CHAPTER 84 INFECTIVE NEUROPATHIES
While most peripheral neuropathies encountered by the clinical neurologist will have toxic or metabolic etiologies, it is very important to consider that the syndrome may be caused by an infection. Both bacterial and viral infections are known to cause peripheral neuropathies, as are the autoimmune sequelae of an infectious process. Most infective neuropathies have a distinctive clinical phenotype that will aid in its recognition, especially in the context of a patient’s medical and social histories. The recognition of an infective neuropathy has profound treatment implications, and may offer the rare opportunity to “cure” a peripheral neuropathy.
LEPROUS NEURITIS
Leprous neuritis is a true bacterial infection of peripheral nerves, and nerve invasion is a major hallmark of the disease. The disease has been known since biblical times, and archeological studies of human remains have documented that leprosy did exist in that era. Recent studies based on single nucleotide polymorphisms of the bacillus have pinpointed the origins of leprosy to East Africa, with subsequent spread to Europe and Asia before being reimported into West Africa by explorers.1 In earlier times, the disease was very common in Europe, especially in Scandinavia and England. In 1200 AD, there were some 200 “lazar” houses in the British Islands. There had been an endemic focus in the United States within 100 miles of the Gulf Coast of Texas, Louisiana, and Florida, and a smaller focus in the Midwest originating from Scandinavian immigrants. Now, there are only a few thousand cases in the United States, almost all of foreign origin.
There has been great recent progress in the control of this disease, which is now most common in underdeveloped countries. Newer methods of treatment, especially the introduction of multiple drug therapy (MDT), have resulted in a dramatic decline in active cases. There are now on the order of 300,000 active cases, whereas there were about 14 million cases 20 years ago.2 Many cases are cleared from the active rolls after therapy is given, without follow-up, but the newer drugs seem to be associated with only a small percentage of resistant or relapse cases. The plan of the World Health Organization (WHO) has been to reduce the caseload to such a low level (1 case per 10,000 population) that further transmission becomes unlikely.3 This idea harkens back to the observation that leprosy disappeared from most of Europe following the loss of more than a third of the population in the wake of the Black Plague. This was attributed to a critical decline in the number of infective cases in population centers.
The organism, Mycobacterium leprae, first seen in 1874 by G.H. Armauer Hansen in Norway, is the first bacterium that was associated with a human disease. M. leprae is less acid-fast than M. tuberculosis and requires the Fite modification for acid-fast staining. The ability to invade human nerves is unique, and recent studies of the bacillus have revealed that the actual site of nerve attachment and invasion is the alpha-dystroglycan moiety of Schwann cell myelin.4 The entire genetic sequence of the bacterium has also recently been elucidated, and demonstrates that the organism is highly specialized as an obligate intracellular human pathogen, and has lost many of the enzymatic functions that other acid-fast bacilli have maintained.5 These data may account for the failure to grow M. leprae on artificial media, and the difficulty to infect animals (mouse footpads and genetically susceptible armadillos). M. leprae has a highly thermosensitive growth rate: optimum cell division occurs once per 12.5 days at 27°C to 32°C. There is no growth at core body temperature, and therefore the viscera and central nervous systems are not involved in this disease. This feature limits leprosy to the skin, the anterior third of the eyes, the upper respiratory tract, and the testes. The incubation period varies from several weeks to several years. Children under 16 appear more susceptible to the disease, as are patients with certain types of immunosuppression. Although acquired immunodeficiency syndrome (AIDS) and leprosy are both very common in certain areas such as Africa, AIDS does not seem to increase the vulnerability or severity of coexistent leprosy. The disease is probably spread by infected nasal droplets (where bacteria remain viable for several days) and prolonged skin-to-skin contact.
Leprosy may manifest as a neuropathy, and a well-informed neurologist is capable of making a definitive bedside diagnosis, even in cases where the disease has been fully treated and only residual neurological deficits remain, or in the more rare patient who is trying to conceal the diagnosis. Early diagnosis is critical because one can expect to halt the progress of the neuropathy, and even obtain some degree of reversal. However, severe deficits may remain in advanced cases even after the patient is successfully treated. Cases may be misdiagnosed at medical centers in developed countries because the possibility of leprosy is not considered.
Although there is only one causative organism, the clinical picture of leprosy is wide and related to the host’s innate immune response to M. leprae. In most human populations, 90% to 95% of individuals are genetically equipped to clear the bacilli completely. When resistance is high but insufficient to entirely prevent the disease, “high resistance” tuberculoid or paucibacillary leprosy occurs. Tissue temperature has only a permissive role in shaping this clinical picture. Rather, local invasion by very few bacilli evokes a vigorous epitheloid granulomatous response, which destroys intracutaneous nerves as the lesion forms. The typical patient shows one or a very few asymmetrically dispersed, hypopigmented, anhydrotic skin lesions (Fig. 84-1). A dry patch with pain and temperature loss coinciding with the skin lesion is the diagnostic picture of paucibacillary leprosy (Fig. 84-2). The skin lesion shows central clearing and a slightly raised edge. The centers of such lesions are often clear of bacilli. For diagnosis, the skin biopsy sample should be taken at the elevated edge. Serial sections may be required to see just one or a few bacilli.
