(Table 72-9) Disorders of hypopigmentation are often classified as either diffuse or localized. The classic example of diffuse hypopigmentation is oculocutaneous albinism (OCA). The most common forms are due to mutations in the tyrosinase gene (type I) or the P gene (type II); patients with type IA OCA have a total lack of enzyme activity. At birth, different forms of OCA can appear similar—white hair, gray-blue eyes, and pink-white skin. However, the patients with no tyrosinase activity maintain this phenotype, whereas those with decreased activity will acquire some pigmentation of the eyes, hair, and skin as they age. The degree of pigment formation is also a function of racial background, and the pigmentary dilution is more readily apparent when patients are compared to their first-degree relatives. The ocular findings in OCA correlate with the degree of hypopigmentation and include decreased visual acuity, nystagmus, photophobia, strabismus, and a lack of normal binocular vision.
|
CAUSES OF HYPOPIGMENTATION |
aAbsence of melanocytes in areas of leukoderma. bNormal number of melanocytes. cPlatelet storage defect and restrictive lung disease secondary to deposits of ceroid-like material or immunodeficiency; due to mutations in β subunit of adaptor protein 3 as well as subunits of biogenesis of lysosome-related organelles complex (BLOC)-1, -2, and -3. dGiant lysosomal granules and recurrent infections. eMinority of patients in a nonreferral setting have systemic abnormalities (musculoskeletal, central nervous system, ocular).
The differential diagnosis of localized hypomelanosis includes the following primary cutaneous disorders: idiopathic guttate hypomelanosis, postinflammatory hypopigmentation, tinea (pityriasis) versicolor, vitiligo, chemical- or drug-induced leukoderma, nevus depigmentosus (see below), and piebaldism (Table 72-10). In this group of diseases, the areas of involvement are macules or patches with a decrease or absence of pigmentation. Patients with vitiligo also have an increased incidence of several autoimmune disorders, including Hashimoto’s thyroiditis, Graves’ disease, pernicious anemia, Addison’s disease, uveitis, alopecia areata, chronic mucocutaneous candidiasis, and the autoimmune polyendocrine syndromes (types I and II). Diseases of the thyroid gland are the most frequently associated disorders, occurring in up to 30% of patients with vitiligo. Circulating autoantibodies are often found, and the most common ones are antithyroglobulin, antimicrosomal, and antithyroid-stimulating hormone receptor antibodies.
|
HYPOPIGMENTATION (PRIMARY CUTANEOUS DISORDERS, LOCALIZED) |
There are four systemic diseases that should be considered in a patient with skin findings suggestive of vitiligo—Vogt-Koyanagi-Harada syndrome, systemic sclerosis, onchocerciasis, and melanoma-associated leukoderma. A history of aseptic meningitis, nontraumatic uveitis, tinnitus, hearing loss, and/or dysacousia points to the diagnosis of the Vogt-Koyanagi-Harada syndrome. In these patients, the face and scalp are the most common locations of pigment loss. The vitiligo-like leukoderma seen in patients with systemic sclerosis has a clinical resemblance to idiopathic vitiligo that has begun to repigment as a result of treatment; that is, perifollicular macules of normal pigmentation are seen within areas of depigmentation. The basis of this leukoderma is unknown; there is no evidence of inflammation in areas of involvement, but it can resolve if the underlying connective tissue disease becomes inactive. In contrast to idiopathic vitiligo, melanoma-associated leukoderma often begins on the trunk, and its appearance, if spontaneous, should prompt a search for metastatic disease. It is also seen in patients undergoing immunotherapy for melanoma, including ipilimumab, with cytotoxic T lymphocytes presumably recognizing cell surface antigens common to melanoma cells and melanocytes, and is associated with a greater likelihood of a clinical response.
There are two systemic disorders (neurocristopathies) that may have the cutaneous findings of piebaldism (Table 72-9). They are Shah-Waardenburg syndrome and Waardenburg syndrome. A possible explanation for both disorders is an abnormal embryonic migration or survival of two neural crest–derived elements, one of them being melanocytes and the other myenteric ganglion cells (leading to Hirschsprung disease in Shah-Waardenburg syndrome) or auditory nerve cells (Waardenburg syndrome). The latter syndrome is characterized by congenital sensorineural hearing loss, dystopia canthorum (lateral displacement of the inner canthi but normal interpupillary distance), heterochromic irises, and a broad nasal root, in addition to the piebaldism. The facial dysmorphism can be explained by the neural crest origin of the connective tissues of the head and neck. Patients with Waardenburg syndrome have been shown to have mutations in four genes, including PAX-3 and MITF, all of which encode DNA-binding proteins, whereas patients with Hirschsprung disease plus white spotting have mutations in one of three genes—endothelin 3, endothelin B receptor, and SOX-10.
In tuberous sclerosis, the earliest cutaneous sign is macular hypomelanosis, referred to as an ash leaf spot. These lesions are often present at birth and are usually multiple; however, detection may require Wood’s lamp examination, especially in fair-skinned individuals. The pigment within them is reduced, but not absent. The average size is 1–3 cm, and the common shapes are polygonal and lance-ovate. Examination of the patient for additional cutaneous signs such as multiple angiofibromas of the face (adenoma sebaceum), ungual and gingival fibromas, fibrous plaques of the forehead, and connective tissue nevi (shagreen patches) is recommended. It is important to remember that an ash leaf spot on the scalp will result in a circumscribed patch of lightly pigmented hair. Internal manifestations include seizures, mental retardation, central nervous system (CNS) and retinal hamartomas, pulmonary lymphangioleiomyomatosis (women), renal angiomyolipomas, and cardiac rhabdomyomas. The latter can be detected in up to 60% of children (<18 years) with tuberous sclerosis by echocardiography.
Nevus depigmentosus is a stable, well-circumscribed hypomelanosis that is present at birth. There is usually a single oval or rectangular lesion, but when there are multiple lesions, the possibility of tuberous sclerosis needs to be considered. In linear nevoid hypopigmentation, a term that is replacing hypomelanosis of Ito and segmental or systematized nevus depigmentosus, streaks and swirls of hypopigmentation are observed. Up to a third of patients in a tertiary care setting had associated abnormalities involving the musculoskeletal system (asymmetry), the CNS (seizures and mental retardation), and the eyes (strabismus and hypertelorism). Chromosomal mosaicism has been detected in these patients, lending support to the hypothesis that the cutaneous pattern is the result of the migration of two clones of primordial melanocytes, each with a different pigment potential.
