Impact of Nontraditional Antiarrhythmic Drugs on Sudden Cardiac Death
Although mortality from coronary artery disease has declined in the United States, sudden cardiac death (SCD) remains a major clinical problem, with a reported range of 184,000 to 462,000 deaths occurring annually.1 Strategies to decrease the incidence of sudden death include preventing underlying structural heart disease, screening for hereditary syndromes and cardiac conditions that predispose persons to sudden death, improving efforts in resuscitation medicine for patients who experience a cardiac arrest (secondary prevention), and identifying patients at high risk for sudden death to provide appropriate intervention.
Potential interventions to prevent sudden death include risk factor and lifestyle modifications, dietary changes, therapy with antiarrhythmic drugs, therapy with other cardiovascular drugs, or device therapy with implantable cardioverter-defibrillators (ICDs). Results of therapy with antiarrhythmic drugs that block sodium or potassium channels for the prevention of sudden death have been mostly disappointing. With the exception of amiodarone, these drugs have had either a neutral or a deleterious effect on mortality.2
Assessing the Impact of Nontraditional Antiarrhythmic Drugs on Sudden Cardiac Death
One difficulty in interpreting the results of clinical studies on SCD is the diversity of potential underlying mechanisms of death, with arrhythmic SCD representing a large fraction of underlying etiologies. This is further complicated by variable definitions of SCD. In some studies, all deaths that occurred up to 24 hours after the onset of symptoms were considered SCDs. This definition is not specific for sudden arrhythmic death because many patients who die from myocardial infarction (MI) within the first 24 hours of symptom onset do so from a cause unrelated to a primary arrhythmia. Thus, in evaluating the effect of nontraditional antiarrhythmic drugs on the incidence of SCD, we will also consider the effects on total mortality, with the understanding that it is possible that some of the mortality reduction may be the result of a decreased incidence of SCD. Studies have suggested that the major mechanisms responsible for sudden arrhythmic death are ventricular tachycardia (VT) and ventricular fibrillation (VF). However, there has been a decreased incidence of VF as a cause of out-of-hospital cardiac arrest and a concomitant increase in non-VF causes.3,4 Therefore, evaluating how therapies alter the occurrence of VT and VF provides more specific insight into the pathophysiological action of these agents to reduce SCD.
One potential model of the pathophysiology of sudden death is shown in Figure 113-1. In this construct, transient factors or triggers interact with spontaneous arrhythmias on an abnormal, underlying anatomic substrate that contributes to electrophysiological conditions leading to SCD. This model recognizes that the triggering beats may be caused by reentry, abnormal automaticity, or triggered activity, and that the electrophysiological substrate could consist of either functional or anatomic reentry. Thus, transient metabolic and autonomic factors that alter electrophysiological properties can modulate the anatomic substrate to heighten the occurrence of sudden death.
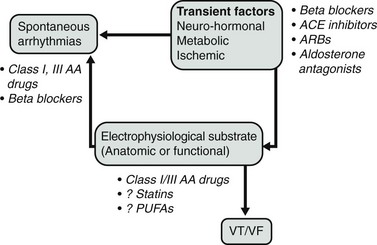
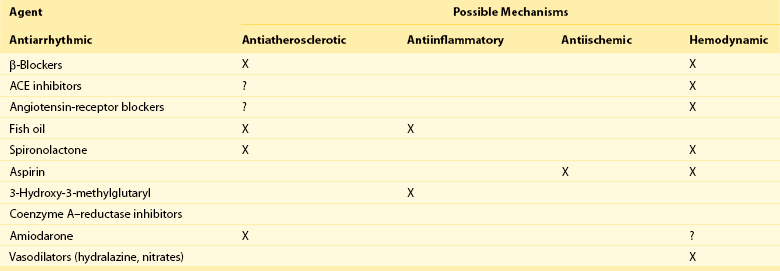
To target the electrophysiological substrate that forms part of the model, it is necessary to identify a specific electrophysiological mechanism and to identify specific ion channels or other targets that are present in the region of the myocardium that predisposes to sudden death. At present, antiarrhythmic drugs target sodium or potassium channels present throughout the myocardium and therefore are not specific for the arrhythmic mechanism (focal or reentrant) or abnormal myocardial region. Drugs such as β-blockers also operate on global regions of the myocardium, but their lack of specificity does not appear to be harmful, but rather is protective. Given the overall safety profile of nontraditional drugs compared with traditional antiarrhythmic drugs, it should not be surprising that nontraditional drugs that act on more global mechanisms of arrhythmogenesis are more effective in preventing sudden death than are drugs that indiscriminately block ion channels throughout the myocardium. Potential target sites for therapies that might reduce sudden death discussed in this chapter are shown in Figure 113-1. Table 113-1 shows interventions that may reduce mortality in patients with heart disease and the drugs that may reduce the incidence of sudden death.
β-Adrenergic Blockers
β-adrenergic blockers (β-blockers) have emerged as important pharmacologic agents for both primary and secondary prevention of SCD (Table 113-2). The mechanism of this class of drugs involves competitive β-adrenergic receptor blockade of sympathetically mediated triggering mechanisms, slowing of the sinus rate, and, possibly, inhibition of excess calcium release by the ryanodine receptor.5,6 Given the presence of β-receptors throughout the heart, cardiac disease contributes to pathophysiological changes in cardiac sympathetic nerves, cardiac responsiveness to sympathetic stimulation, and circulating catecholamines. After MI, for example, there are changes in heart rate variability and baroreflex sensitivity that reflect abnormalities of parasympathetic and sympathetic tone. These abnormalities can also heighten the risk for ventricular arrhythmias.