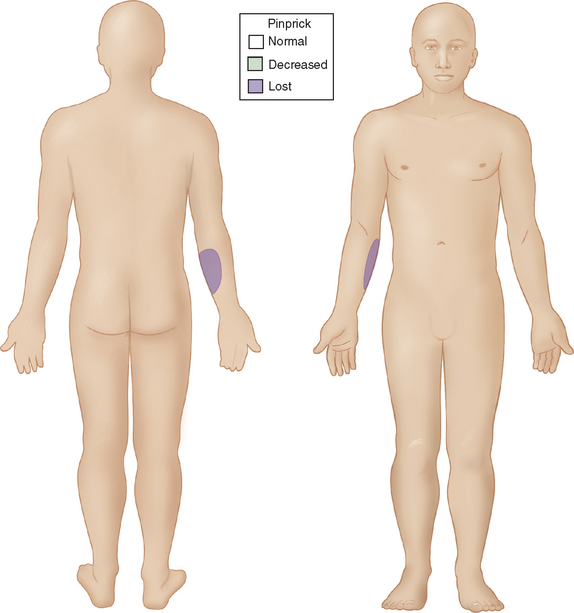
Figure 84-1 Sensory map of tuberculoid leprosy.
(From Sabin TD, Swift TR, Jacobson RR: Leprosy. In: Dyck PJ, Thomas PK, eds: Peripheral Neuropathy. Philadelphia: Elsevier Saunders. 2005, p 2088, Figure 91-3.)
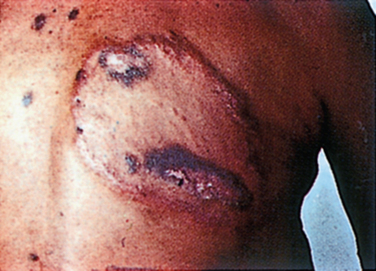
Figure 84-2 Skin lesion of tuberculoid leprosy.
(From Sabin TD, Swift TR, Jacobson RR: Leprosy. In: Dyck PJ, Thomas PK, eds: Peripheral Neuropathy. Philadelphia: Elsevier Saunders. 2005, p 2085, Figure 91-2B.)
When the host has little inborn resistance to the invasion of bacilli, these become widespread and numerous, with hematogenous spread and reproduction in the cooler tissues. Thus, the skin lesions and nerve involvement are roughly symmetric. This form of leprosy is called lepromatous. The palpably enlarged nerves present in this form of leprosy narrow the differential diagnosis. The invasion of intracutaneous nerve networks is manifested by a pattern of temperature-linked sensory loss that starts in the coolest regions of the body, around the breast plate, tip of the nose, lobes and helices of the ears (Fig. 84-3). Once this temperature-linked pattern of sensory loss is discerned, the diagnosis becomes apparent.6 Over the years, the pattern progresses to “less cool” regions and results in very extensive sensory loss, sparing only areas over the carotid arteries, the anterior neck, armpits, inguinal folds, intergluteal folds, and the precordium (Fig. 84-4). The insulating effect of corium of the hands and feet make the tissue temperatures beneath these surfaces somewhat warmer, so that sensory loss tends to occur over the dorsa of hands and feet rather than over the soles and palms. The distribution of skin lesions is also temperature dependent, and roughly approximates the distribution of sensory loss. There are striking examples of small differences in temperature creating distinctive features on the sensory examination. Facial sensation becomes normal at the hairline because of the insulating effect of hair. For example, the old leprosy atlases depict men who followed the Samurai tradition of shaving their heads and experienced leprosy lesions in the scalp; when they let their hair grow back, there was destruction of the follicles except over the warm areas overlying the temporal arteries. The cooler hemiplegic side of a stroke victim showed more extensive sensory loss. Sensory-sparing underbelts, watchbands, and other snug garments have also been documented.
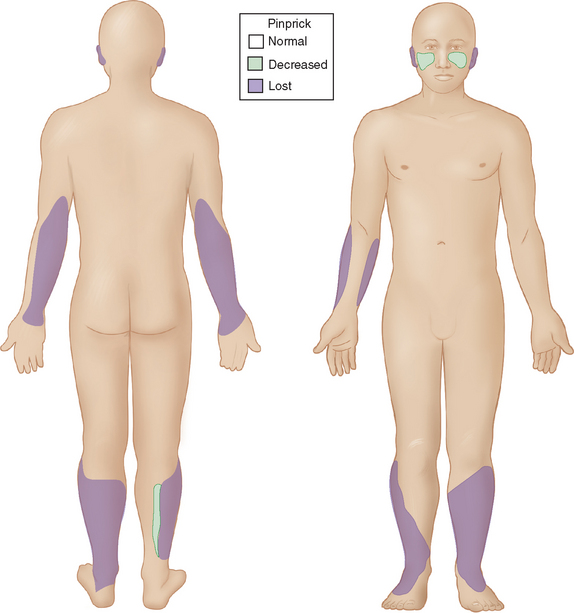
Figure 84-3 Sensory map of early lepromatous leprosy.
(From Sabin TD, Swift TR, Jacobson RR: Leprosy. In: Dyck PJ, Thomas PK, eds: Peripheral Neuropathy. Philadelphia: Elsevier Saunders. 2005, p 2090, Figure 91-5.)
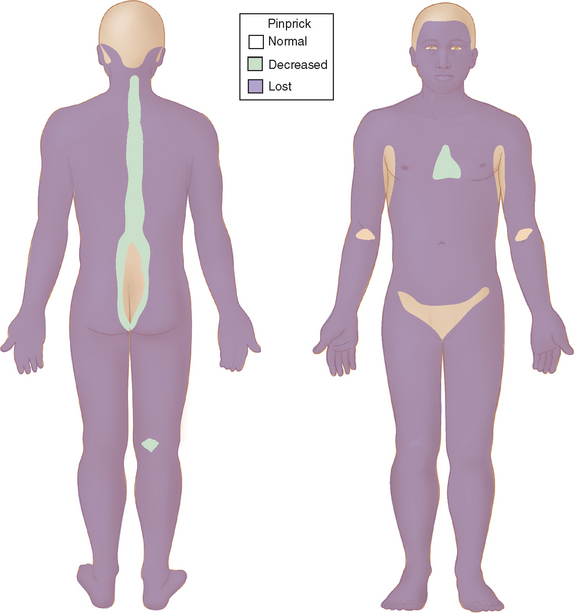
Figure 84-4 Sensory map of chronic lepromatous leprosy.