Localized areas of decreased pigmentation are commonly seen as a result of cutaneous inflammation (Table 72-10) and have been observed in the skin overlying active lesions of sarcoidosis (see “Papulonodular Skin Lesions,” below) as well as in CTCL. Cutaneous infections also present as disorders of hypopigmentation, and in tuberculoid leprosy, there are a few asymmetric patches of hypomelanosis that have associated anesthesia, anhidrosis, and alopecia. Biopsy specimens of the palpable border show dermal granulomas that contain rare, if any, Mycobacterium leprae organisms.
HYPERPIGMENTATION
(Table 72-11) Disorders of hyperpigmentation are also divided into two groups—localized and diffuse. The localized forms are due to an epidermal alteration, a proliferation of melanocytes, or an increase in pigment production. Both seborrheic keratoses and acanthosis nigricans belong to the first group. Seborrheic keratoses are common lesions, but in one rare clinical setting, they are a sign of systemic disease, and that setting is the sudden appearance of multiple lesions, often with an inflammatory base and in association with acrochordons (skin tags) and acanthosis nigricans. This is termed the sign of Leser-Trélat and alerts the clinician to search for an internal malignancy. Acanthosis nigricans can also be a reflection of an internal malignancy, most commonly of the gastrointestinal tract, and it appears as velvety hyperpigmentation, primarily in flexural areas. However, in the majority of patients, acanthosis nigricans is associated with obesity and insulin resistance, although it may be a reflection of an endocrinopathy such as acromegaly, Cushing’s syndrome, polycystic ovary syndrome, or insulin-resistant diabetes mellitus (type A, type B, and lipodystrophic forms).
|
CAUSES OF HYPERPIGMENTATION |
aAlso lentigines. bPolyostotic fibrous dysplasia. cSee also “Papulonodular Skin Lesions.” dLate 1980s.
Abbreviations: LAMB, lentigines, atrial myxomas, mucocutaneous myxomas, and blue nevi; LEOPARD, lentigines, ECG abnormalities, ocular hypertelorism, pulmonary stenosis and subaortic valvular stenosis, abnormal genitalia, retardation of growth, and deafness (sensorineural); NAME, nevi, atrial myxoma, myxoid neurofibroma, and ephelides (freckles); POEMS, polyneuropathy, organomegaly, endocrinopathies, M-protein, and skin changes.
A proliferation of melanocytes results in the following pigmented lesions: lentigo, melanocytic nevus, and melanoma (Chap. 105). In an adult, the majority of lentigines are related to sun exposure, which explains their distribution. However, in the Peutz-Jeghers and LEOPARD (lentigines; ECG abnormalities, primarily conduction defects; ocular hypertelorism; pulmonary stenosis and subaortic valvular stenosis; abnormal genitalia [cryptorchidism, hypospadias]; retardation of growth; and deafness [sensorineural]) syndromes, lentigines do serve as a clue to systemic disease. In LEOPARD syndrome, hundreds of lentigines develop during childhood and are scattered over the entire surface of the body. The lentigines in patients with Peutz-Jeghers syndrome are located primarily around the nose and mouth, on the hands and feet, and within the oral cavity. While the pigmented macules on the face may fade with age, the oral lesions persist. However, similar intraoral lesions are also seen in Addison’s disease, in Laugier-Hunziker syndrome (no internal manifestations), and as a normal finding in darkly pigmented individuals. Patients with this autosomal dominant syndrome (due to mutations in a novel serine threonine kinase gene) have multiple benign polyps of the gastrointestinal tract, testicular or ovarian tumors, and an increased risk of developing gastrointestinal (primarily colon) and pancreatic cancers.
In the Carney complex, numerous lentigines are also seen, but they are in association with cardiac myxomas. This autosomal dominant disorder is also known as the LAMB (lentigines, atrial myxomas, mucocutaneous myxomas, and blue nevi) syndrome or NAME (nevi, atrial myxoma, myxoid neurofibroma, and ephelides [freckles]) syndrome. These patients can also have evidence of endocrine overactivity in the form of Cushing’s syndrome (pigmented nodular adrenocortical disease) and acromegaly.
The third type of localized hyperpigmentation is due to a local increase in pigment production, and it includes ephelides and café au lait macules (CALMs). While a single CALM can be seen in up to 10% of the normal population, the presence of multiple or large-sized CALMs raises the possibility of an associated genodermatosis, e.g., neurofibromatosis (NF) or McCune-Albright syndrome. CALMs are flat, uniformly brown in color (usually two shades darker than uninvolved skin), and can vary in size from 0.5–12 cm. Approximately 80–90% of adult patients with type I NF will have six or more CALMs measuring ≥1.5 cm in diameter. Additional findings are discussed in the section on neurofibromas (see “Papulonodular Skin Lesions,” below). In comparison with NF, the CALMs in patients with McCune-Albright syndrome (polyostotic fibrous dysplasia with precocious puberty in females due to mosaicism for an activating mutation in a G protein [Gsα] gene) are usually larger, are more irregular in outline, and tend to respect the midline.
In incontinentia pigmenti, dyskeratosis congenita, and bleomycin pigmentation, the areas of localized hyperpigmentation form a pattern—swirled in the first, reticulated in the second, and flagellate in the third. In dyskeratosis congenita, atrophic reticulated hyperpigmentation is seen on the neck, trunk, and thighs and is accompanied by nail dystrophy, pancytopenia, and leukoplakia of the oral and anal mucosae. The latter often develops into squamous cell carcinoma. In addition to the flagellate pigmentation (linear streaks) on the trunk, patients receiving bleomycin often have hyperpigmentation overlying the elbows, knees, and small joints of the hand.
Localized hyperpigmentation is seen as a side effect of several other systemic medications, including those that produce fixed drug reactions (nonsteroidal anti-inflammatory drugs [NSAIDs], sulfonamides, barbiturates, and tetracyclines) and those that can complex with melanin (antimalarials) or iron (minocycline). Fixed drug eruptions recur in the exact same location as circular areas of erythema that can become bullous and then resolve as brown macules. The eruption usually appears within hours of administration of the offending agent, and common locations include the genitalia, distal extremities, and perioral region. Chloroquine and hydroxychloroquine produce gray-brown to blue-black discoloration of the shins, hard palate, and face, while blue macules (often misdiagnosed as bruises) can be seen on the lower extremities and in sites of inflammation with prolonged minocycline administration. Estrogen in oral contraceptives can induce melasma—symmetric brown patches on the face, especially the cheeks, upper lip, and forehead. Similar changes are seen in pregnancy and in patients receiving phenytoin.