Table 113-2
Efficacy of Drugs in Reducing Mortality and Preventing Sudden Death
Efficacy of β-Blockers After Myocardial Infarction
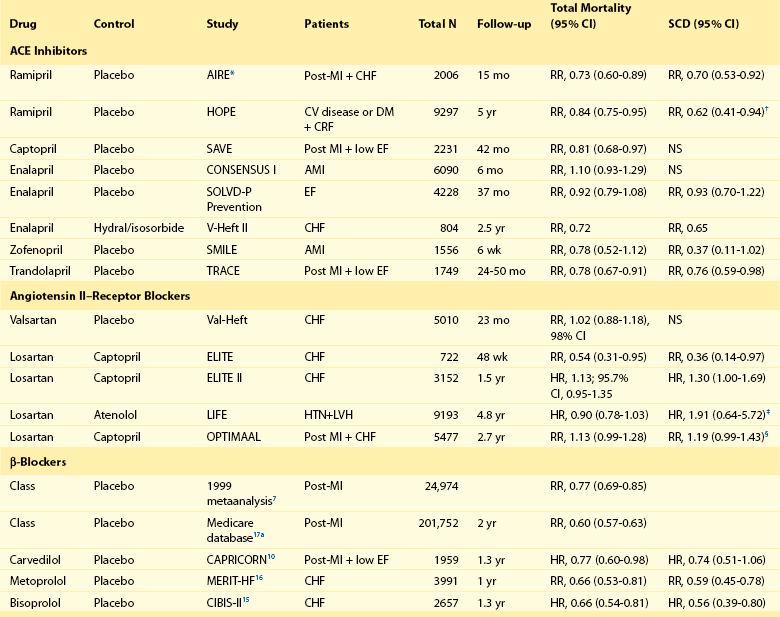
β-Blockers first were recognized as prophylactic agents for preventing sudden death in patients after MI through trials designed to address total mortality reduction. Analysis by Freemantle and colleagues7 of more than 50,000 patients evaluated in numerous randomized clinical trials demonstrated a substantial reduction in total mortality as a result of β-blocker treatment (relative risk [RR], 0.77; 95% confidence interval [CI], 0.69 to 0.85). The largest observational report, including more than 200,000 patients in the Medicare database, supports the role of β-blockers in reducing total mortality.8 Their effect on sudden death was noted in the landmark Beta-Blocker Heart Attack Trial (BHAT), which tested the effect of long-term propranolol therapy in patients after a MI.9 More than 3800 patients were randomized to either propranolol or placebo starting 5 to 21 days after MI and were followed for 2 years. Total mortality was 7.2% in the propranolol group and 9.8% in the placebo group (26% reduction). Sudden cardiac death occurred in 3.3% of the propranolol patients versus 4.6% of the placebo patients (28% reduction). A subset of these patients also had ambulatory electrocardiographic monitoring at baseline and after 6 weeks of therapy. An increase in ventricular arrhythmias over the 6-week period was blunted by propranolol. Moreover, metoprolol therapy has also been shown to reduce total number of deaths, especially SCDs, after MI. The mortality reduction was independent of gender, age, and smoking habits.8 The survival benefit of β-blocker therapy was shown to extend at least 6 years after MI in the Norwegian Multicenter Study Group.8
The benefits of β-blocker therapy have withstood the test of time. In the CAPRICORN study,10 1959 patients who had an MI within 3 to 21 days and an ejection fraction (EF) less than or equal to 40% were randomly assigned to treatment with either carvedilol (n = 975) or placebo (n = 984). Although all-cause mortality was not the primary endpoint, after a mean follow-up of 1.3 years, 12% mortality was seen in patients treated with carvedilol and 15% mortality was seen in those treated with placebo (risk reduction, 23%; 95% CI, 2 to 40).10 Further analysis from this study showed a lower incidence of malignant ventricular arrhythmias in the carvedilol-treated group (0.9% vs. 3.9% in the placebo group; P < .0001).11 In an analysis of the VALIANT registry, Piccini and colleagues12 found that patients presenting with VT/VF in the setting of acute MI had higher mortality if they did not receive β-blocker therapy within 24 hours (RR, 0.28; 95% CI, 0.10 to 0.75; P = .013).
Because clinical trials have demonstrated significant benefits of β-blocker therapy on survival, the vast majority (>90%) of survivors of MI are discharged receiving β-blocker therapy.13 Yet the majority of these patients are treated with doses that are substantially lower than the doses used in clinical trials. The effect of dose on survival benefit has not been definitively demonstrated, although there are reports indicating that low-dose β-blocker therapy does improve survival.
Efficacy of β-Blockers in Patients With Congestive Heart Failure
β-Blockers also are important agents in the prevention of SCD in patients with congestive heart failure. In this population, carvedilol has been shown to reduce mortality by 65%.14 In the Cardiac Insufficiency Bisoprolol Study II (CIBIS-II),15 the estimated annual mortality was 8.8% in patients treated with bisoprolol and 13.2% in those receiving placebo (risk reduction, 34%; 95% CI, 19 to 46). Metoprolol has also been shown to improve survival in patients with congestive heart failure. In the Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF), 3991 patients with class II to IV (96% had class II and III) congestive heart failure (EF <40%) were randomly assigned to treatment with metoprolol (n = 1990) or placebo (n = 2001).16 This trial was also terminated early by the safety committee because of the benefit of β-blocker therapy. The mortality rate was 7.2% per patient-year of follow-up in the metoprolol group versus 11.0% in the placebo group (RR reduction, 34%; 95% CI, 19 to 47). In both trials, prevention of sudden death was an important component of mortality reduction.
