CHAPTER 107 Infectious Enteritis and Proctocolitis and Bacterial Food Poisoning
In Western countries, diarrhea leads to high morbidity with loss of time from school and work. Overall, physicians in the United States are consulted annually for 8.2 million diarrheal episodes.1,2 A long list of complications, some mild and others life-threatening, can accompany infectious diarrhea. Medical costs and loss of productivity resulting from infectious diarrhea amount to more than $23 billion each year in the United States.
Our knowledge of infectious diarrheal disease has expanded exponentially in the past two decades. References 3 to 6 are excellent discussions of various aspects of enteric infections.3–6
SUSCEPTIBILITY TO INTESTINAL INFECTION
HOST DEFENSE FACTORS
Gastric Acidity
Ingested pathogenic bacteria and other pathogens first must survive passage through the stomach to infect the small or large intestine. In this regard, gastric acidity is the first line of defense.7 Most bacterial pathogens are highly susceptible to low pH, and thus exposure to gastric acid significantly reduces the number of viable bacteria after their ingestion. Gastric juice with a pH of less than 4.0 is rapidly bactericidal, whereas bacteria survive prolonged exposure to gastric juice from patients who are achlorhydric.8 In experimental studies of cholera in healthy adults, clinical infection did not develop when as many as 1010 cholera bacteria were ingested, whereas as few as 104 Vibrio cholerae were able to produce disease when organisms were administered with sodium bicarbonate9; even fewer bacteria were necessary to produce clinical illness when organisms were directly instilled into the duodenum. Naturally occurring cholera also occurs more often in achlorhydric patients.10 The gastric barrier also may be important in preventing other enteric infections such as salmonellosis11 and shigellosis.12
Intestinal Motility
Organisms surviving the milieu of the stomach enter the small intestine where normal propulsive motor activity clears them. Some bacteria, including Clostridium difficile, Clostridium perfringens, and heat-stable enterotoxin-producing Escherichia coli, elaborate toxins that impair intestinal motility.13 In experimental animals it is often necessary to restrict intestinal motility with pharmacologic agents or with ligatures to allow enteropathogens to establish infection.14
Intestinal Microflora
The normal intestinal microflora, primarily in the colon, resist colonization of the intestine by newly introduced bacteria. Products elaborated by the resident microflora, including lactic acid and short-chain fatty acids, are toxic to many bacterial pathogens, and when the intestinal microflora are altered in instances such as administration of an antibiotic, colonization resistance is lessened and the host may be more susceptible to intestinal infection (e.g., C. difficile).15 Alteration of intestinal flora by antibiotics also increases susceptibility to salmonellosis.16,17
Mucus
Mucus, in concert with intestinal motility, provides a physical barrier to bacterial proliferation and mucosal colonization. Gastric mucus can act in conjunction with gastric acidity as the first line of enteric defense. Differences in the carbohydrate composition of intestinal mucus between immature and mature rats suggest that this difference plays a role in the reduced host defense of the immature animal.18
Systemic and Local Immune Mechanisms
The mucosa’s antibacterial immune response is quite complex and important in combating enteric pathogens. Secretory antibody in the intestine appears before serum antibody does in response to intestinal infection with Shigella.19 In cholera, there is a better correlation between the level of coproantibody and immune protection than there is with serum antibody and resistance to enteric infection with this pathogen20; however, both mucosal and systemic immune systems provide important protection against pathogenic bacteria. These immune responses may be directed against multiple targets. For example, the immune response against cholera may be directed against the toxin or the bacterium and originate from either the mucosal immune system (secretory IgA) or from the serum (IgG). Regardless, both serum and secretory antibodies exert their protective effects at the intestinal level, even though the serum components are produced outside the intestine.
Others
Breast-feeding also serves as a defense mechanism against bacterial enteropathogens. Breast-fed infants are less susceptible to bacterial diarrhea than are formula-fed infants.21,22 Multiple factors are responsible for this protection. Breast milk contains secretory IgA antibodies against specific enteropathogens that survive passage through the infant’s gastrointestinal tract.23 Other components such as lactoferrin, lysozyme, and lactoperoxidase also have anti-infective properties, and breast milk glycolipids can interfere with toxin or microbial adherence.24
BACTERIAL FACTORS
Adherence
The ability of bacteria to adhere to host mucosal cells is a critical virulence factor in enterotoxin-producing and invasive bacteria as well as in enteroadherent E. coli (EAEC) and enteropathogenic E. coli (EPEC). Bacterial adherence to host mucosal cells may be the predominant virulence factor, as in the case of EPEC; one of two important factors (adherence plus toxin elaboration), as in the case of enterotoxigenic organisms; or only one of several factors required for expression of full pathogenicity, as seen with invasive organisms. Bacteria that cause disease by adhesion alone do not elaborate any of the traditional enterotoxins, but rather they adhere tightly to the mucosa of both the small and large intestine.25 The classic EPEC and the EAEC26 are typical of this group. Other organisms, including enterotoxigenic E. coli (ETEC) and the invasive Salmonella and Shigella species, also must adhere to the intestinal surface to be fully pathogenic.
Studies on the mechanism by which EPEC cause diarrhea show that they attach to the intestinal mucosa in a characteristic manner, producing ultrastructural changes known as attaching-effacing lesions27 that lead to elongation and destruction of microvilli.25,28 This pattern of bacterial binding to enterocytes also has been referred to as attaching and effacing adherence27 and the particular morphologic alteration as pedestal formation (Fig. 107-1).25
The laboratory counterpart of mucosal colonization is adherence in tissue culture to various cell lines such as Hep-2 and HeLa. A characteristic form of localized adherence is observed only with classic EPEC serotypes. These events occur in the following three phases29:
The presence of a plasmid in EPEC serves to increase intimin production; this process is needed for localized adherence to occur.31 EPEC strains with localized adherence produce acute diarrhea when these strains are administered to normal volunteers.32 The role of the eaeA gene as a virulence factor in human EPEC infection has been confirmed in volunteer challenge studies.33
Enterotoxigenic organisms also require expression of bacterial adherence for proliferation of the organisms and colonization, as well as for full expression of toxicity.34 ETEC adhere to the surface of the small bowel epithelium without penetrating the epithelial layer and do so by mechanisms different from those used by EPEC. The most important mechanism by which enterotoxigenic bacteria adhere to the intestinal mucosa is related to specific protein antigens on the surface of the bacterial cell known as pili or fimbriae, also referred to as adherence antigens or colonization factor antigens.35 These pili bind to specific receptor sites on the surface of the intestinal cell via specific ligand-receptor interactions.
Evidence that these colonization factors (e.g., pili and lectins) are important to the pathogenesis of E. coli diarrheal disease in animals is derived from the observations by Moon36 that loss or gain of fimbriae by genetic manipulation results in the loss or gain of the ability to adhere to and colonize the intestine. Adherence not only permits colonization but also can facilitate the delivery of enterotoxin to the epithelium and might even enhance the ability of the organism to elaborate enterotoxin.34,36
Enterotoxin Production
Enterotoxins are polypeptides, secreted by bacteria, that alter intestinal salt and water transport without affecting mucosal morphology.5,37,38 Many organisms elaborate enterotoxins (e.g., V. cholerae, Shigella, ETEC, and Staphylococcus aureus), and several enterotoxins may be elaborated by a single organism. Although most enterotoxins affect the small intestine, the colon also may be a target organ.
Whether the enterotoxin is ingested preformed or is first expressed within the intestinal lumen, the toxin-enterocyte or toxin-colonocyte interaction begins with the binding of the enterotoxin to a specific mucosal receptor. The toxin-receptor interaction increases the concentration of an intracellular mediator, resulting in alteration of salt and water flux. Thus far, three intracellular mediator systems have been shown to be involved in the pathogenesis of enterotoxigenic diarrhea: adenylate cyclase and cyclic adenosine monophosphate (cAMP), the guanylate cyclase and cyclic guanosine monophosphate (cGMP) systems, and intracellular calcium.39,40 Alterations in these mediator systems have similar effects on transport processes to decrease the coupled influx of sodium and chloride and to stimulate the active secretion of chloride from the cell into the intestinal lumen. Other intracellular mediator systems involved in the pathogenesis of bacterial diarrhea include protein kinase C and arachidonic acid metabolites, among others.
Cholera toxin (molecular weight ≈84,000 kDa), which stimulates adenylate cyclase, is a prototypical enterotoxin (Fig. 107-2 and Chapter 99), the toxin-enterocyte interaction of which is well understood. Cholera toxin is composed of an A subunit surrounded by five B subunits that bind the toxin to a ganglioside (GM1) receptor on the brush border membrane of the villus epithelial cell. The A subunit, which consists of two parts (A1 and A2) linked by a disulfide bond, slowly penetrates the brush border membrane and is cleaved into its two component peptides. Reduction of this bond releases the active A1 peptide that traverses the cell to the basolateral membrane, where it stimulates the ribosylation of Gs, the stimulatory subunit of a heterotrimeric G protein. This action results in the irreversible activation of Gs and an increase in cytosolic cAMP. This cAMP in turn activates cAMP-dependent kinases that inhibit NaCl-coupled transport and stimulate chloride secretion.
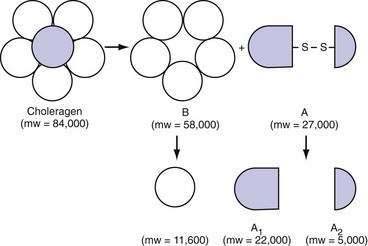
Figure 107-2. Subunit structure of cholera toxin (choleragen) (see text). mw, molecular weight.
(From Fishman PH. Action of cholera toxin: Events on the cell surface. In Field M, Fordtran JS, Schultz SG, editors. Secretory Diarrhea. Bethesda, Md: American Physiological Society; 1980. p 86.)
In addition to cholera toxin, other important enterotoxins are those elaborated by E. coli.41,42 Two classes of E. coli enterotoxins are known: heat-labile (LT) and heat-stable (ST) toxins. LT, which exists in two forms (LT-1 and LT-2),42 is a large molecular-weight protein that causes diarrheal disease similar to, but less severe, than, cholera. The subunit structure and mechanism of action of LT-1 and LT-2 also are similar to those of cholera toxin; although cholera toxin and LT bind to a glycolipid receptor, specifically GM1, an additional glycoprotein receptor might exist for LT.42 Heat-stable enterotoxins (STas) also may be elaborated by E. coli and bind to brush border receptors on enterocytes and colonocytes, a receptor guanylate cyclase, to increase intracellular levels of cGMP. STb is an unrelated enterotoxin elaborated by some E. coli pathogenic for pigs. Other organisms that elaborate highly homologous heat-stable enterotoxins include Yersinia enterocolitica, Citrobacter, and non-O1 vibrios.42
Cytotoxin Production
Cytotoxins are polypeptides that cause cell injury, inflammation, intestinal secretion through inhibition of protein synthesis or via a cascade involving one or more inflammatory mediator substances, and cell death. Examples of organisms that produce cytotoxins include C. difficile (see Chapter 108),15 some EPEC, enterohemorrhagic E. coli (EHEC), and Shigella.43 The mechanisms by which cytotoxins cause cell injury, inflammation, and intestinal secretion are numerous and complex and include inhibition of protein synthesis, disruption of cellular actin and tight junction integrity, mitochondrial damage, and adenosine triphosphate depletion among others.
