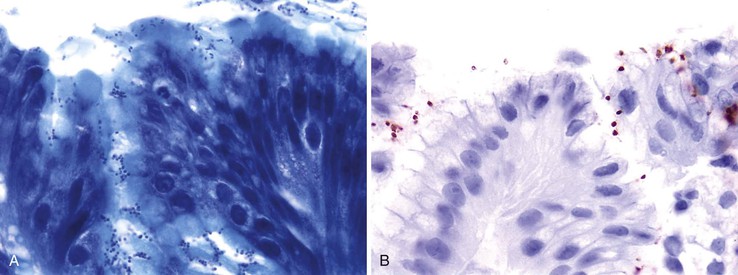
As many as 70% of gastric biopsy specimens from H. pylori–infected subjects reveal organisms by routine H&E staining. In the remaining 30% of cases, a more sensitive stain is usually needed. Many laboratories routinely stain all gastric biopsies, even those without significant inflammation, for H. pylori to ensure identification of organisms.
Commonly used and relatively inexpensive stains, such as Giemsa and Diff-Quik, are adequate for identification of most cases of H. pylori organisms.138,139 The Hp Blue/Hp Yellow stain (Anatech, Battle Creek, Mich.) is designed to make the organisms more discernable by staining the background mucin yellow.140 The yellow counterstain can be eliminated, which results in a cleaner background. The silver-based triple stain simultaneously allows visualization of H. pylori organisms while better featuring the histologic changes in the mucosa (Fig. 15.32; see Figs. 15.6 and 15.11).141 Because this stain involves application of Alcian blue at pH 2.5, it also allows small, inconspicuous foci of intestinal metaplasia to be detected easily (see Fig. 15.11). Immunohistochemical stains for H. pylori increase sensitivity and are particularly useful for detecting coccoid forms of the organism, rare organisms located deep within the glands, and intracytoplasmic organisms.142,143 After H. pylori treatment, organisms may be best visualized with an immunohistochemical stain if chronic gastritis persists.
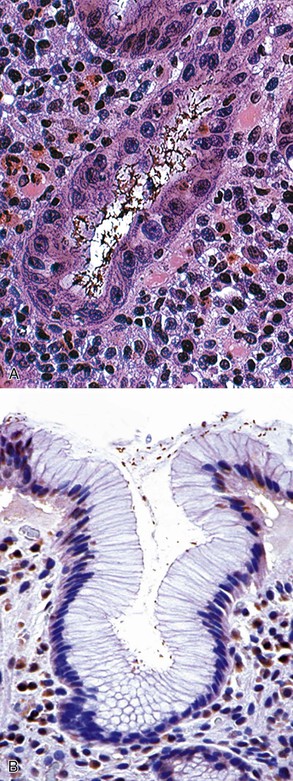
Molecular techniques, specifically PCR, can be used to detect H. pylori organisms in tissue, and claims of higher sensitivity for histologic methods have been published.144,145 However, not all of these studies used immunohistochemistry for H. pylori detection as the reference group, and identification rates varied by technique (e.g., nested versus quantitative PCR). There is no gold standard to serve as the true infection rate to exclude false-positive PCR results, nor is there a clear understanding of the clinical relevance of finding trace bacterial DNA in gastric tissue. Gastroenterologists’ position papers have not specifically recommended use of PCR but do concede that it may be useful if the clinician is prepared to treat a patient with a positive result.146 It seems that the best application for those inclined to use this technique is for cases in which there is a histologic pattern of inflammation suggesting Helicobacter infection but no organisms detectable by immunohistochemistry.
PCR used on gastric tissue has been evaluated for possible identification of antibiotic sensitivity, which could be a valuable clinical application. Most patients are prescribed H. pylori treatment without antibiotic susceptibility testing, but first-line treatment regimens fail to eradicate the organism 20% of the time. The primary culprit appears to be resistance to clarithromycin, which can be significant (about 15% to 20%) in certain populations. Resistance has been related to point mutations in bacterial 23S rRNA, and genotypic (PCR) evaluation of resistance is more sensitive than phenotypic (culture) testing. However, PCR-based testing correlates less well than culture with treatment outcomes and therefore has a low predictive value.147
Helicobacter heilmannii Infection
More than 50 species of Helicobacter have been described, but only a few cause gastritis in humans. They include Helicobacter felis, Helicobacter fennelliae, Helicobacter cinaedi, and Helicobacter heilmannii. Among these, H. heilmannii (formerly known as Gastrospirillum hominis) is the most common,148 with an estimated prevalence of 1% of all human Helicobacter infections.149–151 In some rural areas in eastern Europe, infection with H. heilmannii is more common, supporting the hypothesis that it is acquired by zoonotic transmission.152 The organisms are 5 to 9 µm long (twice as long as H. pylori) and have five to seven spirals.
Although H. heilmannii organisms are easily visualized with any of the common histochemical H. pylori stains (Fig. 15.33), their detection may be difficult because they tend to be less numerous and more focally distributed than H. pylori. Polyclonal immunohistochemical stains for H. pylori also react with H. heilmannii, as do some commercially available monoclonal stains. H. heilmannii can be missed by those who rely exclusively on these immunostains for their diagnosis.