Efficacy of β-Blockers in Secondary Prevention of Sudden Death
β-Blockers also demonstrate efficacy in patients known to have significant ventricular tachyarrhythmias. Clinical information from the Antiarrhythmics Versus Implantable Defibrillators (AVID) trial registry provides support for the role of β-blocker therapy in patients with sustained ventricular tachyarrhythmias,8 where there was an approximately 50% reduction in adjusted relative risk of mortality because of β-blocker therapy. β-Blockers have been shown to reduce the incidence of ventricular tachyarrhythmias in patients with ICDs.8 The overwhelming majority of the study populations in all of these reports were patients with coronary artery disease. Limited data suggest that β-blocker therapy in patients with nonischemic dilated cardiomyopathy and ICDs is associated with a marked reduction in appropriate therapy for ventricular tachyarrhythmias.8 In a multivariate analysis,8 β-blocker therapy was associated with a 0.15 relative risk (95% CI, 0.05-0.45; P < .0007) of appropriate ICD therapy.
Although β-blockers appear to be highly effective in reducing the risk of SCD, it is noteworthy that there still are other therapies that can provide incremental benefit when added to β-blocker therapy. In both the European Myocardial Infarct Amiodarone Trial (EMIAT) and the Canadian Amiodarone Myocardial Infarction Arrhythmia Trial (CAMIAT),8 there appeared to be an interaction between the use of β-blockers and amiodarone; specifically, amiodarone therapy had a greater effect on mortality reduction in patients receiving β-blockers. In CAMIAT, there was a 29% reduction in relative risk related to amiodarone therapy (versus placebo) in patients who were receiving concomitant therapy with β-blockers.8 Thus, although β-blocker therapy appears to be an important agent in the strategy of preventing SCD, monotherapy with β-blockers is likely not sufficient for the prevention of SCD in susceptible populations.8
Renin-Angiotensin-Aldosterone System
Angiotensin-Converting Enzyme Inhibitors
There are several possible antiarrhythmic actions of ACE inhibitors. In patients with or without heart failure, hypokalemia has been shown to be an important risk factor for ventricular arrhythmias. ACE inhibitors can raise serum potassium levels, which may lead to a possible beneficial effect on the myocardial substrate, minimizing arrhythmic risk.8 These results have not been consistent in other studies. However, ACE inhibitors have several direct and indirect effects on the autonomic nervous system that could modify the risk of ventricular arrhythmias.8 They enhance baroreflex sensitivity, thereby reducing sympathetic and increasing parasympathetic tone. Improvements in hemodynamics can also lead to a decrease in circulating catecholamines. Adverse ventricular remodeling, a complex process involving fibrosis and dilatation of the failing heart, has been shown to have adverse effects on the conduction system, including increasing risk for ventricular arrhythmias.8 Treatment with an ACE inhibitor can limit adverse remodeling, including reductions in left ventricular mass, and this, secondarily, may lead to reductions in ventricular arrhythmias and a lower incidence of sudden death. It is also possible that SCD risk may be contributed to by ACE pathways that are, in part, genetically determined.17
The Acute Infarction Ramipril Efficacy Study (AIRE) and the Trandolapril Cardiac Evaluation (TRACE) study are two placebo-controlled trials that did demonstrate significant reductions in SCD.8 In the AIRE study, 2006 patients who had a recent MI and clinical evidence of heart failure were randomly assigned to treatment with ramipril (5 mg daily) or placebo. At 15 months, the ramipril group showed a 27% reduction in total mortality. There was also a 30% reduction in SCD (12.3% to 8.9%). In the TRACE study, 1749 patients with an acute MI and EF less than or equal to 35% were randomly assigned to treatment with trandolapril or placebo to evaluate the effects of ACE inhibitor therapy in the early postinfarction period in patients with left ventricular dysfunction. At follow-up of 24 to 50 months, there was a 25% decrease in cardiovascular deaths and a 24% reduction in SCD (RR, 0.76; 95% CI, 0.59 to 0.98; P = .03). A metaanalysis of intermediate- or long-term post-MI ACE inhibitor trials (including AIRE and RACE) involving 15,104 patients8 showed an overall reduction in SCD with an odds ratio of 0.80 (95% CI, 0.70 to 0.92). There were also similar significant reductions in total and cardiovascular mortality. In the Heart Outcomes Prevention Evaluation (HOPE) study, more than 9000 patients with vascular disease or diabetes mellitus and cardiac risk factors were randomly assigned to treatment with ramipril or placebo.8 After 5 years, there was a 26% reduction in cardiovascular mortality and a 37% reduction in cardiac arrest. In a substudy of HOPE,8 during a median follow-up of 4.5 years, the composite outcome of unexpected death, documented arrhythmic death, or resuscitated cardiac arrest was reduced by 21% in patients without clinical heart failure or overt left ventricular systolic dysfunction randomized to ramipril therapy compared with those randomized to placebo.
No randomized, controlled trials of an ACE inhibitor versus placebo in patients with chronic heart failure have shown a reduction in SCD. The three major trials that have reported results for sudden death are the Cooperative New Scandinavian Enalapril Survival Study (CONSENSUS), the Studies of Left Ventricular Dysfunction (SOLVD)-Treatment, and SOLVD-Prevention.8 They did not show significant reduction in SCD. A meta-analysis of 32 RCTs (n = 7105) in patients with congestive heart failure8 reported that treatment with an ACE inhibitor reduced the rate of SCD nonsignificantly from 5.6% in the control group to 4.7% (odds ratio [OR], 0.91; 95% CI, 0.73 to 1.12) while reducing all-cause mortality from 21.9% in the control group to 15.8% (OR, 0.77; 95% CI, 0.67 to 0.88). In the Second Veterans Administration Vasodilator–Heart Failure Trial (V-HeFT II), 804 men with New York Heart Association (NYHA) class I or III heart failure were randomly assigned to treatment with enalapril (20 mg daily) or the combination of hydralazine (300 mg daily) and isosorbide dinitrate (160 mg daily).8 At 2 years, the enalapril group had a 28% reduction in total mortality. This effect was because of a 38% decrease in the incidence of SCD. It is possible that the beneficial effect of enalapril was caused by an increase in sudden death in the hydralazine–isosorbide arm. Nevertheless, ACE inhibitors confer mortality and sudden death reduction in susceptible populations.