Mucosal Invasion
The mechanism of mucosal invasion involves invasion of enterocytes or colonocytes by the infecting organisms with subsequent intracellular multiplication, resulting in cell injury and possibly cell death. Shigella species are classic examples of invasive enteropathogens. Salmonella species, Campylobacter jejuni, Y. enterocolitica, and some (enteroinvasive) strains of E. coli invade intestinal cells and pass into the lamina propria, where they elicit an inflammatory response and cause mucosal ulceration.4,6
Unlike enterotoxigenic organisms that favor colonization of the small intestine, invasive organisms primarily, but not exclusively, colonize the colon. In salmonellosis, the ileum is colonized in addition to the colon; in shigellosis, the small intestine is colonized transiently early in the course of the disease when watery diarrhea rather than dysentery is the predominant symptom.44 Subsequently, colonization occurs in the colon and bloody diarrhea ensues. In the cases of Shigella and Salmonella species, the ability to invade the gastrointestinal mucosa is of primary importance in establishing the enteric infection.45,46
Bacterial invasion alone is not sufficient to establish disease; other properties of invading organisms also are required. In the case of Shigella species, the organisms must multiply intracellularly. Thus, strains of Shigella flexneri that can invade but cannot multiply do not cause disease when fed to a susceptible host.45 Intracellular multiplication of Shigella organisms also involves lateral spread to adjacent intestinal cells and their death. In the cases of Salmonella species and Y. enterocolitica, however, the organisms penetrate into the lamina propria and can disseminate to extraintestinal sites. As a consequence of mucosal invasion and of the intramucosal multiplication of the organisms, an acute inflammatory reaction develops and mucosal ulceration can occur. Gross ulceration of the colonic mucosa commonly occurs in shigellosis, which accounts for dysenteric stools, but it is much less common with Salmonella and Yersinia infections. Yersinia infection more commonly manifests with microscopic and minute ulcerations involving both the ileum and colon.
CLASSIFICATION OF BACTERIAL DIARRHEA
DIAGNOSIS OF INFECTIOUS DIARRHEAL DISEASE
EVALUATION OF THE PATIENT
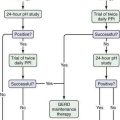
The initial step in the diagnostic evaluation of a patient with acute diarrhea should be a thorough history and physical examination, the goals of which are to identify patients who may be at risk of severe illness or susceptible to complications and those who will benefit from specific therapy. Most patients simply need rehydration therapy. Consideration of the patient’s general health, severity and duration of illness, and the setting in which the illness was acquired should enable the clinician to determine who needs further evaluation (Fig. 107-3).
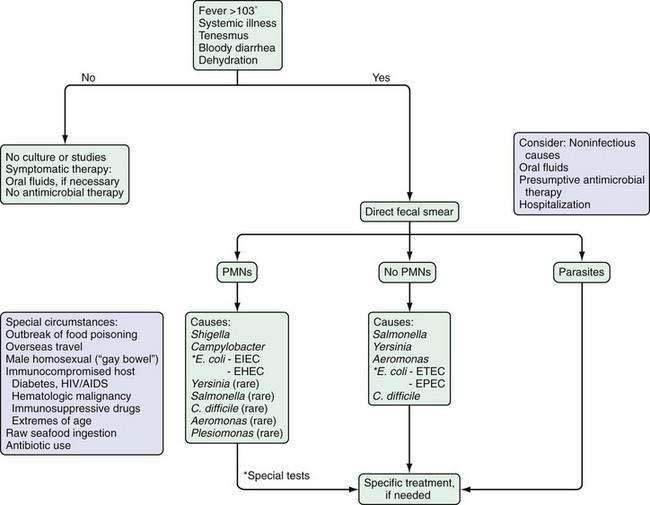
Patients who are debilitated, malnourished, or immunocompromised and those who have severe comorbid illnesses are at increased risk for complications of diarrhea and infection. They can require hospitalization and early diagnostic tests. Other patients who also require a more-aggressive approach include those with systemic signs and evidence of an inflammatory diarrhea, illness lasting more than three to four days, a history or physical examination suggesting a disease process that will benefit from specific therapy, and infection with certain specific organisms (Table 107-1).6
Table 107-1 Pathogens Indicating Need for Antimicrobial Therapy in Patients with Infectious Diarrhea
| Bacterial Infection |
From Park SI, Giannella RA. Approach to the adult patient with acute diarrhea. Gastroenterol Clin North Am 1993;22:483.
Because the number of conditions that cause acute and chronic diarrhea is large, a useful method is to classify the diarrheal illness into one of two clinical syndromes: a watery, noninflammatory diarrheal syndrome and an inflammatory diarrheal syndrome (Table 107-2); a subgroup of the latter is the proctitis diarrheal syndrome. Categorization into one of these syndromes limits the number of potential causes and diagnostic tests that need to be considered. This classification can usually be made on clinical grounds and with simple, inexpensive diagnostic tests.
Table 107-2 Characteristics That Help Distinguish Inflammatory from Noninflammatory Diarrhea
| CHARACTERISTIC | INFLAMMATORY DIARRHEA | NONINFLAMMATORY DIARRHEA |
|---|---|---|
| Clinical presentation | Bloody, small-volume diarrhea; lower quadrant cramps; patients may be febrile and toxic | Large-volume, watery diarrhea; patients may have nausea, vomiting, cramps |
| Site of involvement | Colon | Small intestine |
| Diagnostic evaluation | Indicated | Indicated only if the patient is severely volume depleted or appears toxic |
| Fecal leukocytes | Present | Absent |
| Causes | Shigella spp., Salmonella spp., Entamoeba histolytica, Campylobacter spp., Yersinia spp., invasive Escherichia coli, Clostridium difficile | Viruses, Vibrio spp., Giardia lamblia, enterotoxigenic E. coli, enterotoxin-producing bacteria, food-borne gastroenteritis |
From Park SI, Giannella RA. Approach to the adult patient with acute diarrhea. Gastroenterol Clin North Am 1993; 22:483-97.
Inflammatory Diarrhea
Patients with inflammatory diarrhea usually present with numerous small-volume stools that may be mucoid, grossly bloody, or both. Such patients may appear toxic and usually are febrile. Abdominal cramping may be severe. Because of the small stool volumes, these patients are less likely to be dehydrated than those with noninflammatory diarrhea. Physical findings might point to a specific diagnosis (Table 107-3).
Table 107-3 Clinical Finding(s) That Suggest the Causative Organisms for Some Inflammatory Diarrheas
| FINDING | CAUSATIVE ORGANISMS |
|---|---|
| Hemolytic-uremic syndrome | Shigella spp., enterohemorrhagic E. coli |
| Reactive arthritis | Salmonella spp., Shigella spp., Campylobacter spp., Yersinia spp. |
| Peritoneal signs | C. difficile, enterohemorrhagic E. coli |
| Right lower quadrant tenderness | Yersinia spp. |
| Thyroiditis, pericarditis, glomerulonephritis | Yersinia spp. |
From Park SI, Giannella RA. Approach to the adult patient with acute diarrhea. Gastroenterol Clin North Am 1993; 22:483.
Organisms causing inflammatory diarrheas usually affect the colon and either invade or elaborate cytotoxins, resulting in an acute inflammatory reaction with disruption of the epithelial barrier, mucus, red blood cells, and white blood cells in the stool (Table 107-2). Microbes causing this syndrome include Shigella, Campylobacter, EHEC, C. difficile, Salmonella, Yersinia, and Entamoeba histolytica. Fecal leukocytes (or positive stool lactoferrin test) indicate an acute inflammatory process, and sheets of polymorphonuclear leukocytes (PMNs) usually indicate colitis. The acute inflammatory diarrheal syndrome also can have an noninfectious etiology, for example, ulcerative colitis, Crohn’s disease, radiation or ischemic colitis, and diverticulitis. Table 107-4 lists the organisms that may be associated with the presence of fecal leukocytes.47
Table 107-4 Fecal Leukocytes in Intestinal Infections
| Usually Present |
DAEC, diffusely adhering Escherichia coli; EAEC, enteroaggregative E. coli; EHEC, enterohemorrhagic E. coli; EIEC, enteroinvasive E. coli; EPEC, enteropathogenic E. coli; ETEC, enterotoxigenic E. coli.
LABORATORY DIAGNOSIS
After taking an adequate medical history and performing a physical examination, the clinician should be able to classify the acute diarrheal illness as inflammatory or noninflammatory; to determine its severity and whether complications are likely; and to determine if diagnostic testing is needed and, if so, which tests should be used. In general, most episodes of acute diarrheal illness in the United States are self-limited; diagnostic testing may be kept to a minimum, and treatment is aimed at preventing dehydration. Investigations should be performed only if it is likely the result will influence management and outcome. Diagnostic testing should be reserved for patients with severe illness, including large-volume dehydrating diarrhea, severe abdominal pain, or a prolonged course (i.e., longer than three days); patients with bloody stools, systemic symptoms such as fever higher than 101°F, or prostration; patients with history of recent travel to high-risk areas; and patients at high risk for complications because of extremes of age, diabetes mellitus, and immunocompromise such as acquired immunodeficiency syndrome (AIDS), hematologic malignancy, or use of immunosuppressive medications (see Fig. 107-3).
Fecal Leukocytes
A particularly useful technique to focus the differential diagnosis is microscopic examination of the stool for PMNs (see Table 107-4). Invasive pathogens that primarily affect the colon, such as Shigella and Campylobacter, produce a “sea of polys,” as well as red blood cells. The toxigenic organisms, viruses, and food-poisoning bacteria cause a watery stool that harbors few formed elements. Stool tests for lactoferrin or calprotectin (proteins made by PMNs) in fecal specimens are available and provide a rapid and sensitive alternative to microscopy for identifying PMNs and, by inference, inflammatory diarrhea.48
Stool Cultures
Stool cultures are ordered too frequently. In most microbiology laboratories, routine stool cultures are processed for Shigella, Salmonella, and Campylobacter. Other enteric pathogens such as Yersinia, Vibrio, and E. coli O157:H7 are not sought routinely. Therefore, if clinical suspicion is high, the microbiology department needs to be notified to search for these pathogens. Because of sporadic shedding of pathogens (nontyphoidal Salmonella spp., Salmonella typhi) and because most episodes of acute diarrhea are caused by viruses, undetectable pathogens, or noninfectious causes, stool cultures are not usually positive. At Massachusetts General Hospital, the isolation rate of bacterial pathogens from 2000 fecal cultures in 1980 was 2.4%.49 In patients with severe diarrhea requiring hospitalization, the bacterial isolation rate from feces is somewhat higher, ranging from 27% to 43%,50,51 and up to 58% in a study that used more-advanced techniques.52 Even in patients hospitalized for dysentery, the rate of positivity for microbiologic diagnosis is only 40% to 60%. In community patients with severe acute gastroenteritis (more than four fluid stools per day, lasting at least three days and with at least one associated symptom), the yield of a stool culture and ova and parasite examination increased to 88%.53 In outbreaks of gastroenteritis in the United States, only one half of the cases have a confirmed etiology, of which two thirds are bacterial in origin. These figures suggest that many cases of acute diarrhea are caused by unidentified pathogens.