In the diffuse forms of hyperpigmentation, the darkening of the skin may be of equal intensity over the entire body or may be accentuated in sun-exposed areas. The causes of diffuse hyperpigmentation can be divided into four major groups—endocrine, metabolic, autoimmune, and drugs. The endocrinopathies that frequently have associated hyperpigmentation include Addison’s disease, Nelson syndrome, and ectopic ACTH syndrome. In these diseases, the increased pigmentation is diffuse but is accentuated in sun-exposed areas, the palmar creases, sites of friction, and scars. An overproduction of the pituitary hormones α-MSH (melanocyte-stimulating hormone) and ACTH can lead to an increase in melanocyte activity. These peptides are products of the proopiomelanocortin gene and exhibit homology; e.g., α-MSH and ACTH share 13 amino acids. A minority of patients with Cushing’s disease or hyperthyroidism have generalized hyperpigmentation.
The metabolic causes of hyperpigmentation include porphyria cutanea tarda (PCT), hemochromatosis, vitamin B12 deficiency, folic acid deficiency, pellagra, and malabsorption, including Whipple’s disease. In patients with PCT (see “Vesicles/Bullae,” below), the skin darkening is seen in sun-exposed areas and is a reflection of the photoreactive properties of porphyrins. The increased level of iron in the skin of patients with type 1 hemochromatosis stimulates melanin pigment production and leads to the classic bronze color. Patients with pellagra have a brown discoloration of the skin, especially in sun-exposed areas, as a result of nicotinic acid (niacin) deficiency. In the areas of increased pigmentation, there is a thin, varnish-like scale. These changes are also seen in patients who are vitamin B6 deficient, have functioning carcinoid tumors (increased consumption of niacin), or take isoniazid. Approximately 50% of the patients with Whipple’s disease have an associated generalized hyperpigmentation in association with diarrhea, weight loss, arthritis, and lymphadenopathy. A diffuse, slate-blue to gray-brown color is seen in patients with melanosis secondary to metastatic melanoma. The color reflects widespread deposition of melanin within the dermis as a result of the high concentration of circulating melanin precursors.
Of the autoimmune diseases associated with diffuse hyperpigmentation, biliary cirrhosis and systemic sclerosis are the most common, and occasionally, both disorders are seen in the same patient. The skin is dark brown in color, especially in sun-exposed areas. In biliary cirrhosis, the hyperpigmentation is accompanied by pruritus, jaundice, and xanthomas, whereas in systemic sclerosis, it is accompanied by sclerosis of the extremities, face, and, less commonly, the trunk. Additional clues to the diagnosis of systemic sclerosis are mat and periungual telangiectasias, calcinosis cutis, Raynaud’s phenomenon, and distal ulcerations (see “Telangiectasias,” above). The differential diagnosis of cutaneous sclerosis with hyperpigmentation includes the POEMS (polyneuropathy; organomegaly [liver, spleen, lymph nodes]; endocrinopathies [impotence, gynecomastia]; M-protein; and skin changes) syndrome. The skin changes include hyperpigmentation, induration, hypertrichosis, angiomas, clubbing, and facial lipoatrophy.
Diffuse hyperpigmentation that is due to drugs or metals can result from one of several mechanisms—induction of melanin pigment formation, complexing of the drug or its metabolites to melanin, and deposits of the drug in the dermis. Busulfan, cyclophosphamide, 5-fluorouracil, and inorganic arsenic induce pigment production. Complexes containing melanin or iron plus the drug or its metabolites are seen in patients receiving minocycline, and a diffuse, blue-gray, muddy appearance within sun-exposed areas may develop, in addition to pigmentation of the mucous membranes, teeth, nails, bones, and thyroid. Administration of amiodarone can result in both a phototoxic eruption (exaggerated sunburn) and/or a slate-gray to violaceous discoloration of sun-exposed skin. Biopsy specimens of the latter show yellow-brown granules in dermal macrophages, which represent intralysosomal accumulations of lipids, amiodarone, and its metabolites. Actual deposits of a particular drug or metal in the skin are seen with silver (argyria), where the skin appears blue-gray in color; gold (chrysiasis), where the skin has a brown to blue-gray color; and clofazimine, where the skin appears reddish brown. The associated pigmentation is accentuated in sun-exposed areas, and discoloration of the eye is seen with gold (sclerae) and clofazimine (conjunctivae).
VESICLES/BULLAE
(Table 72-12) Depending on their size, cutaneous blisters are referred to as vesicles (<1 cm) or bullae (>1 cm). The primary autoimmune blistering disorders include pemphigus vulgaris, pemphigus foliaceus, paraneoplastic pemphigus, bullous pemphigoid, gestational pemphigoid, cicatricial pemphigoid, epidermolysis bullosa acquisita, linear IgA bullous dermatosis (LABD), and dermatitis herpetiformis (Chap. 73).
|
CAUSES OF VESICLES/BULLAE |
aIntraepidermal. bSubepidermal. cAssociated with gluten enteropathy. dAssociated with inflammatory bowel disease. eDegeneration of cells within the basal layer of the epidermis can give impression split is subepidermal. fAlso systemic. gIn adults, associated with renal failure and immunocompromised state.
Vesicles and bullae are also seen in contact dermatitis, both allergic and irritant forms (Chap. 71). When there is a linear arrangement of vesicular lesions, an exogenous cause or herpes zoster should be suspected. Bullous disease secondary to the ingestion of drugs can take one of several forms, including phototoxic eruptions, isolated bullae, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) (Chap. 74). Clinically, phototoxic eruptions resemble an exaggerated sunburn with diffuse erythema and bullae in sun-exposed areas. The most commonly associated drugs are doxycycline, quinolones, thiazides, NSAIDs, voriconazole, and psoralens. The development of a phototoxic eruption is dependent on the doses of both the drug and ultraviolet (UV)-A irradiation.