Angiotensin-Receptor Blockers
Physiologically active levels of angiotensin II, a potent vasoconstrictor, persist despite long-term therapy with ACE inhibitors and can also be formed by non–ACE-dependent pathways. Possible arrhythmogenic mechanisms of angiotensin II include release of neurohormonal agents (including norepinephrine, aldosterone, and endothelin), as well as increased conduction velocity and shorter refractory periods in cardiac myocytes.18 Furthermore, unlike ACE inhibitors, angiotensin II receptor blockers (ARBs) do not increase bradykinin levels, which also can increase norepinephrine levels.19 Elevated catecholamine levels theoretically could result in more ventricular arrhythmias and sudden death. Therefore, direct blockade of angiotensin II receptors might further reduce morbidity and mortality in patients requiring blockade of the renin-angiotensin-aldosterone system. Both ARBs as well as ACE inhibitors increase serum potassium levels, which may provide a potential protective mechanism against ventricular arrhythmias.
ARBs have mostly been compared with ACE inhibitors in several heart failure or hypertension trials. In the Evaluation of Losartan in the Elderly (ELITE) study,8 722 patients with NYHA Class II-IV heart failure and EF less than or equal to 40% were randomly assigned to treatment with losartan or captopril. At 48 weeks, the losartan group had a 45% reduction in total mortality. There was also a decrease in the number of deaths attributable to SCD (5 vs. 14 patients) with a relative risk reduction of 36%. However, in the much larger ELITE II study, 3152 patients (with heart failure and EF ≤ 40%) were randomly assigned to treatment with losartan or captopril.8 At 1.5 years, there were trends toward reductions in overall mortality and sudden death with captopril compared with losartan, but no statistically significant differences in all-cause mortality, sudden death, or resuscitated arrests between the two treatment groups were noted. In the Optimal Trial in Myocardial Infarction with the Angiotensin II Antagonist Losartan (OPTIMAAL), losartan and captopril were also studied in patients with heart failure after an MI.8 Although not a primary endpoint of the study, the results for the secondary endpoint of sudden cardiac death or resuscitated cardiac arrest were 9% for the losartan compared with 7% in the captopril group (RR, 1.19; 95% CI, 0.98 to 1.43; P = .07).
Recent studies have evaluated the role of ARBs in special populations. Candesartan was shown to reduce sudden death and death from worsening heart failure in patients with symptomatic heart failure in the CHARM study.20 Losartan was shown to afford better protection than atenolol against cardiac death from arrhythmias (14 deaths in the losartan group vs. 30 deaths in the atenolol group; P = .027) in diabetic patients in the LIFE study.21 Thus, ARBs appear to reduce mortality and may even reduce the risk of SCD, and they appear to be well-tolerated medications without some of the potential side effects of ACE inhibitors.
Aldosterone-Receptor Antagonists
Neurohormonal suppression of the renin-angiotensin-aldosterone system by ACE inhibitors alone is incomplete. Aldosterone can continue to exert harmful effects on the cardiovascular system in patients with heart failure. Aldosterone promotes sodium retention, magnesium and potassium wasting, sympathetic activation, parasympathetic inhibition, myocardial and vascular fibrosis, baroreceptor dysfunction, and vascular damage and impairment of arterial compliance. Animal models have provided insight into the potential mechanisms of arrhythmogenesis in both murine models of mineralcorticoid overexpression, leading to lethal ventricular arrhythmias22 and canine models of heart failure, in which eplerenone administration reduced susceptibility to life-threatening arrhythmias.23
Spironolactone, an aldosterone-receptor antagonist and potassium-sparing diuretic, has gained acceptance for the treatment of severe congestive heart failure. In the Randomized Aldactone Evaluation Study (RALES), 1663 patients with NYHA class III-IV heart failure and an EF less than or equal to 35% were randomly assigned to treatment with spironolactone (25 mg daily) or placebo.24 After a mean follow-up of 24 months, cardiac death was reduced by 31%. This decline was the result of a 36% decrease in death from progressive heart failure and a 29% reduction in SCD. A selective aldosterone blocker, eplerenone, was evaluated in the EPHESUS study25: 6632 patients with an EF less than 40% who had an MI within 30 days, and who had clinical signs of heart failure, were randomly assigned to eplerenone treatment or standard treatment. The eplerenone group had a 15% reduction in all-cause mortality (P < .008) and a significant reduction in cardiac deaths (RR, 0.83; 95% CI, 0.72 to 0.94; P < .005).25 Similar benefits were demonstrated in patients with an EF less than or equal to 30% after MI.26 Specifically, SCD was reduced by 33% compared with placebo, and there was a 58% relative risk reduction for SCD (P = .008) within 30 days after MI.26 Benefits of aldosterone blockade were extended beyond the post-MI setting to the chronic heart failure population. Zannad et al. reported a 24% reduction in risk of death in the chronic heart failure population (hazard ratio [HR], 0.76; 95% CI, 0.62 to 0.93; P = .008).27 A recent large meta-analysis supports the hypothesis that aldosterone-receptor blockade affords protection against SCD, reporting a 23% lower odds of experiencing SCD compared with controls (OR, 0.77; 95% CI, 0.66 to 0.89; P = .001).28
There are several possible mechanisms for a therapeutic effect of aldosterone-receptor antagonists. One possible explanation is that aldosterone seems to enhance positive cardiac remodeling. Fibroblasts and inflammatory cells invade the perivascular space of damaged vessels, leading to fibrosis. These changes can have adverse effects on the mechanical function, vasodilatory reserve, and electrical system of the heart. A recent substudy of the RALES trial evaluated serum markers of collagen synthesis, which has been shown to correlate with morphologic evidence of cardiac fibrosis.29 Spironolactone decreased the levels of these markers at 6 months, whereas the placebo group levels of these markers remained constant. Several studies have shown that non–potassium-sparing diuretics are associated with a dose-dependent increase in SCD. Modification of the electrophysiological substrate is another potential mechanism of aldosterone blockade. In a canine model of heart failure induced by rapid ventricular pacing, eplerenone attenuated increases in refractory periods and dispersion of refractoriness, as well as ventricular tachyarrhythmia induction.30 Thus, aldosterone-receptor blockers also seem to provide mortality and possibly sudden death reduction in susceptible heart failure groups through a variety of pathways.
