CHAPTER 3 Inflammatory Basis of Spinal Pain
THE INTERVERTEBRAL DISC
Disc biomechanics
In general, tissue failure occurs because the loads to which they are exposed as stresses generated exceed the strength of the tissue. These can be tensile, compressive, or shear forces contributing to the damage. Stokes and Greenapple demonstrated strains of 6–10% during extremes of flexion and axial rotation in lumbar disc fibers.1 The strains were greater in the posterolateral areas than in the anterior regions. While pure axial compression, even in testing at very high loads, does not cause herniation of the nucleus pulposus, cyclic loading can cause annular tears that may eventually lead to disc herniation. Discs are known to exhibit creep, relaxation, and hysteresis. In these studies, the amount of hysteresis was shown to increase with load and decrease with age. These studies also demonstrate that nondegenerative discs creep less slowly than degenerative discs. This may indicate that there is less physiologic elasticity in degenerative discs.
INFLAMMATION
Celsus described the four principal effects of acute inflammation nearly 2000 years ago (Table 3.1). These include redness from acute dilatation of small blood vessels within the area. Heat, or warmth, is usually seen only in the peripheral parts of the body such as the skin. It is also due to the increased blood flow or hyperemia through the region from vascular dilatation. Swelling results from edema which is the accumulation of fluid in the extravascular space and from the physical mass of the inflammatory cells migrating to the area. Pain is one of the best-known features of acute inflammation and it results partly from the stretching and distortion of tissues due to the edema in the area. Chemical mediators of acute inflammation including bradykinin, the prostaglandins, and serotonin are also known to induce pain. Loss of function is a well-known consequence of inflammation added by Virchow to the list originated by Celsus. Movement of an inflamed area is consciously and reflexively inhibited by pain, while severe swelling or local muscle spasm may limit movement of the area.
Table 3.1 Celsus’s original description of the characteristic signs of inflammation
| Erythema (rubor) |
| Warmth (calor) |
| Pain (dolor) |
| (Loss of function was added by Virchow) |
The spread of the inflammatory response following injury to a small area of tissue suggests that chemical substances are released from the injured tissues spreading out to uninjured areas. These chemicals are called endogenous mediators and contribute to the vasodilatation, de-margination of neutrophils, chemotaxis, and increased vascular permeability. Chemical mediators released from the cells include histamine, which is probably the best-known chemical mediator in acute inflammation. It causes vascular dilatation in the immediate transient phase of increased vascular permeability. This substance is stored in mast cells, basophils and eosinophils, as well as platelets. Histamine released from those sites is stimulated by complement components C3a and C5a, and by lysosomal proteins released from neutrophils. Lysosomal compounds are released from neutrophils and include cationic proteins that may increase vascular permeability and neutral proteases, which may activate complement. Prostaglandins are a group of long-chain fatty acids derived from arachidonic acid and synthesized by many cell types. Some prostaglandins potentiate the increase in vascular permeability caused by other compounds. Part of the antiinflammatory activity of drugs such as aspirin and nonsteroidal antiinflammatory drugs (NSAIDs) is attributable to inhibition of one of the enzymes involved in prostaglandin synthesis. Leukotrienes are also synthesized from arachidonic acid, especially in neutrophils, and appear to have vasoactive properties. SRS-A (slow-reacting substance of anaphylaxis) involved in type I hypersensitivity is a mixture of leukotrienes. Serotonin (5-hydroxytryptamine) is present in high concentration in mast cells and platelets and is a potent vasoconstrictor. Lymphokines are a family of chemical messengers released by lymphocytes. Aside from their major role in type IV sensitivity, lymphokines also have vasoactive or chemotactic properties.
The process of inflammation
Inflammation is a complex, stereotypical reaction of the body in response to damage of cells in vascularized tissues. In avascular tissue such as the normal cornea or within the disc space, true inflammation does not occur. The cardinal signs of inflammation presented earlier, including redness, swelling, heat, pain and deranged function, have been known for thousands of years. The inflammatory response can be divided temporally into hyperacute, acute, subacute, and chronic inflammation. The response can be based on the degree of tissue damage, such as superficial or profound, or on the immunopathological mechanisms such as allergic, or inflammation mediated by cytotoxic antibodies, or inflammation mediated by immune complexes, or delayed-type hypersensitivity reactions. As presented earlier, the development of inflammatory reactions is controlled by cytokines, by products of the plasma enzyme systems (complement, the coagulation system, the kinin and fibrinolytic pathways), by lipid mediators (prostaglandins and leukotrienes) released from different cells, and by vasoactive mediators released from mast cells, basophils, and platelets. These inflammatory mediators controlling different types of reactions differ from one another. Fast-acting mediators such as the vasoactive amines and the products of the kinin system modulate the immediate response. Later, newly synthesized mediators such as leukotrienes are involved in the accumulation and activation of other cells. Once the leukocytes have arrived at the site of inflammation, they release mediators that control the later accumulation and activation of other cells. However, it is important to realize that in inflammatory reactions initiated by the immune system the ultimate control is exerted by the antigen itself, in the same way as it controls the immune response itself. For this reason, the cellular accumulation at the site of a chronic infection or in an autoimmune reaction is quite different from that at sites where the antigenic stimulus is rapidly cleared.