Toxic epidermal necrolysis is characterized by bullae that arise on widespread areas of tender erythema and then slough. This results in large areas of denuded skin. The associated morbidity, such as sepsis, and mortality rates are relatively high and are a function of the extent of epidermal necrosis. In addition, these patients may also have involvement of the mucous membranes and respiratory and intestinal tracts. Drugs are the primary cause of TEN, and the most common offenders are aromatic anticonvulsants (phenytoin, barbiturates, carbamazepine), sulfonamides, aminopenicillins, allopurinol, and NSAIDs. Severe acute graft-versus-host disease (grade 4), vancomycin-induced LABD, and the acute syndrome of apoptotic pan-epidermolysis (ASAP) in patients with lupus can also resemble TEN.
In erythema multiforme (EM), the primary lesions are pink-red macules and edematous papules, the centers of which may become vesicular. In contrast to a morbilliform exanthem, the clue to the diagnosis of EM, and especially SJS, is the development of a “dusky” violet color in the center of the lesions. Target lesions are also characteristic of EM and arise as a result of active centers and borders in combination with centrifugal spread. However, target lesions need not be present to make the diagnosis of EM.
EM has been subdivided into two major groups: (1) EM minor due to herpes simplex virus (HSV) and (2) EM major due to HSV; Mycoplasma pneumoniae; or, occasionally, drugs. Involvement of the mucous membranes (ocular, nasal, oral, and genital) is seen more commonly in the latter form. Hemorrhagic crusts of the lips are characteristic of EM major and SJS as well as herpes simplex, pemphigus vulgaris, and paraneoplastic pemphigus. Fever, malaise, myalgias, sore throat, and cough may precede or accompany the eruption. The lesions of EM usually resolve over 2–4 weeks but may be recurrent, especially when due to HSV. In addition to HSV (in which lesions usually appear 7–12 days after the viral eruption), EM can also follow vaccinations, radiation therapy, and exposure to environmental toxins, including the oleoresin in poison ivy.
Induction of SJS is most often due to drugs, especially sulfonamides, phenytoin, barbiturates, lamotrigine, aminopenicillins, nonnucleoside reverse transcriptase inhibitors (e.g., nevirapine), and carbamazepine. Widespread dusky macules and significant mucosal involvement are characteristic of SJS, and the cutaneous lesions may or may not develop epidermal detachment. If the latter occurs, by definition, it is limited to <10% of the body surface area (BSA). Greater involvement leads to the diagnosis of SJS/TEN overlap (10–30% BSA) or TEN (>30% BSA).
In addition to primary blistering disorders and hypersensitivity reactions, bacterial and viral infections can lead to vesicles and bullae. The most common infectious agents are HSV (Chap. 216), varicella-zoster virus (Chap. 217), and S. aureus (Chap. 172).
Staphylococcal scalded-skin syndrome (SSSS) and bullous impetigo are two blistering disorders associated with staphylococcal (phage group II) infection. In SSSS, the initial findings are redness and tenderness of the central face, neck, trunk, and intertriginous zones. This is followed by short-lived flaccid bullae and a slough or exfoliation of the superficial epidermis. Crusted areas then develop, characteristically around the mouth in a radial pattern. SSSS is distinguished from TEN by the following features: younger age group (primarily infants), more superficial site of blister formation, no oral lesions, shorter course, lower morbidity and mortality rates, and an association with staphylococcal exfoliative toxin (“exfoliatin”), not drugs. A rapid diagnosis of SSSS versus TEN can be made by a frozen section of the blister roof or exfoliative cytology of the blister contents. In SSSS, the site of staphylococcal infection is usually extracutaneous (conjunctivitis, rhinorrhea, otitis media, pharyngitis, tonsillitis), and the cutaneous lesions are sterile, whereas in bullous impetigo, the skin lesions are the site of infection. Impetigo is more localized than SSSS and usually presents with honey-colored crusts. Occasionally, superficial purulent blisters also form. Cutaneous emboli from gram-negative infections may present as isolated bullae, but the base of the lesion is purpuric or necrotic, and it may develop into an ulcer (see “Purpura,” below).
Several metabolic disorders are associated with blister formation, including diabetes mellitus, renal failure, and porphyria. Local hypoxemia secondary to decreased cutaneous blood flow can also produce blisters, which explains the presence of bullae over pressure points in comatose patients (coma bullae). In diabetes mellitus, tense bullae with clear sterile viscous fluid arise on normal skin. The lesions can be as large as 6 cm in diameter and are located on the distal extremities. There are several types of porphyria, but the most common form with cutaneous findings is porphyria cutanea tarda (PCT). In sun-exposed areas (primarily the face and hands), the skin is very fragile, with trauma leading to erosions mixed with tense vesicles. These lesions then heal with scarring and formation of milia; the latter are firm, 1- to 2-mm white or yellow papules that represent epidermoid inclusion cysts. Associated findings can include hypertrichosis of the lateral malar region (men) or face (women) and, in sun-exposed areas, hyperpigmentation and firm sclerotic plaques. An elevated level of urinary uroporphyrins confirms the diagnosis and is due to a decrease in uroporphyrinogen decarboxylase activity. PCT can be exacerbated by alcohol, hemochromatosis and other forms of iron overload, chlorinated hydrocarbons, hepatitis C and HIV infections, and hepatomas.
The differential diagnosis of PCT includes (1) porphyria variegata—the skin signs of PCT plus the systemic findings of acute intermittent porphyria; it has a diagnostic plasma porphyrin fluorescence emission at 626 nm; (2) drug-induced pseudoporphyria—the clinical and histologic findings are similar to PCT, but porphyrins are normal; etiologic agents include naproxen and other NSAIDs, furosemide, tetracycline, and voriconazole; (3) bullous dermatosis of hemodialysis—the same appearance as PCT, but porphyrins are usually normal or occasionally borderline elevated; patients have chronic renal failure and are on hemodialysis; (4) PCT associated with hepatomas and hemodialysis; and (5) epidermolysis bullosa acquisita (Chap. 73).