Modulators of Cholesterol and Inflammation
3-Hydroxy-3-Methylglutaryl-Coenzyme A–Reductase Inhibitors
Although the benefit of statin therapy in patients with coronary artery disease has been well-established in multiple large-scale randomized clinical trials, only a few studies have focused on the effect of statins on ventricular tachyarrhythmias and effects on sudden death risk reduction in patients with coronary artery disease or in patients with nonischemic cardiomyopathy. Statins are a group of lipid-lowering drugs that are 3-hydroxy-3-methylglutaryl-coenzyme A–reductase inhibitors. As a class, they may indirectly exert an antiarrhythmic benefit by reducing the overall ischemic burden by prevention of plaque progression, and reducing ischemia and infarction. However, statins have been shown to have beneficial effects beyond prevention of plaque progression. They can modulate endothelial function, signaling pathways for inflammation, endothelial nitric oxide synthesis, plasminogen, endothelin-1, platelet activation, angiotensin II–receptor regulation, oxidative stress, sympathetic nerve activity, left ventricular mass regression, and left ventricular reverse remodeling. Statins may also have direct antiarrhythmic property by modulating sarcolemmal ion channel function. A recent study suggested that reconstituted high-density lipoprotein cholesterol infusion shortens cardiac repolarization and thereby provides an alternative plausible mechanism for a reduction in sudden cardiac death.31
A metaanalysis of 10 randomized controlled trials involving 22,275 patients who had coronary artery disease reported that statins reduced the rate of SCD from 3.8% in the control group to 3.0% in patients treated with statins (HR, 0.81; 95% CI, 0.71-0.93) after 4.4 years.32 A potential antiarrhythmic benefit of statins was first suggested in 2 secondary prevention trials. In the Scandinavian Simvastatin Survival Study (4S), 4444 patients with coronary artery disease and hypercholesterolemia were randomly assigned to receive treatment with simvastatin or placebo.8 After 5.4 years, there was a 30% decrease in total mortality. Data on instantaneous death and death occurring within 1 hour of symptom onset were supplied but not statistically assessed. There were 63 sudden deaths in the placebo group, whereas the simvastatin group had 37. In the Long-term Intervention with Pravastatin in Ischemic Heart Disease (LIPID) study, 9014 patients with coronary artery disease and hypercholesterolemia were randomly assigned to receive treatment with pravastatin or placebo.8 After 6.1 years, there was a 22% reduction in total mortality with pravastatin. There were 211 sudden deaths in the placebo group and 182 in the simvastatin group. These results were not statistically analyzed. However, they suggest a possible antiarrhythmic benefit, but because of the length of follow-up of these trials, differences in sudden death could also be a result of the ability of statins to prevent the progression of atherosclerosis.
Other studies have suggested that statin therapy is associated with a reduction in ventricular tachyarrhythmias or appropriate ICD therapy in patients with coronary artery disease.33,34 Post hoc analyses of the AVID and MADIT-II patients with coronary artery disease treated with an ICD33,35 demonstrated 40% and 28% reductions, respectively, in VT and VF episodes in the patients treated with statins. The benefit of statins may not be limited to those patients with ischemic heart disease. Retrospective analyses36–38 in patients with nonischemic dilated cardiomyopathy have also shown a reduction in sudden death or mortality associated with statin use. In the DEFINITE study, of 458 patients with nonischemic dilated cardiomyopathy (randomized to medical therapy vs. medical therapy plus an ICD), 110 were being treated with statins.36 Statin use was associated with a significant reduction in total mortality (adjusted HR, 0.23; 95% CI, 0.09 to 0.58; P = .002). Only one arrhythmic death was noted in the 110 patients treated with statins, and 18 were noted in the 348 patients not treated with statins. Interestingly, there was no difference in the incidence of appropriate shocks in those treated compared with those not treated with statin therapy. Patients in MADIT-II were evaluated for the effect of statin use on the combined endpoint of ICD therapy for VT/VF or cardiac death and for the end point of VT/VF alone. The cumulative rate of ICD therapy for VT/VF or cardiac death was significantly reduced in those with more than or equal to 90% statin usage compared with those with lower statin usage (P = .01). A recent study37 evaluated the association between statin therapy on the risk of appropriate defibrillator therapy for fast VT/VF or death and appropriate ICD shocks among 821 patients with nonischemic cardiac myopathy (NICM) enrolled in the MADIT-CRT trial. Multivariate analysis showed that time-dependent statin therapy was independently associated with a significant 77% reduction in the risk of fast VT/VF or death (P < .001) and with a significant 46% reduction in the risk of appropriate ICD shocks (P = .01). Consistent with these findings, the cumulative probability of fast VT/VF or death at 4 years’ follow-up was significantly lower among patients who were treated with statins (11%) compared with study patients who were not treated with statins (19%; P = .006 for the overall difference during follow-up).