Inflammatory cells
Neutrophils
Neutrophils, macrophages, endothelial, and other cells produce two types of free radicals. The first type is represented by reactive oxygen intermediates that are formed in neutrophils by the activity of NADPH oxidase. The second type includes reactive nitrogen intermediates such as nitric oxide. Reactive nitrogen intermediates have been of some interest in low back-associated pain. These are sometimes called reactive oxynitrogen intermediates. The pathway by which they are originated is an oxidative process in which short-lived nitric oxide is derived from the guanidino nitrogen in the conversion of L-arginine to L-citrulline. This reaction is catalyzed by nitric oxide synthase (NOS) and, like the respiratory burst, it involves oxygen uptake. Three distinct isoforms of nitric oxide synthase representing three distinct gene products have been isolated and purified. The three isoforms vary considerably in their subcellular location, structure, kinetics, regulation, and hence functional roles. Two of the enzymes are constantly present and termed constitutive NOS (cNOS). The endothelial cNOS is mostly membrane bound and formed only in endothelial cells. The neuronal cNOS was identified in cytosol or central and peripheral neurons. The third isoform is an inducible form that is not present in resting cells. Cytokines are a potent stimulus for iNOS production or suppression. Those with an apparent stimulating effect include IFN-γ, IL-1, IL-6, TGF-α, GM-CSF, and PAF (platelet activating factor) while suppression has been observed by IL-4, IL-8, IL-10, TGF-β, PDGF (platelet derived growth factor), and MDF (macrophage deactivating factor). Cytokines are basic regulators of all neutrophil functions. Many of them, including somatesthetic growth factors and pyogens, have shown to be potent neutrophil priming agents. Neutrophils are also capable of de novo synthesis and secretion of small amounts of some cytokines including IL-1, IL-6, IL-8, TNF-α and GM-CSF.
Mediators of inflammation
Products of the complement system
Complement is a complex system containing more than 30 different glycoproteins present in the serum in the form of components, factors, or other regulators, and on the surface of different cells in the form of receptors. The components of the classical pathway are numbered 1–9 and in prefix by the letter ‘C.’ All these pathways use C5–C9 that form the membrane attack complex (MAC). Activation of each of the components results from the proteolytic cleavage event in a cascade mechanism. The complement system influences the activity of numerous cells, tissues, and physiologic mechanism of the body. The result of cytotoxic complement reaction may be beneficial or harmful to the body. The complement system is a potent mechanism for initiating and amplifying inflammation. This is mediated through fragments of complement components. Tissue injury following ischemic infarction may also cause complement activation and abundant deposition of membrane attack complex may be readily seen in tissue following ischemic injury.
BIOCHEMISTRY OF DISC DEGENERATION
MMPs, cytokines, and nitrous oxide
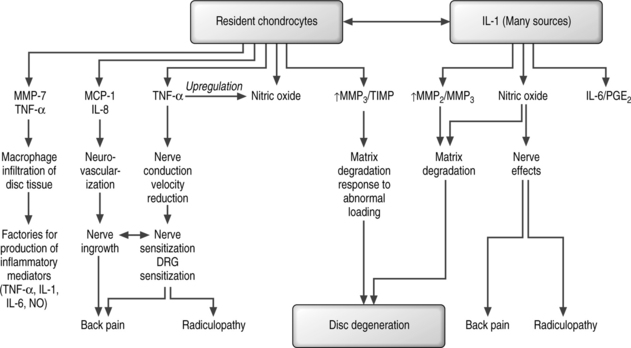
MMPs come in several different varieties. The most commonly investigated ones in terms of intervertebral disc degeneration have been MMP2 (gelatinase-α) and MMP3 (stromelysin). Kang investigated stromelysin production as well as production of nitric oxide IL-6 and PGE2, comparing 18 herniated lumbar discs with 8 control discs obtained from patients undergoing anterior surgery for scoliosis and burst fractures.2 Kang examined gelatinase, stromelysin, as well as collagenase activity. His group found a nearly sixfold increase in gelatinase among the herniated disc samples compared to the controls. Collagenase production was absent in the control subjects and nonsignificantly elevated in the herniated discs. Caseinase (or stromelysin – MMP3) showed an approximately fourfold increase in the herniated samples compared with the control discs. This early finding and the activity of MMPs in herniated disc samples was interesting, especially in the case of caseinase (stromelysin) which is known to degrade the core protein of cartilage proteoglycans. The progressive loss of these proteoglycans within the nucleus pulposus is believed to be one of the central reasons behind its desiccation and failure to retain its water content. The high levels found in the herniated discs probably repre sent the levels found in the degenerative discs compared to the lower level of MMP activity in the normal discs. It is likely that the smaller or lower activity of the MMPs in the normal discs reflects a basal amount of MMP activity responsible for ongoing remodeling of the disc architecture. The high MMP production in the herniated discs is likely a result of the increased inflammatory mediators produced within the discs or in the immediate area of the discs because of the inflammation.
