[level-membership-for-pediatrics-category]
Chapter 104 Inflammation and Immunity
Systemic Inflammatory Response Syndrome, Sepsis, Acute Lung Injury, and Multiple Organ Failure
The Inflammation/Coagulation/Immune Dysfunction/Dysregulated Metabolism Hypothesis
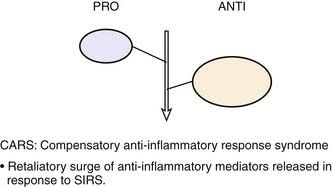
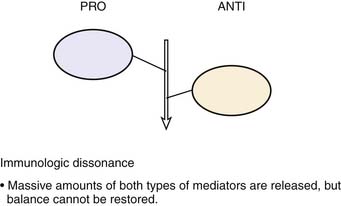
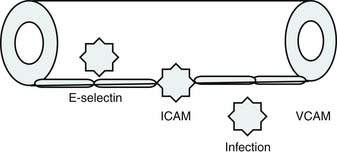
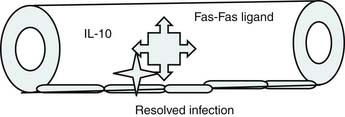
Roger Bone popularized the notion that systemic inflammation led to the development of sepsis syndrome and MOF in the 1990s. According to this theory, an initial proinflammatory cytokine response to foreign or self-antigen caused SIRS, a systemic inflammatory response characterized by two of four criteria: tachycardia, tachypnea, fever, and leukocytosis. This syndrome was thought to be mediated by increased interleukin (IL)-1 and tumor necrosis factor (TNF)-α levels (Figure 104-1). It could be blunted by nonsteroidal antiinflammatory drugs and hence was prostaglandin mediated. A systemic inflammatory response was called sepsis if related to infection and SIRS if related to surgery, pancreatitis, or other noninfectious processes. This syndrome was self-limited when antibiotics were used to kill the infection or time allowed recovery from surgery, pancreatitis, and other noninfectious causes. This proinflammatory response was turned off by an antiinflammatory response led by the antiinflammatory cytokine IL-10. When IL-10 resulted in reduction of IL-1 and TNF-α to normal control values, sepsis or SIRS stopped and organ failure did not occur (Figure 104-2). In patients who developed severe sepsis and MOF, the compensatory anti-inflammatory response was unsuccessful in turning off inflammation. Instead, these patients had persistent elevation of the cytokine IL-6 along with IL-10. Bone coined the term immunologic dissonance to describe this process. He viewed MOF as an inflammatory condition in which the normal antiinflammatory response was unable to turn off the proinflammatory process (Figure 104-3).
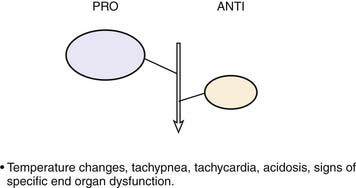
Figure 104–1 SIRS shows proinflammation self-limited by antiinflammation and eradication of the source of inflammation.
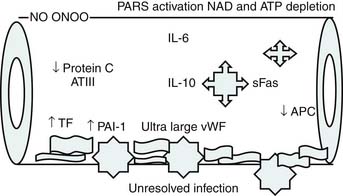
Proulx and colleagues1 defined pediatric MOF syndromes as primary and secondary. Primary MOF occurred in 90% of children and was present at the time of admission, and secondary MOF occurred in 10% of children and was not present until 7 days after admission.1 To evaluate the role of inflammation in pediatric sepsis-induced MOF, this group defined children as having no MOF (NMOF) if they had two-organ failure at presentation but never reached three-organ failure, resolved MOF (RMOF) if they had three-organ failure at admission that resolved to two-organ or less at 48 hours, persistent MOF (PMOF) if they had three or more organ failures at presentation that did not resolve by 48 hours, and sequential MOF (SMOF) if they had acute lung injury/adult respiratory distress syndrome at admission and then progressed to hepatorenal failure/dysfunction.2 Similar to Proulx’s classification, children in the latter investigation with SMOF represented 10% of the population. The mortality rates with NMOF, RMOF, PMOF, and SMOF were 2%, 8%, 35%, and 50%, respectively. The authors then investigated Bone’s hypothesis in this categories and reported that patients with NMOF had an increase in IL-10 with subsequent turning off of inflammation; however, those with PMOF or SMOF had persistently high IL-6 and IL-10 levels—the hallmark of Bone’s “immunologic dissonance.”2
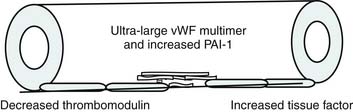
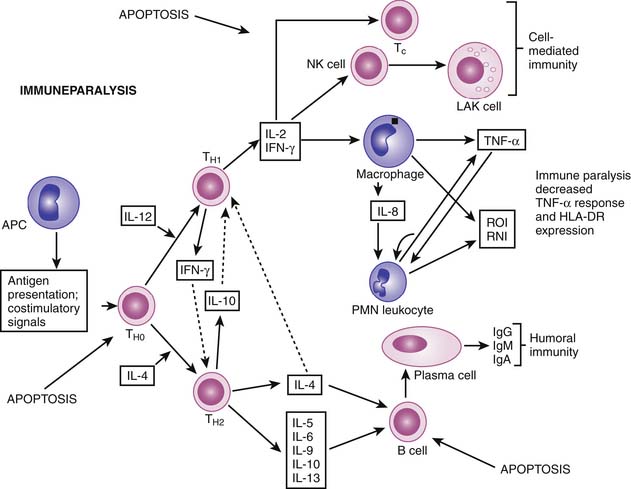