(From Sabin TD, Swift TR, Jacobson RR: Leprosy. In: Dyck PJ, Thomas PK, eds: Peripheral Neuropathy. Philadelphia: Elsevier Saunders. 2005, p 2091, Figure 91-6.)
The major named nerves, which run closest to the surface of the body, are also affected in a predictable fashion (Fig. 84-5). Thus, the deep peroneal branch to the extensor digitorum brevis of the foot and the ulnar nerve proximal to the olecranon groove are involved early. The median nerve segment from the wrist crease to where it is covered by the forearm flexor muscle group is affected later, with further sensory loss and paralysis of median-innervated intrinsic muscles. Foot drop ensues when the peroneal nerve is affected as it circles around the head of the fibula. Multiple branches of the facial nerve are involved, particularly affecting the medial corrugators of the forehead, the small twigs to the orbicularis oculi and to the muscles composing the nasolabial folds, so as to create a highly diagnostic, patchy paralysis described by Monrad-Krohn.7 If one actually severed the ulnar, median, and peroneal nerves at the sites affected by leprosy, the corresponding deep tendon reflexes would remain intact. The preservation of deep tendon reflexes in the presence of significant peripheral neuropathy is yet another very helpful diagnostic feature.
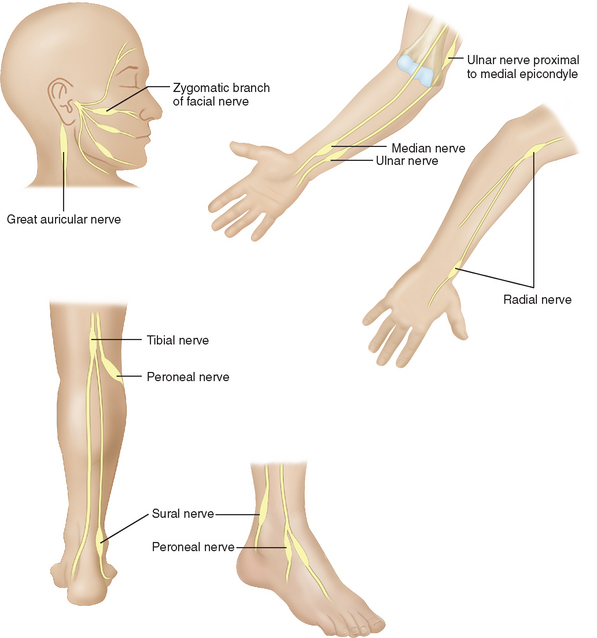
Figure 84-5 The most commonly affected nerves in leprosy.
(From Sabin TD, Swift TR, Jacobson RR: Leprosy. In: Dyck PJ, Thomas PK, eds: Peripheral Neuropathy. Philadelphia: Elsevier Saunders. 2005, p 2089, Figure 91-4.)
Although there are many intermediate forms between these polar types of leprosy, complex interplay between the two themes—tissue temperature and natural immunity of the host—can always be discerned. These cases are labeled dimorphous, borderline, or intermediate. Modern leprosy workers tend to divide all cases into paucibacillary and multibacillary for purposes of management. Among the high-resistance dimorphous cases, there is a group called pure neural leprosy. These patients have no skin lesions but extensive nerve damage, and they can be very difficult to diagnosis, even on sophisticated neurological services. A nerve biopsy may be helpful (the sural or cutaneous branches of the radial nerve are good sites), but a fascicular biopsy at a site of clinical involvement will have the best yield. Polymerase chain reaction techniques can also help in this situation.8
Alterations in immune response, known as leprosy reactions, occur in some patients. In high-resistance cases, there may be a sudden increase in cell-mediated immunity causing acute inflammation of existing lesions and rapid onset of deficits in neighboring and subjacent nerves that had already been invaded by bacilli. The nerve damage will be permanent unless this situation is rapidly recognized and treated with steroids. In multibacillary patients, there is an entity called erythema nodosum leprosum. Here, the circulating immune response goes awry and produces what resembles an autoimmune disease. Features of an Arthus reaction, including consumption of complement and arteritis, are present. Although there are systemic symptoms, the brunt of the damage occurs in the area of greatest bacillary density. Except for the distribution, the erythema nodosum looks the same as in other systemic diseases. Rapid, widespread nerve pain and dysfunction results from vasculitis of the vasa nervorum. This is a situation that must also be quickly recognized. The treatment of choice is thalidomide, but steroids can be used if thalidomide is contraindicated or not available.
When early diagnosis is made, most people require only antibacterial treatment. The most effective antibacterial regimen for M. leprae includes the drugs dapsone, clofazimine, rifampin, ofloxacin, and minocycline. The course of antibacterial therapy depends on whether the patient has the paucibacillary or multibacillary forms of leprosy. Paucibacillary patients receive 100 mg of dapsone daily with rifampin 600 mg monthly for 6 months. Multibacillary patients are given the same combination for 1 year, in addition to clofazimine 50 mg daily, with a monthy 300-mg dose of clofazimine. Shorter regimens used by WHO also make the patient noninfectious in a few days and have an extremely low relapse rate for both types of disease.9 The addition of rifampin has made the greatest difference in the antibacterial treatment of leprosy since the introduction of dapsone in 1940.