IL-1 is known to have a positive modulating response on the MMPs. In the presence of a high IL-1 concentration and a low or relatively low TIMP concentration, the degradative enzymes may be expected to flourish. In a follow-up study to this article, Kang et al. reported on the effect of interleukin-1β on control and herniated discs using samples from the lumbar and cervical spine.3 They showed significantly elevated MMP production in the form of gelatinase and stromelysin by normal nondegenerated disc specimens after the addition of IL-1β. The basal levels of gelatinase and stromelysin were already increased in the lumbar and cervical degenerative disc specimens and the addition of IL-1β to these cultures did not significantly increase them. Collagenase activity was not detected.
An interesting control in this last study was the use of L-NMA (n-monomethyl-L-arginine) to block endogenously produced NO. Cells were cultured (control and diseased) in the presence of L-NMA in order to study the effects of endogenously produced nitrous oxide on the other mediators. When L-NMA was added to the nondegenerate control specimens that had been stimulated with IL-1β, the production of gelatinase was significantly decreased, but not the production of stromelysin. When this same effect was studied in herniated lumbar discs that were stimulated with IL-lβ, both gelatinase and stromelysin were significantly reduced. Interestingly, the same study done on the herniated cervical discs stimulated with IL-1β had no significant effect on gelatinase or stromelysin.3
Several other authors have studied MMPs and their association with intervertebral disc degeneration. Fujita et al. studied autopsy specimens of degenerative discs.4 They first discovered serine elastases with high activity in the endplate and nucleus pulposus of degenerative discs. Another group using a monoclonal antibody against MMP3, found the MMP3-positive cell ratio was significantly correlated with the magnetic resonance imaging grade of intervertebral disc degeneration. The MMP3-positive cell ratio observed in prolapsed lumbar intervertebral discs was significantly higher than in nonprolapsed discs. The same study used an anti-TIMP1 monoclonal antibody to demonstrate the normal presence of MMP3 and TIMP1 together in the degenerative intervertebral discs and hypothesized that an imbalance between MMP3 and TIMP may induce degeneration.
IL-1 is a known mediator of mesenchymal cells and probably has a central role in disc degeneration. It is one of the key inflammatory mediators and it has been found in mononuclear cells responding to disc herniations. The studies on human disc tissue have had difficulty demonstrating IL-1β in the intervertebral disc tissues, but when disc cells were stimulated with lipopolysaccharide, elevated levels of IL-1β were found. Both MMP2 (gelatinase) and MMP3 (stromelysin) respond to IL-1. In an experiment using ovine disc cells, Shen et al. demonstrated the ability of IL-1 to enhance the in vitro production of MMP2 and MMP3 by cells of the nucleus pulposus.5 However, the active form of MMP3 predominated over the active form of MMP2 in this model of IL-1 activation. This suggests that, in the presence of IL-1 as an inflammatory mediator, MMP3 may be more intimately involved with ongoing intervertebral disc degeneration than is MMP2.
Therefore, the MMPs appear to be key factors in disc degeneration (Fig. 3.1). They are the active form of the enzymes that they produce, and are capable of degrading constituents of the extracellular matrix and basement membrane at physiologic pH values. Substrates for these MMPs are present in abundance in the disc: collagens II and III are substrates for MMP1, MMP8 and MMP13, as well as their proteoglycans and other minor collagens which are substrates for MMP2 and MMP9. Compared with healthy discs, degenerative discs have been noted to have higher activities of not only MMP3 and MMP7, but also TIMP1. MMP3 activity has been correlated to the size of osteophytes present in disc degeneration. Inhibitors of MMPs have been found in low levels and are constitutively expressed. TIMP2 appears to be released by most cell types within the discs, whereas TIMP1 appears to be exclusively overexpressed in discs with degenerative disease. These expressions of MMPs and TIMPs have also been measured in spines with presumed abnormal biomechanical loading characteristics such as those with scoliosis. Handa et al. showed that proteoglycans and inhibitors of MMPs were produced in increased amounts under hydrostatic conditions when loads were increased to within a normal range.6 Taking these loads to abnormally high pressures resulted in decreased proteoglycan production and an increased production of MMP3.
Much of the work involving the study of MMP activation and measurement has been done using tissue obtained from patients operated on for herniated discs. Therefore, many of these publications include the fact that the assay was done on disc tissue that had been presumably exposed to some type of a burst of inflammation after exposure to the epidural space. It has been proposed that patients with herniated, sequestered, or noncontained herniations may have a more severe inflammatory reaction and pain response. Nygaard et al. looked at 37 patients undergoing surgery for lumbar disc herniation.7 They divided the patients into those who had a bulging disc, a contained or incomplete herniation, or a noncontained or sequestered free disc fragment. Unfortunately, they were unable to recruit enough patients with bulging discs to investigate this phenomenon statistically. In looking at the two groups with the largest number of patients, including the contained herniation group which had 25 members and the noncontained herniation group which had 9 members, there was a significant difference in the mean concentrations of LTB4, with the noncontained group having almost double the concentration versus the contained herniation group. As well, thromboxane B2 was significantly higher in the noncontained versus the contained herniation group. Although the measured concentration of these two proinflammatory cytokines was lower in the bulging disc, their numbers were too small to be included in the statistical analysis. This study seems to support the theory that there are different inflammatory characteristics of different degrees of disc herniations.