TOXIGENIC PATHOGENS
VIBRIO CHOLERAE
Microbiology
Toxigenic V. cholerae that agglutinates in O1 antiserum is the main cause of epidemic cholera. There are two major biotypes of V. cholerae O1: classic and El Tor. The latter strain is responsible for the current pandemic that began in 1961. El Tor vibrios are somewhat hardier than others in nature. Clinical disease is similar with both biotypes, although on average, El Tor infections are milder. The major serotypes associated with clinical disease are Inaba and Ogawa; a rare third type is Hikojima. The El Tor Inaba type was responsible for the 1991 outbreak in South America. There also are unique O1 cholera strains (e.g., V. parahaemolyticus and V. vulnificus) that cause endemic disease along the Gulf Coast of the United States.54
A newly described toxigenic non-O1 strain, now designated V. cholerae O139 Bengal, was responsible for an epidemic that started in southern India and Bangladesh in late 1992 and spread rapidly to many countries in Southeast Asia.55,56 This strain was classified as a new serogroup because it did not react with antisera to the previously identified 138 serogroups.56
Cholera Toxin
All wild strains of V. cholerae, including O139, elaborate the same enterotoxin, a protein molecule with a molecular weight of 84,000 kDa (see Fig. 107-2).57 The structural genes for the cholera toxin are encoded by a filamentous bacteriophage.58 Like the diphtheria toxin, the cholera toxin is composed of two types of subunits. Each toxin molecule contains five B subunits that encircle a single A subunit. The B subunit is responsible for binding to the receptor on the mucosa. The A subunit is responsible for activation of adenylate cyclase located on the basolateral cellular membrane (see earlier). A second 10 to 30 kDa LT, zonula occludens toxin, has been described that alters intestinal permeability by acting on intestinal epithelial cell tight junctions.59
Epidemiology
For many centuries, the Bay of Bengal had been considered the “cradle of cholera.” Western countries were relatively free of cholera epidemics until the 19th century, but since then, with the worldwide spread of the disease, six pandemics (across continents) have been reported. We are currently in the seventh pandemic, which started in 1961 in Indonesia and then made its way to the Philippines, Hong Kong, Japan, Korea, Thailand, India, Pakistan, and the Middle East, passing across the African continent to engulf the entire region, and, in 1991, spreading to South America. Although the overall number of cases of cholera in Latin America has subsided since 1991, outbreaks of V. cholerae have continued to occur sporadically throughout sub-Saharan Africa. During 1999, more than 200,000 cases of cholera were reported from Africa, accounting for 81% of the global total of cholera cases.60
Cholera occurs sporadically along the Gulf Coast of the United States, primarily in Texas and Louisiana.61 Among the millions of American travelers to endemic areas in foreign countries, only 41 imported cases of cholera were reported in the United States from 1961 to 1990, and none was associated with secondary spread. The epidemic in South America resulted in 151 cases of cholera in the United States: 26 cases in 1991, 103 in 1992, and 22 in 1993; only one death was reported.62
The South American epidemic that began in Peru in January 1991 reached more than one million cases in its first three years. From 15,000 to 20,000 cases of cholera were reported each week during the peak of the epidemic, for a national incidence of 1 : 1000 persons. Unboiled drinking water, unwashed fruits or vegetables, and food or water from street vendors were implicated risk factors in this explosive outbreak.63,64
The epidemic of V. cholerae O139 Bengal that began in southern India and Bangladesh in late 1992 affected adults predominantly.56 The clinical features of infection with the O139 Bengal strain were virtually indistinguishable from infection caused by V. cholerae O1.65,66
Pathogenesis
Attachment of V. cholerae to the intestinal mucosa is mediated by various surface components, including a fimbrial colonization factor known as toxin-coregulated pilus. The toxin-coregulated pilus attachment protein might play an important role in producing naturally occurring protective antibodies against V. cholerae.67
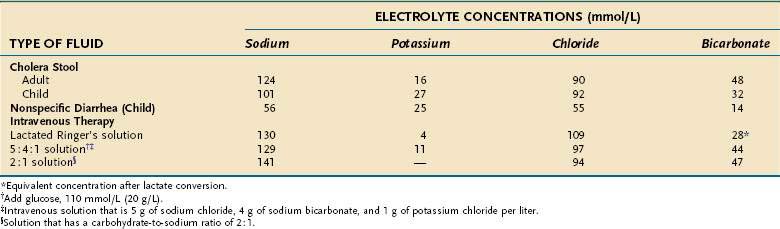
Despite the derivation of the term cholera (Greek: chole, bile), the appearance of choleric stools resembles rice water; that is, the stool has lost all pigment and becomes a clear fluid with small flecks of mucus. The electrolyte composition (Table 107-5) is isotonic with plasma, and the effluent has a low protein concentration. On microscopic examination there are no inflammatory cells, only small numbers of shed mucosal cells.
Clinical Features
Like many other infectious diseases, there is a spectrum of clinical manifestations with V. cholerae, from an asymptomatic carrier state to a desperately ill patient with severe dehydration. The initial stage is characterized by vomiting and abdominal distention and is followed rapidly by diarrhea that accelerates over the next few hours to frequent large volumes of rice-water stools. All the clinical symptoms and signs can be ascribed to fluid and electrolyte losses. Patients present with profound dehydration and hypovolemic shock, usually leading to kidney failure. The stool is isotonic with plasma, although there is an inordinate loss of potassium and bicarbonate, with resultant hypokalemic acidosis (see Table 107-5). Mild fever may be present, but there are no signs of sepsis.
Immunologic Responses
The susceptibility of adults in areas endemic for the O139 Bengal strain of cholera indicates that the afflicted populations are immunologically naive and that prior exposure to V. cholerae O1 does not provide cross-protective immunity. Nevertheless, volunteer challenge studies indicate that an initial infection with O139 Bengal provides protection against recurrent disease.65
Treatment
Treatment of acute cholera is based on the physiologic principles of restoring fluid and electrolyte balance and maintaining intravascular volume. These objectives can be accomplished with intravenous solutions or oral fluids that contain electrolytes in isotonic concentrations (see Table 107-5). Particular attention is paid to administration of bicarbonate and potassium, which are lost excessively in choleric stool. Various oral rehydration solutions (ORSs) have been developed for treating mild to moderate cases; ORS is especially useful in developing countries (Table 107-6).68
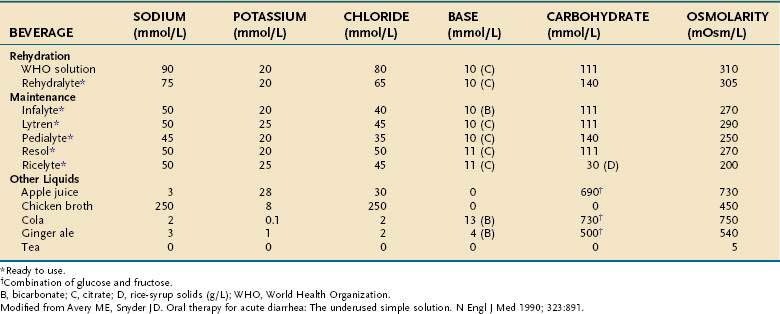
Antimicrobial agents are useful ancillary measures to treat cholera because their use leads to reductions in stool output, duration of diarrhea, fluid requirements, and Vibrio excretion.69 Tetracycline is recommended at a dose of 40 mg/kg per day orally up to a maximum of 4 g/day in four divided doses for two days; there is no proven value in lengthening the duration of treatment to four days. Single-dose therapy with ciprofloxacin results in a successful clinical response in 94% of patients infected with V. cholerae.70,71 As a result of rising rates of resistance, tetracycline and doxycycline often are less effective than the fluoroquinolones.71,72 Alternative drugs include trimethoprim-sulfamethoxazole (TMP-SMX) and furazolidone.
OTHER VIBRIO SPECIES
In addition to the cholera vibrios, at least nine other vibrios have important pathogenic significance.61,73,74 These strains represent a diverse group of organisms that are morphologically and biochemically identical to V. cholerae but that do not agglutinate with the O group antiserum of the three cholera serotypes.74,75 The non-O1 cholera vibrios produce several toxins and cause a wider range of infection than do the cholera vibrios, including watery diarrhea, dysentery, wound infections, ear infections, and septicemia.61,74
Strains within the same species can produce different enterotoxins, cytotoxins, and hemolysins. The diversity of toxin production is matched by the diversity of clinical symptoms: diarrhea ranges from watery dehydrating diarrhea to frank dysentery; some strains penetrate the intestinal mucosa and produce bacteremia; others have been incriminated in wound infections after exposure to ocean water or handling raw seafood.76
In East Asia, non-O1 cholera vibrios have been associated mainly with severe dehydrating diarrhea. In Peru, serogroups O10 and O12 were isolated from patients with liquid diarrhea associated with mild to moderate dehydration.77 In the United States, reported cases of disease caused by non-O1 cholera vibrios include wound and ear infections, septicemia, and infections of the lung and biliary tract.74 The most common antecedent history is consumption of raw oysters within the previous 72 hours. In outbreaks, there is a high attack rate, with incubation periods that range from as short as six to 12 hours to as long as three days. A one-week course of diarrheal illness is common. Because the gastrointestinal disease is self-limited and relatively benign in the United States, antibiotics are not recommended; however, septicemia, wound infections, and deep organ infections should be treated with appropriate antibiotics.
The incidence of Vibrio intestinal infections was studied among participants at an antimicrobial conference in New Orleans, many of whom had consumed raw oysters. Of 479 persons surveyed, 11% had a positive stool culture for vibrios, mainly V. parahaemolyticus, and approximately one third of those with a positive culture had diarrhea. Samples of local seafood, especially oysters, were found to harbor five different species of vibrios.78 In the Chesapeake Bay area, the annual incidence of Vibrio infections related to consuming seafood is estimated to be 1.6 per 100,000 persons.79
Vibrio parahaemolyticus
Pathogenic strains of V. parahaemolyticus produce a number of other toxins, including a lethal toxin that also is hemolytic. In some studies, these organisms produce an enterotoxin that causes fluid accumulation in the rabbit ileal loop model and a cytotoxic toxin that causes damage to HeLa cells. Some strains have the ability to invade the intestinal mucosa and cause bacteremia in experimental animals.80
Clinical Features
The diversity in toxins and virulence mechanisms is reflected in the variation in symptoms and signs observed in outbreaks in the United States.81,82 Explosive watery diarrhea is the cardinal manifestation in more than 90% of the cases. Abdominal cramps, nausea, vomiting, and headaches are common. Fever and chills occur in 25% to 50% of cases. Clinically, this illness resembles that produced by nontyphoidal Salmonella; however, in some cases a bloody dysenteric syndrome is observed, with fecal leukocytes and superficial mucosal ulcerations seen on sigmoidoscopic examination.
Additional Vibrio Species
V. vulnificus is perhaps the most important noncholera Vibrio species in the United States because of its severity of illness, especially in patients with underlying liver disease. V. vulnificus can be acquired as a wound infection in people swimming in salt water or by direct consumption of seafood, usually raw oysters; the mortality rate of resulting septicemia is 50%. Because this infection can be fatal in patients with underlying liver disease, such persons should be warned to avoid eating raw seafood, especially oysters.83
Vibrio mimicus acquires its name from its similarity to cholera vibrios, even in producing an enterotoxin that resembles cholera toxin.84 The organism has been isolated from patients in the United States with diarrhea, septicemia, or wound infections.73 Vibrio hollisae, also known as enteric EF-13, is a rare isolate from stool and, occasionally, blood cultures. Vibrio furnissii is found in Asia, but its most celebrated outbreak was on an air flight from Tokyo to Seattle, during which 23 passengers developed severe diarrhea, resulting in one death and two hospitalizations.