HUMAN IMMUNODEFICIENCY VIRUS
Human immunodeficiency virus (HIV) is associated with diverse peripheral nervous system complications (Table 84-1). The specific type of peripheral nervous system syndrome is dictated by the severity of the HIV infection, as determined by CD4+ cell counts.10 Prospective studies indicate that the incidence of symptomatic peripheral nervous system complications due to HIV is around 3%, and complications are more common in more advanced cases.11 If peripheral nervous system complications are assessed by clinical symptoms and electrophysiological tests, the incidence in prospective studies is much higher, at least 30%, and advanced HIV infection again poses a higher risk. Assessing the epidemiology of HIV-related peripheral nervous system complications is confounded by the fact that several common antiretroviral drugs used to treat HIV are themselves toxic to peripheral nerves.
TABLE 84-1 Peripheral Nervous System Complications From Human Immunodeficiency Virus Infection
HIV seroconversion may be accompanied by an acute peripheral nervous system process, most commonly unilateral but also bilateral facial nerve palsies.12 Guillain-Barré syndrome is known to occur at the time of HIV seroconversion, and Guillain-Barré syndrome may be the presenting feature of HIV infection. The Guillain-Barré syndrome associated with HIV is clinically and electrophysiologically identical to non-HIV Guillain-Barré syndrome,13 and is characterized by acute flaccid paralysis with diminution or loss of reflexes. However, instead of the albuminocytological dissociation of typical Guillain-Barré syndrome, the cerebrospinal fluid in HIV-associated Guillain-Barré syndrome often shows a pleocytosis. HIV serology may be initially negative for several weeks following seroconversion. If clinical suspicion for HIV is high, HIV viral load can be used for the diagnosis in the acute setting. The therapeutic approach and therapeutic response is similar in HIV-associated Guillain-Barré syndrome and in idiopathic Guillain-Barré syndrome.
The peripheral nervous system complications of early HIV infection, with CD4 cell counts above 500 cells/μL, are similar to the complications of HIV seroconversion, and are dominated by facial nerve palsies and Guillain-Barré syndrome. However, these peripheral nervous system processes are seen in a small minority of HIV patients.10
In moderately advanced HIV infection, with CD4 cell counts between 200 and 500 cells/μL, peripheral nervous system complications are due to immune dysregulation.10 Chronic inflammatory demyelinating polyneuropathy (CIDP) is known to occur in HIV, and, like Guillain-Barré syndrome, CIDP in HIV is clinically and electrophysiologically identical to non-HIV CIDP. Again, a mild mononuclear cerebrospinal fluid pleocytosis is often seen. Pathologically, CIDP in both HIV and non-HIV forms is characterized by macrophage-mediated demyelination. Although there are no therapeutic trials with HIV-infected CIDP patients, it is believed that they respond to treatment like non-HIV CIDP patients.14 Corticosteroids, plasmapheresis, and intravenous immunoglobulin are the usual therapies for CIDP. When immunosuppressant treatment is utilized, prophylaxis against opportunistic infections is warranted.10
Mononeuritis multiplex caused by vasculitis is known to occur with HIV infection of all severities, but especially in moderately advanced cases. The clinical syndrome is characterized by a progressive accumulation of individual nerve lesions, leading to multifocal sensory and motor derangement, with electrophysiological evidence of multifocal axonal neuropathies. Axonal degeneration is the most common pathological finding on nerve biopsy, with perivascular or endoneurial inflammatory infiltration. Although the etiology of mononeuritis multiplex in HIV is unknown, it may be due to the deposition of HIV-related antigen and antibody immune complexes in perineural vessels. Treatment with immunotherapy should be reserved for patients with disabling deficits, as all therapies have the potential of worsening the underlying immunodeficiency, and milder cases are known to improve spontaneously. When the degree of immunodeficiency from HIV is profound (CD4+ cell count less than 100) and mononeuritis multiplex is present, the suspicion of cytomegalovirus infection should be high, and these cases should be treated aggressively with ganciclovir, foscarnet, or both.15 Cytomegalovirus infection is discussed in further detail later.
Diffuse infiltrative lymphocytosis syndrome is characterized by CD8+ cell infiltration of visceral organs, resulting in enlarged salivary glands and sicca syndrome.16 Peripheral nerves can be infiltrated, resulting in polyneuropathy. This is usually a painful distal symmetric sensory polyneuropathy, which develops subacutely over weeks. Electrophysiological studies reflect an axonal process. Nerve biopsy reveals diffuse CD8+ cell infiltration of the epineurium and endoneurium. Highly active antiretroviral therapy is effective in diffuse infiltrative lymphocytosis syndrome.17
Several recent case reports have described motor neuron disease in the setting of HIV infection, with the potential for clinical improvement after the initiation of highly active retroviral therapy.18,19 These cases are of great interest because they may help us understand the basic mechanisms of motor neuron diseases in general.
Approximately one third of patients with AIDS will develop a clinically significant distal symmetric polyneuropathy (DSP).20 DSP is the most common peripheral nervous system complication of HIV infection, and is present on autopsy in nearly all patients who die from AIDS.21 Risk factors for the development of DSP include elevated HIV viral load, low CD4+ cell counts, and exposure to neurotoxic antiretroviral medications.