One of the other paradoxes in the delineation of an inflammatory response for disc herniation has to do with the atypical cellular response when compared to inflammation occurring at other places in the body. Neutrophils are the sine qua non of acute inflammation; however, they have really only been found in noncontained or sequestered disc fragments where neovascularization may be occurring. Most of the cellular elements that have been identified and are proposed to be the source or factories for most of the inflammatory cytokines are macrophages. Gronblad,8 Nojara,9 Yasuma,10 and Haro11 have identified macrophages as well as vascular proliferation in the granulation tissue of herniated discs. Haro additionally found that inflammatory cells were more abundant in the noncontained group of disc herniations than in the contained group. Inflammatory cells are known to act in an autocrine or paracrine type fashion with regard to their effect on resident cells in the inflammatory process. This must also be true for the degenerative disc. The intervertebral disc, which is normally nourished through diffusion, can become neovascularized to some extent after exposure to the epidural space. These discs display granulation tissue with macrophage and T-lymphocyte infiltration not observed in healthy discs. Haro et al. have proposed that the natural resorption of a herniated disc appears to occur by a vascularization-mediated process and is correlated with macrophage infiltration.
It is also known that chondrocytes replace proteoglycans within the nucleus pulposus and these cells have been proposed to play a very important role in the inflammatory process in regards to production of abnormal types of collagen as well as MMPs and TIMPs in response to abnormal loading characteristics. Haro et al. reported their results in a co-culture system of chondrocytes and macrophages and demonstrated a marked upregulation of MMP3 by disc chondrocytes with the addition of macrophages to the culture system. This resulted in eventual resorption of the disc through macrophage action. They further used MMP-null mice to determine that the production of MMP3 by the chondrocytes was required for macrophage infiltration in disc resorption. In a more recent study, this same group has shown that the production of MMP7 by macrophages was found to be required for infiltration into disc tissue through a mechanism involving the release of soluble TNF-α.12
The support for macrophage-mediated cellular response in herniated disc tissue is also supported by another study by Haro. While macrophage invasion appears to accompany and participate in the inflammatory response, the likely end to this is reabsorption of the herniated disc tissue. Groups have proposed neovascularization of these disc tissues as the means by which this happens. Previous studies have shown that resorption may be mediated by neovascularization as measured through Gd-DTPA MRI. Komori showed that the tendency of these herniated disc tissues to spontaneously resorb was proportional to the degree of Gd-DTPA enhancement, which suggests that the resorption was mediated by a vascular event.13 Haro and his group have shown that, in an in vitro co-culture system they have used previously, an increase in macrophage VEGF protein (vascular endothelial growth factor) and mRNA expression was observed after they exposed disc tissue to the co-culture.14 They found TNF-α was required for induction of VEGF protein and conclude that this may be one mechanism for resorption for herniated disc tissue.
Further evidence for the involvement of the macrophage and its importance is shown in the paper by Burke et al.15 This group studied the production of monocyte chemoattractant protein-1 (MCP1) and interleukin-8 (IL-8) by intervertebral discs removed after surgery. Burke found that MCP1 and IL-8 were detected in both the control and herniated disc specimens and that the noncontained herniated samples contained higher levels of these chemokines than those with an intact anulus. They proposed that the MCP1 production attracts the macrophages while IL-8 may influence the angiogenesis or the neovascularization that is seen in these samples. Although the stimulus for MCP1 in this in vitro experiment was not investigated and is as yet unknown, this may represent a physiologic mechanism for initiation of macrophage infiltration after disc prolapse and the process of disc resorption. IL-8 was also strongly influenced by the noncontained morphology of these samples. In addition to the angiogenic properties of IL-8, it is also chemotactic for T cells that have been identified in the chronic inflammatory filtrate around disc herniations.