Bone’s untimely death prevented him from further pursuing the mechanisms by which immunologic dissonance could mediate organ injury. However, Proulx et al. first reviewed the autopsy bank at their hospital and noted that children who died from sepsis-induced MOF exhibited two unsuspected findings compared with children who died of other causes: persistent or unrecognized infection and thrombotic and bleeding complications with organ infarctions. Accordingly, they pursued the notion that immunologic dissonance might lead to an inability to mediate coagulation homeostasis as well as an inability to clear infection. Proinflammatory cytokines including TNF-α and IL-1 induce endothelial activation in vitro with an increased prothrombotic and antifibrinolytic state and expression of adhesion molecules (Figures 104-4 to 104-6). Patients with PMOF had increased tissue factor and plasminogen activator-1 activity as well as circulating adhesion molecules, intercellular adhesion molecule, and vascular adhesion molecule consistent with the hypothesis that immunologic dissonance was associated with an activated prothrombotic/antifibrinolytic endothelium.3,4 IL-6 also inhibits ADAMTS-13, the von Willebrand factor (vWF) multimer-cleaving protease, in vitro. PMOF patients also had deficient ADAMTS-13 activity compared with NMOF patients, consistent with increased risk of vWF multimer–mediated thrombosis.5 Increased IL-10 is the hallmark of the TH2 antiinflammatory cytokine response. When the TH2 response is dominant, the TH1 response can be turned off, a state known as immune paralysis (Figure 104-7). These patients have a diminished ex vivo ability to secrete TNF-α from endotoxin-stimulated monocytes or macrophages. Without this ability, infections cannot be terminated. PMOF patients exhibited immune paralysis more frequently than did NMOF patients.6
Cytokines and nitric oxide also inhibit cytochrome P450 activity and mitochondrial respiration in vitro. One study group found that patients with PMOF had reduced cytochrome P450 (10-fold) activity compared with patients with NMOF (twofold).7 Immunologic dissonance in PMOF was associated with endotheliopathy, immune paralysis, and poor metabolism, but how was PMOF different from SMOF? The authors determined that patients with SMOF were more likely to have viral or lymphoproliferative disease with very high soluble Fas ligand (sFasL) levels. In vitro, sFasL levels greater than 500 pg/mL cause hepatocyte necrosis. Children with MOF with sFasL levels greater than 500 pg/mL demonstrated liver injury at autopsy.8 The mechanism of injury in SMOF was different than in patients who presented with MOF (Figure 104-8).
In this decade, investigators from the United Kingdom have investigated the inflammation/immune dysregulation hypothesis in SIRS/sepsis, hypothesizing that reduced complement activity increases the risk of SIRS/sepsis.9 Mannose-binding lectin (MBL) is the first complement component released and is required for the first 12 hours of complement activity. Approximately 30% of children are MBL deficient. Complement factor H inhibits complement activity. The complement H factor H Y402H polymorphism reduces this complement inhibition activity. Using genotype analyses, these investigators have demonstrated that children at risk for SIRS/sepsis more commonly have deficiencies in MBL production and a reduction in this H factor polymorphism. These two polymorphisms together lead to an even greater risk of SIRS/sepsis than either alone. The interpretation is that diminished complement function prevents rapid clearance of antigen, leading to the SIRS/sepsis syndrome. The inflammation hypothesis is alive and well in SIRS, sepsis, severe sepsis, and MOF.