An analysis of the SCD-HeFT study38 showed that the mortality reduction related to statin use was identical in those with ischemic heart disease as in those with nonischemic dilated cardiomyopathy (HRs, 0.69 and 0.67, respectively). The mechanism for possible antiarrhythmic effects of statins and the reduction in sudden death in patients with nonischemic cardiomyopathy remains unclear. In patients with coronary artery disease, statins have been reported to improve regulation of coronary arterial tone and nitric oxide–mediated endothelial function, inhibit cell proliferation, stabilize atherosclerotic plaques, exert antiinflammatory properties, and provide antioxidant effects. The mechanism by which statins reduce ventricular tachyarrhythmias may relate indirectly to one or more of these effects. For example, the antioxidant and antiproliferative effects of statins may play a role in plaque stabilization and thus contribute to an antiarrhythmic effect by reducing ischemia-related ventricular tachyarrhythmias. An antiischemic effect also is plausible in patients with nonischemic cardiomyopathy, because a substantial proportion of patients with nonischemic cardiomyopathy are found to have coronary artery disease at autopsy. Finally, small studies that have identified improvements in EF and exercise capacity related to statin therapy support the notion that statins may have beneficial effects on left ventricular remodeling. Thus, it is most likely that statin therapy exerts multiple beneficial effects in patients with nonischemic cardiomyopathy through not only its lipid-lowering effects but also its antiinflammatory, antioxidant, and autonomic effects. Although these data provide strong support for the possibility that statins reduce the incidence of arrhythmic sudden death, a recent metaanalysis39 did not demonstrate such a benefit. Twenty-nine trials of statin versus control (N = 113,568 participants) were included. Statin therapy did not significantly reduce the risk of ventricular tachyarrhythmia (OR, 1.02; 95% CI, 0.84 to 1.25; P = .87) or of cardiac arrest (OR, 1.05; 95% CI, 0.76 to 1.45; P = .84) but was associated with a significant 10% reduction in sudden cardiac death (OR, 0.90; 95% CI, 0.82 to 0.97; P = .01). Results were not altered by the inclusion of eight trials (41,452 participants) of intensive versus standard-dose statin regimens.39 This large metaanalysis supports a modest beneficial effect of statins on SCD and does not support a substantial protective effect of statins on ventricular arrhythmias.
Polyunsaturated Fatty Acids
Several prospective cohort studies have shown an association between omega-3 PUFA use and SCD in patients with and without known coronary disease. In the DART trial, men who had recently survived an MI were randomly assigned to one of 8 groups of dietary intervention.40 In the group assigned to receive an increase in fatty fish consumption, at 2 years there was a 33% relative decrease in deaths resulting from coronary heart disease, whereas there was a nonsignificant increase in the incidence of nonfatal MI. The mortality benefit was mostly seen in the first 6 months. There were eight SCDs in the experimental group and none in the control group. In the Nurse’s Health Study,41 food frequency questionnaires in 76,763 participating women were used to assess α-linolenic acid intake, which was inversely related to the risk of SCD but not nonfatal MI. The GISSI-Prevenzione study, a multicenter, open-label, randomized trial, tested 11,323 patients who had a recent MI.42 At an average of 42 months of follow-up, SCD occurred in 2.7% of control subjects and 2% of patients receiving omega-3 PUFAs (P < .001), and it accounted for 59% of the total mortality benefit. Importantly, there were borderline differences in sudden death percentages by 3 months, with no change in plasma lipid levels. Additional substudies found that the reduction in sudden death related to omega-3 PUFAs was greater in patients with left ventricular dysfunction after MI43 and in chronic heart failure.44 Bucher et al.45 conducted a meta-analysis of randomized, controlled trials of omega-3 PUFAs in coronary heart disease. In their analysis, total mortality, fatal MI, and sudden death were significantly decreased by intake of omega-3 PUFAs, whereas there was a trend toward reduction in nonfatal MI. Finally, Siscovik et al.46 and Albert et al.47 have reported that the consumption of fatty acids reduces the risk of sudden death, supporting the hypothesis that these compounds provide antiarrhythmic properties.
Although PUFAs seem to be effective in reducing SCD in post-MI patients and in patients with chronic heart disease with left ventricular dysfunction, 3 trials using PUFAs in patients with ICDs have shown mixed results.48–50 The initial trial showed that a subgroup of PUFA-treated patients had more frequent ICD discharges compared with the placebo group, suggesting that these supplements may be proarrhythmic in certain subgroups.48 In another trial, Leaf et al.49 found a trend for lower risk for the combined end point of ICD discharge and death from any cause (28%; P = .057) in the group randomized to PUFAs, with risk reduction of close to 40% (P = .03) when adjusting for probable episodes of malignant arrhythmias and compliance. A third trial (and the largest) showed no significant differences between PUFAs and placebo in patients with ICD, but in a subgroup of patients who had a prior MI, the PUFA group had a trend toward benefit (P = .09).50 Whether these results from highly selected populations translate to other subgroups at risk for ventricular arrhythmias, such as ischemia-induced VF, are not known. Overall, although evidence from in vitro studies, animal experiments, and some human studies remains compelling, confirmation of any clinically relevant antiarrhythmic effects of omega-3 PUFA has remained elusive. It is also unclear whether such benefits, if present, are because of direct effects on myocyte electrophysiology or more indirect effects such as improvements in myocardial energetics, autonomic tone, reduction in local inflammatory processes, or other pathways.51
Despite the initial reports suggesting the possible arrhythmic benefit of PUFAs, recent studies have questioned these initial findings. A recent canine model calls into question the prior reports suggesting a benefit of dietary omega-3 PUFAs on sudden death in the ischemia setting.52 This study reported that dietary omega-3 PUFAs did not prevent ischemia-induced VF and actually increased the risk of susceptibility in both noninfarcted dogs and low-risk post-MI dogs.52 Moreover, a large meta-analysis by Rizos et al. reported that PUFAs were not associated with lower risk of all-cause mortality, cardiac death, sudden death, myocardial infarction, or stroke.53 All things considered, there is now discordance of evidence from observational studies and prospective studies that modest PUFA consumption, compared with no PUFA consumption, reduces mortality from chronic heart disease and possibly from sudden death.