EXANTHEMS
(Table 72-13) Exanthems are characterized by an acute generalized eruption. The most common presentation is erythematous macules and papules (morbilliform) and less often confluent blanching erythema (scarlatiniform). Morbilliform eruptions are usually due to either drugs or viral infections. For example, up to 5% of patients receiving penicillins, sulfonamides, phenytoin, or nevirapine will develop a maculopapular eruption. Accompanying signs may include pruritus, fever, eosinophilia, and transient lymphadenopathy. Similar maculopapular eruptions are seen in the classic childhood viral exanthems, including (1) rubeola (measles)—a prodrome of coryza, cough, and conjunctivitis followed by Koplik’s spots on the buccal mucosa; the eruption begins behind the ears, at the hairline, and on the forehead and then spreads down the body, often becoming confluent; (2) rubella—the eruption begins on the forehead and face and then spreads down the body; it resolves in the same order and is associated with retroauricular and suboccipital lymphadenopathy; and (3) erythema infectiosum (fifth disease)—erythema of the cheeks is followed by a reticulated pattern on the extremities; it is secondary to a parvovirus B19 infection, and an associated arthritis is seen in adults.
|
CAUSES OF EXANTHEMS |
aPrimary infection in infants and reactivation in the setting of immunosuppression.
Abbreviations: CMV, cytomegalovirus; HHV, human herpesvirus; HIV, human immunodeficiency virus.
Both measles and rubella can occur in unvaccinated adults, and an atypical form of measles is seen in adults immunized with either killed measles vaccine or killed vaccine followed in time by live vaccine. In contrast to classic measles, the eruption of atypical measles begins on the palms, soles, wrists, and ankles, and the lesions may become purpuric. The patient with atypical measles can have pulmonary involvement and be quite ill. Rubelliform and roseoliform eruptions are also associated with Epstein-Barr virus (5–15% of patients), echovirus, coxsackievirus, cytomegalovirus, adenovirus, dengue virus, and West Nile virus infections. Detection of specific IgM antibodies or fourfold elevations in IgG antibodies allow the proper diagnosis, but polymerase chain reaction (PCR) is gradually replacing serologic assays. Occasionally, a maculopapular drug eruption is a reflection of an underlying viral infection. For example, ~95% of the patients with infectious mononucleosis who are given ampicillin will develop a rash.
Of note, early in the course of infections with Rickettsia and meningococcus, prior to the development of petechiae and purpura, the lesions may be erythematous macules and papules. This is also the case in chickenpox prior to the development of vesicles. Maculopapular eruptions are associated with early HIV infection, early secondary syphilis, typhoid fever, and acute graft-versus-host disease. In the last, lesions frequently begin on the dorsal hands and forearms; the macular rose spots of typhoid fever involve primarily the anterior trunk.
The prototypic scarlatiniform eruption is seen in scarlet fever and is due to an erythrogenic toxin produced by bacteriophage-containing group A β-hemolytic streptococci, most commonly in the setting of pharyngitis. This eruption is characterized by diffuse erythema, which begins on the neck and upper trunk, and red follicular puncta. Additional findings include a white strawberry tongue (white coating with red papillae) followed by a red strawberry tongue (red tongue with red papillae); petechiae of the palate; a facial flush with circumoral pallor; linear petechiae in the antecubital fossae; and desquamation of the involved skin, palms, and soles 5–20 days after onset of the eruption. A similar desquamation of the palms and soles is seen in toxic shock syndrome (TSS), in Kawasaki disease, and after severe febrile illnesses. Certain strains of staphylococci also produce an erythrotoxin that leads to the same clinical findings as in streptococcal scarlet fever, except that the anti-streptolysin O or -DNase B titers are not elevated.
In toxic shock syndrome, staphylococcal (phage group I) infections produce an exotoxin (TSST-1) that causes the fever and rash as well as enterotoxins. Initially, the majority of cases were reported in menstruating women who were using tampons. However, other sites of infection, including wounds and nasal packing, can lead to TSS. The diagnosis of TSS is based on clinical criteria (Chap. 172), and three of these involve mucocutaneous sites (diffuse erythema of the skin, desquamation of the palms and soles 1–2 weeks after onset of illness, and involvement of the mucous membranes). The latter is characterized as hyperemia of the vagina, oropharynx, or conjunctivae. Similar systemic findings have been described in streptococcal toxic shock syndrome (Chap. 173), and although an exanthem is seen less often than in TSS due to a staphylococcal infection, the underlying infection is often in the soft tissue (e.g., cellulitis).
The cutaneous eruption in Kawasaki disease (Chap. 385) is polymorphous, but the two most common forms are morbilliform and scarlatiniform. Additional mucocutaneous findings include bilateral conjunctival injection; erythema and edema of the hands and feet followed by desquamation; and diffuse erythema of the oropharynx, red strawberry tongue, and dry fissured lips. This clinical picture can resemble TSS and scarlet fever, but clues to the diagnosis of Kawasaki disease are cervical lymphadenopathy, cheilitis, and thrombocytosis. The most serious associated systemic finding in this disease is coronary aneurysms secondary to arteritis. Scarlatiniform eruptions are also seen in the early phase of SSSS (see “Vesicles/Bullae,” above), in young adults with Arcanobacterium haemolyticum infection, and as reactions to drugs.
URTICARIA
(Table 72-14) Urticaria (hives) are transient lesions that are composed of a central wheal surrounded by an erythematous halo or flare. Individual lesions are round, oval, or figurate and are often pruritic. Acute and chronic urticaria have a wide variety of allergic etiologies and reflect edema in the dermis. Urticarial lesions can also be seen in patients with mastocytosis (urticaria pigmentosa), hypo- or hyperthyroidism, and systemic-onset juvenile idiopathic arthritis (Still’s disease). In both juvenile- and adult-onset Still’s disease, the lesions coincide with the fever spike, are transient, and are due to dermal infiltrates of neutrophils.
|
CAUSES OF URTICARIA AND ANGIOEDEMA |
aA small minority develop anaphylaxis. bAlso systemic. cAcquired angioedema can be idiopathic, associated with a lymphoproliferative disorder, or due to a drug, e.g., angiotensin-converting enzyme (ACE) inhibitors.
The common physical urticarias include dermatographism, solar urticaria, cold urticaria, and cholinergic urticaria. Patients with dermatographism exhibit linear wheals following minor pressure or scratching of the skin. It is a common disorder, affecting ~5% of the population. Solar urticaria characteristically occurs within minutes of sun exposure and is a skin sign of one systemic disease—erythropoietic protoporphyria. In addition to the urticaria, these patients have subtle pitted scarring of the nose and hands. Cold urticaria is precipitated by exposure to the cold, and therefore exposed areas are usually affected. In occasional patients, the disease is associated with abnormal circulating proteins—more commonly cryoglobulins and less commonly cryofibrinogens. Additional systemic symptoms include wheezing and syncope, thus explaining the need for these patients to avoid swimming in cold water. Autosomal dominantly inherited cold urticaria is associated with dysfunction of cryopyrin. Cholinergic urticaria is precipitated by heat, exercise, or emotion and is characterized by small wheals with relatively large flares. It is occasionally associated with wheezing.