Vibrio fluvialis, previously designated as enteric group EF-6, has been isolated from patients with severe watery diarrhea in Asia and the coastal United States.73,74,79 The isolates produced a range of toxins, including an enterotoxin similar to classic cholera toxin. The organism is found only rarely in other parts of the world, including the United States. Bacteremia caused by Vibrio metschnikovii has been described in a limited number of cases and may be more common in patients with an underlying disease.85 Vibrio alginolyticus is a rare cause of wound or ear infections and gastroenteritis.73 Vibrio damsela is encountered rarely in wound infections.
AEROMONAS SPECIES
Aeromonas species are ubiquitous environmental organisms found principally in fresh and brackish water, especially in the summer months. These Gram-negative organisms often are mistaken for coliforms in the laboratory and, as a result, reported incidence rates are falsely low. Aeromonas species are divided into two groups: psychrophilic (Greek: psychros, cold) aeromonads, which grow optimally at temperatures ranging from 22°C to 28°C, and mesophilic aeromonads, which grow best between 35°C and 37°C.43 Psychrophilic strains usually are isolated from environmental water sources and fish; Aeromonas salmonicida is the most common strain in this group. Based on their phenotypic features, the mesophilic aeromonads are grouped into three complexes: Aeromonas hydrophila, Aeromonas caviae, and Aeromonas veronii. All three of these Aeromonas species have been associated with human infection.86,87 Aeromonas strains produce an array of toxins, including heat-labile enterotoxin, hemolysin, and cytotoxin.88
Epidemiology
Aeromonas infections often are associated with drinking untreated water, such as well water or spring water.89 Several studies have reported a high frequency of isolation of the organism from the stools of children with diarrhea; for example, the incidence of Aeromonas isolations in Western Australia was 10.2% in more than 1000 cases of childhood diarrhea, compared with 0.06% in control subjects.90 Other studies have found a high carrier rate in healthy people, with a range of 0.7% to 3.2% and up to 27% in Thailand. The high carrier rate has raised some question about the pathogenicity of Aeromonas.91
Clinical Features
Aeromonas has long been recognized as a cause of wound infections after swimming in fresh or brackish water and of bacteremic or deep organ infections in immunocompromised hosts. In recent years, however, most isolates have come from intestinal infections. There is a range of illness, from mild diarrhea, seen mostly in children, to more-severe cases that can require hospitalization. In a study from Western Australia, 22% of patients had blood and mucus in their stools, and one third required hospitalization for severe illness.90 Most cases resolved within one week, but 37% of these children had symptoms for two or more weeks. In adults, chronic diarrhea is even more common, lasting an average of 42 days in the United States.89
Treatment
These organisms are consistently resistant to β-lactam antibiotics, such as penicillin, ampicillin, and first- or second-generation cephalosporins.92 In fact, some cases of Aeromonas diarrhea have been activated apparently by prior treatment with ampicillin. Aeromonas species tend to be sensitive to TMP-SMX, third-generation cephalosporins, fluoroquinolones, tetracycline, and chloramphenicol. There is no convincing evidence that mild cases are improved by antibiotic treatment, but the duration of a chronic infection may be shortened by appropriate use of these drugs.
PLESIOMONAS SHIGELLOIDES
Plesiomonas shigelloides also is a member of the family Vibrionaceae but is isolated less often than Aeromonas in the United States.86,91,93 Most cases have been associated with consumption of raw oysters or recent travel to Mexico or Asia.94 Diarrhea ranges from mild and watery to severe colitis with visible blood. Abdominal pain often is prominent. Antibiotic sensitivity is similar to that of Aeromonas, but little information is available on the efficacy of treatment.
ESCHERICHIA COLI
E. coli are major components of the normal intestinal microflora in humans and animals. Although most strains are relatively harmless in the bowel, others possess virulence factors that are related to diarrheal disease. At least six types of E. coli intestinal pathogens have been recognized (Table 107-7). Their virulence factors include toxin production, adherence to epithelial cells, and invasiveness, each encoded by specific genetic elements (plasmids or chromosomal genes) that determine pathogenicity.

Enteropathogenic Escherichia coli
Severe epidemics of diarrhea raged in neonatal nurseries for decades, starting in the 1920s; although uncommon in recent years, such outbreaks had a high mortality. Approximately 14 serotypes were associated epidemiologically with neonatal diarrhea, including the well-known types O55, O111, and O119.26
These organisms adhere to the mucosal surface of the small and large intestine and cause dissolution of the glycocalyx and flattening and dissolution of the microvilli.25,28 This results in a form of localized adherence resulting in an attaching and effacement lesion (see Fig. 107-1). (This process is discussed earlier in “Bacterial Factors Involved in Intestinal Infection.”) The mechanisms by which this attachment results in intestinal secretion is not understood clearly, but they include alterations in enterocyte tyrosine kinase activity and intracellular calcium.
EPEC adherence factor is contained within a plasmid of the EPEC and has been used to construct a probe that, in turn, has been used to rapidly identify EPEC strains in stools of patients with diarrhea.95 In a study from São Paulo, Brazil, E. coli adherence factor-positive classic EPEC was found in 26% of children with acute diarrhea; these organisms were the most common pathogens isolated from the children, exceeding rotavirus isolations in frequency.96 EPEC strains are less-common causes of diarrhea in industrialized countries, but they seem to be important pathogens in many developing countries, especially in children in their first two years of life.97,98
Resistance to antimicrobial drugs is common in E. coli adherence factor–positive classic EPEC strains.96 Because most of these infections appear to be self-limited, there is no indication for antibiotic treatment, although nonabsorbable antibiotics such as neomycin have been used in the past for neonates with severe EPEC diarrhea.
Enterotoxigenic Escherichia coli
Inspired by the discoveries in cholera, investigators directed their attention to E. coli as a cause of acute toxigenic diarrheal disease. Originally in India, and thereafter in many parts of the world, strains of E. coli were found to elaborate an enterotoxin similar to that of V. cholerae.99 ETEC is a group of E. coli distinct from EPEC serotypes. ETEC infections mostly are sporadic but can cause large outbreaks.
Mechanism of Infection
Two types of enterotoxins are produced by ETEC.29 The LT is a protein that is destroyed by heat and acid and has a molecular weight of approximately 80,000 kDa. It acts pathophysiologically like cholera toxin by activating adenylate cyclase, thereby causing secretion of fluid and electrolytes into the small intestinal lumen. LT also shares antigenic components with cholera toxin. The second toxin is heat stable (ST) and is able to withstand heating to 100°C. This toxin has a low molecular weight of approximately 2000 kDa and activates guanylate cyclase; the resultant increase in cGMP induces intestinal secretion from both the small and large intestine. ST is really a family of toxins; the forms that cause disease most commonly in humans are ST1a and ST1b (which differ from each other by a few amino acid residues). ETEC strains elaborate LT only, ST only, or both LT and ST. These toxins cause diarrhea in humans, and similar types of toxigenic E. coli also cause dehydrating diarrhea in domestic animals, including pigs, cows, and sheep.
Epidemiology
ETEC infections are acquired from other humans; animal strains of ETEC are host-specific. The major vehicles of infection appear to be contaminated foods and beverages. Infection occurs primarily in children, and the highest incidence is in the tropics. There have been varying reports of ETEC infection in the United States, with high incidences in Chicago and Dallas and low figures in other American cities and Canada. In developing countries, the frequency of ETEC infection in children has varied from 15% to 50% of all diarrheal episodes. ETEC is the most common cause of diarrhea in travelers from North America and Northern Europe to areas of the developing world where diarrheal disease is prevalent. ETEC also has become the leading bacterial etiology of gastroenteritis outbreaks on cruise ships; water stored at overseas ports is the probable source of these ETEC infections.100
Clinical Features
ETEC infections are among the most common causes of diarrhea in children living in developing countries and travelers to these regions.97 There is nothing distinctive about the clinical presentation of ETEC diarrhea. The incubation period is 24 to 48 hours, and the disease often begins with upper intestinal distress, followed shortly by watery diarrhea. The infection can be mild, with only a few loose bowel movements, or quite severe, mimicking cholera, with severe dehydration and even rice-water stools. Indeed, the initial demonstration of such toxigenic diarrhea came from studies in Calcutta of a serious form of diarrheal disease called acute undifferentiated diarrhea. Affected patients were admitted to the cholera ward until it was determined that vibrios were not present in their stools. ST-only strains cause a milder attack of diarrhea than do LT-producing strains, but ST-affected patients have more vomiting and constitutional complaints.101
Immunity
Antibodies to the enterotoxins and colonization factors occur in persons infected with ETEC. It appears that people residing in areas at high risk for ETEC infection acquire some mucosal immunity over time.29 Thus, the risk that ETEC diarrhea would develop in students at a college in Mexico depended on their country of origin; those from South America had a relatively low risk of ETEC diarrhea, whereas those from North America had a high risk.102
Treatment
Most patients with ETEC diarrhea have only mild dehydration, but even small amounts of intestinal purging can have serious consequences in children and older people. The stool electrolyte losses in ETEC diarrhea are similar to those in cholera, and fluid replacement should follow the same principles. Although these organisms often are sensitive to many antimicrobial drugs, including TMP-SMX and quinolones, resistant isolates are increasingly encountered.100 Studies of patients with acute traveler’s diarrhea have demonstrated shortening of the duration of diarrhea when effective antimicrobial therapy is initiated early in the course of illness.103 Nevertheless, because most episodes of ETEC diarrhea are self-limited, treatment with antibiotics generally is not necessary.
Enteroinvasive Escherichia coli
Originally described in Asia, enteroinvasive E. coli (EIEC) is recognized as a rare cause of dysentery. During 1971, there was an EIEC outbreak in the United States that was related to contaminated imported cheese.104 Most episodes of EIEC infections are characterized by watery diarrhea; some patients experience a dysenteric syndrome that manifests as bloody mucoid diarrhea, tenesmus, fever, and intestinal cramps, with multiple PMNs in stool. EIECs have been recognized in at least eight E. coli serogroups, most of which are related biochemically and antigenically to Shigella. Other similarities to Shigella include the ability to invade epithelial cells and the production of two toxins, a Shiga-like toxin (STX) and an enterotoxin. Diagnosis of EIEC in a routine bacteriologic laboratory is difficult and generally impractical. Surveys of EIEC in the United States have shown low isolation rates, except in a few celebrated outbreaks. Low rates of infection have been observed in some less-developed countries,97 although in Thailand the organism is common in children with diarrhea.105
Enterohemorrhagic Escherichia coli
Acute hemorrhagic colitis, which first was recognized in two separate outbreaks in Michigan and Ohio in 1982,106 has been associated mainly with a specific serotype of E. coli, O157:H7. This organism is estimated to be responsible for 0.6% to 2.4% of all cases of diarrhea and 15% to 36% of cases of hemorrhagic colitis in Canada, the United Kingdom, and the United States.107,108 The spectrum of disease associated with E. coli O157:H7 includes bloody diarrhea, which is seen in up to 95% of patients, nonbloody diarrhea, hemolytic-uremic syndrome (HUS), and thrombotic thrombocytopenic purpura (TTP). Currently, the class of EHEC includes more than 100 different serotypes.108
Epidemiology
EHEC has become the most commonly isolated pathogen from the stools of patients with bloody diarrhea in the United States.109 EHEC disease is most common in northern climates such as in Massachusetts, Minnesota, and the Pacific Northwest, but it occurs throughout the United States. It also is well known in Canada, Great Britain, and throughout Europe. Infections occur sporadically or in large outbreaks. The leading vehicle of infection is hamburger meat, although outbreaks have been associated with precooked meat patties, roast beef, salami, fresh-pressed apple cider, lettuce, alfalfa sprouts, and unpasteurized milk.29,110–113 Water-borne outbreaks also have been associated with contaminated swimming pools and other recreational water bodies, well water, and municipal water systems.29 Person-to-person transmission probably has played a role in outbreaks in daycare centers, and nursing homes.107,114,115 Infection rates vary seasonally, and peak incidence is from June to September.