Definitions and Scoring
As previously noted, SIRS is defined by two of the following four criteria: tachypnea, tachycardia, fever, and leukocytosis. Sepsis is defined as SIRS plus suspected or documented infection. Severe sepsis is defined as sepsis plus organ failure. Acute lung injury is defined by bilateral pulmonary infiltrates and a PaO2/FIO2 ratio less than 300. MOF or multiple organ dysfunction syndrome is defined by failure of more than one organ. Many definitions exist for defining organ failure. Wilkinson and Pollack10 published the first definitions in pediatrics, which were subsequently modified by Proulx and then again by Doughty.2 Goldstein and colleagues11 subsequently developed two more sets of definitions. Doughty et al.2 developed the organ failure index to score organ failure. This index is a simple integer score that assigns 1 point for each organ failure. Leteurtre and colleagues12 reported two more MOF scores, the Pediatric Multiple Organ Dysfuction (PEMOD) and the Pediatric Logistic Organ Dysfunction (PELOD) scores.12 The PEMOD score assigns 1 to 4 points per organ failure depending on severity. The PELOD score logistically weights these scores in the hope of allowing for prognostication. It remains unclear whether the PELOD score can be used in this manner.
Outcomes
In the United States, survival in the setting of SIRS/sepsis is almost universal. Odetola and colleagues13 reported that survival in severe sepsis is 96%, 98% overall in previously healthy children and 92% in children with chronic diseases. Typpo and colleagues14 reported that survival from MOF is 90%. Survival rates decrease as the number of organ failures increase.
Multiple Organ Failure/Dysfunction Phenotypes, Respective Biomarkers, and Therapies
Thrombocytopenia-Associated Multiple Organ Failure
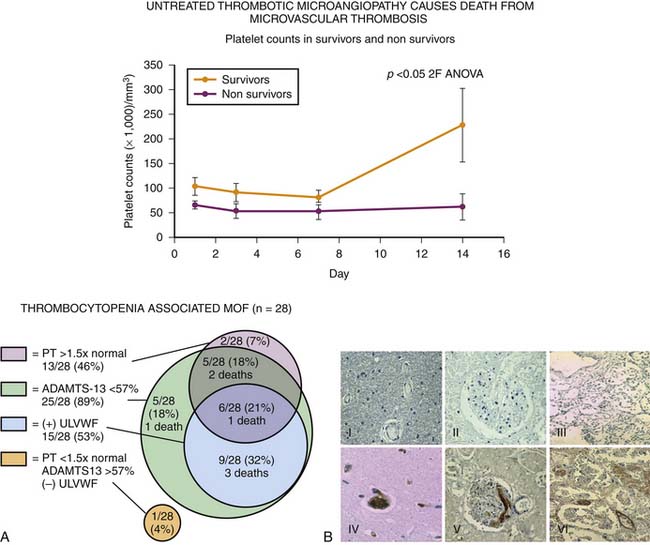
New-onset thrombocytopenia is a harbinger of thrombotic microangiopathy in MOF, especially when accompanied by increased lactate dehydrogenase and reduced renal function (Figure 104-9). Until the current decade, thrombotic microangiopathy of critical illness was believed to be mediated by fibrin, but more recent work suggests that vWF multimer–associated platelet thrombi are an unrecognized cause. If ADAMTS-13 activities (vWF-cleaving protease) are less than 57% of control and vWF multimer antigen levels are increased, this form of thrombosis should be highly suspected. Intensive plasma exchange can be required to reverse this process because it removes the thrombogenic ultralarge vWF multimers and replenishes ADAMTS-13 (vWF-cleaving protease) activity.5,15
Immune Paralysis and Lymphoid Depletion Syndrome
Adult and neonatal investigators noted the associations among lymphocyte apoptosis, lymphoid depletion, and death from nosocomial sepsis. When dendritic cells, monocytes, or macrophages ingest these apoptotic bodies, a profound TH2 response ensues, with immune paralysis and death associated with uneradicated infection (Figure 104-10). Pediatric studies corroborate these findings with immunoparalysis and prolonged lymphopenia associated with nosocomial infection and death.6 In transplant recipients, rapid tapering of immune suppressants can restore monocyte function and reverse immune paralysis. In non-immune-suppressed patients, low-dose subcutaneous granulocyte-macrophage colony-stimulating factor (GM-CSF) can reverse immune paralysis. If patients have hypogammaglobulinemia, intravenous immunoglobulin (IVIG) can be given. If the patient is severely lymphopenic, antimicrobial prophylaxis can be considered.