Whereas urticarias are the result of dermal edema, subcutaneous edema leads to the clinical picture of angioedema. Sites of involvement include the eyelids, lips, tongue, larynx, and gastrointestinal tract as well as the subcutaneous tissue. Angioedema occurs alone or in combination with urticaria, including urticarial vasculitis and the physical urticarias. Both acquired and hereditary (autosomal dominant) forms of angioedema occur (Chap. 376), and in the latter, urticaria is rarely, if ever, seen.
Urticarial vasculitis is an immune complex disease that may be confused with simple urticaria. In contrast to simple urticaria, individual lesions tend to last longer than 24 h and usually develop central petechiae that can be observed even after the urticarial phase has resolved. The patient may also complain of burning rather than pruritus. On biopsy, there is a leukocytoclastic vasculitis of the small dermal blood vessels. Although many cases of urticarial vasculitis are idiopathic in origin, it can be a reflection of an underlying systemic illness such as lupus erythematosus, Sjögren’s syndrome, or hereditary complement deficiency. There is a spectrum of urticarial vasculitis that ranges from purely cutaneous to multisystem involvement. The most common systemic signs and symptoms are arthralgias and/or arthritis, nephritis, and crampy abdominal pain, with asthma and chronic obstructive lung disease seen less often. Hypocomplementemia occurs in one- to two-thirds of patients, even in the idiopathic cases. Urticarial vasculitis can also be seen in patients with hepatitis B and hepatitis C infections, serum sickness, and serum sickness–like illnesses (e.g., due to cefaclor, minocycline).
PAPULONODULAR SKIN LESIONS
(Table 72-15) In the papulonodular diseases, the lesions are elevated above the surface of the skin and may coalesce to form larger plaques. The location, consistency, and color of the lesions are the keys to their diagnosis; this section is organized on the basis of color.
|
PAPULONODULAR SKIN LESIONS ACCORDING TO COLOR GROUPS |
aIf multiple with childhood onset, consider Gardner syndrome. bMay have darker hue in more darkly pigmented individuals. cSee also “Hyperpigmentation.”
Abbreviation: MEN, multiple endocrine neoplasia.
WHITE LESIONS
In calcinosis cutis, there are firm white to white-yellow papules with an irregular surface. When the contents are expressed, a chalky white material is seen. Dystrophic calcification is seen at sites of previous inflammation or damage to the skin. It develops in acne scars as well as on the distal extremities of patients with systemic sclerosis and in the subcutaneous tissue and intermuscular fascial planes in DM. The latter is more extensive and is more commonly seen in children. An elevated calcium phosphate product, most commonly due to secondary hyperparathyroidism in the setting of renal failure, can lead to nodules of metastatic calcinosis cutis, which tend to be subcutaneous and periarticular. These patients can also develop calcification of muscular arteries and subsequent ischemic necrosis (calciphylaxis). Osteoma cutis, in the form of small papules, most commonly occurs on the face of individuals with a history of acne vulgaris, whereas plate-like lesions occur in rare genetic syndromes (Chap. 82).
SKIN-COLORED LESIONS
There are several types of skin-colored lesions, including epidermoid inclusion cysts, lipomas, rheumatoid nodules, neurofibromas, angiofibromas, neuromas, and adnexal tumors such as tricholemmomas. Both epidermoid inclusion cysts and lipomas are very common mobile subcutaneous nodules—the former are rubbery and drain cheeselike material (sebum and keratin) if incised. Lipomas are firm and somewhat lobulated on palpation. When extensive facial epidermoid inclusion cysts develop during childhood or there is a family history of such lesions, the patient should be examined for other signs of Gardner syndrome, including osteomas and desmoid tumors. Rheumatoid nodules are firm 0.5- to 4-cm nodules that favor the extensor aspect of joints, especially the elbows. They are seen in ~20% of patients with rheumatoid arthritis and 6% of patients with Still’s disease. Biopsies of the nodules show palisading granulomas. Similar lesions that are smaller and shorter-lived are seen in rheumatic fever.
Neurofibromas (benign Schwann cell tumors) are soft papules or nodules that exhibit the “button-hole” sign; that is, they invaginate into the skin with pressure in a manner similar to a hernia. Single lesions are seen in normal individuals, but multiple neurofibromas, usually in combination with six or more CALMs measuring >1.5 cm (see “Hyperpigmentation,” above), axillary freckling, and multiple Lisch nodules, are seen in von Recklinghausen’s disease (NF type I) (Chap. 118). In some patients, the neurofibromas are localized and unilateral due to somatic mosaicism.
Angiofibromas are firm pink to skin-colored papules that measure from 3 mm to a few centimeters in diameter. When multiple lesions are located on the central cheeks (adenoma sebaceum), the patient has tuberous sclerosis or multiple endocrine neoplasia (MEN) syndrome, type 1. The former is an autosomal disorder due to mutations in two different genes, and the associated findings are discussed in the section on ash leaf spots as well as in Chap. 118.
Neuromas (benign proliferations of nerve fibers) are also firm, skin-colored papules. They are more commonly found at sites of amputation and as rudimentary supernumerary digits. However, when there are multiple neuromas on the eyelids, lips, distal tongue, and/or oral mucosa, the patient should be investigated for other signs of the MEN syndrome, type 2b. Associated findings include marfanoid habitus, protuberant lips, intestinal ganglioneuromas, and medullary thyroid carcinoma (>75% of patients; Chap. 408).