Virulence Factors
EHEC strains possess at least two virulence factors: an adherence mechanism causing attachment-effacement lesions similar to those seen with EPEC (see earlier) and two Shiga-like toxins (STX cytotoxins I and II).107,108,116 The toxins, which are identified either in stool samples or from culture of the organism itself, cause characteristic lesions in tissue culture lines such as Vero cells and HeLa cells. Some EHEC strains produce only STX I or II, whereas others produce both toxins. Most strains of E. coli O157:H7 possess the eaeA gene, which is associated with intimate attachment to the intestinal mucosa, as in EPEC (see earlier). They also produce enterohemolysin and are capable of using both heme and hemoglobin, a property that may enhance their virulence.117 E. coli O157:H7 toxins can result in colitis that resembles ischemic colitis because they can cause endothelial damage, platelet aggregation, and microvascular fibrin thrombi.107,108,118,118a
Clinical Features
Examination of the colon by endoscopy demonstrates a segmental colitis (i.e., friable inflamed mucosa with patchy erythema, edema, and superficial ulcerations). The process usually is most evident in the right colon (Fig. 107-4), but virtually any part of the colon may be affected, just as with idiopathic ischemic colitis. Plain films of the abdomen might show subepithelial edema and hemorrhage (thumbprinting), usually in the ascending and transverse colon. Leukocytosis with a shift to the left usually is present, but anemia is uncommon unless infection is complicated by the development of HUS or TTP.107 Microscopic examination of the stool reveals red and white blood cells in low to moderate amounts. The median duration of diarrhea is three to eight days, with longer durations in children and persons with bloody diarrhea.107
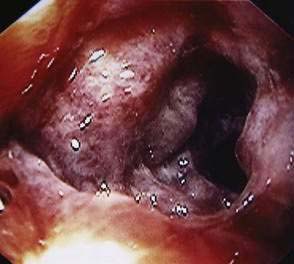
A striking association has been noted between intestinal infection with EHEC and HUS. In Minnesota, the incidence of HUS increased progressively during the 1980s to a current rate of 2.0 cases per 100,000 child-years. E. coli O157:H7 was isolated in 46% of children presenting with HUS. Risk factors for HUS include age younger than five years, attendance at a large daycare center, presence of bloody diarrhea, and a high white blood cell count.110 A study from the British Isles showed that 95% of the cases of HUS had a prodromal diarrheal illness. The disease was seen most commonly in the summer. Most EHEC strains were O157:H7 or O157:H− (H not able to be typed) and approximately 30% of the isolates belonged to nine other serogroups of E. coli.119,120 HUS is characterized by acute renal failure, microangiopathic hemolytic anemia, and thrombocytopenia.
Diagnosis
The chances of obtaining a positive culture in stool depend on the time between the onset of symptoms and collection of the stool. Within two days of onset, virtually all stool specimens from EHEC-infected patients are positive for EHEC, whereas after seven days only one third are positive.121 In contrast, other studies have found that the median duration of excretion of EHEC is 17 to 29 days, with some patients shedding the bacterium for as long as 124 days.29,122,123
One report detailed use of peroxidase-labeled antibody directed against whole E. coli O157:H7, and subsequent immunohistochemical staining to identify the organisms on archival paraffin block tissue sections from patients with hemorrhagic colitis and ischemic colitis.118a Positive staining was found in three of 11 cases of ischemic colitis, suggesting an etiologic role for E. coli O157:H7 in some cases of presumably idiopathic ischemic colitis.
Treatment
The desire to treat EHEC infections is understandable because of the presence of bloody diarrhea and concern that complications such as HUS will develop. Several reports, however, have raised concern that the risk of HUS is increased by antimicrobial therapy. In a murine model, certain antibiotics, notably ciprofloxacin, caused enhanced STX production by E. coli O157:H7 in vitro via the induction of bacteriophage encoded genes; this occurrence was associated with an increased death rate in antibiotic-treated mice.124
Antimicrobial therapy in humans does not appear to provide much benefit and might even be harmful. A randomized, controlled trial of TMP-SMX in children with E. coli O157:H7 enteritis found no effect of therapy on the duration of symptoms, pathogen excretion, or incidence of HUS.125 One prospective cohort study identified 71 children with acute E. coli O157:H7 gastroenteritis, of whom only 9 had been treated with antibiotics. However, 5 of the 10 children in whom HUS developed had received either TMP-SMX or a cephalosporin.126 In this study, antibiotic therapy was associated with a significantly increased risk of HUS, but this conclusion has been challenged by others.127,128
Thorough cooking of ground beef is an important preventive measure.
Enteroaggregative Escherichia coli
Unlike the attaching and effacing adherence to Hep-2 cells seen with EPEC, some E. coli strains have been observed to adhere in an aggregative pattern, with the bacteria clumping to the cell surface in a stacked-brick pattern.29 Although some investigations have implicated enteroaggregative E. coli (EAEC) as a cause of acute and persistent diarrhea in children in developing countries,29,129 other investigations have failed to find a significant association with diarrhea.96,97,130 Up to one third of children infected with EAEC have grossly bloody diarrhea. EAEC has been associated with diarrhea in patients infected with HIV,131 and EAEC has been shown to be a cause of traveler’s diarrhea.132
Volunteer challenge studies with different strains of EAEC have yielded mixed results, suggesting that certain strains are more virulent than others.133 As yet, there have been no studies documenting the need for or efficacy of treatment of EAEC infections. EAEC include numerous serogroups that largely are distinct from those of EPEC. Certain serotypes such as O44:H18 appear to be more pathogenic than others.
There have been no controlled trials of therapy for EAEC infections in children. One study of HIV-positive patients with diarrhea caused by EAEC found a 50% reduction in stool output, fewer intestinal symptoms, and microbiologic eradication of the organism during treatment with ciprofloxacin.134 Similarly, ciprofloxacin therapy of EAEC resulted in a reduction of the duration of diarrhea in patients with traveler’s diarrhea.132
Diffusely Enteroadherent Escherichia coli
Another type of adherent E. coli is diffuse adhering E. coli (DAEC), which adheres to tissue culture cells in a diffuse pattern. The role of these organisms in diarrheal disease is unclear, but they may be a cause of acute or persistent diarrhea in children.132a
INVASIVE PATHOGENS
SHIGELLA SPECIES
Epidemiology
Measurements of inoculum size in volunteers reveal that 105 organisms produce an attack rate of 75%,139 but increasing the inoculum size above this number does not increase the attack rate. There is not a good dose-response curve with Shigella (in contrast with Salmonella); indeed, dysentery can be produced with as few as 200 bacteria. The ability of Shigella species to survive in acidic conditions might account for the small inoculum that can produce disease.140 Person-to-person transmission, facilitated by the low infective dose, accounts for rapid spread of Shigella in daycare centers and among people living in conditions of poor hygiene. These factors also explain the high frequency of dysentery among male homosexuals.
Pathogenicity
The major site of attack of Shigella is the colon, although scattered ulcerations can be seen in the terminal ileum as well. Invasion by Shigella is associated with a constellation of virulence factors that are related to various stages of invasion and lead eventually to death of the intestinal epithelial cell, focal ulcers, and inflammation of the lamina propria. These virulence factors are encoded by both chromosomal and plasmid genes, all of which are needed for the full expression of virulence. All virulent Shigella, as well as EIEC, contain large 120- to 140-megadalton plasmids, which are related to outer membrane proteins. Various loci encode for an invasion plasmid antigen (ipa); invasion factors (inv); and a series of vir proteins that are involved in cell regulation mechanisms within the cell.141 Having penetrated the mucosal surface, the organisms multiply within the epithelial cells and extend the infected area by cell-to-cell transfer of bacilli. Shigellae rarely penetrate beyond the intestinal mucosa and generally do not invade the bloodstream; however, bacteremia can occur in malnourished children and immunocompromised hosts.
The initial lesions are confined to the epithelial layer, and the local inflammatory response is severe, consisting of PMNs and macrophages. There is edema, formation of microabscesses, loss of goblet cells, degeneration of normal cellular architecture, and ulceration of mucosa. These events give rise to the characteristic clinical picture of bloody, mucopurulent diarrhea. As the disease progresses, the lamina propria is involved extensively with the inflammatory response. Crypt abscess formation is a nonspecific but prominent feature (Fig. 107-5).
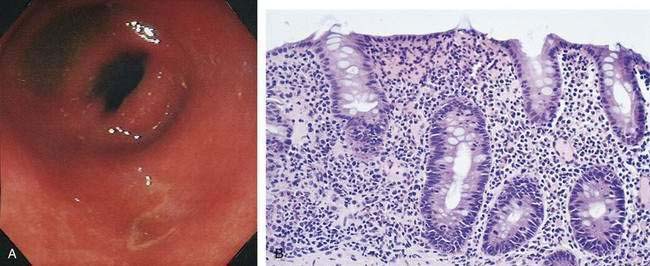
Cytotoxins
Initially, only S. dysenteriae 1 was known to elaborate an enterotoxin. This toxin, first identified by Shiga, has been shown to display a variety of biologic effects, depending on the experimental model used, including cytotoxicity, neurotoxicity in mice (seizures), and enterotoxicity (secretion of fluid and electrolytes); the neurotoxic properties can contribute to the seizures seen in some children with shigellosis. Shiga toxin, which is a 75-kDa protein composed of two subunits, inhibits protein synthesis by irreversible inactivation of the 60S ribosomal subunit.142 Inhibition of protein synthesis causes cytotoxicity and cell death. A toxin with similar antigenic and physiologic effects has been found in strains of S. flexneri and S. sonnei.143
Clinical Features
The classic presentation of bacillary dysentery is cramping abdominal pain, rectal burning, and fever, associated with multiple small-volume, bloody, mucoid bowel movements142; this full array of symptoms, however, is not seen in all patients. The most constant findings are lower abdominal pain and diarrhea. Fever is present in approximately 40% of patients, and the typical dysenteric stool, consisting of blood and mucus, is present in only one third. Approximately one third of patients only have diarrhea without dysentery.