Viral/Lymphoproliferative Disease–Associated Sequential or Liver-Associated Multiple Organ Failure Syndrome
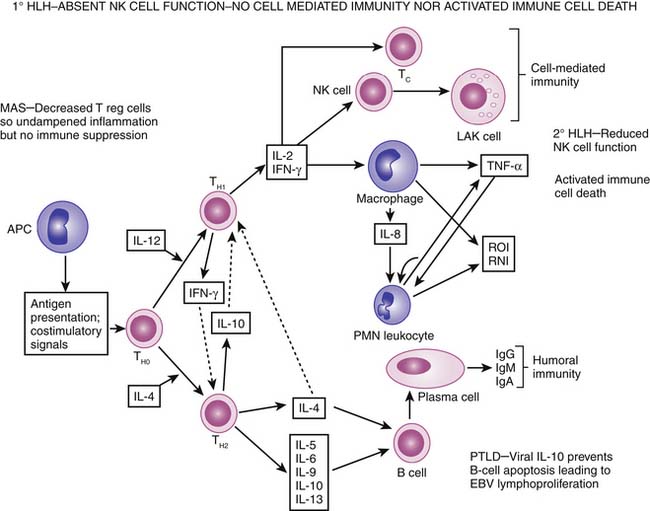
Viral/lymphoproliferative disease associated with SMOF or liver-associated MOF represents only 10% of MOF, but this diagnosis carries the worst prognosis (Figure 104-11). Liver injury is associated with high sFasL levels (>500 pg/mL).8 In transplant recipients, the most common cause is posttransplant lymphoproliferative disease. Epstein-Barr virus intercalates the viral IL-10 genome into B cells, preventing activated immune cell death and promoting lymphoproliferation. This is treated by stopping immune suppression, administering the B-cell monoclonal antibody rituximab, and administering interferon-α if necessary. In nontransplant patients with a family history of a similar disease, primary hemophagocytic lymphohistiocytosis (HLH) must be considered. These patients have absent natural killer (NK)/cytotoxic T-lymphocyte (CTL) cell activity secondary to mutations in perforin signaling, rendering them unable to kill viruses and cancer cells. Furthermore, these patients cannot direct programmed cell death in activated immune cells. These patients are treated with chemotherapy and bone marrow transplantation to kill activated immune cells and replenish functioning NK/CTL cells. In patients with rheumatologic disease, this syndrome may be caused by macrophage activation syndrome (MAS) as a result of decreased T-regulatory cell activity (see Chapter 98). These patients respond well to pulse steroids. Patients with infection-associated or secondary HLH will not have the rash, arthralgias, or serositis of MAS or the family history of primary HLH. These patients have decreased but not absent NK/CTL cell function. Therefore they can kill viruses and cancer cells and induce activated immune cell death, but at a slower rate. These patients respond well to eradication of infection with supportive therapy. In addition, IVIG, plasma exchange, and interferon-α have been reported as successful therapies.16
Therapeutic Approaches to Systemic Inflammatory Response Syndrome, Sepsis, Acute Lung Injury, and Multiple Organ Failue
A patient with SIRS can be distinguished from a patient with sepsis by history. An antecedent history of surgery, pancreatitis, or trauma is supportive of SIRS. The prototype for SIRS can be found in the patient with a spinal fusion or the postoperative cardiac surgery patient. Tissue injury induces the inflammatory complement/cytokine-driven response of tachycardia, tachypnea, fever, and leukocytosis. If the systemic inflammatory response worsens or occurs de novo several days after the antecedent insult, sepsis should be considered and infection suspected. SIRS is treated by watchful waiting, fluid resuscitation if hypovolemia is suspected, and temperature control as required. Acute lung injury should be treated with supplemental oxygen as needed. SIRS is thought to be a self-limited response that can be prolonged in the presence of decreased complement production found in one third of the population who have MBL and complement H polymorphisms. Of note, the rate of tachycardia and tachypnea decreases with time. If sepsis is suspected, antibiotics should be given.