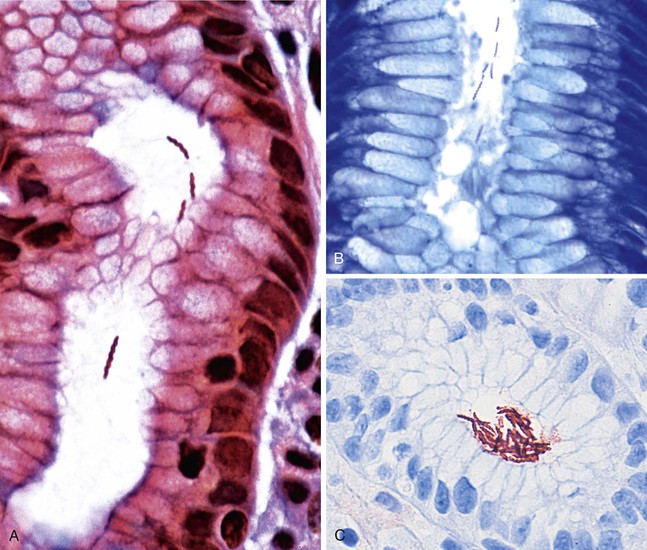
Gastritis caused by H. heilmannii differs from H. pylori gastritis. It is more common in children and usually milder and patchier, with fewer erosions and ulcers. The inflammation tends to be more circumscribed, mainly affecting the antrum, although cases of severe corpus active gastritis may be seen. The diagnosis rests on the morphologic recognition of the organisms, although differentiation of H. heilmannii from H. felis is not possible by light microscopy. These organisms have not been cultured successfully in vitro, and the specific antibiotic susceptibility of H. heilmannii has therefore not been well studied. Clinically, H. heilmannii patients are treated identically to those with H. pylori infection, and rates of successful outcomes are similar.153
Similar to H. pylori, H. heilmannii infection has been linked to the development of gastric adenocarcinoma154 and MALT lymphoma. Regression of lymphoma after successful eradication therapy has been reported.155,156
Natural History and Complications
H. pylori is a lifelong infection, and if left untreated, it may progress from nonatrophic to atrophic gastritis and, depending on its phenotype, may cause peptic ulcer disease, adenocarcinoma, and primary MALT lymphoma. The relative risk of each of these conditions varies greatly in different populations and is related to bacterial, environmental, and genetic factors. More than 90% of gastric MALT lymphomas are diagnosed in patients with chronic H. pylori infection, and more than 75% will regress for extended periods with successful eradication therapy for the infection.155,157–159 Gastric carcinoma and lymphoma are discussed in Chapters 25 and 31, respectively.
Gastric Ulcers
Gastric ulcer, in contrast to erosion, is defined by the loss of the entire mucosa, including the muscularis mucosae. Ulcers may extend deep into the submucosa and even muscularis propria. Ulcers typically begin as erosions, but not all erosions progress to ulceration. Ulcers may be acute or chronic. Peptic ulcers are considered chronic and are most often solitary. They may occur in any portion of the GI tract that is exposed to acid peptic juices. Approximately 98% of peptic ulcers occur in the stomach and duodenum, with an incidence ratio of approximately 1 : 4.
Etiology
H. pylori infection is a major cause of peptic ulcers not associated with NSAIDs or the Zollinger-Ellison syndrome.160,161 H. pylori is detected in the stomach of almost all patients with duodenal peptic ulcer and in more than 90% of gastric ulcer patients who are not NSAID users. Eradication of H. pylori facilitates healing of peptic ulcers and essentially prevents their recurrence. In contrast, more than 80% of patients treated only with acid inhibitors experience recurrence within 1 year.162
Benign and malignant tumors may cause gastric ulceration. Malignant ulcers occur most often with adenocarcinoma and stromal tumors and less commonly with lymphomas, metastases, and direct extension of an adjacent extragastric malignancy. In immunocompromised patients, cytomegalovirus and histoplasmosis (rare) may cause gastric ulceration.163–165
Pathologic Features
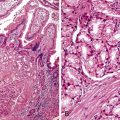
Acute stress ulcers are usually multiple. They are more than 0.5 cm in diameter and form well-delineated craters of various depths. Fibrin mixed with an acute inflammatory exudate and necrotic debris is usually found within the ulcer bed. Because these ulcers often bleed, blood clots may fill the ulcer crater.69,166 The pathologist usually sees an abrupt transition between the necrotic edges of the ulcer and adjacent mucosa (Fig. 15.34). The surrounding mucosa may be normal, or it may show features of the underlying chronic gastritis, depending on the cause of the ulcer.
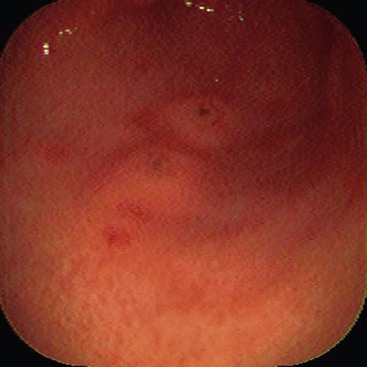
Most peptic ulcers occur on the lesser curvature of the stomach in the antrum close to the incisura angularis. Gastric ulcers that occur in the greater curvature and in other parts of the proximal stomach are more likely to be related to chronic NSAID use than H. pylori infection.167,168 Peptic ulcers are typically sharply demarcated lesions, which may appear erythematous, edematous, and only slightly elevated above the level of the surrounding mucosa (Fig. 15.35). They are usually small (0.5 to 2.0 cm in diameter), but some can be more than 3.0 cm in diameter. Large (i.e., giant) ulcers may be misdiagnosed endoscopically as malignant.169,170
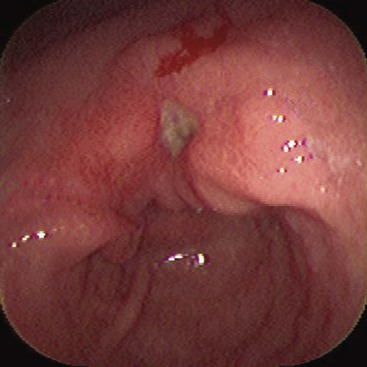
Perforating ulcers penetrate into adjacent structures, most commonly the pancreas. The mucosa surrounding ulcers may appear normal or show a radiating pattern to the rugal folds endoscopically, unless the ulcer is complicated by extensive fibrosis. This feature is useful in differentiating benign from malignant ulcers because malignancies usually show heaped-up mucosal borders and irregular rugal folds. However, ulcers associated with malignant lymphomas are often flat or only slightly elevated, frequently multiple, and may reach large sizes (10 to 12 cm in diameter).171
Microscopically, the mucosa surrounding peptic ulcers typically shows chronic active inflammation and marked regenerative changes, increased mitoses, mucin depletion, and foveolar hyperplasia—features that can mimic dysplasia (Fig. 15.36). Features that support regeneration include evidence of surface maturation, mitotic activity confined to the deeper foveolar regions, and nuclei without loss of polarity that have even chromatin, thin nuclear membranes, and uniform, central punctate nucleoli. A gradual transition from atypical to normal epithelium favors regeneration.