Nerve biopsies are not routinely performed in DSP. When obtained, nerve biopsies show distal axonal degeneration, affecting both unmyelinated and myelinated axons.21 Perivascular mononuclear infiltrates may be present.
The pathogenesis of DSP in HIV infection is not known. As direct neuronal infection is not believed to occur, recent research has focused on indirect mechanisms. Attention has been directed toward the HIV-1 envelope protein gp120. Current hypotheses propose indirect neurotoxicity from gp120 via chemokine receptors on Schwann cells and neurons.14
As with other painful neuropathies, treatment is largely symptomatic. There is some evidence that highly active antiretroviral therapy may mildly improve DSP.22,23 Symptomatic treatments have been based on drugs used in other painful sensory neuropathies (especially diabetic polyneuropathy), including amitriptyline, gabapentin, and lamotrigine. There is no evidence to suggest that any one of these agents is specifically effective in the DSP of HIV. Narcotics may be needed in refractory cases. As with all other aspects of HIV management, careful attention to drug-drug interactions is important when choosing a symptomatic therapy.
Several nucleoside analogue reverse transcriptase inhibitors are known to cause a toxic polyneuropathy, and this has been a frequent confounder in the study of DSP. The so-called “d-drugs”—ddC (dideoxycytidine), ddI (didanosine), and d4T (stavudine)—are all known to cause a dose-dependent toxic polyneuropathy.24 The clinical syndrome is identical to DSP, and usually develops after several months of treatment with a d-drug. Distal numbness and painful dysesthesias develop, and exam reveals distal sensory abnormalities with diminished or abolished ankle jerks. Electrophysiological studies demonstrate a distal sensory-predominant axonal polyneuropathy. Serum lactate levels may help in determining if a painful sensory polyneuropathy is drug-related or due to the HIV infection itself.25 Offending drugs should be stopped in all patients with DSP. “Coasting” may occur, whereby the patient’s symptoms continue or worsen for 6 to 8 weeks after removal of the toxic drug.10
Due to similar modes of transmission, co-infections with syphilis, hepatitis C, and human T-lymphotropic virus type 1 (HTLV-1) should be considered in the evaluation of peripheral nervous system syndromes in the HIV-infected patient, as they may act as confounders.10
CYTOMEGALOVIRUS
Cytomegalovirus lumbosacral polyradiculopathy is a severe infectious cauda equina syndrome that affects patients with severe immunosuppression, usually patients with HIV and CD4 counts less than 50 cells/μL. The clinical presentation consists of a rapidly progressive flaccid areflexic paraparesis with urinary retention.26 Weakness of the lower extremities progresses over days and may be asymmetrical. Although there may be complaints of numbness, particularly in the perianal region, objective sensory findings are uncommon. Rarely, the syndrome manifests as a polyradiculomyelitis, with a thoracic spinal level and Babinski responses. The prognosis of patients with cytomegalovirus lumbosacral polyradiculopathy is very poor, even with optimal therapy, and the condition is uniformly fatal if left untreated.26
In severely immunosuppressed patients who present with the above syndrome, cerebrospinal fluid examination is of critical importance, typically showing a polymorphonuclear pleocytosis of 50 to 1000 cells/mm3, usually with elevated protein and low glucose.26,27 However, if cerebrospinal fluid does not demonstrate the expected polymorphonuclear pleocytosis, polymerase chain reaction is required for definitive diagnosis.28 While cytomegalovirus can be cultured from cerebrospinal fluid, polymerase chain reaction of cerebrospinal fluid is more sensitive, and will be positive in more than 90% of patients with cytomegalovirus lumbosacral polyradiculopathy. In immunosuppressed patients with the clinical syndrome and cerebrospinal fluid profile of cytomegalovirus lumbosacral polyradiculopathy, empiric treatment should begin before a definitive diagnosis of cytomegalovirus in cerebrospinal fluid is obtained, as test results are often delayed. If needed, evidence of cytomegalovirus infection should be sought in another organ system.29
As with any patient presenting with a cauda equina syndrome, magnetic resonance imaging (MRI) should be obtained. MRI in cytomegalovirus lumbosacral polyradiculopathy may reveal thickened nerve roots and enhancement with gadolinium contrast.30
Treatment of cytomegalovirus lumbosacral polyradiculopathy involves intravenous administration of ganciclovir, foscarnet, or both.31 The major dose-related medication side effects are neutropenia with ganciclovir and nephrotoxicity with foscarnet. In HIV patients, antiretroviral therapy should be optimized.
In the evaluation of suspected cytomegalovirus lumbosacral polyradiculopathy in a patient with advanced HIV, cerebrospinal fluid should be screened for both cytology and syphilis serology.14 This is because lymphomatous meningitis and syphilis can mimic the presentation and cerebrospinal fluid profile of cytomegalovirus polyradiculopathy.10
Cytomegalovirus can present as a mononeuritis multiplex in HIV patients with CD4+ counts less than 50 cells/μL.15 Symptoms begin as sensory complaints in the distribution of a named nerve, and progress to asymmetrical weakness. Electrophysiological studies demonstrate a multifocal axonal process. Cerebrospinal fluid pleocytosis is present only in a minority of patients, but cytomegalovirus polymerase chain reaction is positive in at least 90% of patients. The prognosis for cytomegalovirus mononeuritis multiplex is better than that of cytomegalovirus polyradiculopathy, as most patients improve after treatment with ganciclovir or foscarnet.