In addition to TNF activation of or paracrine/autocrine effects governing MMP production, TNF-αγ has long been regarded to be a key player in mediating the sensitization of nerve roots by material from the nucleus pulposus, and other effects such as edema, intervascular coagulation, reduction in blood flow, and the splitting of myelin. TNF-αγ is known to be released from the chondrocyte resident cells in the nucleus pulposus. In a local application of TNF-αγ, it induced a reduction in nerve conduction velocity in a porcine experiment done by Aoki et al.16 In this study, applications of interleukin-1β and interferon-γ induced a very small reduction of nerve velocity compared with epidural fat. In a follow-up study to this, Olmarker and Rydevik demonstrated that local blockers to TNF-αγ prevented the reduction of nerve conduction velocity and seemed to limit the nerve fiber injury and intercapillary thrombus formation, as well as the intraneural edema seen in the absence of the inhibitor.17 These authors have suggested that TNF-αγ inhibitors may be important therapeutically in the future. Presently, synthesis of TNF-αγ can be blocked with systemic corticosteroids, IL-10, TGF-βγ or by other drugs such as chlorpromazine, pentoxifylline, or ciclosporin. However, these drugs are non-specific inhibitors and may result in side effects that would be undesirable. Presently, there are anti-TNF agents being used in the treatment of rheumatoid arthritis. The first of these, infliximab (Remicade), was quickly followed by etanercept (Enbrel). Recently, a monoclonal antibody against TNF-αγ, adalimumab (Humira), has been released. These agents are not presently approved for treatment of sciatic pain, but have given sufferers of rheumatoid arthritis a further dimension for their treatment.
Another potent inflammatory mediator that is also induced by TNF-αγ is nitric oxide. Nitric oxide is a particularly interesting compound in that it has been shown to act in various ways depending on the tissues that in which it resides. In bone, mechanical stress affects intracellular cyclic AMP, calcium, and PGE2 levels, as well as having effects on matrix synthesis. It has been demonstrated that nitric oxide is a key mediator of these processes. Articular chondrocytes have been shown to produce large amounts of nitric oxide. As described in the preceding sections introducing the inflammatory process, nitric oxide is produced in several forms including the inducible form that is present in chondrocytes. Kang et al. first showed the spontaneous production of nitric oxide from human lumbar discs and that this production was higher in herniated discs than normals.2 In a follow-up study, Kang et al. examined the effects of IL-1β on normal and herniated disc tissue. They found that the addition of IL-1βγ caused a significant increase in the production of nitric oxide as well as IL-6 and PGE2.3 While these inflammatory mediators were sharply increased in both normal and herniated disc tissue, the interesting point to this paper was that MMP production did not change in the herniation disc material, while the normal disc showed a sharp increase in the production of MMPs. It is also noted by this group that endogenously produced nitric oxide had a large inhibitory effect on IL-6.
PUTTING IT ALL TOGETHER
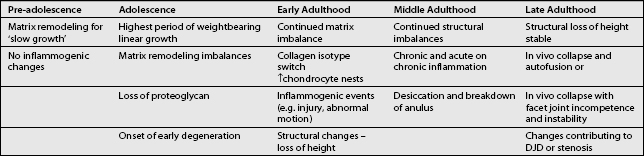
The inflammatory basis for intervertebral disc degeneration likely begins at or around the time of puberty when linear growth accelerates. It is possible that the rapid growth rate seen during this time outstrips the ability of the intervertebral disc to remodel effectively, leading to imbalances in MMP and TIMP concentrations. This may be further enhanced by increased diffusional demands for nutrition and a less than desirable pH balance within the disc (Table 3.2).
1 Stokes I, Greenapple DM. Measurement of surface deformation of soft tissue. J Biomech. 1985;18:107.
2 Kang JD, Georgescu HI, McIntyre-Larkin L, et al. Herniated lumbar intervertebral discs spontaneously produce matrix metalloproteinases, nitric oxide, interleukin-6, and prostaglandin E2. Spine. 1996;21(3):271-277.
3 Kang JD, Stefanovic-Racic M, McIntyre LA, et al. Toward a biochemical understanding of human intervertebral disc degeneration and herniation: contributions of nitric oxide, interleukins, prostaglandin E2, and matrix metalloproteinases. Spine. 1997;22(10):1065-1073.
4 Fujita K, Nakagawa T, Hirabayashi K, et al. Neutral proteinases in human intervertebral disc: role in degeneration and probable origin. Spine. 1993;18(13):1766-1773.
5 Shen B, Melrose J, Ghosh P, et al. Induction of matrix metalloproteinase-2 and -3 activity in ovine nucleus aragose gel culture by interleukin-1β: a potential pathway of disc degeneration. Eur Spine J. 2003;12:66-75.
6 Handa T, Ishihara H, Ohshima H, et al. Effects of hydrostatic pressure on matrix synthesis and matrix metalloproteinase production in the human lumbar intervertebral disc. Spine. 1997;22(10):1085-1091.
7 Nygaard ØP, Mellgren SI, Østerud B. The inflammatory properties of contained and noncontained lumbar disc herniation. Spine. 1997;22(21):2484-2488.
8 Gronblad M, Virri J, Ronkko S, et al. Type (group II) phospholipase A2 and inflammatory cells in macroscopically normal, degenerated, and herniated human lumbar disc tissues. Spine. 1996;21(22):2531-2538.
9 Nojara. Marseilles: ISSLS Presentation; 1993.
10 Yasuma T, Arai K, Yamauchi Y. The histology of lumbar intervertebral disc herniation. The significance of small blood vessels in the extruded tissue. Spine. 1993;18(13):1761-1765.