An extensive list of extraintestinal complications of various bacterial enterocolitides including bacillary dysentery is presented in Table 107-8.144 Many patients complain of respiratory symptoms, such as cough and coryza, although pneumonia is rare. In young children, hypoglycemia can occur, and several neurologic findings can dominate the clinical picture, even before the diarrheal symptoms. Meningismus and seizures can occur with shigellosis, although there is no direct involvement of the central nervous system.145,146 The meningismus and seizures have been related to the high fever, but they also can occur when the fever is not extraordinarily high. During the acute phase of disease, HUS may occur.147 Thrombocytopenia and a severe leukemoid reaction also have been reported.148 A rash (rose spots) can occur during the acute phase of shigellosis.
Table 107-8 Complications and Manifestations of Bacterial Enterocolitis
| Campylobacter fetus |
* Associated with human leukocyte antigen B27.
Modified from Wolf D, Giannella RA. Antibiotic therapy for bacterial enterocolitis: A comprehensive review. Am J Gastroenterol 1993; 88:1667.
After an acute attack of dysentery, usually two to three weeks after onset, arthritis can appear. Joint pain or effusion usually is asymmetrical and involves large joints. Joint complaints are present by themselves, not necessarily with other signs of Reiter’s syndrome, and usually occur in patients who are positive for human leukocyte antigen (HLA)-B27; autoantibodies to this antigen cross-react with Shigella proteins, thereby resulting in circulating antibody-antigen complexes.149
Diagnosis
A subacute presentation of dysentery can masquerade as ulcerative colitis (see later). The patient might have endured bloody diarrhea, cramps, and rectal pain for two to four weeks, and sigmoidoscopic findings are indistinguishable from those of idiopathic ulcerative colitis (see Fig. 107-5) A colonic biopsy may be helpful to differentiate the two150: With dysentery, the inflammatory process is acute (PMNs) and involves the entire lamina propria; also, colonic glands are straight and without signs of regeneration. In ulcerative colitis, inflammation is chronic (lymphocytes) and involves mainly the lower one third of the lamina propria; colonic glands show signs of regeneration such as branching. Two major differences between dysentery and idiopathic ulcerative colitis are a positive stool culture for Shigella and dramatic improvement in symptoms after treatment of patients with dysentery with appropriate antimicrobial agents. When in doubt, treatment for shigellosis is recommended.
Deaths are rare in healthy persons, particularly adults, with bacillary dysentery; mortality usually is seen in young, often malnourished children or in debilitated patients—either the elderly or those with an immunodeficiency disease. A decreased level of consciousness and documented seizures are associated with a poor outcome in children.145
Treatment
The following general principles apply to the therapeutic approach to bacillary dysentery:
Fluid and Electrolyte Therapy
Most patients with dysentery can be managed with oral rehydration. High-volume diarrhea is seen occasionally with shigellosis and can result in severe dehydration and hypovolemia. Intravenous fluid replacement is indicated in this situation and also when severe vomiting prevents oral replacement. Fluid losses can be replaced within a few hours by intravenous solutions, and oral replacement should be encouraged as soon as possible (see Table 107-6). Antidiarrheal remedies generally are unhelpful, and some believe they might even aggravate bacillary dysentery. Kaolin and pectate and other water-binding agents do not diminish stool volume or frequency.
Antimicrobial Agents
Ampicillin was previously the preferred antibiotic, with TMP-SMX as an alternative choice; however, many strains, if not most, in the United States and abroad now are resistant to these antibiotics. The quinolone antibiotics, such as ciprofloxacin, ofloxacin, and norfloxacin, are highly active in vitro against Shigella and are the drugs of choice. Single-dose therapy with 1 g of ciprofloxacin is as effective as two doses or a five-day standard regimen in patients with Shigella infection; however, single-dose therapy proved less effective than multiple-dose regimens for patients with S. dysenteriae 1.151 Problems with using quinolones in the treatment of Shigella include the high cost of the drugs and concern about cartilage damage in young children. Nalidixic acid is an alternative therapeutic agent that has produced good results, although resistance develops rapidly with widespread use of this drug.152
Because there is now increasing evidence of the skeletal safety of quinolones in children,153 these drugs are being studied increasingly in pediatric populations. With single-dose pefloxacin therapy of infected children during an outbreak of multidrug-resistant S. dysenteriae 1 in Burundi, 91% of treated children became symptom free by day five; the remainder were substantially improved.154 None of the children experienced any joint problems during the four-week period of follow-up. Similarly, a double-blind trial of pivmecillinam compared with ciprofloxacin suspension for childhood shigellosis found that ciprofloxacin resulted in clinical responses in 80% of children, with no associated arthropathy.155
Although early animal and human volunteer studies indicated that the use of antimotility agents in the treatment of invasive diarrhea might lead to prolonged fever and pathogen carriage, a recent study has challenged this dictum. Treatment of dysenteric patients with a combination of the synthetic antidiarrheal agent loperamide and ciprofloxacin resulted in a significantly shortened duration of diarrhea and decreased number of stools when compared with ciprofloxacin alone.156 The use of loperamide did not lead to prolonged fever or excretion of the pathogenic bacilli.
Antibiotics for shigellosis must be absorbed from the bowel to reach organisms within the intestinal wall and lamina propria, and the only effective delivery system is the bloodstream.157 Nonabsorbable drugs, such as neomycin, kanamycin, paromomycin, colistin, and polymyxin, are clinically ineffective, despite in vitro sensitivity. Intravenous cefamandole also has proved disappointing. Curiously, amoxicillin, which is well absorbed and achieves higher serum levels than ampicillin, is not effective therapy for shigellosis.158
Mild diarrhea and cramps can continue for days to weeks after treatment of bacillary dysentery, even when the organism is no longer present and the acute episode seems to have passed. These symptoms are not necessarily a cause for alarm, because the bowel might have sustained severe mucosal injury that requires time for repair. Approximately 10% of patients with shigellosis, however, may be left with these symptoms chronically, a condition called postinfection irritable bowel syndrome.159
NONTYPHOIDAL SALMONELLOSIS
Microbiology
For convenience in the laboratory, a series of serogroups, the Kauffmann-White serotypes, was developed based on shared antigens among the most common Salmonella types. Ninety percent of Salmonella species pathogenic for humans falls into groups A to E, which contain 40 serotypes. The application of newer molecular methods to the taxonomy of Salmonella has revealed that all serotypes of Salmonella belong to one species that includes seven subspecies, which can be differentiated with biochemical tests. To avoid confusion with previous nomenclature, the new species Salmonella enterica has been proposed.160 Using this approach, the typhoid bacillus would be named S. enterica subspecies enterica serotype typhi. Because this lengthy name is cumbersome, however, simpler acceptable versions are S. typhi or S. enterica serotype typhi.
Epidemiology
Salmonella is one of the great food-borne infections.2 The major route of passage is by the five Fs: flies, food, fingers, feces, and fomites. The disease can cause large outbreaks, which often are associated with common-source routes of spread. A typical setting is an institutional supper or barbecue. Community outbreaks can persist for several months. For example, Riverside, California, experienced an epidemic involving 16,000 persons that raged for months and was related to a contaminated municipal water supply.
Although approximately 45,000 cases of salmonellosis are reported annually, these numbers reflect vast under-reporting, and it is estimated that 1.4 million cases of Salmonella food poisoning occur each year.2 The two most common serotypes in the United States are Salmonella enteritidis and Salmonella typhimurium.
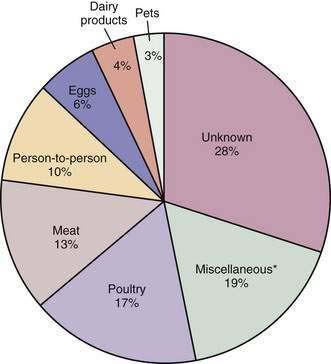
Nonhuman reservoirs play a crucial role in the transmission of the disease. In 500 outbreaks investigated over a 10-year period, almost 50% were related to animals or animal products. Poultry, meats, eggs, and dairy products were involved most often when a causative product was identified (Fig. 107-6).
Pathogenic Mechanisms
A series of pathogenic factors, each controlled by plasmids or chromosomal loci, are required for a Salmonella strain to be fully pathogenic. Specific plasmids encode for bacterial spread from Peyer’s patches to other sites in the body161,162 or the ability to survive within macrophages after phagocytosis.163 The outer membrane lipopolysaccharide and the Vi antigen are additional virulence factors. Another virulence factor imparts the ability of salmonellae to elicit transepithelial signaling to neutrophils,164 which contribute to cell damage and secretion. Finally, Salmonella strains produce enterotoxins that can play a role in diarrhea.161,165,166
In experimental animals, the number of bacteria required to produce infections can be reduced by pretreating the animals with antibiotics. Antibiotic exposure in humans also increases susceptibility to Salmonella infection.167 In addition, reduced or absent gastric acid is known to increase susceptibility to infection, because acid in the stomach kills many of the challenge organisms.140,141
Predisposing Conditions
A number of conditions increase the risk of salmonellosis (Table 107-9). The relationship between sickle cell anemia and Salmonella osteomyelitis is well known. Indeed, several forms of hemolytic anemia predispose to this infection, including malaria, bartonellosis, and louse-borne relapsing fever. The presumed mechanism of increased susceptibility is blockage of the reticuloendothelial system by macrophages that have ingested breakdown products of red blood cells, thereby reducing their ability to phagocytose salmonellae.168 Patients with sickle cell anemia also have a decreased capacity to opsonize salmonellae because of defective activation of the alternative complement pathway.169
Table 107-9 Conditions That Predispose to Salmonella Infection
AIDS, acquired immunodeficiency syndrome.
Neoplastic disease is associated with an increased risk of salmonellosis. Leukemia, lymphoma, and disseminated malignancy predispose patients to bloodstream invasion by this organism.170 Use of glucocorticoids, chemotherapy, or radiation therapy also is associated with Salmonella sepsis. In AIDS patients, persistent Salmonella bacteremia, only temporarily yielding to antibiotic therapy, is related to the profound suppression of cell-mediated immunity.171 Gastric surgery appears to be an important predisposing condition in the development of Salmonella infection because destruction of the gastric acid barrier enhances the host’s susceptibility to infection.7 Schistosomiasis also is associated with invasive salmonellosis.172 Ulcerative colitis also can predispose to Salmonella infection and the carrier state.173
Clinical Features
Five clinical syndromes are seen with Salmonella (Table 107-10), including gastroenteritis, noted in 75% of Salmonella infections; bacteremia, with or without gastrointestinal involvement, seen in approximately 10% of cases; typhoidal or enteric fever, seen with all typhoid and paratyphi strains and in approximately 8% of other Salmonella infections; localized infections (e.g., bones, joints, meninges, and blood vessels), seen in approximately 5%; and a carrier state in asymptomatic people (the organism usually is harbored in the gallbladder).174
Table 107-10 Relative Frequencies of the Clinical Syndromes of Salmonella Infection
| SYNDROME | FEATURES | FREQUENCY (%) |
|---|---|---|
| Gastroenteritis | Characterized by mild to severe and dehydrating (dysenteric) colitis | 75 |
| Bacteremia | With or without gastroenteritis, endocarditis, arteritis, or AIDS | 5-10 |
| Typhoid fever (“enteric fever”) | With or without gastroenteritis | 5-10 |
| Localized infection | May involve meninges, bones and joints, wounds, gallbladder, and may form abscesses | |
| Carrier state (>1 yr) | — | <1 |
AIDS, acquired immunodeficiency syndrome.