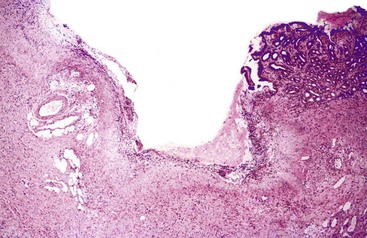
The base or crater of peptic ulcers consists of necroinflammatory debris, granulation tissue, and fibrosis with chronic inflammation from the luminal surface to the deep portions of the gastric wall. Adjacent to ulcers may be found a substantial lymphoid infiltrate arranged in follicles, in some cases mimicking malignant lymphoma.104 At the ulcer’s base, blood vessels proliferate, often showing prominent inflammation and arteritis obliterans. When disrupted, these arteries may bleed profusely.
Complications
The most common complications of peptic ulcer disease, in order of frequency, are hemorrhage, perforation, and obstruction. Approximately one third of patients experience at least one of these complications during the course of disease. Pyloric channel ulcers are most commonly associated with complications, particularly obstruction. Bleeding occurs when ulcers erode underlying blood vessels.
Dieulafoy Lesion
A Dieulafoy lesion refers to a prominent, large-caliber artery that may protrude into the gut lumen and cause recurrent and sometimes massive hemorrhage when the overlying mucosa becomes ulcerated. These lesions have been described in all parts of the GI tract, but they are most common at the gastroesophageal junction and in the lesser curve of the stomach. Dieulafoy lesions account for the 0.5% to 14% of patients in whom acute upper GI bleeding develops. Treatment options include endoscopic injection, cautery, ligation, embolization, and surgery.172,173
Ulcers may perforate adjacent organs such as the pancreas or liver. Chronic organization of a perforated ulcer may result in an inflammatory pseudotumor that clinically mimics a malignant tumor. Rarely, perforated ulcers may form a fistula with the small intestine, transverse colon, or gallbladder. Gastric outlet obstruction may develop as a result of distortion and narrowing of the pyloric area because of fibrosis, edema, and smooth muscle spasm. It occurs almost exclusively in patients with long-standing peptic ulcers of the pyloric channel or duodenum. Surgical repair or endoscopic pyloric dilation is often necessary to alleviate the symptoms of gastric outlet obstruction.172,173
Helicobacter pylori–Negative Chronic Gastritis
Although no studies documenting this phenomenon have been published, there is a common perception among pathologists practicing in the Western world that the incidence of chronic active gastritis with no detectable H. pylori organisms (i.e., H. pylori–negative chronic active gastritis) is increasing.20,174 The often cited 1990s axiom that even a small number of polymorphonuclear neutrophils in a gastric biopsy is an indicator of active H. pylori infection is no longer applicable.175 Although no formal hypothesis has been put forward and tested, commonly offered explanations include antibiotic therapy administered to treat other infections, the masking effect of PPIs, and inadequate sampling or suboptimal staining techniques.
Just as we used to think that chronic active gastritis was virtually synonymous with H. pylori infection, we also thought that no H. pylori infection occurred without active gastritis. We were wrong in both cases. When anti–H. pylori immunohistochemical stains for all gastric biopsies are routinely used, organisms are occasionally found in unexpected histologic settings, such as antral mucosa with reactive gastropathy and no active inflammation, antral mucosa with inactive inflammation (Fig. 15.37), fundic mucosa with only minimal subepithelial lymphocytes or plasma cells and no neutrophils, or almost normal mucosa. Having been misled by such cases, we advocate the preemptive use of an immunohistochemical stain in all gastric biopsies. Those who cannot or choose not to routinely use immunohistochemical or histochemical stains for the detection of H. pylori must decide when to employ the stains based on patient history, histology, and personal experience.
Regardless of the method used, it is important to consider the circumstances that can result in chronic active inflammation of the gastric mucosa in the absence of visible organisms. Two possibilities are a true H. pylori gastritis in which the organisms are undetectable and injuries or disease states unrelated to Helicobacter infection that are characterized by chronic active infiltrates in the gastric mucosa.
In addition to H. heilmannii gastritis, it is difficult or impossible to visualize Helicobacter organisms in other circumstances. One is a biopsy specimen with extensive intestinal metaplasia. Although we have shown significant exceptions to the concept that Helicobacter organisms adhere exclusively to native gastric epithelium and are not found on areas of intestinal metaplasia,176,177 for practical purposes, this notion can be considered correct. A biopsy specimen that consists primarily or exclusively of intestinalized gastric mucosa is inadequate to evaluate the possibility of H. pylori infection. If the biopsy specimen is the only one available, the pathologist should document this point in a comment and suggest that other samples be obtained.
Helicobacter organisms rarely colonize necrotic tissue and regenerative epithelium. The edges of an ulcer, even in a background of severe H. pylori gastritis, are unlikely to show organisms. If specimens from other areas are available, the issue can be easily resolved; unfortunately, the pathologist often receives only one specimen labeled “gastric ulcer—rule out H. pylori.” In these cases, the pathologist can exclude neoplasm but not H. pylori and is therefore obliged to suggest more extensive sampling or the performance of a noninvasive test.
Although we discourage the use of the expression sampling error, which implies that the endoscopist has taken the specimens from the wrong sites, sampling does matter because not all areas of an infected stomach contain H. pylori. In 1994, Genta and Graham evaluated the relative yield of biopsies taken from different sites of proven H. pylori–infected stomachs from untreated subjects.19 A combination of two specimens (one each from the antrum and corpus) showed organisms in more than 95% of the patients. At that time, the sampling site might have had only a very minor role in the ability to detect H. pylori organisms. However, the results of this study may no longer be valid, because almost all patients undergoing an upper endoscopy are on PPIs, which may alter the dynamics of bacterial colonization and gastric inflammation. Supporting this notion, data from Lash and co-workers for more than 400,000 patients showed a doubling of Helicobacter detection with sampling of at least two biopsies each from the antrum and corpus.178
Proton Pump Inhibitor Use
PPIs, some of the most commonly prescribed GI medications worldwide, have become available over the counter in the United States and in many other countries. PPIs are highly effective for chronic gastroesophageal reflux disease, for prevention of gastric damage associated with NSAIDs, and for dyspepsia.179,180 After they are recommended, most people continue taking them indefinitely.