11 Haro H, Shinomiya K, Komori H, et al. Upregulated expression of chemokines in herniated nucleus pulposus resorption. Spine. 1996;21:1647-1652.
12 Haro H, Crawford HC, Fingleton B, et al. Matrix metalloproteinase-7-dependent release of tumor necrosis-αγ in a model of herniated disc resorption. J Clin Invest. 2000;105(2):143-150.
13 Komori H, Okawa A, Haro H, et al. Contrast-enhanced magnetic resonance imaging in conservative management lumbar disc herniation. Spine. 1998;23(1):67-73.
14 Haro H, Kato T, Komori H, et al. Vascular endothelial growth factor (VEGF)-induced angiogenesis in herniated disc resorption. J Orthop Res. 2002;20(3):409-415.
15 Burke JG, Watson RWG, McCormack D, et al. Spontaneous production of monocyte chemoattractant protein-1 and interleukin-8 by the human lumbar intervertebral disc. Spine. 2002;27(13):1402-1407.
16 Aoki Y, Rydevik B, Kikuchi S, et al. Local application of disc-related cytokines on spinal nerve roots. Spine. 2002;27(15):1614-1617.
17 Olmarker K, Rydevik B. Selective inhibition of tumor necrosis factor-α prevents nucleus pulposus-induced thrombus formation, intraneural edema, and reduction of nerve conduction velocity. Spine. 2001;26(8):863-869.
Adams MA, Hutton WC. 1981 Volvo Award in Basic Science. Prolapsed intervertebral disc. A hyperflexion injury. Spine. 1982;7(3):184-191.
Ahn SH, Cho YW, Ahn MW, et al. mRNA expression of cytokines and chemokines in herniated lumbar intervertebral discs. Spine. 2002;27(9):911-917.
An HS, Thonar EJ-MA, Masuda K. Biological repair of intervertebral disc. Spine. 2003;28(15):S86-S92.
Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines-CXC and CC chemokines. Adv Immunol. 1994;55:97-179.
Brisby H, Byröd G, Olmarker K, et al. Nitric oxide as a mediator of nucleus pulposus-induced effects on spinal nerve roots. J Orthop Res. 2000;18(5):815-820.
Burke JG, Watson RWG, McCormack D, et al. Intervertebral discs which cause low back pain secrete high levels of proinflammatory mediators. J Bone Joint Surg Br. 2002;84(2):196-201.
Burke JG, Watson RWG, Conhyea D, et al. Human nucleus pulposus can respond to a pro-inflammatory stimulus. Spine. 2003;28(24):2685-2693.
Caron JP, Fernandes JC, Martel-Pelletier J, et al. Chondroprotective effect of intraarticular injections of interleukin-1 receptor antagonist in experimental osteoarthritis. Suppression of collagenase-1 expression. Arthritis Rheum. 1996;39(9):1535-1544.
Collier S, Ghosh P. The role of plasminogen in interleukin-1 mediated cartilage degradation. J Rheumatol. 1988;15(7):1129-1137.
Cooper RG, Freemont AJ, Hoyland JA, et al. Herniated intervertebral disc-associated periradicular fibrosis and vascular abnormalities occur without inflammatory cell infiltration. Spine. 1995;20(5):591-598.
Coppes MH, Marani E, Thomeer RTWM, et al. Innervation of ‘painful’ lumbar discs. Spine. 1997;22(20):2342-2350.
Dayer JM, de Rochemonteix B, Burrus B, et al. Human recombinant interleukin-1 stimulates collagenase and prostaglandin E2 production by human synovial cells. J Clin Invest. 1986;77:645-648.
Dean DD, Martel-Pelletier J, Pelletier JP, et al. Evidence for metalloproteinase and metalloproteinase inhibitor imbalance in human osteoarthritic cartilage. J Clin Invest. 1989;84:678-685.
DiPasquale G, Caccese R, Pasternak R, et al. Proteoglycan- and collagen-degrading enzymes from human interleukin 1-stimulated chondrocytes from several species: proteoglycanase and collagenase inhibitors as potentially new disease-modifying antiarthritic agents (42416). Proc Soc Exp Biol Med. 1986;183(2):262-267.
Doers TM, Kang JD. The biomechanics and biochemistry of disc degeneration. Curr Opin Orthop. 1999;10:117-121.
Doita M, Kanatani T, Ozaki T, et al. Influence of macrophage infiltration of herniated disc tissue on the production of matrix metalloproteinases leading to disc resorption. Spine. 2001;26(14):1522-1527.
Edwards DR, Murphy G, Reynolds JJ, et al. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. EMBO J. 1987;6(7):1899-1904.
Evans CH, Watkins SC, Stefanovic-Racic M. Nitric oxide and cartilage metabolism. Methods Enzymol. 1996;269:75-88.
Eyre DR, Muir H. Types I and II collagens in intervertebral disc. Interchanging radial distributions in anulus fibrosus. Biochem J. 1976;157:267-270.
Eyre DR, Muir H. Quantitative analysis of types I and II collagens in human intervertebral discs at various ages. Biochim Biophys Acta. 1977;492:29-42.