References
1. Goldberger, JJ, Buxton, AE, Cain, M, et al. Risk stratification for arrhythmic sudden cardiac death: identifying the roadblocks. Circulation. 2011; 123:2423–2430.
2. Piccini, J, Berger, JS, O’Connor, CM. Amiodarone for the prevention of sudden cardiac death: a meta-analysis of randomized controlled trials. Eur. Heart. 2009; 30:1245–1253.
3. Hulleman, M, Berdowski, J, de Groot, JR, et al. Implantable cardioverter-defibrillators have reduced the incidence of resuscitation for out-of-hospital cardiac arrest caused by lethal arrhythmias. Circulation. 2012; 126:815–821.
4. Ilkhanoff, L, Goldberger, JJ. Out-of-hospital cardiac arrest: getting beyond the tip of the iceberg. Circulation. 2012; 126:793–796.
5. Reiken, S, Wehrens, XHT, Vest, JA, et al. β-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation.. 2003; 107:2459–2466.
6. Anderson, JL, Adams, CD, Antman, EM, et al. ACC/AHA 2007 Guidelines for the Management of Patients with Unstable Angina/Non-ST Elevation Myocardial Infarction. J Am Coll Cardiol. 2007; 50:1–157.
7. Freemantle, N, Cleland, J, Young, P, et al. Beta blockade after myocardial infarction: Systematic review and meta regression analysis. BMJ. 1999; 318:1730–1737.
8. Zipes, DP, Jalife, J. Cardiac electrophysiology: from cell to bedside, ed 5. Philadelphia: Saunders; 2014.
9. A randomized trial of propranolol in patients with acute myocardial infarction, I: mortality results. JAMA. 1982; 247:1707–1714.
10. Dargie, HJ. Effect of carvedilol on outcome after myocardial infarction in patients with left-ventricular dysfunction: the CAPRICORN randomized trial. Lancet. 2011; 357:1385–1390.
11. McMurray, J, Kober, L, Robertson, M, et al. Antiarrhythmic effect of carvedilol after acute myocardial infartion: results of the Carvedilol Post-Infarct Survival Control in Left Ventricular Dysfunction (CAPRICORN) trial. J Am Coll Cardiol. 2005; 45:525–530.
12. Piccini, JP, Hranitzky, PM, Kilaru, R, et al. Relation of mortality to failure to prescribe beta blockers acutely in patients with sustained ventricular tachycardia and ventricular fibrillation following acute myocardial infarction (from the VALsartan In Acute myocardial iNfarcTion [VALIANT] Registry). Am J Cardiol. 2008; 102:1427–1432.
13. Goldberger, J, Bonow, R, Cuffe, M, et al. Beta-blocker use following myocardial infarction: Low prevalence of evidence-based dosing. Am Heart J. 2010; 160:435–442.
14. Packer, M, Bristow, M, Cohn, J, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N Engl J Med. 1996; 334:1349–1355.
15. Lechat, P, Brunhuber, K, Hofmann, R, et al. The cardiac insufficiency bisoprolol study II (CIBIS-II): A randomized trial. Lancet. 1999; 353:9–13.
16. MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL randomized intervention trial in congestive heart failure (MERIT-HF). Lancet. 1999; 353:2001–2007.
17. Jong, P, Demers, C, McKelvie, R, et al. Angiotensin receptor blockers in heart failure: Meta-analysis of randomized controlled trials. J Am Coll Cardiol. 2002; 39:463–470.
17a. Gottlieb, S, McCarter, R, Vogel, R. Effect of beta-blockade on mortality among high-risk and low-risk patients after myocardial infarction. N Engl J Med. 1998; 339:489–497.
18. Gavras, I, Gavras, H. The antiarrhythmic potential of angiotensin II antagonism: Experience with losartan. Am J Hypertens. 2002; 13:512–517.
19. Minisi, A, Thames, M. Distribution of left ventricular sympathetic afferents demonstrated by reflex responses to transmural myocardial ischemia and to intracoronary and epicardial bradykinin. Circulation. 1993; 87:240–246.
20. Solomon, SD, Wang, D, Finn, P, et al. Effect of candesartan on cause-specific mortality in heart failure patients: the Candesartan in Heart failure Assessment of Reduction in Mortality and morbidity (CHARM) program. Circulation. 2004; 110:2180–2183.
21. Lindholm, LH, Dahlof, B, Edelman, JM, et al. Effect of losartan on sudden cardiac death in people with diabetes: data from the LIFE study. Lancet. 2003; 362:619–620.
22. Ouvrard-Pascaud, A, Sainte-Marie, Y, Benitah, JP, et al. Conditional mineralcorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation. 2005; 111:3025–3033.
23. Stambler, BS, Laurita, KR, Shroff, SC, et al. Aldosterone blockade attenuates development of an electrophysiological substrate associated with ventricular tachyarrhythmias in heart failure. Heart Rhythm. 2009; 6:776–783.
24. Pitt, B, Zannad, F, Remme, W, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure: Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999; 341:709–717.
25. Pitt, B, Remme, W, Zannad, F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003; 348:1309–1321.
26. Pitt, B, Gheorghiade, M, Zannad, F, et al. Evaluation of eplerenone in the subgroup of EPHESUS patients with baseline left ventricular ejection fraction ≤ 30%. Eur J Heart Fail. 2006; 8:295–301.
27. Zannad, F, McMurray, JJ, Krum, H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011; 364:11–21.
28. Bapoje, SR, Bahia, A, Hokanson, JE, et al. Effects of mineralcorticoid receptor antagonists on the risk of sudden cardiac death in patients with left ventricular systolic dysfunction: a meta-analysis of randomized controlled trials. Circ Heart Fail. 2013; 6:166–173.