LYME DISEASE
The pathognomonic finding in Lyme borreliosis is erythema migrans, the characteristic rash. Erythema migrans starts as a small erythematous macule or papule at the site of the tick bite, which expands in size over days or weeks. As the rash expands, the border of the lesion remains erythematous while the center clears, leading to a characteristic “bull’s eye” appearance. The rash may be accompanied by fever or malaise. Days to weeks after the primary infection, multifocal erythema migrans may occur in the setting of spirochetal dissemination via the blood-stream (Fig. 84-6). It is at this point that the spirochetes may spread to multiple organ systems, causing a systemic illness with fever, malaise, headache, myalgias and arthralgias. Erythema migrans is neither pruritic nor painful, and thus may be ignored or unnoticed by patients. However, as this skin finding is pathognomonic of the disease, a history of prior rashes should be vigorously sought in cases of suspected Lyme disease, as 80% of Lyme patients have this finding in early infection.32
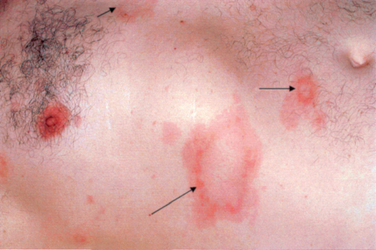
Figure 84-6 Disseminated erythema migrans approximately 1 month after tick bite.
(From Said G: Lyme disease. In: Dyck PJ, Thomas PK, eds: Peripheral Neuropathy. Philadelphia: Elsevier Saunders. 2005, p 2111, Figure 92-1.)
Painful radiculoneuritis is the other classic peripheral nervous system syndrome of Lyme disease. The syndrome is characterized by the acute presentation of severe radiating dermatomal pain. The affected dermatome may involve limb or trunk. After several weeks from the onset of pain, dermatomal weakness, hyporeflexia, and sensory loss develop. This syndrome can mimic a typical compressive radiculopathy. Thus, when evaluation of radiculopathy does not reveal a compressive etiology, Lyme borreliosis should be considered in patients from endemic areas. The radiculoneuritis of Lyme disease may be electrophysiologically indistinguishable from a compressive radiculopathy as well.33
Laboratory diagnosis of Lyme disease rests on demonstration of an antibody response to the spirochete. Culture of the organism from blood and cerebrospinal fluid has been disappointing. Multiple laboratory techniques have been devised in an attempt to improve upon the laboratory diagnosis of Lyme disease. The multitude of laboratory approaches (many of which have not undergone rigorous scientific review) has led governmental agencies in the United States, Canada, and Europe to publish guidelines for the use and interpretation of appropriate serological tests.34
To minimize the above problems, and to maximize the diagnostic accuracy of serological testing, current recommendations in the United States are that the initial screening test for Lyme disease be an enzyme-linked immunosorbent assay (ELISA). If the ELISA is borderline-positive or positive, a Western blot should be obtained. Criteria have been published for the laboratory interpretation of Western blot analysis, based on high specificity.35 It is not recommended that Western blots be given diagnostic significance when the ELISA is normal. Polymerase chain reaction testing is not recommended for routine diagnosis.
The prognosis for appropriately treated Lyme disease is excellent. Early Lyme disease, where the only manifestations are dermatological, is appropriately treated with doxycycline 100 mg by mouth twice a day or amoxicillin 500 mg by mouth three times a day for 14 to 21 days. Some experts advocate also treating the cranial nerve syndromes of Lyme disease with oral antibiotics, unless a lymphocytic meningitis is present, in which case intravenous therapy is recommended. Intravenous therapy is indicated for all Lyme infections with radiculitis or meningitis. Intravenous therapy generally consists of ceftriaxone 2 g daily intravenously for 14 to 28 days.36
VARICELLA ZOSTER VIRUS
Zoster sine herpete is a syndrome identical to postherpetic neuralgia, with burning dermatomal pain. However, there is no antecedent rash. There are cases where VZV DNA has been detected in the cerebrospinal fluid of such patients using polymerase chain reaction.37 Resolution of chronic pain has occurred with high-dose acyclovir at ten to fifteen mg per kilogram intravenously every 8 hours for 2 weeks.
HEPATITIS C VIRUS
The most common neuropathy associated with hepatitis C is a distal symmetrical sensory polyneuropathy, which may be associated with purpura of the skin. Electrophysiologically and pathologically, the neuropathy is axonal. The pathological mechanism appears to be ischemia due to vasculitis of vaso nervorum.38
The optimal treatment of peripheral nervous system syndromes in the setting of hepatitis C is not known. Immunosuppression, either with corticosteroids or cytotoxic agents, has been attempted, as has immunomodulation with plasma exchange. Interferon α, in an attempt to treat the underlying viral infection, has also been tried. The efficacy of the above therapies for peripheral nervous system disease is uncertain, and these treatments have been associated with worsening of the overall clinical syndrome.39
HUMAN T-LYMPHOTROPIC VIRUS TYPE 1
HTLV-1 is a common cause of endemic spastic paraparesis in tropical areas. Although generally overshadowed by signs and symptoms referable to the central nervous system, a distal symmetric sensory polyneuropathy has been described in isolation.40 The optimal treatment for this infection is not yet defined.