If SIRS/sepsis progresses to organ failure or MOF, it is no longer a self-limited disease. Further support and interventions are needed (Box 104-1). If acute lung injury progresses, intubation and mechanical ventilation will be needed. Intubation with ketamine as an induction agent in this setting is antiinflammatory and will not interfere with the adrenal stress response. Pulmonary capillary and epithelium leak will result in indirect ARDS, and the patient will need positive end-expiratory pressure to facilitate adequate oxygenation. Protective lung ventilation is recommended. Use of high lung volumes results in increased systemic inflammation and immune paralysis. For this reason, an effective tidal volume of no more than 8 mL/kg/breath is recommended. Cardiovascular management should be goal directed in a time-sensitive manner. Fluid and inotrope administration targeting capillary refill of less than 2 seconds and normal blood pressure in the emergency department is associated with a 40% reduction in mortality rate.17 Fluid, inotrope, and blood resuscitation directed to attaining superior vena cava oxygen saturation greater than 70% reduced the incidence of MOF (renal and neurologic failure in particular) and mortality rate fourfold in a randomized controlled trial.18 The use of antibiotics and antitoxins in a time-sensitive and appropriate manner is paramount. In a retrospective cohort study involving 14 North American adult intensive care units, septic patients received effective antibiotics on average 6 hours after the onset of hypotension. Administration of an effective antimicrobial within the first hour of documented hypotension was associated with a survival rate of 79.9%. However, for each subsequent hour delay in administering an effective antimicrobial, average survival decreased by 7.6%.19
Box 104–1 Nonspecific Therapies to Prevent and Resolve MOF
ACCM, American College of Critical Care Medicine; PALS, Pediatric Advanced Life Support.
Unresolving MOF should evoke suspicion that the source of infection is not being eradicated. Persistent inflammation with thrombosis and infection is the cause of PMOF. Careful review of the minimum inhibitory concentration profile of the infection should be performed and antibiotics with the lowest possible minimum inhibitory concentration should be used. If the infection is toxin producing, clindamycin should be given to stop production and IVIG administered to neutralize the toxic effects. The nidus of infection may need to be removed surgically. Barie and colleagues20 found that surgical sepsis mortality rate was 96% with failed nidus removal compared with only 4% with successful removal. Primary or acquired immune deficiency syndromes can also be the cause of uneradicated infection. Primary immunodeficiencies are usually suspected in children younger than 5 years but can present at any time in life. Initial screening for these syndromes includes white blood cell count, lymphocyte subset, complement level, gammaglobulin level, and tests for chronic granulomatous disease. Secondary immune deficiencies include severe neutropenia (absolute neutrophil count <500) and severe graft-versus-host disease (grade III and above). Neutropenia is most commonly related to bone marrow suppression induced by chemotherapy. Infection eradication and subsequent resolution of MOF occur only after restoration of the neutrophil counts. The pathobiology of graft-versus-host disease is one of donor T cells being directed against host epithelial cells. This results in denudation of the epithelial cell barrier with secondary bacterial and fungal infections. Treatment is difficult but is directed at stopping T cell–mediated epithelial destruction without concomitantly stopping the host’s ability to kill infection. This is the most difficult MOF syndrome to treat.
Hemolysis is another cause of unresolving MOF. Free hemoglobin becomes hemoglobin peroxidase when exposed to superoxide and hydrogen peroxide generated by activated white blood cells. Hemoglobin peroxidase can lead to endothelial cell injury, microvascular constriction secondary to NO scavenging, and when bound to haptoglobin reduction in viable macrophages.21 The prototype of this form of MOF is found in children with sepsis who are on poorly functioning extracorporeal membrane oxygenation (ECMO) support. If the ECMO circuit induces excessive hemolysis (free hemoglobin >10 μg/L), MOF can ensue. The approach should be to attempt to stop ongoing hemolysis by changing oxygenator circuits, adding a second venous cannula, and reducing ECMO flow. In hemolytic disease such as sickle cell anemia, plasma exchange can help reduce hemolysis and resolve MOF.