Most patients who undergo endoscopy are taking PPIs, and gastroenterologists consider any attempt to discontinue the medication for 2 weeks before the procedure futile. As a consequence, most gastric biopsies show a presumed PPI effect: focally dilated oxyntic glands with flattened or hypertrophic parietal cells protruding into the lumen.54,181 Patients with H. pylori gastritis may have additional PPI-related changes, especially a milder intensity of antral inflammation accompanied by a greater intensity in the corpus, few or no detectable organisms in the antrum, and a peculiar redistribution of H. pylori organisms in the deeper portions of the oxyntic glands, even reaching the intracellular canaliculi of the parietal cells182–187 (Fig. 15.38). PPI use can cause false-negative results for urea breath tests.
Recent Antibiotic Treatment
Innumerable persons receive antibiotic treatments for a variety of verified or suspected infections; among the most common are dental procedures and ear, throat, and urinary tract infections. Because these treatments are usually not coordinated with gastroenterologists, procedures may be done on patients who recently received or are still on antibiotics. Some of these antibiotics affect H. pylori, but when used outside of a coordinated triple or quadruple therapy, they rarely cure the infection. They may, however, temporarily decrease the bacterial load and make the infection, which is still active and capable of inducing inflammation, visually undetectable. We suspect that these incidental antibiotic treatments are one of the most common causes of H. pylori–negative gastritis.
Antibiotics may also be prescribed by the gastroenterologist. Because almost no gastroenterologist performs endoscopy in a patient treated for H. pylori infection earlier than 3 to 6 months after completion of therapy, biopsies from these patients are likely to reveal one of two findings. If the therapy was successful (i.e., H. pylori infection was eradicated), the gastric mucosa shows various degrees of chronic gastritis, possibly some lymphoid follicles, and no active inflammation.18,188 If the therapy failed, a recrudescence occurs, and the chronic active gastritis is often more severe than before the unsuccessful therapeutic trial. Biopsies after eradication treatment are an exceedingly uncommon source of H. pylori–negative active gastritis.
Causes of Chronic Active Gastritis Unrelated to Helicobacter Infection
In some biopsy specimens, the features of reactive gastropathy are accompanied by those of chronic gastritis with or without foci of active inflammation. Because the features of H. pylori gastritis may overlap with those of reactive gastropathy (and the two types may coexist), a careful search for H. pylori organisms is imperative.
The two causes can usually be discerned. In contrast to H. pylori gastritis, the chronic infiltrate of reactive gastropathy is loose, does not form a subepithelial band (formerly designated superficial gastritis), and rarely has significant numbers of plasma cells. Any polymorphonuclear neutrophils tend to be confined to the lamina propria, sparing the glandular epithelium, where they are often associated with eosinophils. Intraepithelial neutrophils, aggregates of neutrophils, and regenerative epithelium are usually found only in the vicinity of erosions, which are often revealed by additional sections into the block.
Focally Active Gastritis
The finding of individual or few gastric foveolae or glands discretely surrounded and infiltrated by lymphocytes, macrophages, plasma cells, and occasional neutrophils in a background of normal gastric mucosa has been referred to as focally enhanced gastritis (Fig. 15.39).17 Although the meaning of the word enhanced in this context remains puzzling to many pathologists, the term has become tacitly accepted.
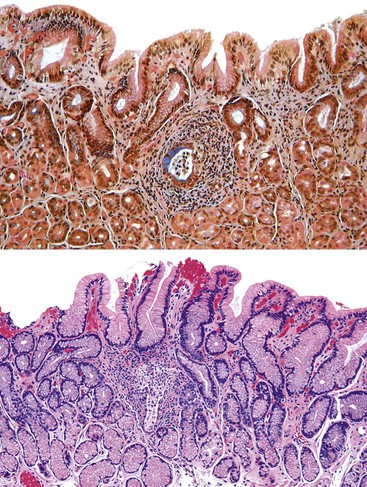
Focal and often intense gastritis has been described in a high percentage of patients with inflammatory bowel disease, most notably children with Crohn’s disease, but it can be seen in many patients with no inflammatory bowel disease.189–191 H. pylori infection can also be patchy, although typically with a less striking contrast between inflamed and adjacent, normal-appearing mucosa than seen with inflammatory bowel disease. Nevertheless, when focal gastritis is seen, an effort should be made to detect organisms. If none is found, a measured comment can be made indicating that one of the possible causes is inflammatory bowel disease elsewhere in the GI tract.
Other Infections
Other bacteria, fungi, and viruses may infect the gastric mucosa, but only rarely do they produce the histopathologic appearance of chronic active gastritis. Rather, they typically elicit a more mixed inflammatory infiltrate that is less superficial, as in the case of cytomegalovirus gastritis (Fig. 15.40). Unless specific clinical information is available (e.g., an immunosuppressed patient, a case of disseminated aspergillosis), a search for infectious agents other than Helicobacter species in the gastric mucosa is likely to be laborious and, ultimately, unrewarding.
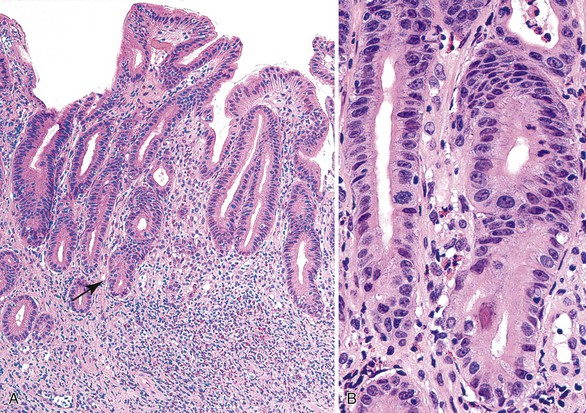
Reported Conclusions
After the pathologist has reviewed the slides, restained, and used alternate techniques, a well-worded report should state that no H. pylori organisms were found after a meticulous search, despite the characteristic histologic appearance. These reports, however, are unlikely to gratify an inquisitive clinician, who will be left wondering what should be done.