Adnexal tumors are derived from pluripotent cells of the epidermis that can differentiate toward hair, sebaceous, apocrine, or eccrine glands or remain undifferentiated. Basal cell carcinomas (BCCs) are examples of adnexal tumors that have little or no evidence of differentiation. Clinically, they are translucent papules with rolled borders, telangiectasias, and central erosion. BCCs commonly arise in sun-damaged skin of the head and neck as well as the upper trunk. When a patient has multiple BCCs, especially prior to age 30, the possibility of the nevoid basal cell carcinoma syndrome should be raised. It is inherited as an autosomal dominant trait and is associated with jaw cysts, palmar and plantar pits, frontal bossing, medulloblastomas, and calcification of the falx cerebri and diaphragma sellae. Tricholemmomas are also skin-colored adnexal tumors but differentiate toward hair follicles and can have a wartlike appearance. The presence of multiple tricholemmomas on the face and cobblestoning of the oral mucosa points to the diagnosis of Cowden disease (multiple hamartoma syndrome) due to mutations in the phosphatase and tensin homolog (PTEN) gene. Internal organ involvement (in decreasing order of frequency) includes fibrocystic disease and carcinoma of the breast, adenomas and carcinomas of the thyroid, and gastrointestinal polyposis. Keratoses of the palms, soles, and dorsal aspect of the hands are also seen.
PINK LESIONS
The cutaneous lesions associated with primary systemic amyloidosis are often pink in color and translucent. Common locations are the face, especially the periorbital and perioral regions, and flexural areas. On biopsy, homogeneous deposits of amyloid are seen in the dermis and in the walls of blood vessels; the latter lead to an increase in vessel wall fragility. As a result, petechiae and purpura develop in clinically normal skin as well as in lesional skin following minor trauma, hence the term pinch purpura. Amyloid deposits are also seen in the striated muscle of the tongue and result in macroglossia.
Even though specific mucocutaneous lesions are present in only ~30% of the patients with primary systemic (AL) amyloidosis, the diagnosis can be made via histologic examination of abdominal subcutaneous fat, in conjunction with a serum free light chain assay. By special staining, amyloid deposits are seen around blood vessels or individual fat cells in 40–50% of patients. There are also three forms of amyloidosis that are limited to the skin and that should not be construed as cutaneous lesions of systemic amyloidosis. They are macular amyloidosis (upper back), lichen amyloidosis (usually lower extremities), and nodular amyloidosis. In macular and lichen amyloidosis, the deposits are composed of altered epidermal keratin. Early-onset macular and lichen amyloidosis have been associated with MEN syndrome, type 2a.
Patients with multicentric reticulohistiocytosis also have pink-colored papules and nodules on the face and mucous membranes as well as on the extensor surface of the hands and forearms. They have a polyarthritis that can mimic rheumatoid arthritis clinically. On histologic examination, the papules have characteristic giant cells that are not seen in biopsies of rheumatoid nodules. Pink to skin-colored papules that are firm, 2–5 mm in diameter, and often in a linear arrangement are seen in patients with papular mucinosis. This disease is also referred to as generalized lichen myxedematosus or scleromyxedema. The latter name comes from the induration of the face and extremities that may accompany the papular eruption. Biopsy specimens of the papules show localized mucin deposition, and serum protein electrophoresis plus immunofixation electrophoresis demonstrates a monoclonal spike of IgG, usually with a λ light chain.
YELLOW LESIONS
Several systemic disorders are characterized by yellow-colored cutaneous papules or plaques—hyperlipidemia (xanthomas), gout (tophi), diabetes (necrobiosis lipoidica), pseudoxanthoma elasticum, and Muir-Torre syndrome (sebaceous tumors). Eruptive xanthomas are the most common form of xanthomas and are associated with hypertriglyceridemia (primarily hyperlipoproteinemia types I, IV, and V). Crops of yellow papules with erythematous halos occur primarily on the extensor surfaces of the extremities and the buttocks, and they spontaneously involute with a fall in serum triglycerides. Types II and III result in one or more of the following types of xanthoma: xanthelasma, tendon xanthomas, and plane xanthomas. Xanthelasma are found on the eyelids, whereas tendon xanthomas are frequently associated with the Achilles and extensor finger tendons; plane xanthomas are flat and favor the palmar creases, neck, upper trunk, and flexural folds. Tuberous xanthomas are frequently associated with hypertriglyceridemia, but they are also seen in patients with hypercholesterolemia and are found most frequently over the large joints or hand. Biopsy specimens of xanthomas show collections of lipid-containing macrophages (foam cells).
Patients with several disorders, including biliary cirrhosis, can have a secondary form of hyperlipidemia with associated tuberous and plane xanthomas. However, patients with plasma cell dyscrasias have normolipemic plane xanthomas. This latter form of xanthoma may be ≥12 cm in diameter and is most frequently seen on the upper trunk or side of the neck. It is important to note that the most common setting for eruptive xanthomas is uncontrolled diabetes mellitus. The least specific sign for hyperlipidemia is xanthelasma, because at least 50% of the patients with this finding have normal lipid profiles.
In tophaceous gout, there are deposits of monosodium urate in the skin around the joints, particularly those of the hands and feet. Additional sites of tophi formation include the helix of the ear and the olecranon and prepatellar bursae. The lesions are firm, yellow in color, and occasionally discharge a chalky material. Their size varies from 1 mm to 7 cm, and the diagnosis can be established by polarized light microscopy of the aspirated contents of a lesion. Lesions of necrobiosis lipoidica are found primarily on the shins (90%), and patients can have diabetes mellitus or develop it subsequently. Characteristic findings include a central yellow color, atrophy (transparency), telangiectasias, and a red to red-brown border. Ulcerations can also develop within the plaques. Biopsy specimens show necrobiosis of collagen and granulomatous inflammation.
In pseudoxanthoma elasticum (PXE), due to mutations in the gene ABCC6, there is an abnormal deposition of calcium on the elastic fibers of the skin, eye, and blood vessels. In the skin, the flexural areas such as the neck, axillae, antecubital fossae, and inguinal area are the primary sites of involvement. Yellow papules coalesce to form reticulated plaques that have an appearance similar to that of plucked chicken skin. In severely affected skin, hanging, redundant folds develop. Biopsy specimens of involved skin show swollen and irregularly clumped elastic fibers with deposits of calcium. In the eye, the calcium deposits in Bruch’s membrane lead to angioid streaks and choroiditis; in the arteries of the heart, kidney, gastrointestinal tract, and extremities, the deposits lead to angina, hypertension, gastrointestinal bleeding, and claudication, respectively.