Even in clinical settings without sophisticated research tests, patients with PMOF can be phenotyped (Table 104-1). New-onset thrombocytopenia with MOF, increase lactate dehydrogenase with or without schistocytes, and renal dysfunction likely reflect a thrombotic microangiopathy that resides within the disseminated intravascular coagulation/thrombotic thrombocytopenic purpura (DIC/TTP) continuum. This complex coagulopathy can be treated with plasma infusion or exchange, protein C, antithrombin III, pentoxifylline, prostacyclin, and/or heparin. New or persistent bacterial or fungal infections in a patient with MOF who does not have evidence of primary or secondary immune deficiency or an uneradicated nidus of infection is highly suggestive of immune paralysis/lymphoid depletion syndrome. Immune suppressant therapies should be rapidly tapered in these patients. Dexamethasone in particular should be stopped and transitioned to hydrocortisone. GM-CSF can be given subcutaneously at 125 μg/m2 daily subcutaneously to reverse immunoparalysis. If immunoglobulin G levels are less than 500 mg/dL, IVIG should be considered. If the CD4 count is low, antifungal, antiviral, and antiprotozoal prophylaxis can be considered. Patients with SMOF that begins with ARDS and progresses to hepatorenal syndrome should be recognized and treated accordingly. If the child has EBV-related posttransplant lymphoproliferative disease (high EBV titers), rapid immune suppression tapering is recommended along with rituximab and IVIG therapy. If the ferritin level is higher than 500 μg/L, then MAS (rheumatologic signs), secondary hemochromatosis, or the HLH syndrome should be considered. History and physical exam findings of rash, serositis, and arthralgias support MAS and should be treated with pulse steroids. A long-term history of transfusions, a ferritin level greater than 1000 μg/L, and an iron-binding capacity less than 50% suggests secondary hemochromatosis. These patients should be treated with iron chelation therapy. These patients are prone to iron-loving Aspergillus infection during chelation therapy. Appropriate antifungal coverage is recommended in these children. In the absence of rheumatologic or secondary hemochromatosis disease, HLH should be considered. A positive family history should invoke evaluation for primary HLH. These patients should have no NK cell cytotoxicity activity and specific perforin mutations. Treatment includes chemotherapy followed by bone marrow transplantation. In patients without a family history who have bacterial/fungal infection, secondary HLH is a possibility. These patients have reduced NK cell activity but no perforin mutations. Successful treatment for this group of patients has included interferon-α plus IVIG, steroids plus plasma exchange, and supportive care with eradication of infection. Cancer patients with absent NK cell function and HLH have responded to the IL-2 receptor antibody daclizumab.
| Phenotype | Diagnosis | Therapy |
|---|---|---|
| Thrombocytopenia-associated MOF |
LDH, Lactate dehydrogenase; HLA, human leukocyte antigen; PTLD, posttransplantation lymphoproliferative disorder.
1. Proulx F., Fayon M., Farrell C.A., et al. Epidemiology of sepsis and multiple organ dysfunction syndrome in children, Chest. 1996;109(4):1033-1037.
2. Doughty L.A., Kaplan S.S., Sasser H., Carcillo J. The compensatory anti-inflammatory cytokine IL-10 response in pediatric sepsis-induced multiple organ failure, Chest. 1998;113(6):1625-1631.
3. Green J., Doughty L., Kaplan S.S., et al. The tissue factor and plasminogen activator inhibitor type 1 response in pediatric sepsis-induced multiple organ failure, Thromb Haemost. 2002;87(2):218-223.
4. Whalen M.J., Doughty L.A., Carlos T.M., et al. Carcillo JA Intercellular adhesion molecule-1 and vascular adhesion molecule-1 are increased in children with sepsis induced multiple organ failure, Crit Care Med. 2000;28(7):2600-2607.
5. Nguyen T.C., Han Y.Y., Kiss J.E., et al. Intensive plasma exchange therapy increases ADAMTS 13 an dreverses organ dysfunction in children with thrombocytopenia associated MOF, Crit Care Med. 2008;36(10):2878-2887.
6. Felmet K.A., Hall M.W., Clark R.S., Jaffe R. Carcillo JA Prolonged lymphopenia, lymphoid depletion, and hypoprolactinemia in children with nosocomial sepsis and MOF. J Immunol. 2005;174(6):3765-3772.
7. Carcillo J.A., Doughty L.A., Kofos D., et al. Cytochrome P450 mediated drug metabolism is reduced in children with sepsis-induced MOF, Intensive Care Med. 2003;29(6):980-984.
8. Doughty L., Clark R.S., Kaplan S.S., Sasser H. Carcillo J sFas and sFasL and pediatric sepsis induced MOF, Pediatr Res. 2002;52(6):922-927.
9. Agbeko RS, Fidler KJ, Allen ML, Wilson P, Klein NJ, Peters MJ: Genetic variability in complement activation modulates the systemic inflammatory response syndrome in children, Pediatr Crit Care Med 11(5):561–567, 2010.
10. Wilkinson J.D., Pollack M.M., Ruttiman U.E., et al. Outcome of pediatric patients with multiple organ system failure. Crit Care Med. 1986;14(4):271-274.
11. Goldstein B., Giroir B., Randolph A. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunctions. Pediatr Crit Care Med. 2005;6(1):2-8.
12. Leteurtre S., Martinot A., Duhamel A., et al. Development of a pediatric multiple organ dysfunction score: use of two strategies. Med Decision Making. 1999;19(4):399-410.
