Inborn errors of metabolism
Patterns of inheritance
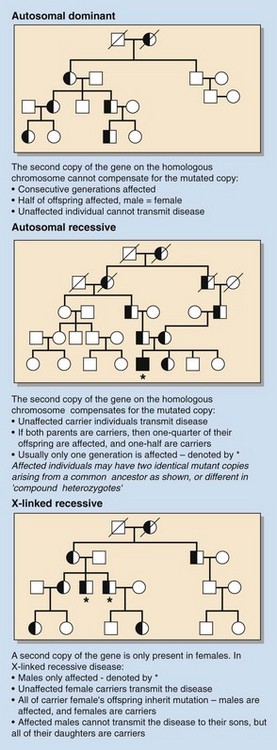
Inborn errors can be autosomal (involving a chromosome other than X or Y) or X-linked, and the genetic defect can be either dominant or recessive. In dominant disorders, everyone who carries the gene is affected by the disease, so every affected individual has at least one affected parent. If the defective gene is recessive, it will be silent unless both copies (maternal and paternal) of the gene carry the mutation, i.e. affected individuals must be homozygous; parents carrying only one copy of the affected gene (heterozygotes) are carriers and are not clinically affected. These patterns of inheritance are illustrated in Figure 80.1.
Mechanisms of disease
Inborn errors of metabolism can manifest clinically in various ways:





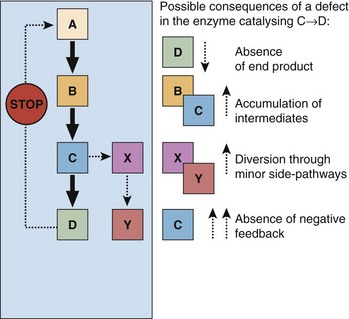
These are shown in Figure 80.2.
Clinical diagnosis
 parents are cousins (so-called consanguinous mating)
parents are cousins (so-called consanguinous mating)
 history of unexplained premature death in an older sibling
history of unexplained premature death in an older sibling
 onset of symptoms following change in feeding regimen
onset of symptoms following change in feeding regimen

 unusual smell (see Table 80.1).
unusual smell (see Table 80.1).
Table 80.1
Inborn errors of metabolism associated with characteristic smells due to volatile organic intermediates
| Inborn error of metabolism | Smell |
| Maple syrup urine disease | Maple syrup |
| Phenylketonuria | Musty |
| Isovaleric acidaemia | Sweaty feet or cheese |
| Trimethylaminuria | Fish |
| Hypermethioninaemia | Cabbage |
One useful classification of inborn errors includes both clinical and laboratory features (Table 80.2).
Table 80.2
Classification of inborn errors of metabolism on basis of clinical and laboratory features
| Presentation | Most likely diagnoses |
| ‘Intoxication’, ketoacidosis (blood H+ not ↑↑) | Maple syrup urine disease (amino acid disorder) |
| ‘Intoxication’, ketoacidosis | Organic acid disorders |
| ‘Energy deficiency’, lactic acidosis | Congenital lactic acidoses |
| ‘Intoxication’, high ammonia, no ketoacidosis | Urea cycle defects |
| ‘Energy deficiency’, no metabolic disturbance | Peroxisomal disorders Non-ketotic hyperglycinaemia |
| Storage disorders, no metabolic disturbance | Lysosomal storage diseases |
| Hypoglycaemia, hepatomegaly, abnormal LFTs | Glycogen storage diseases |
Laboratory diagnosis
Clearly if there is a clinical basis for suspecting a particular inborn error of metabolism, specific investigations should be requested. For example, the presence of cataracts should make one suspect galactosaemia, for which the appropriate investigation is measurement of galactose-1-phosphate uridyl transferase in red blood cells. More often, however, there are no specific features. Routine laboratory investigations may help point the direction of further investigations by suggesting particular groups of metabolic disorders (see Table 80.3). In the acute situation, in the absence of clues, the following investigations should always be considered, and performed urgently if indicated:
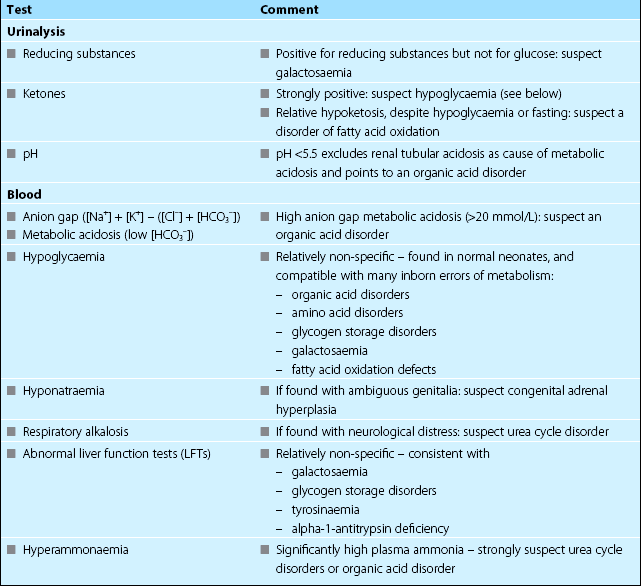
 Plasma ammonia. Indicated particularly when there is neurological distress/intoxication; grossly elevated levels are most frequently due to urea cycle disorders.
Plasma ammonia. Indicated particularly when there is neurological distress/intoxication; grossly elevated levels are most frequently due to urea cycle disorders.



 Clinical note
Clinical note