Calling the clinician and explaining the circumstances that led to an equivocal diagnosis is probably the best way to avoid being perceived as a timid pathologist. Before discussing a case, however, it is necessary to be fully informed. If the pathologist can elicit a history of recent incidental antibiotic treatment, he or she can suggest a plausible explanation for H. pylori–negative chronic active gastritis. The pathologist can indicate that the inadequately treated infection may reemerge and that a new set of biopsies after a few weeks may show organisms. A tactfully delivered reminder on the effects of PPIs may also help to persuade clinicians to start regularly sampling the antrum and the corpus. It is important not to leave the impression that H. pylori–negative chronic active gastritis is a final diagnosis without need for further workup.
Autoimmune Gastritis
Clinical Features
Autoimmune gastritis is a type of corpus-restricted, chronic atrophic gastritis associated with serum anti–parietal cell and anti–intrinsic factor antibodies with resultant intrinsic factor deficiency with or without anemia.192,193 Autoimmune gastritis does not cause specific clinical manifestations until a critical decrease has occurred in the parietal cell mass, beyond which anemia develops. Years before the onset of anemia, patients may show various degrees of hypochlorhydria, hypergastrinemia, and loss of pepsin and pepsinogen secretion.
Achlorhydria, which is a direct result of destruction of acid-producing parietal cells, typically occurs in the most advanced stage of disease. However, hypochlorhydria may occur in patients with a large number of preserved parietal cells, which suggests that there may be a possible role for anti–proton pump antibodies or inhibitory lymphokines released by inflammatory cells in the pathogenesis of this disease. Patients with atrophy of the corpus and achlorhydria have a high level of hypergastrinemia that tends to correlate with disease severity.194,195 Damage to chief cells leads to a reduction in pepsin activity in gastric juice and in the level of pepsinogen in serum. The finding of a low pepsinogen I level (<20 ng/mL) is a sensitive and specific indicator for corpus atrophy.194,196
Iron deficiency anemia or pernicious anemia develops in many patients with autoimmune gastritis, and achlorhydria is a major contributor to the pathogenesis of anemia. Gastric acid is important for absorption of nonheme iron, which supplies at least two thirds of the nutritional iron supply in most Western diets.197 Pernicious anemia, which results from loss of intrinsic factor production by parietal cells, is usually preceded by corpus-restricted chronic atrophic gastritis (usually end stage) and reduced or absent acid secretion of at least 10 years’ duration.198,199 Autoimmune gastritis is a risk factor for hyperplastic and dysplastic polyps, carcinoma, and endocrine tumors. Polyps are detected in 20% to 40% of patients with pernicious anemia; they are mostly sessile, less than 2 cm in diameter, and often multiple. Most are hyperplastic, but as many as 10% contain foci of dysplasia.200,201 Gastric cancers associated with pernicious anemia are mostly intestinal type and arise from intestinal metaplasia, suggesting that carcinoma in autoimmune gastritis likely develops through a metaplasia–dysplasia–carcinoma pathway.202
Epidemiology
Pernicious anemia is an uncommon disease with a reported prevalence rate of approximately 1%, even among older people and in high-incidence regions of the world. However, autoimmune gastritis may be underdiagnosed because most patients have microcytic or macrocytic anemia and are treated with iron, folate, and cobalamin without undergoing a thorough investigation of the underlying cause. However, targeted studies suggest an overall prevalence of 2%, with a peak of 4% to 5% among elderly women.203
Pathogenesis
The cause of autoimmune gastritis is unknown. One theory suggests the disease may be initiated by H. pylori infection. A high prevalence of antibodies with specificity for gastric mucosal antigens has been reported for patients with H. pylori–associated gastritis.204,205 Twenty percent of H. pylori–positive individuals have autoantibodies that react with the canaliculi of parietal cells, which are a primary antibody target in autoimmune gastritis. Studies with cloned T cells from H. pylori–infected patients and patients with autoimmune atrophic gastritis have identified molecular mimicry between H. pylori and hydrogen and potassium receptors and ATPase, suggesting that infection may stimulate T cells that target parietal cells.83 These studies provide support for the concept of a cross-reactive mechanism between H. pylori organisms and gastric epithelial antigens that may be responsible for or at least participate in the pathogenesis of autoimmune gastritis.206 The role of immunoglobulin G4 (IgG4) in autoimmune gastritis may be similar to that in autoimmune pancreatitis, and a study found IgG4-immunoreactive plasma cells in gastric biopsies to be 100% specific (albeit not highly sensitive) for autoimmune atrophic gastritis and pernicious anemia.207
Pathologic Features
Grossly, the mucosa of the corpus in patients with autoimmune gastritis is usually thinner than normal and shows a reduction or complete absence of rugal folds. Fine submucosal vessels are usually easily recognizable on endoscopic examination in advanced cases. Hyperplastic polyps are also common in advanced-stage disease.
Microscopically, the main pathologic features of uncomplicated autoimmune gastritis are diffuse corpus-restricted chronic atrophic gastritis with some degree of intestinal metaplasia. In the absence of comorbidities, such as concurrent H. pylori infection or bile reflux, the antrum is typically normal. This pattern of involvement is characteristic of patients with advanced disease and is found in patients with pernicious anemia. Patients without pernicious anemia but with anti–parietal cell antibodies have a spectrum of atrophic changes that range from minimal to severe and diffuse atrophy of the oxyntic mucosa, and they have some degree of ECL cell proliferation (see Table 15.3).