Fox SW, Chambers TJ, Chow JW. Nitric oxide is an early mediator of the increase in bone formation by mechanical stimulation. Am J Physiol. 1996;270:E955-E960.
Franson RC, Saal JS, Saal JA. Human disc phospholipase A2 is inflammatory. Spine. 1992;17(6S):S129-S132.
Freemont AJ, Peacock TE, Goupille P, et al. Nerve ingrowth into diseased intervertebral disc in chronic back pain. Lancet. 1997;350(9072):178-181.
Freemont AJ, Watkins A, Maitre CL, et al. Current understanding of cellular and molecular events in intervertebral disc degeneration: implications for therapy. J Pathol. 2002;196(4):374-379.
Gaetani P, Rodriguez y Baena R, Riva C, et al. Collagenase-1 and stromelysin distribution in fresh human herniated intervertebral disc: a possible link to the in vivo inflammatory reactions. Neurol Res. 1999;21(7):677-681.
Goupille P, Jayson MIV, Valat JP, et al. Matrix metalloproteinases: the clue to intervertebral disc degeneration. Spine. 1998;23(14):1612-1626.
Goupille P, Jayson MIV, Valat JP, et al. The role of inflammation in disk herniation-associated radiculopathy. Semin Arthritis Rheum. 1998;28(1):60-71.
Grabowski PS, Wright PK, Van’t Hof RJ, et al. Immunolocalization of inducible nitric oxide synthase in synovium and cartilage in rheumatoid arthritis and osteoarthritis. Br J Rheumatol. 1997;36:651-655.
Grange L, Gaudin P, Trocme C, et al. Intervertebral disk degeneration and herniation: the role of metalloproteinases and cytokines. Joint Bone Spine. 2001;68(6):547-553.
Habtemariam A, Gronblad M, Virri J, et al. A comparative immunohistochemical study of inflammatory cells in acute-stage and chronic-stage disc herniations. Spine. 1998;23(20):2159-2165.
Hashizume H, Kawakami M, Nishi H, et al. Histochemical demonstration of nitric oxide in herniated lumbar discs. Spine. 1997;22(10):1080-1084.
Häuselmann HJ, Oppliger L, Michel BA, et al. FEBS Letts. 1994;352:361-364.
Horwitz AL, Hance AJ, Crystal RG. Granulocyte collagenase: selective digestion of type I relative to type III collagen. Proc Natl Acad Sci USA. 1977;74(3):897-901.
Igarashi T, Kikuchi S, Shubayev V, et al. 2000 Volvo Award Winner in Basic Science Studies. Exogenous tumor necrosis factor-alpha mimics nucleus pulposus-induced neuropathology. Spine. 2000;25(23):2975-2980.
Kääpä E, Han X, Holm S, et al. Collagen synthesis and types I, III, IV, and VI collagens in an animal model of disc degeneration. Spine. 1995;20(1):59-66.
Kanemoto M, Hakuda S, Komiya Y, et al. Immunohistochemical study of matrix metalloproteinase-3 and tissue inhibitor of metalloproteinase-1 human intervertebral discs. Spine. 1996;21(1):1-8.
Kanerva A, Kommonen B, Gronblad M, et al. Inflammatory cells in experimental intervertebral disc injury. Spine. 1997;22(23):2711-2715.
Koch H, Reinecke JA, Meijer H, et al. Spontaneous secretion of interleukin 1 receptor antagonist (IL-1Ra) by cells isolated from herniated lumbar discal tissue after discectomy. Cytokine. 1998;10(9):703-705.
Kokkonen SM, Kurunlahti M, Tervonen O, et al. Endplate degeneration observed on magnetic resonance imaging of the lumbar spine. Spine. 2002;27(20):2273-2278.
Lefebvre V, Peeters-Joris C, Vaes G. Modulation by interleukin 1 and tumor necrosis factor-α of production of collagenase, tissue inhibitor of metalloproteinases and collagen types in differentiated and dedifferentiated articular chondrocytes. Biochim Biophys Acta. 1990;1052:366-378.
Liu GZ, Ishihara H, Osada R, et al. Nitric oxide mediates the change of proteoglycan synthesis in the human lumbar intervertebral disc in response to hydrostatic pressure. Spine. 2001;26(2):134-141.
Liu J, Roughley PJ, Mort JS. Identification of human intervertebral disc stromelysis and its involvement in matrix degradation. J Orthop Res. 1991;9(4):568-575.
Lotz M, Guerne PA. Interleukin-6 induces the synthesis of tissue inhibitor of metalloproteinases-1/erythroid potentiating activity (TIMP-1/EPA). J Biol Chem. 1991;266(4):2017-2020.
Maroudas A, Stockwell A, Nachemson A, et al. Factors involved in the nutrition of the human lumbar intervertebral disc: cellularity and diffusion of glucose in vitro. J Anat. 1975;120(1):113-130.