13. Odetola F.O., Gebremanian A., Freed G.L. Patient and hospital correlates of clinical outcomes and resource utilization in severe pediatric sepsis. Pediatrics. 2007;119(3):487-494.
14. Typpo K.V., Petersen N.J., Hamilton D.M. et al: Day 1 multiple organ dysfunction syndrome is associated with poor functional outcome and mortality in the pediatric intensive care unit. Pediatr Crit Care Med. 2009;10(5):562-570.
15. Bongers T.M., Emonts M., de Maat M.P., et al. Reduced ADAMTS 13 in children with severe meningococcal sepsis is associated with severity and outcome. Thromb Haemost. 103(6), 2010.
16. Castillo L., Carcillo J. Secondary hemophagocytic lympho histiocytosis and severe sepsis/systemic inflammatory response syndrome/multiple organ dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med. 2009;10(3):387-392.
17. Han Y.Y., Carcillo J.A., Dragotta M.A., et al. Early reversal of pediatric-neonatal septic shock by community physicians is associated with improved outcome. Pediatrics. 2003;112(4):793-798.
18. de Oliveira C.F., de Oliveira D.S., Gottschald A.F., et al. ACCM/PALS hemodynamic support guidelines for pediatric septic shock: an outcomes comparison with and without monitoring central venous oxygen saturation. Intensive Care Med. 2008;34(6):1065-1076.
19. Kumar A., Roberts D., Wood K.E., et al. The duration of hypotension before initiation of effective antimicrobial therapy is critical determinant of survival in human septic shock. Crit Care Med. 2006;34(6):1589-1596.
20. Barie P.S., Williams M.D., McCollam J.S., et al. Benefit/risk profile of drotrecogin alpha in surgical patients with severe sepsis. An J Surg. 2004;188(3):212-220.
21. Kapralov A., Vlasova II, Feng W., et al. Peroxidase activity of hemoglobin haptoglobin complexes: covalent aggregation and oxidative stress in plasma and macrophages. J Biol Chem. 2009;284(44):30395-30407.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 104 Inflammation and Immunity
Systemic Inflammatory Response Syndrome, Sepsis, Acute Lung Injury, and Multiple Organ Failure
The Inflammation/Coagulation/Immune Dysfunction/Dysregulated Metabolism Hypothesis
Roger Bone popularized the notion that systemic inflammation led to the development of sepsis syndrome and MOF in the 1990s. According to this theory, an initial proinflammatory cytokine response to foreign or self-antigen caused SIRS, a systemic inflammatory response characterized by two of four criteria: tachycardia, tachypnea, fever, and leukocytosis. This syndrome was thought to be mediated by increased interleukin (IL)-1 and tumor necrosis factor (TNF)-α levels (Figure 104-1). It could be blunted by nonsteroidal antiinflammatory drugs and hence was prostaglandin mediated. A systemic inflammatory response was called sepsis if related to infection and SIRS if related to surgery, pancreatitis, or other noninfectious processes. This syndrome was self-limited when antibiotics were used to kill the infection or time allowed recovery from surgery, pancreatitis, and other noninfectious causes. This proinflammatory response was turned off by an antiinflammatory response led by the antiinflammatory cytokine IL-10. When IL-10 resulted in reduction of IL-1 and TNF-α to normal control values, sepsis or SIRS stopped and organ failure did not occur (Figure 104-2). In patients who developed severe sepsis and MOF, the compensatory anti-inflammatory response was unsuccessful in turning off inflammation. Instead, these patients had persistent elevation of the cytokine IL-6 along with IL-10. Bone coined the term immunologic dissonance to describe this process. He viewed MOF as an inflammatory condition in which the normal antiinflammatory response was unable to turn off the proinflammatory process (Figure 104-3).
Figure 104–1 SIRS shows proinflammation self-limited by antiinflammation and eradication of the source of inflammation.