Three pathologic phases can be identified during the course of autoimmune gastritis in corpus mucosa: early, florid, and end stage. The early phase is characterized by diffuse or multifocal, dense lymphocytic and plasma cell infiltration involving the entire thickness of the lamina propria and often mixed with eosinophils and mast cells. Patchy destruction of individual oxyntic glands by lymphocytes may be seen at this stage (Fig. 15.41). The pathologist may see patchy pseudopyloric metaplasia and hypertrophic changes of the remaining parietal cells. Hypertrophic changes indicate a high level of functional stimulation, which may be related to hypergastrinemia and can be found in the parietal cells of patients with autoimmune atrophic gastritis, those who have received PPI therapy, and a subset of patients with peptic ulcer disease.56,208
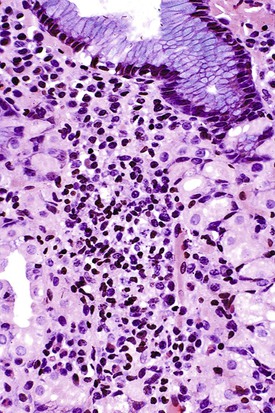
In the early phase of autoimmune gastritis, intestinal metaplasia rarely occurs and is often focal. At this stage, various degrees of chronic and active inflammation involve the full thickness of the lamina propria with some degree of loss of oxyntic glands. Although the histology may suggest the cause, the diagnosis at this stage requires demonstration of circulating anti–parietal cell autoantibodies, a test rarely performed in routine clinical practice.209
The florid phase of autoimmune gastritis is characterized by marked atrophy of oxyntic glands, diffuse lymphoplasmacytic infiltration of the lamina propria, and normal or reduced thickness of the mucosa with a relative increase in the thickness of the foveolar component. Pseudopyloric metaplasia is often extensive, and intestinal metaplasia tends to be more prominent than in the early phase. At this stage of the disease, the pathologic features of autoimmune atrophic gastritis are sufficiently distinctive, particularly if the antrum is normal. However, demonstration of antibodies directed against parietal cell and intrinsic factor antigens is necessary for confirmation.
The end stage of disease is characterized by a marked reduction in oxyntic glands, foveolar hyperplasia with elongation and microcystic change, hyperplastic polyp formation, and increasing degrees of pseudopyloric, pancreatic, and intestinal metaplasia. The muscularis mucosae may be thickened threefold to fourfold (Fig. 15.42). At this stage, parietal cells are difficult to detect, and inflammation is usually minimal or absent, although scattered lymphoid aggregates may persist.
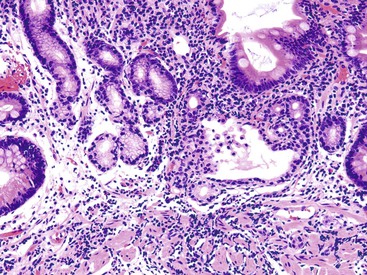
During the florid and end stages, hypochlorhydria and achlorhydria cause physiologic hypergastrinemia, which stimulates ECL cell proliferation. ECL cell proliferation may also occur in patients with Zollinger-Ellison syndrome, multiple endocrine neoplasia syndromes, and H. pylori–associated, multifocal atrophic gastritis. Solcia and colleagues proposed a reproducible, albeit arbitrary, classification that helps to distinguish normal from dysplastic and overtly malignant ECL cell proliferations. For example, an intramucosal carcinoid tumor is defined as an expansile or infiltrative endocrine growth that is more than 0.5 mm in diameter. Invasive carcinoids are those in which tumor invades the submucosal space (see Table 15.3 and Chapter 29).53 Although ECL cell carcinoids may arise during the florid phase, they are found most commonly in patients with end-stage disease.202 Carcinoid tumors associated with ECL cell hyperplasia occur in 5% to 8% of patients with autoimmune gastritis and severe hypergastrinemia, and they account for 70% to 80% of all gastric carcinoid tumors. These tumors are relatively innocuous and are associated with a more than 95% 5-year survival rate, which sharply contrasts with the less common solitary, sporadic type of carcinoid tumors that are biologically more aggressive (<35% 5-year survival rate).210 It is important to convey to clinicians the clinical and pathologic context in which a gastric carcinoid is diagnosed.
The antral mucosa in autoimmune gastritis may be normal or show mild, chronic inflammation with small foci of intestinal metaplasia similar to that observed in the age-matched general population. However, most patients have the characteristic features of reactive gastropathy, with foveolar hyperplasia and mucin depletion of the surface and foveolar epithelium. Hyperplasia of gastrin cells caused by achlorhydria is often seen in the antrum. Concurrent H. pylori infection may involve the antrum and cause neutrophilic inflammation in addition to chronic gastritis.
Special Forms of Gastritis
Special forms of gastritides include inflammatory conditions of the stomach that are of uncertain origin, such as reactive and vascular gastropathies, although they are well characterized topographically and morphologically.
Lymphocytic Gastritis
Clinical Features
Lymphocytic gastritis is characterized by large numbers of mature lymphocytes that infiltrate the surface and foveolar epithelium. Although this pattern of gastritis has reportedly been seen in 1% to 4% of patients who undergo upper endoscopy, in our experience with more than 500,000 patients with gastric biopsies, the prevalence was less than 0.3%. Lymphocytic gastritis is most commonly diagnosed in the fifth decade of life, and it affects women more than men, especially when it is associated with celiac sprue.211
Lymphocytic gastritis was initially thought to correspond to an entity described by François Moutier in 1945 as varioliform gastritis, a nodular form of gastritis characterized by central depressions (i.e., octopus sucker gastritis).101,102 However, lymphocytic gastritis may also be observed in patients with celiac disease, Ménétrier disease, H. pylori infection, and, less commonly, lymphocytic or collagenous colitis, and NSAID use. Lymphocytic gastritis is essentially a pathologic reaction pattern without specificity for any particular disease. In one pediatric study of patients with celiac disease, the degree of duodenal and gastric intraepithelial lymphocytosis decreased concurrently with withdrawal of gluten from the patients’ diet. Histologic evidence of lymphocytic gastritis has been reported for patients with endoscopically normal gastric mucosa.211–214
Patients with endoscopic varioliform lymphocytic gastritis have rapid weight loss, anorexia, hypoproteinemia, hypoalbuminemia, and peripheral edema, suggesting a protein-losing gastroenteropathy.215 Because this entity has not been described again in the past 2 decades and remains largely unknown to gastroenterologists, it may be the expression of a severe, perhaps acute form of H. pylori infection.