Martel-Pelletier J, McCollum R, Fujimoto N, et al. Excess of metalloproteases over tissue inhibitor of metalloprotease may contribute to cartilage degradation in osteoarthritis and rheumatoid arthritis. Lab Invest. 1994;79(6):807-815.
Matrisian LM. Metalloproteinases and their inhibitor in matrix remodeling. Trends Genet. 1990;6(4):121-125.
Meachim G, Cornah MS. Fine structure of juvenile human nucleus pulposus. J Anat. 1970;107(2):337-350.
Melrose J, Ghosh J, Taylor TKF. Neutral proteinases of the human intervertebral disc. Biochim Biophys Acta. 1987;923:483-495.
Melrose J, Ghosh J, Taylor TKF, et al. The serine proteinase inhibitory proteins of the human intervertebral disc: their isolation, characterization and variation with aging and degeneration. Matrix. 1992;12:456-470.
Miyamoto H, Saura R, Harada T, et al. The role of cyclooxygenase-2 and inflammatory cytokines in pain induction of herniated lumbar intervertebral disc. Kobe J Med Sci. 2000;46:13-28.
Mort JS, Dodge GR, Roughley PJ, et al. Direct evidence for active metalloproteinases mediating matrix degradation in interleukin 1-stimulated human articular cartilage. Matrix. 1993;13:95-102.
Nagano T, Yonenobu K, Miyamoto S, et al. Distribution of the basic fibroblast growth factor and its receptor gene expression in normal and degenerated rat intervertebral discs. Spine. 1995;20(18):1972-1978.
Ng SCS, Weiss JB, Quennel R, et al. Abnormal connective tissue degrading enzyme patterns in prolapsed intervertebral discs. Spine. 1986;11(7):695-701.
Olmarker K, Larsson K. Tumor necrosis factor alpha and nucleus pulposus-induced nerve root injury. Spine. 1998;23(23):2538-2544.
Özaktay AC, Cavanaugh JM, Asik I, et al. Dorsal root sensitivity to interleukin-1 beta, interleukin-6 and tumor necrosis factor in rats. Eur Spine J. 2002;11(5):467-475.
Park JB, Kim KW, Han CW, et al. Expression of Fas receptor on disc cells in herniated lumbar disc tissue. Spine. 2001;26(2):142-146.
Park JB, Chang H, Kim YS. The pattern of interleukin-12 and T-helper types 1 and 2 cytokine expression in herniated lumbar disc tissue. Spine. 2002;27(19):2125-2128.
Pearce RH, Mathieson JM, Mort JS, et al. Effect of age on the abundance and fragmentation of link protein of the human intervertebral disc. J Orthop Res. 1989;7(6):861-867.
Pendás AM, Knäuper V, Puente XS, et al. Identification and characterization of a novel human matrix metalloproteinase with unique structural characteristics, chromosomal location, and tissue distribution. J Biol Chem. 1997;272(7):4281-4286.
Roberts S, Menage J, Duance V, et al. 1991 Volvo Award in Basic Sciences. Collagen types around the cells of the intervertebral disc and cartilage end plate: an immunolocalization study. Spine. 1991;16(9):1030-1038.
Saal JS, Franson RC, Dobrow R, et al. High levels of inflammatory phospholipase A2 activity in lumbar disc herniations. Spine. 1990;15(7):674-678.
Sakurai H, Kohsaka H, Liu MF, et al. Nitric oxide production and inducible nitric oxide synthase expression in inflammatory arthritides. J Clin Invest. 1995;96:2357-2363.
Sedowofia KA, Tomlinson IW, Weiss JB, et al. Collagenolytic enzyme systems in human intervertebral disc. Their control, mechanism, and their possible role in the initiation of biomechanical failure. Spine. 1982;7(3):213-222.
Shinmei M, Kikuchi T, Yamagishi M, et al. The role of interleukin-1 on proteoglycan metabolism of rabbit anulus fibrosus cells cultured in vitro. Spine. 1988;13(11):1284-1290.
Specchia N, Pagnotta A, Toesca A, et al. Cytokines and growth factors in the protruded intervertebral disc of the lumbar spine. Eur Spine J. 2002;11(2):145-151.
Stadler J, Stefanovic-Racic M, Billiar TR, et al. Articular chondrocytes synthesize nitric oxide in response to cytokines and lipopolysaccharide. J Immunol. 1991;147:3915-3920.
Takahashi H, Suguro T, Okazima Y, et al. Inflammatory cytokines in the herniated disc of the lumbar spine. Spine. 1996;21(2):218-224.
Tengblad A, Pearce RH, Grimmer BJ. Demonstration of link protein in proteoglycan aggregates from human intervertebral disc. Biochem J. 1984;222(1):85-92.
Tolonen J, Grönblad M, Virri J, et al. Oncoprotein c-Fos and c-Jun immunopositive cells and cell clusters in herniated intervertebral disc tissue. Eur Spine J. 2002;11(5):452-458.
Willburger RE, Wittenberg RH. Prostaglandin release from lumbar disc and facet joint tissue. Spine. 1994;19(18):2068-2070.