Proulx and colleagues1 defined pediatric MOF syndromes as primary and secondary. Primary MOF occurred in 90% of children and was present at the time of admission, and secondary MOF occurred in 10% of children and was not present until 7 days after admission.1 To evaluate the role of inflammation in pediatric sepsis-induced MOF, this group defined children as having no MOF (NMOF) if they had two-organ failure at presentation but never reached three-organ failure, resolved MOF (RMOF) if they had three-organ failure at admission that resolved to two-organ or less at 48 hours, persistent MOF (PMOF) if they had three or more organ failures at presentation that did not resolve by 48 hours, and sequential MOF (SMOF) if they had acute lung injury/adult respiratory distress syndrome at admission and then progressed to hepatorenal failure/dysfunction.2 Similar to Proulx’s classification, children in the latter investigation with SMOF represented 10% of the population. The mortality rates with NMOF, RMOF, PMOF, and SMOF were 2%, 8%, 35%, and 50%, respectively. The authors then investigated Bone’s hypothesis in this categories and reported that patients with NMOF had an increase in IL-10 with subsequent turning off of inflammation; however, those with PMOF or SMOF had persistently high IL-6 and IL-10 levels—the hallmark of Bone’s “immunologic dissonance.”2
Bone’s untimely death prevented him from further pursuing the mechanisms by which immunologic dissonance could mediate organ injury. However, Proulx et al. first reviewed the autopsy bank at their hospital and noted that children who died from sepsis-induced MOF exhibited two unsuspected findings compared with children who died of other causes: persistent or unrecognized infection and thrombotic and bleeding complications with organ infarctions. Accordingly, they pursued the notion that immunologic dissonance might lead to an inability to mediate coagulation homeostasis as well as an inability to clear infection. Proinflammatory cytokines including TNF-α and IL-1 induce endothelial activation in vitro with an increased prothrombotic and antifibrinolytic state and expression of adhesion molecules (Figures 104-4 to 104-6). Patients with PMOF had increased tissue factor and plasminogen activator-1 activity as well as circulating adhesion molecules, intercellular adhesion molecule, and vascular adhesion molecule consistent with the hypothesis that immunologic dissonance was associated with an activated prothrombotic/antifibrinolytic endothelium.3,4 IL-6 also inhibits ADAMTS-13, the von Willebrand factor (vWF) multimer-cleaving protease, in vitro. PMOF patients also had deficient ADAMTS-13 activity compared with NMOF patients, consistent with increased risk of vWF multimer–mediated thrombosis.5 Increased IL-10 is the hallmark of the TH2 antiinflammatory cytokine response. When the TH2 response is dominant, the TH1 response can be turned off, a state known as immune paralysis (Figure 104-7). These patients have a diminished ex vivo ability to secrete TNF-α from endotoxin-stimulated monocytes or macrophages. Without this ability, infections cannot be terminated. PMOF patients exhibited immune paralysis more frequently than did NMOF patients.6
Cytokines and nitric oxide also inhibit cytochrome P450 activity and mitochondrial respiration in vitro. One study group found that patients with PMOF had reduced cytochrome P450 (10-fold) activity compared with patients with NMOF (twofold).7 Immunologic dissonance in PMOF was associated with endotheliopathy, immune paralysis, and poor metabolism, but how was PMOF different from SMOF? The authors determined that patients with SMOF were more likely to have viral or lymphoproliferative disease with very high soluble Fas ligand (sFasL) levels. In vitro, sFasL levels greater than 500 pg/mL cause hepatocyte necrosis. Children with MOF with sFasL levels greater than 500 pg/mL demonstrated liver injury at autopsy.8 The mechanism of injury in SMOF was different than in patients who presented with MOF (Figure 104-8).
In this decade, investigators from the United Kingdom have investigated the inflammation/immune dysregulation hypothesis in SIRS/sepsis, hypothesizing that reduced complement activity increases the risk of SIRS/sepsis.9 Mannose-binding lectin (MBL) is the first complement component released and is required for the first 12 hours of complement activity. Approximately 30% of children are MBL deficient. Complement factor H inhibits complement activity. The complement H factor H Y402H polymorphism reduces this complement inhibition activity. Using genotype analyses, these investigators have demonstrated that children at risk for SIRS/sepsis more commonly have deficiencies in MBL production and a reduction in this H factor polymorphism. These two polymorphisms together lead to an even greater risk of SIRS/sepsis than either alone. The interpretation is that diminished complement function prevents rapid clearance of antigen, leading to the SIRS/sepsis syndrome. The inflammation hypothesis is alive and well in SIRS, sepsis, severe sepsis, and MOF.
Definitions and Scoring
As previously noted, SIRS is defined by two of the following four criteria: tachypnea, tachycardia, fever, and leukocytosis. Sepsis is defined as SIRS plus suspected or documented infection. Severe sepsis is defined as sepsis plus organ failure. Acute lung injury is defined by bilateral pulmonary infiltrates and a PaO2/FIO2 ratio less than 300. MOF or multiple organ dysfunction syndrome is defined by failure of more than one organ. Many definitions exist for defining organ failure. Wilkinson and Pollack10
[/not-level-membership-for-pediatrics-category]