DIPHTHERIA
Infection with Corynebacterium diphtheriae can result in a distinct, delayed polyneuropathy. Days to weeks after the local diphtheria infection, numbness of the pharynx and bulbar weakness develop. Paralysis of the soft palate and inability of the pupils to accommodate are particularly suggestive of diphtheria. Diaphragmatic paralysis may also occur, requiring ventilatory support. Diphtheritic polyneuropathy typically has a biphasic course.41 After bulbar symptoms have been present for several weeks, motor and sensory symptoms appear in limbs, leading to a flaccid paralysis. The limb symptoms of diphtheria may persist for many months, and may recover incompletely. There may also be dysautonomia that is not explained by the myocarditis.42
It is unclear whether administration of diphtheria antitoxin provides any clinical benefit when given after 2 or 3 days of infection. Penicillin can be used to treat the acute infection. Corticosteroids are not effective in preventing diphtheritic polyneuropathy.43
GUILLAIN-BARRÉ SYNDROME
The classic cerebrospinal fluid finding in Guillain-Barré syndrome is albuminocytological dissociation—that is, increased protein concentration without pleocytosis. However, increased protein is not demonstrated in all cases of Guillain-Barré syndrome, especially early in the illness. When pleocytosis is present and the electrophysiology is demyelinating, HIV and Lyme disease should be considered. When pleocytosis is present and the electrophysiology is axonal and purely motor, a poliomyelitis syndrome should be considered. Although in the past the polio virus was the main cause of poliomyelitis, other viruses can also cause this phenotype, including the emerging pathogen West Nile virus.44
Immunomodulation via plasmapheresis and intravenous immunoglobulin have both been shown to hasten recovery in AIDP. Neither modality is clearly superior to the other, and the decision for treatment is often based on availability and the likelihood of adverse events. Both modalities are generally safe, albeit expensive. The primary risks of plasmapheresis are related to the placement and maintenance of the large-bore pheresis catheter, and include pneumothorax, sepsis, and bacterial endocarditis. Risks of intravenous immunoglobulin include arterial thrombotic events, renal failure, and aseptic meningitis.
The working hypothesis on the pathogenesis of the Guillain-Barré syndromes has been the concept of molecular mimicry. Several weeks after a systemic infection, epitopes of peripheral nerve shared with the infective agent are attacked, leading to the clinical syndrome. Most attention has been focused on Campylobacter jejuni, Mycoplasma pneumonia, and the herpesviruses, including cytomegalovirus, as the most common infective triggers. Because the Guillain-Barré syndrome occurs in the postinfective state, there is usually no clinical need to determine the antecedent infective agent or to use antibiotic or antiviral therapies. Evidence suggests that the genetic polymorphism of the infective agent determines the clinical phenotype of the Guillain-Barré syndrome. For example, the genetic polymorphism of Campylobacter jejuni correlates with the clinical variant of Guillain-Barré syndrome.45
Brew BJ. The peripheral nerve complications of human immunodeficiency virus (HIV) Infection. Muscle Nerve. 2003;28:542-552.
Halperin JJ. Lyme disease and the peripheral nervous system. Muscle Nerve. 2003;28:133-143.
Koga M, Takahashi M, Masuda M, et al. Campylobacter gene polymorphism as a determinant of clinical features of Guillain-Barré syndrome. Neurology. 2005;65:1376-1381.
Nemni R, Sanvito L, Quattrini A, et al. Peripheral neuropathy in hepatitis C virus infection with and without cryoglobulinaemia. J Neurol Neurosurg Psychiatry. 2003;74:1267-1271.
Sabin TD, Swift TR, Jacobson RR. Leprosy. In: Dyck PJ, Thomas PK, editors. Peripheral Neuropathy. Philadelphia: Elsevier Saunders; 2005:2081-2108.
1 Monot M, Honore N, Garnier T, et al. On the origin of leprosy. Science. 2005;308:936-937.
2 World Health Organization. Global leprosy situation. 2005. Wkly Epidemiol R. 2005;80:289-295.
3 Lockwood DN, Suneetha S. Leprosy: too complex a disease for a simple elimination paradigm. Bull World Health Organ. 2005;83:230-235.
4 Rambukkana A, Zanazzi G, Tapinos, et al. Contact-dependent demyelination of Mycobacterium leprae in the absence of immune cells. Science. 2002;296:927-931.
5 Vissa VD, Brennan PJ. The genome of Mycobacterium leprae: a minimal mycobacterial gene set. Genome Biol. 2001;2:1023.1-1023.8.
6 Sabin TD. Neurologic features of lepromatous leprosy. Am Fam Phys. 1971;4:84-94.
7 Monrad-Krohn GH. The Neurological Aspect of Leprosy. Cristiana, Norway: Jacob Dybwad, 1923.
8 Jadhav RS, Kamble RR, Shinde VS, et al. Use of reverse transcription polymerase chain reaction for the detection of Mycobacterium leprae in the slit-skin smears of leprosy patients. Indian J Lepr. 2005;77:116-127.
9 World Health Organization. A Guide to Leprosy Control, 2nd ed. Geneva: World Health Organization, 1988.
10 Brew BJ. The peripheral nerve complications of human immunodeficiency virus (HIV) Infection. Muscle Nerve. 2003;28:542-552.
11 Alliegro MB, Petrucci A, Arpino C, et al. Peripheral neuropathies among patients with HIV infection. J Neurol Neurosurg Psychiatry. 1998;64:414-415.
12 Wechsler AF, Ho DD. Bilateral Bell’s palsy at the time of HIV seroconversion. Neurology. 1989;39:747-748.
13 Cornblath DR, McArthur JC, Kennedy PG, et al. Inflammatory demyelinating peripheral neuropathies associated with human T-cell lymphotropic virus type III infection. Ann Neurol. 1987;21:32-40.
14 Höke A, Cornblath DR. Peripheral neuropathies in human immunodeficiency virus infection. In: Dyck PJ, Thomas PK, editors. Peripheral neuropathy. Philadelphia: Elsevier Saunders; 2005:2129-2145.