The most common syndrome caused by Salmonella is gastroenteritis. The incubation period is usually six to 48 hours but can last as long as seven to 12 days. Initial symptoms are nausea and vomiting, followed by abdominal cramps and diarrhea. The diarrhea usually lasts three or four days and is accompanied by fever in approximately 50% of persons. Diarrhea can vary from a few loose stools to dysentery with grossly bloody and mucopurulent feces to a cholera-like syndrome, in patients who are hypochlorhydric or achlorhydric.11,175 Persistent fever or specific findings on physical examination suggest bacteremia or focal infection. Salmonella bacteremia is similar to sepsis caused by other Gram-negative bacteria. Recurrent Salmonella bacteremia is seen in patients with AIDS.171
Once the organism invades the bloodstream, almost any organ can become involved, (e.g., meningitis, arteritis, endocarditis, osteomyelitis, wound infections, septic arthritis, and focal abscesses).174
Patients can become chronic carriers (defined as persistence for longer than one year) of nontyphoidal Salmonella. The overall carrier rate is between 2 : 1000 and 6 : 1000 infected persons. Children, especially neonates, and persons older than 60 years of age become carriers more commonly than do others. Also, structural abnormalities in the biliary tract (e.g., cholelithiasis) or the urinary tract (e.g., nephrolithiasis) predispose to and perpetuate the carrier state.176
Salmonella Colitis
Involvement of the colon in the course of Salmonella gastroenteritis probably is common. Although most patients with Salmonella present with mild diarrhea and watery bowel movements, colonic involvement can dominate the clinical picture; toxic megacolon and perforation due to Salmonella can occur.177 Patients with Salmonella colitis typically have diarrhea for 10 to 15 days before the diagnosis is established. In contrast, patients with the usual form of gastroenteritis are symptomatic for five days or less. In the colonic form, diarrhea is more persistent, even though the organism might have disappeared from the feces on clinical presentation. Bowel movements are grossly bloody in approximately one half of the patients with Salmonella colitis. Sigmoidoscopic findings include hyperemia, granularity, friability, and ulcerations. Rectal biopsy specimens reveal mucosal ulcerations, hemorrhage, crypt abscesses, straight glands, and a PMN infiltration that involves the lamina propria. Barium enema films confirm these findings and usually show pancolitis.
Treatment
Although many antibiotics have been used to treat nontyphoidal Salmonella gastroenteritis, all have failed to alter the rate of clinical recovery. In fact, antibiotic therapy increases the frequency and duration of intestinal carriage of these organisms.178 A review of 12 randomized trials found no differences in the duration of illness, diarrhea, or fever between patients treated with antibiotics and those treated with placebo.179 Relapses were more common in those treated with antimicrobial agents, as were adverse drug reactions. Thus antimicrobial therapy should not be used in most cases of Salmonella gastroenteritis.
Despite this general rule, antibiotics should be used when Salmonella gastroenteritis complicates certain conditions (Table 107-11), such as lymphoproliferative disorders, malignant disease, immunosuppressed states (AIDS and congenital or acquired disorders), organ transplantation, known or suspected abnormalities of the cardiovascular system (e.g., prosthetic heart valves, vascular grafts, aneurysms, and rheumatic or congenital valvular heart disease), foreign bodies implanted in the skeletal system, hemolytic anemia, extreme ages of life, and pregnancy. In addition, patients with Salmonella gastroenteritis should be treated with antibiotics when they exhibit findings of severe sepsis including high fever, rigors, hypotension, decreased kidney function, and systemic toxicity.
Table 107-11 Indications for Antibiotic Therapy in Salmonella Gastroenteritis
AIDS, acquired immunodeficiency syndrome.
If a decision is made to initiate therapy, the choice of drug may be problematic because of high levels of antibiotic resistance to ampicillin or TMP-SMX; currently, a fluoroquinolone is the drug of first choice. For patients with strains sensitive to ampicillin or TMP-SMX, these agents can be used (Table 107-12). The quinolones, particularly ciprofloxacin, have shown good results in patients with enteric fever180 and in chronic carriers.181 Ciprofloxacin therapy in patients with uncomplicated Salmonella gastroenteritis, however, has led to a high relapse rate that is associated with more-prolonged fecal excretion of salmonellae than seen in placebo-treated control subjects.178 As might be expected, resistance to ciprofloxacin has been observed during therapy.182
Table 107-12 Antibiotic Therapy for Bacterial Enteropathogens in Adults
| ORGANISM OR SYNDROME | RECOMMENDED ANTIBIOTIC(S) | ALTERNATIVE ANTIBIOTIC(S) |
|---|---|---|
| Shigella Species | ||
| Shigella infection | Ciprofloxacin 500 mg twice daily × 3 days | |
Note: All antibiotics are administered orally unless otherwise indicated.
AIDS, acquired immunodeficiency syndrome; IV, intravenous; TMP-SMX, trimethoprim-sulfamethoxazole.
* Bacteremia is treated for 10-14 days; endocarditis and osteomyelitis are treated for ≥4-6 wk.
† Enteroaggregative and diffusely enteroadherent E. coli are omitted from this table because these types are defined in research laboratories and are not diagnosed in routine clinical practice.
From Giannella RA. Treatment of acute infectious diarrhea. In Wolfe M, editor. Therapy of Digestive Disorders: A Companion to Sleisinger and Fordtran’s Gastrointestinal and Liver Disease. 2nd ed. London: Elsevier; 2006.
Since the turn of the 21st century there has been an increase in the isolation of quinolone-resistant Salmonella isolates in Europe.183 The use of quinolones for veterinary use has been blamed for this rise in resistance. Resistance to ceftriaxone of a Salmonella strain from livestock also has been observed in the United States.184 As a consequence of the increasing levels of drug resistance, both domestically and internationally, antimicrobial therapy of Salmonella infections must be limited to high-risk patients and should be based on sensitivity testing.
TYPHOID FEVER
Pathogenic Mechanisms
The pathologic events of typhoid fever are initiated in the intestinal tract after oral ingestion of typhoid bacilli.16 The organism penetrates the small intestinal mucosa and makes its way rapidly to the lymphatics, the mesenteric nodes, and the bloodstream. There is a paucity of local inflammatory findings, which explains the lack of intestinal symptoms at this early stage. This sequence of events is in marked contrast to that of other forms of salmonellosis and shigellosis, in which intestinal findings are prominent at the onset.
Epidemiology
Improvements in environmental sanitation have reduced the incidence of typhoid fever in industrialized nations. Approximately 400 to 500 cases occur each year in the United States, chiefly in young people. Large-scale epidemics of typhoid occur on a regular basis in developing countries and usually are traced to contaminated food that is imported from an endemic area or to contaminated water supplies.185
Because S. typhi cohabits exclusively with humans, the appearance of a case could indicate the presence of a carrier. An investigation by public health authorities should be instituted to determine the source and the presence of other cases. As they are discovered, chronic carriers are registered with the health authorities, and the particular microorganism is phage-typed so that it can be traced in the event of an outbreak. Registered carriers represent only some of the potential reservoir, however, and do not take into account imported cases of typhoid, which represent more than 70% of the acute infections in the United States.186
Clinical Features
In its classic form and without treatment, typhoid fever lasts about four weeks and evolves in a manner consistent with the pathologic events. The illness is described traditionally as a series of one-week stages, although this pattern may be altered in mild cases and by antibiotic treatment.187 The incubation period generally is seven to 14 days, with wide variations.
The preeminent complications are intestinal hemorrhage and perforation.188 These events are most likely to occur in the third week and during convalescence and are not related to the severity of the disease; they tend to occur in the same patient, with bleeding serving as a harbinger of possible perforation. Bleeding may be sudden and severe or a slow ooze. Before the availability of antibiotics, the incidence of hemorrhage was as high as 20%; it is less common since specific treatment has become available. Approximately 3% of patients with typhoid fever experience intestinal perforation, most commonly in the ileum.189 Onset of perforation may be sudden, with signs of an acute abdomen, or there may be a leak of intraluminal contents to form an abscess in the lower quadrant or pelvis, producing a more insidious course.
Carrier State
After six weeks, approximately 50% of typhoid victims still shed organisms in their feces. This figure declines progressively with time, and after three months only 5% to 10% are excreters; by one year the frequency is 1% to 3%.190 The chronic carrier is identified by positive stool cultures for S. typhi at least one year after the acute episode or, in some cases, positive stool cultures without a documented history of disease. The probability of spontaneously aborting the carrier state is highly unlikely after this time. Chronic carriers are more common in older age groups, women (a 3 : 1 ratio of women to men), and persons with biliary disease. The organism usually is harbored in the gallbladder, although occasionally it is carried in the large intestine without involvement of the biliary tract.
Diagnosis
The diagnosis of typhoid fever is established by isolating the organism. Blood culture is the primary diagnostic test, is positive in 90% of patients during the first week, and remains positive for several weeks thereafter if the patient is not treated. Bone marrow culture also has a high yield, even in treated patients.191 Stool cultures become positive in the second and third weeks. Sampling duodenal contents by a string test yields a positive culture in 70% of patients. By the third week, urine cultures reveal the organism in approximately 25% of patients. The titer of agglutinins against somatic (O) antigen (Widal test) rises during the second and third weeks of illness. A serum O titer of 1 : 80 or more in a nonimmunized person is suggestive of typhoid fever, and a titer of 1 : 320 usually is diagnostic in the appropriate clinical setting; a fourfold rise in titer provides stronger evidence. Although the H antigen is less specific than the O titer and is likely to be elevated from prior immunization or by infection with other enteric bacteria, an initial H antigen titer of 1 : 640 is strongly suggestive of typhoid fever. There are many false-positive and occasional false-negative Widal reactions, so that diagnosis based on a rise in titer alone is tenuous. Other serologic tests have become available and permit rapid diagnosis of typhoid fever with a higher sensitivity and specificity relative to blood culture than the Widal test; PCR assays for the diagnosis of S. typhi have been developed.
Treatment
Drug resistance, mediated by plasmids, occurs among typhoid bacilli. Most strains are susceptible to chloramphenicol and ampicillin, although notable epidemics with strains resistant to either or both of these drugs have been reported in recent years. Hence, a great effort should be made in each case to isolate the organism and perform drug susceptibility tests. Antibiotic treatment is listed in Table 107-12.
Chloramphenicol has high activity against most clinical isolates of typhoid bacilli. The response to therapy is remarkably constant, and defervescence regularly occurs three to five days after treatment is begun.188 The clinical condition improves within one to two days, with decreased toxemia and slowly declining fever. In adults, chloramphenicol should be given in a total daily dose of 2 g, administered in four equally divided doses by mouth. Occasionally, in very sick patients it may be necessary to give the drug by the intravenous route in the same total daily dose. Oral medication can be given upon clinical improvement. Chloramphenicol is well absorbed from the intestinal tract but is rather poorly absorbed from intramuscular sites; the intramuscular route is to be avoided. The duration of treatment is two weeks; prolongation of this treatment does not reduce the frequency of complications or carriers. Intestinal perforation and hemorrhage can occur during apparently successful treatment. Relapse can follow an otherwise uneventful course and should be treated with the same drug as was used initially.
Ampicillin has been recommended as alternative therapy but it has been disappointing in comparison with chloramphenicol.187,192 Amoxicillin, a closely related drug, provides better absorption and increased efficacy. Several studies have shown that amoxicillin, in doses of 4 g/day in four divided doses, has good activity. TMP-SMX also has been used with good results in typhoid fever.