Almost all intraepithelial lymphocytes in lymphocytic gastritis are CD8+ T cells, similar to those found in the duodenal mucosa of celiac disease patients. An allergic or autoimmune pathogenesis has been proposed. This hypothesis is supported by the increasingly apparent association between lymphocytic gastritis and celiac disease.211 Some investigators have postulated a relationship between lymphocytic gastritis and Ménétrier disease.216,217
Pathologic Features
On gross examination, most patients with lymphocytic gastritis (including those with celiac disease) have scattered superficial erosions in the corpus or antrum or, in our experience, a normal-appearing gastric mucosa.
Microscopically, the severity of inflammatory changes in lymphocytic gastritis varies widely. In mild cases, there is only a minor increase in chronic inflammation without neutrophils. At the other extreme, marked chronic inflammation may be seen in the lamina propria that is occasionally associated with surface erosions. The lamina propria infiltrate is composed mainly of lymphocytes, with scattered plasma cells, eosinophils, and mast cells. In lymphocytic gastritis, more than 25 intraepithelial lymphocytes per 100 epithelial cells is typical, although counting is usually unnecessary (Fig. 15.43).211,218 The histologic features are readily distinguishable from those of typical H. pylori gastritis (see Figs. 15.4, 15.6, 15.10), in which only rarely are more than 5 or 6 intraepithelial lymphocytes per 100 epithelial cells found, and the lymphocytes are usually more irregularly distributed. In cases with prominent neutrophils, other disorders such as H. pylori gastritis should be considered. Other features of lymphocytic gastritis include degenerative epithelial changes, mucin depletion, foveolar hyperplasia, and increased mitoses. The type and intensity of these associated changes may be related to the underlying cause of the lymphocytic infiltration.
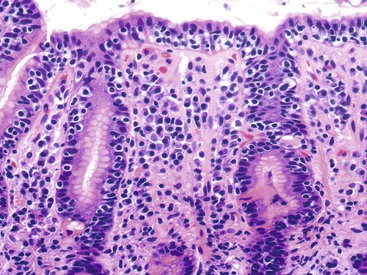
Collagenous Gastritis
Collagenous gastritis is a rare entity that is histologically similar to collagenous colitis and is characterized by a distinct subepithelial collagenous band and nonspecific inflammatory infiltrate that may or may not be associated with increased intraepithelial lymphocytosis (Fig. 15.44). Similar to collagenous colitis, the collagen band typically is 10 to 25 µm thick in well-oriented sections, has an irregular contour, and demonstrates a ragged interface with the underlying lamina propria. Capillaries are usually entrapped by the fibrous band. One study reported intestinal metaplasia, ECL cell hyperplasia, and epithelial atypia (considered indefinite for dysplasia) in a case of collagenous gastritis and postulated an unproven risk of carcinoma.219
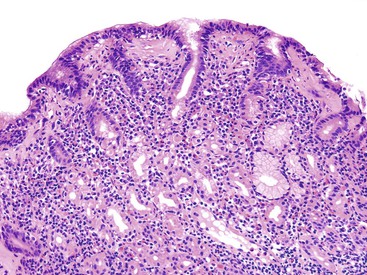
Most collagenous gastritis patients can be segregated into two groups: children and young adults with severe anemia, endoscopically nodular mucosa, and disease limited to the stomach and adults with chronic, watery diarrhea and associated collagenous colitis. One report described a case associated with lymphocytic gastritis and celiac disease.220–224
The differential diagnosis includes fibrosis associated with autoimmune gastritis, healed ulcer, or radiation therapy in which the collagen deposition is usually more diffuse and not specifically subepithelial. Similarly, patients with scleroderma have fibrosis at all levels of the mucosa, and it may involve deeper levels of the bowel wall. As in the colon, the subsurface basement membrane may be prominent in some preparations and often is accentuated by basal epithelial cytoplasm. Paying attention to the nature of the fibrosis helps the clinician to avoid an overdiagnosis.
Eosinophilic Gastritis
Normal Eosinophil Counts in the Gastric Mucosa
Although most observers consider the finding of a few eosinophils in the lamina propria common, the updated Sydney System posits that intraepithelial eosinophils in gastric mucosa are always abnormal.175 The limited information available regarding the normal range of gastric mucosal eosinophils was derived from infrequent and often uncontrolled case series, isolated case reports, and brief sections in textbooks of GI pathology.225–228
In the Kalixanda study, Talley and colleagues found a mean eosinophil count of 11 eosinophils per 5 high-power fields (HPFs) in biopsies from the cardia, body, and antrum of asymptomatic adult volunteers from Northern Sweden.227 DeBrosse and co-workers found peak eosinophil counts of 8 eosinophils/HPF in antral biopsies and 11 eosinophils/HPF in oxyntic mucosal biopsies from 19 children.226 Lwin and colleagues enumerated eosinophils in the lamina propria of patients from a wide age range and from diverse geographic areas in the United States with no known history of relevant GI disease and whose gastric biopsies were diagnosed as unremarkable.27 The mean eosinophil count for 135 normal patients (age range, 4 to 81 years) was 4 eosinophils/HPF (±4 standard deviations [SD]), equivalent to 15 ± 17 SD eosinophils/mm2 (range, 0 to 110 eosinophils/mm2). There were no significant differences between the counts in biopsies from the antrum and corpus and no significant variation by age or geographic location. These findings essentially agreed with those of DeBrosse and colleagues and Talley and co-workers.226,227
Causes of Gastric Mucosal Eosinophilia
Increased numbers of eosinophils in the lamina propria have been documented in a variety of conditions, including infection with Anisakis spp.,229 Strongyloides stercoralis,230 or H. pylori231–233 and after H. pylori treatment.18 Other associations include drug-induced injury,59 Crohn’s disease,27 pyloric obstruction,234,235 tumors,236 connective tissue diseases, hematopoietic disorders, food allergy, and rare eosinophil-associated GI disorders.26
Idiopathic Eosinophilic Gastritis
Idiopathic (isolated) eosinophilic gastritis is a poorly characterized condition. Although rare cases have been reported sporadically in the literature,237–241 H. pylori has monopolized the efforts of gastric researchers since the mid-1980s, and the few descriptions of eosinophilia in the gastric mucosa have been related to current or treated H. pylori infection.18
Lwin and colleagues reported a series of 60 patients with gastric mucosal eosinophil counts of 50 to more than 400 eosinophils/HPF (i.e., 200 to more than 1000 eosinophils/mm2).27 Epigastric pain was the most common reason for endoscopy in adults and children, followed by reflux disease and dysphagia. The most common endoscopic findings were a normal stomach or erythema and gastritis with or without erosions.