29. Zannad, F, Alla, F, Dousset, B, et al. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Circulation. 2000; 102:2700–2706.
30. Stambler, BS, Laurita, KR, Shroff, SC, et al. Aldosterone blockade attenuates developmentof an electrophysiological substrate associated with ventricular tachyarrhythmias in heart failure. Heart Rhythm. 2009; 6:776–783.
31. Den Ruijter, HM, Franssen, R, Verkerk, AO, et al. Reconstituted high-density lipoprotein shortens cardiac repolarization. J Am Coll Cardiol. 2011; 58:40–44.
32. Levantesi, G, Scarano, M, Marfisi, R, et al. Meta-analysis of effect of statin treatment on risk of sudden death. Am J Cardiol. 2007; 100:1644–1650.
33. Vyas, A, Guo, H, Moss, A, et al. Reduction in ventricular tachyarrhythmias with statins in the Multicenter Automatic Defibrillator Implantation Trial (MADIT)-II. J Am Coll Cardiol. 2006; 47:769–773.
34. Chiu, J, Abdelhadi, R, Chung, M, et al. Effect of statin therapy on risk of ventricular arrhythmia among patients with coronary artery disease and an implantable cardioverter defibrillator. Am J Cardiol. 2005; 95:490–491.
35. Mitchell, L, Powell, J, Gillis, A, et al. Are lipid-lowering drugs also antiarrhythmic drugs? An analysis of the Antiarrhythmics versus Implantable Defibrillators (AVID) trial. J Am Coll Cardiol. 2003; 42:81–87.
36. Goldberger, JJ, Subacius, H, Schaechter, A, et al. DEFINITE Investigators. Effects of statin therapy on arrhythmic events and survival in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol. 2006; 48:1228–1233.
37. Buber, J, Goldenberg, I, Moss, AJ, et al. Reduction in life-threatening ventricular tachyarrhythmias in statin-treated patients with nonischemic cardiomyopathy enrolled in the MADIT-CRT (Multicenter Automatic Defibrillator Implantation Trial with Cardiac Resynchronization Therapy). J Am Coll Cardiol. 2012; 60:749–755.
38. Dickinson, M, Ip, J, Olshansky, B, et al. Statin use was associated with reduced mortality in both ischemic and nonischemic cardiomyopathy and in patients with implantable defibrillators: Mortality data and mechanistic insights from the Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT). Am Heart J. 2007; 153:573–578.
39. Rahimi, K, Majoni, W, Merhi, A, et al. Effect of statins on ventricular arrhythmia, cardiac arrest, and sudden cardiac death: a meta-analysis of published and unpublished evidence from randomized trials. Eur Heart J. 2012; 33:1571–1581.
40. Burr, M, Fehily, A, Gilbert, J, et al. Effects of changes in fat, fish, and fiber intakes on death and reinfarction: Diet and reinfarction trial (DART). Lancet. 1989; 757–761.
41. Albert, C, Oh, K, Whang, W, et al. Dietary alpha-linolenic acid intake and risk of sudden cardiac death and coronary heart disease. Circulation. 2005; 112:3232–3238.
42. Marchioli, R, Barzi, F, Bomba, E, et al. Early protection against sudden death by n-3 polyunsaturated fatty acids after myocardial infarction. Circulation. 2002; 105:1903–1997.
43. Macchia, A, Levantesi, G, Franzosi, M, et al. Left ventricular systolic dysfunction, total mortality, and sudden death in patients with myocardial infarction treated with n-3 polyunsaturated fatty acids. Eur J Heart Fail. 2005; 7:904–909.
44. Tavazzi, L, Maggioni, AP, Marchioli, R, et al. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomized, double-blind, placebo-controlled trial. Lancet. 2008; 372:1223–1230.
45. Bucher, H, Hengstler, P, Schindler, C, et al. N-3 polyunsaturated fatty acids in coronary heart disease: A meta-analysis of randomized controlled trials. Am J Med. 2002; 112:298–304.
46. Siscovick, DS, Raghunathan, TE, King, I, et al. Dietary intake and cell membrane levels of long-chain n-3 polyunsaturated fatty acids and the risk of primary cardiac arrest. JAMA. 1995; 274:1373.
47. Albert, CM, Campos, H, Stampfer, MJ, et al. Blood levels of long-chain n-3 fatty acids and the risk of sudden death. N Engl J Med. 2002; 346:1113.
48. Raitt, MH, Connor, WE, Morris, C, et al. Fish oil supplementation and risk of ventricular tachycardia and ventricular fibrillation in patients with implantable defibrillators: a randomized controlled trial. JAMA. 2005; 293:2884–2891.
49. Leaf, A, Albert, CM, Josephson, M, et al. Prevention of fatal arrhythmias in high-risk subjects by fish oil n-3 fatty acid intake. Circulation.. 2005; 112:2762–2768.
50. Brouwer, IA, Zock, PL, Camm, AJ, et al. Effect of fish oil on ventricular tachyarrhythmia and death in patients with implantable cardioverter defibrillators: the Study on Omega-3 Fatty Acids and Ventricular Arrhythmia (SOFA) randomized trial. JAMA. 2006; 295:2613–2619.
51. Mozaffarian, D, Wu, JHY. Omega-3 fatty acids and cardiovascular disease: effects on risk factors, molecular pathways, and clinical events. J Am Coll Cardiol. 2011; 58:2047–2067.
52. Billman, GE, Carnes, CA, Adamson, PB, et al. Dietary omega-3 fatty acids and susceptibility to ventricular fibrillation: lack of protection and a proarrhythmic effect. Circ Arrhythm Electrophysiol. 2012; 5:553–560.
53. Rizos, EC, Ntzani, EE, Bika, E, et al. Association between omega-3 fatty acid supplementation and risk of major cardiovascular disease events: a systematic review and meta-analysis. JAMA. 2012; 308:1024–1033.