The advent of plasmid-mediated multidrug resistance and newer potentially more effective antimicrobial agents, such as the quinolones and third-generation cephalosporins, has led to a reevaluation of the treatment of typhoid fever. The fluoroquinolones, ciprofloxacin and ofloxacin, have been found to be highly effective therapy for infections caused by multidrug-resistant S. typhi and S. paratyphi. Long-term fecal carriage of S. typhi is rare in patients treated with quinolones. A 10- to 14-day course of a quinolone has proved highly effective for treating enteric fever, with cure rates consistently close to 100%.72 The only exception has been norfloxacin, which has provided slightly lower cure rates of 83% to 90%.72 Defervescence generally occurs within three to five days of initiating therapy.
The optimal length of fluoroquinolone therapy for typhoid fever has not been fully elucidated. A number of studies have shown that courses of therapy ranging from seven to 14 days provide a high degree of success. Courses of therapy shorter than five to six days have been associated with unacceptable levels of failure.72 The duration of fever before treatment, severity of infection at the time of presentation, and time to defervescence are factors that must be considered when determining the duration of fluoroquinolone therapy in a patient with enteric fever. The frequency of resistance of S. typhi to ciprofloxacin has been increasing gradually, especially in the Indian subcontinent, central Asia, and Vietnam.185,193,194
Third-generation cephalosporins, such as cefotaxime, ceftriaxone, and cefoperazone, also have been used successfully to treat typhoid fever; courses as short as three days have been shown to be as effective as the usual 10- to 14-day regimens.195,196 By contrast, one trial that compared a five-day course of ofloxacin with a seven-day course of cefixime found that the median fever clearance time was significantly longer and the rate of treatment failure was higher in children treated with cefixime.197
Glucocorticoids are administered for severe toxemia and fever and can produce a dramatic response in patients with profound sepsis.198 Glucocorticoids should be given in high doses—for example, 60 mg/day of prednisone divided into four doses—and tapered rapidly over the following three days. The wide experience with glucocorticoid treatment has failed to show any adverse effects, although the potential for masking intestinal perforation always is present. Glucocorticoids should be reserved for patients with severe toxicity.
Studies have emphasized the importance of aggressive surgical intervention in typhoid fever.189,199,200 Indications for surgery include progressive peritoneal signs or formation of abscesses. Closure of the intestinal perforation coupled with broad-spectrum antibiotics has resulted in reduced mortality from this dreaded complication200; the ileum may be riddled with multiple perforations, and resection may be required.
A chronic carrier who has been discharging S. typhi for longer than one year can be treated with antimicrobials in an attempt to eliminate the infection. The quinolone antibiotics, including ciprofloxacin and norfloxacin, have become the treatment of choice in eradicating the carrier state.201 Reappearance of the carrier state after such treatment generally is associated with gallbladder disease. Cholecystectomy eliminates the carrier state in 85% of carriers with gallstones or chronic cholecystitis, but it is recommended only for persons whose profession is incompatible with the typhoid carrier state, such as food handlers and health care providers.
Vaccines
Two types of vaccine are available currently. The acetone-inactivated injectable vaccine affords 55% to 85% protection for two to five years. A live attenuated S. typhi strain, Ty21a, which is given by mouth, produced 96% protection in an initial field trial in Alexandria, Egypt,202 although subsequent studies showed less-impressive results.203,204 Because of its low toxicity and ease of administration, this vaccine should be used for travelers to high-risk areas, particularly in the developing world.
CAMPYLOBACTER SPECIES
The incidence and importance of human Campylobacter gastroenteritis have been recognized increasingly in recent years. It is estimated that 4% to 11% of all cases of diarrhea in the United States are caused by C. jejuni, and the isolation of Campylobacter often exceeds that of Salmonella and Shigella.205
Epidemiology
Transmission to humans appears to occur most commonly from infected animals and their food products. The reservoir for Campylobacter is enormous, because many animals can be infected, including cattle, sheep, swine, birds (poultry and others), and dogs. Most human infections are related to consumption of improperly cooked or contaminated foods. Just as for Salmonella, chicken seems to be the major source, accounting for 50% to 70% of infections.206
Clinical Features
The incubation period is 24 to 72 hours after organisms are ingested, but it can extend as long as 10 days. There is a wide spectrum of clinical illness, from frank dysentery to watery diarrhea to asymptomatic excretion.207 Diarrhea and fever are almost invariable (90%). Abdominal pain is usually present (70%), and the patient may note bloody stools (50%). Constitutional symptoms such as headache, myalgia, backache, malaise, anorexia, and vomiting are common. Stool examination suggests colitis on the basis of fecal leukocytes and occult blood.207 Endoscopy might reveal an inflammatory colitis (Figs. 107-7 and 107-8). The duration of illness usually is less than one week, although symptoms can persist for two weeks or more, and relapses occur in as many as 25% of patients. Prolonged carriage of Campylobacter for two to 10 weeks after the onset of illness occurs in 16% of patients.208
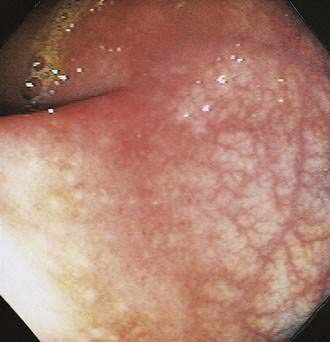
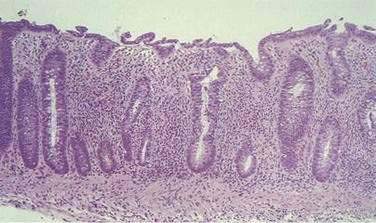
Infections rarely are complicated by gastrointestinal hemorrhage, toxic megacolon, pancreatitis, cholecystitis, HUS, bacteremia, meningitis, and purulent arthritis.207 Postinfection complications include reactive arthritis, usually in patients with the HLA-B27 phenotype, Guillain-Barré syndrome,207,209 and immunoproliferative small intestinal disease.210
Treatment
Antibiotic treatment is listed in Table 107-12.
Although C. jejuni is sensitive to erythromycin in vitro, three controlled therapeutic trials with this drug have shown no effect on the clinical course compared with placebo.211 One study showed some clinical benefit when the antibiotic was started within three days of the onset of symptoms. If therapy is delayed beyond four days, it yields no clinical improvement; fecal excretion of the organism, however, is reduced by erythromycin. The fluoroquinolone antibiotics, such as ciprofloxacin, also are active against these organisms.212
Resistance to fluoroquinolones has been observed during the course of treatment for Campylobacter diarrhea,213,214 and such resistance is a major problem in some parts of the developing world. A study of U.S. military personnel in Thailand found that 50% of isolates were resistant to ciprofloxacin, whereas none was resistant to azithromycin.215 A large study of human Campylobacter isolates in Minnesota found a rise in quinolone resistance from 1.3% to 10.2% between 1992 and 1998.216 Factors associated with quinolone resistance of Campylobacter species include foreign travel and local patterns of fluoroquinolone use, especially if these agents are used in animal husbandry.216,217 In locales where quinolone resistance is common, azithromycin has been shown to be superior to ciprofloxacin in decreasing the excretion of Campylobacter species and equivalent in terms of reducing the duration of symptoms.215
YERSINIA ENTEROCOLITICA
Y. enterocolitica causes a spectrum of clinical illnesses from simple gastroenteritis to invasive ileitis and colitis.218 It is a non–lactose-fermenting, urease-positive, Gram-negative rod. More than 50 serogroups and five biotypes have been identified.218 Pathogenic mechanisms include the ability to invade epithelial cells and the production of a heat-stable enterotoxin, which is elaborated at 25°C but not at 37°C. Not all strains have these pathogenic properties.
The organism targets the epithelium overlying Peyer’s patches for invasion, after which it proliferates within the follicles and spreads to the lamina propria. The abilities to attach to and penetrate epithelial cells are determined by the inv gene, which encodes a 103-kDa protein known as invasin.219
Epidemiology
Yersinia gastroenteritis has been reported more often in Scandinavian and other European countries than in the United States. Several epidemics have been related to the consumption of contaminated milk and ice cream. The organism can be isolated from many animals, including puppies, cats, cows, pigs, chickens, and horses. Animals, either as pets or food sources, are believed to be involved in the transmission of this disease.220
The serotypes most commonly involved in Scandinavia and Europe are O:3 and O:9, and Canada has many serotype O:3 isolates.221 Most of the isolates in the United States are serotype O:8. Serogroups O:8 and O:5,27 have been responsible for most episodes of invasive disease in the United States.
Clinical Features
Several clinical syndromes have been described with Yersinia and tend to vary with the age of the patient and the underlying disease state.218,222 Enterocolitis is the most common clinical condition and accounts for two thirds of all reported cases. This illness occurs most commonly in children younger than five years.219,222 Presentation is nonspecific, with fever, abdominal cramps, and diarrhea, usually lasting one to three weeks. Microscopic examination of stool reveals leukocytes and red blood cells in most instances. Profuse watery diarrhea, possibly related to the enterotoxin, also can occur. Diarrhea can persist for several weeks and can raise the possibility of inflammatory bowel disease. Radiologic findings, particularly in prolonged cases, are most intense in the terminal ileum and can resemble those of Crohn’s disease223; most patients, however, have normal findings on endoscopy, intestinal biopsy, and barium studies.221
In children older than five years, mesenteric adenitis and associated ileitis have been described. Accompanying symptoms include nausea, vomiting, and oral aphthous ulcers. Affected children often undergo a laparotomy, at which time enlarged mesenteric nodes and an ulcerated ileitis are observed. Clinically, the condition may be confused with acute appendicitis, although ultrasonography can be useful in separating these processes.224 Yersinia is less likely to cause severe disease in adults, in whom acute diarrhea may be followed two to three weeks later by joint symptoms and rash (erythema nodosum or erythema multiforme), reminiscent of Reiter’s syndrome. Reactive polyarthritis occurs in 2% of patients with yersiniosis, usually in persons seropositive for HLA-B27. Yersinia antigens can be detected in synovial fluid cells225 and in intestinal mucosal biopsies, and specific IgA antibodies are found in the blood.226
The diagnosis of yersiniosis is established by culture of stool or body fluids. Because the organism resembles coliforms and therefore is easily misdiagnosed on the culture plate, the laboratory should be advised that this infection is clinically suspected. Serologic tests have proved useful in Europe and Canada227 but have not provided much help for the cases reported in the United States.228
Treatment
Y. enterocolitica strains are susceptible to several antimicrobial agents, including chloramphenicol, gentamicin, tetracycline, TMP-SMX, and fluoroquinolones, but they are resistant to penicillins and cephalosporins. There is no substantial evidence that antibiotics alter the course of the gastrointestinal infection218,228; indeed, the diagnosis often is established late in the course when the patient is already improving spontaneously. Antibiotics should be used in more severe intestinal infections, particularly those masquerading as appendicitis. For the chronic relapsing form of diarrhea, antibiotic therapy has not proved useful. Septicemia in the immunocompromised patient is associated with a high mortality, with no apparent benefit from antibiotics, although treatment is mandatory in this setting. Antibiotic treatment is listed in Table 107-12.