15 Roullet E, Assueurus V, Gozlan J, et al. Cytomegalovirus multifocal neuropathy in AIDS: analysis of 15 consecutive cases. Neurology. 1994;44:2174-2182.
16 Moulignier A, Autheir FJ, Baudrimont M, et al. Peripheral neuropathy in human immunodeficiency virus–infected patients with the diffuse infiltrative lymphocytosis syndrome. Ann Neurol. 1997;41:438-445.
17 Olney RK. HIV-Associated neuropathies. In: Cros D, editor. Peripheral Neuropathy. Philadelphia: Lippincott Williams & Wilkins; 2001:197-212.
42 Idiaquez J. Autonomic dysfunction in diphtheritic neuropathy. J Neurol Neurosurg Psychiatry. 1992;55:159-161.
43 Thisyakorn U, Wongvanich J, Kumpeg V. Failure of corticosteroid therapy to prevent diphtheritic myocarditis or neuritis. Pediatr Infect Dis. 1984;3:126-128.
44 Jeha LE, Sila CA, Lederman RJ, et al. West Nile virus infection: a new acute paralytic illness. Neurology. 2003;61:55-59.
45 Koga M, Takahashi M, Masuda M, et al. Campylobacter gene polymorphism as a determinant of clinical features of Guillain-Barré syndrome. Neurology. 2005;65:1376-1381.
18 Moulignier A, Maoulonguet A, Pialoux G, et al. Reversible ALS-like disorder in HIV infection. Neurology. 2001;57:995-1001.
19 MacGowen DLJ, Scelsa SN, Waldron M. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology. 2001;57:1094-1097.
20 Tagliati M, Grinnel J, Godbold J, et al. Peripheral nerve function in HIV infection. Arch Neurol. 1999;56:84-89.
21 Griffin JW, Crawford TO, Tyor WR, et al. Predominately sensory neuropathy in AIDS: distal axonal degeneration and unmyelinated fiber loss. Neurology. 1991;41(Suppl 1):374.
22 Markus R, Brew BJ. HIV-1 peripheral neuropathy and combination antiretroviral therapy. Lancet. 1998;352:1906-1907.
23 Martin C, Solders G, Sonnerborg A, et al. Antiretroviral therapy may improve sensory function in HIV-infected patients: a pilot study. Neurology. 2000;54:2120-2127.
24 Simpson DM, Tagliati M. Nucleoside analogue-associated peripheral neuropathy in human immunodeficiency virus infection. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;9:153-161.
25 Brew BJ, Tisch S, Law M. Lactate concentrations distinguish between nucleoside neuropathy and HIV neuropathy. AIDS. 2003;17:1094-1096.
26 Anders HJ, Goebel FD. Cytomegalovirus polyradiculopathy in patients with AIDS. Clin Infect Dis. 1998;27:345-352.
27 So YT, Olney RK. Acute lumbosacral polyradiculopathy in acquired immunodeficiency syndrome: experience in 23 patients. Ann Neurol. 1994;5:53-58.
28 Miller RF, Fox JD, Thomas P, et al. Acute lumbosacral polyradiculopathy due to cytomegalovirus in advanced HIV disease: cerebrospinal fluid findings in 17 patients. J Neurol Neurosurg Psychiatry. 1996;61:456-460.
29 Brew BJ. Infective causes of peripheral neuropathy In advanced HIV disease. In: Brew BJ, editor. HIV Neurology. New York: Oxford University Press; 2001:206.
30 Talpos D, Tien RD, Hesselink JR. Magnetic resonance imaging of AIDS-related polyradiculopathy. Neurology. 1991;41:1995-1997.
31 Anders HJ, Weiss N, Bogner JR, et al. Ganciclovir and foscarnet efficacy in AIDS-related polyradiculopathy. J Infect. 1998;36:29-33.
32 Steere AC. Lyme disease. N Engl J Med. 2001;345:115-125.
33 Krishnamurthy KB, Liu GT, Logigian EL. Acute Lyme neuropathy presenting with polyradicular pain, abdominal protrusion, and cranial neuropathy. Muscle Nerve. 1993;16:1261-1264.
34 CDC. Notice to readers: Caution regarding testing for Lyme disease. MMWR. 2005;54:125.
35 Dressler F, Whalen JA, Reinhardt BN, et al. Western blotting in the serodiagnosis of Lyme disease. J Infect Dis. 1993;167:392-400.
36 Wormser GP, Nadelman RB, Dattwyler RJ, et al. Practice guidelines for the treatment of Lyme disease. Clin Infect Dis. 2000;31(Suppl 1):1-14.
37 Gilden DH, Wright RR, Schneck SA, et al. Zoster sine herpete, a clinical variant. Ann Neurol. 1994;35:530-533.
38 Nemni R, Sanvito L, Quattrini A, et al. Peripheral neuropathy in hepatitis C virus infection with and without cryoglobulinaemia. J Neurol Neurosurg Psychiatry. 2003;74:1267-1271.
39 Ammandola A, Sampaolo S, Ambrosone L, et al. Peripheral neuropathy in hepatitis-related mixed cryoglobulinemia: electro-physiologic follow-up study. Muscle Nerve. 2005;31:382-385.
40 Leite AC, Silva MT, Alamy AH, et al. Peripheral neuropathy in HTLV-I infected individuals without tropical spastic paraparesis/HTLV-I-associated myelopathy. J Neurol. 2004;251:877-881.
41 Logina I, Donaghy. Diphtheritic polyneuropathy: a clinical study and comparison with Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1999;67:433-438.