Practical Suggestions for Diagnosis
Epithelial eosinophilic infiltration (Fig. 15.45, A) is a common finding, particularly if a sufficient number of sections are examined. Sheets of eosinophils (see Fig. 15.45, B) are seen in more than one half of specimens from patients with eosinophilic gastritis. Eosinophils tend to surround the foveolae and infiltrate the epithelium, but they typically do not spill into the lumen to form eosinophilic pit abscesses (see Fig. 15.45, B). Involvement of the muscularis mucosae or submucosa may occur. Although reactive epithelial changes similar to those found in chemical gastropathy are common, neither foveolar hyperplasia nor intestinal metaplasia is characteristic.
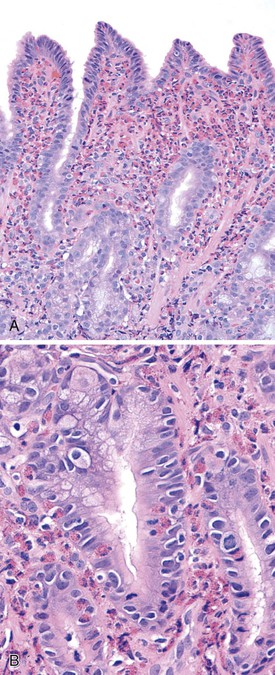
We recommend that the term histologic eosinophilic gastritis be used for the diagnosis of patients with gastric biopsies that show at least 30 eosinophils/HPF (on microscopes equipped with wide-lens oculars) in at least five separate HPFs (focusing on the most concentrated fields) and who have no known cause of eosinophilia. If H. pylori organisms are detected, the diagnosis of eosinophilic gastritis can be established only if the mucosal eosinophilia persists several months after successful eradication.
When regenerative epithelial changes are observed in a gastric biopsy with significant eosinophilic infiltrates that fall below the recommended quantitative threshold, we suggest a diagnosis of reactive gastropathy with increased eosinophils. The diagnosis should include a comment mentioning the possibility of eosinophilic gastritis, among other differential diagnoses deemed appropriate in the context of the patient’s clinical situation.27
Granulomatous Gastritis
Granulomatous gastritis is a nonspecific histologic pattern of disease characterized by multiple granulomas in the gastric mucosa (Fig. 15.46, A and B). The morphologic appearance of the granulomas does not provide useful clues about their cause, except when foreign material, acid-fast bacilli, or fungal forms are identified; the latter two entities occasionally cause central necrosis or caseation. In most cases, the cause cannot be determined without specific clinical and laboratory information.
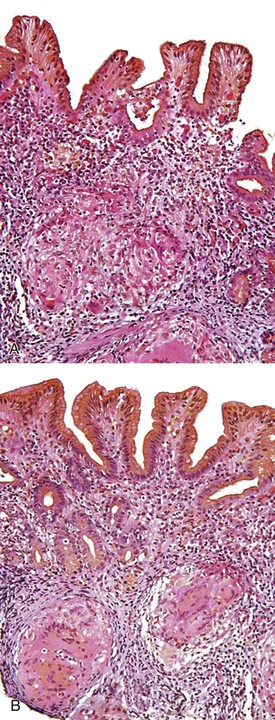
Granulomas may be found in the gastric mucosa of patients with many types of infectious, inflammatory, and neoplastic diseases, and they may be seen in otherwise healthy individuals. The endoscopic appearance of the stomach may be normal, or it may exhibit characteristics of the specific disease entity that caused the granulomas to form. Possible causes of granulomas in the gastric mucosa are provided in Box 15.1.
Mycobacterial and Fungal Infections
Worldwide, tuberculosis is the most common cause of granulomatous disease of the GI tract. However, the stomach is rarely affected. Primary gastric tuberculosis has been reported mostly in developing countries with a high prevalence of Mycobacterium tuberculosis infection.242–245 In almost all cases, gastric tuberculosis manifests as a large, nonhealing ulcer. A few cases of primary infection of the stomach by the dimorphic fungus Histoplasma capsulatum have been reported. Similar to those with gastric tuberculosis, these patients often have signs and symptoms of a large gastric ulcer.246–248
Helicobacter pylori Infection
Unexplained granulomas may be found in less than 10% of patients with H. pylori gastritis, and the role of this organism in causing granulomas is unknown and controversial.249 Because granulomas in the stomach are not rare and may also be seen in healthy individuals, the discovery of granulomas in patients with H. pylori gastritis may represent a coincidental finding. Alternatively, granulomas in this setting may indicate a second disorder, such as a drug reaction, another type of infection, or foreign body.
Parasites
The nematodes most commonly associated with the formation of gastric granulomas are Anisakis spp. Early lesions show interstitial edema accompanied by loose, predominantly eosinophilic inflammation, and late lesions may show overt eosinophilic microabscesses. Well-preserved larvae are often detected. In late lesions, the most common findings are foreign body granulomas, which are sometimes associated with fragments of helminthic cuticles.28,250 Rarely, fragments of larvae interpreted as Strongyloides stercoralis have been observed in gastric granulomas.251,252 These conditions are discussed further in Chapter 4.