Adnexal tumors that have differentiated toward sebaceous glands include sebaceous adenoma, sebaceous carcinoma, and sebaceous hyperplasia. Except for sebaceous hyperplasia, which is commonly seen on the face, these tumors are fairly rare. Patients with Muir-Torre syndrome have one or more sebaceous adenoma(s), and they can also have sebaceous carcinomas and sebaceous hyperplasia as well as keratoacanthomas. The internal manifestations of Muir-Torre syndrome include multiple carcinomas of the gastrointestinal tract (primarily colon) as well as cancers of the larynx, genitourinary tract, and breast.
RED LESIONS
Cutaneous lesions that are red in color have a wide variety of etiologies; in an attempt to simplify their identification, they will be subdivided into papules, papules/plaques, and subcutaneous nodules. Common red papules include arthropod bites and cherry hemangiomas; the latter are small, bright-red, dome-shaped papules that represent a benign proliferation of capillaries. In patients with AIDS (Chap. 226), the development of multiple red hemangioma-like lesions points to bacillary angiomatosis, and biopsy specimens show clusters of bacilli that stain positive with the Warthin-Starry stain; the pathogens have been identified as Bartonella henselae and Bartonella quintana. Disseminated visceral disease is seen primarily in immunocompromised hosts but can occur in immunocompetent individuals.
Multiple angiokeratomas are seen in Fabry disease, an X-linked recessive lysosomal storage disease that is due to a deficiency of α-galactosidase A. The lesions are red to red-blue in color and can be quite small in size (1–3 mm), with the most common location being the lower trunk. Associated findings include chronic renal disease, peripheral neuropathy, and corneal opacities (cornea verticillata). Electron photomicrographs of angiokeratomas and clinically normal skin demonstrate lamellar lipid deposits in fibroblasts, pericytes, and endothelial cells that are diagnostic of this disease. Widespread acute eruptions of erythematous papules are discussed in the section on exanthems.
There are several infectious diseases that present as erythematous papules or nodules in a lymphocutaneous or sporotrichoid pattern, i.e., in a linear arrangement along the lymphatic channels. The two most common etiologies are Sporothrix schenckii (sporotrichosis) and the atypical mycobacterium Mycobacterium marinum. The organisms are introduced as a result of trauma, and a primary inoculation site is often seen in addition to the lymphatic nodules. Additional causes include Nocardia, Leishmania, and other atypical mycobacteria and dimorphic fungi; culture of lesional tissue will aid in the diagnosis.
The diseases that are characterized by erythematous plaques with scale are reviewed in the papulosquamous section, and the various forms of dermatitis are discussed in the section on erythroderma. Additional disorders in the differential diagnosis of red papules/plaques include cellulitis, polymorphous light eruption (PMLE), cutaneous lymphoid hyperplasia (lymphocytoma cutis), cutaneous lupus, lymphoma cutis, and leukemia cutis. The first three diseases represent primary cutaneous disorders, although cellulitis may be accompanied by a bacteremia. PMLE is characterized by erythematous papules and plaques in a primarily sun-exposed distribution—dorsum of the hand, extensor forearm, and upper trunk. Lesions follow exposure to UV-B and/or UV-A, and in higher latitudes, PMLE is most severe in the late spring and early summer. A process referred to as “hardening” occurs with continued UV exposure, and the eruption fades, but in temperate climates, it will recur in the spring. PMLE must be differentiated from cutaneous lupus, and this is accomplished by observation of the natural history, histologic examination, and direct immunofluorescence of the lesions. Cutaneous lymphoid hyperplasia (pseudolymphoma) is a benign polyclonal proliferation of lymphocytes in the skin that presents as infiltrated pink-red to red-purple papules and plaques; it must be distinguished from lymphoma cutis.
Several types of red plaques are seen in patients with systemic lupus, including (1) erythematous urticarial plaques across the cheeks and nose in the classic butterfly rash; (2) erythematous discoid lesions with fine or “carpet-tack” scale, telangiectasias, central hypopigmentation, peripheral hyperpigmentation, follicular plugging, and atrophy located on the face, scalp, external ears, arms, and upper trunk; and (3) psoriasiform or annular lesions of subacute cutaneous lupus with hypopigmented centers located primarily on the extensor arms and upper trunk. Additional mucocutaneous findings include (1) a violaceous flush on the face and V of the neck; (2) photosensitivity; (3) urticarial vasculitis (see “Urticaria,” above); (4) lupus panniculitis (see below); (5) diffuse alopecia; (6) alopecia secondary to discoid lesions; (7) periungual telangiectasias and erythema; (8) EM-like lesions that may become bullous; (9) oral ulcers; and (10) distal ulcerations secondary to Raynaud’s phenomenon, vasculitis, or livedoid vasculopathy. Patients with only discoid lesions usually have the form of lupus that is limited to the skin. However, up to 10% of these patients eventually develop systemic lupus. Direct immunofluorescence of involved skin, in particular discoid lesions, shows deposits of IgG or IgM and C3 in a granular distribution along the dermal-epidermal junction.
In lymphoma cutis, there is a proliferation of malignant lymphocytes in the skin, and the clinical appearance resembles that of cutaneous lymphoid hyperplasia—infiltrated pink-red to red-purple papules and plaques. Lymphoma cutis can occur anywhere on the surface of the skin, whereas the sites of predilection for lymphocytomas include the malar ridge, tip of the nose, and earlobes. Patients with non-Hodgkin’s lymphomas have specific cutaneous lesions more often than those with Hodgkin’s disease, and, occasionally, the skin nodules precede the development of extracutaneous non-Hodgkin’s lymphoma or represent the only site of involvement (e.g., primary cutaneous B cell lymphoma). Arcuate lesions are sometimes seen in lymphoma and lymphocytoma cutis as well as in CTCL. Adult T cell leukemia/lymphoma that develops in association with HTLV-1 infection is characterized by cutaneous plaques, hypercalcemia, and circulating CD25+ lymphocytes. Leukemia cutis has the same appearance as lymphoma cutis, and specific lesions are seen more commonly in monocytic leukemias than in lymphocytic or granulocytic leukemias. Cutaneous chloromas (granulocytic sarcomas) may precede the appearance of circulating blasts in acute myelogenous leukemia and, as such, represent a form of aleukemic leukemia cutis.
Sweet syndrome is characterized by pink-red to red-brown edematous plaques that are frequently painful and occur primarily on the head, neck, and upper (and, less often, lower) extremities. The patients also have fever, neutrophilia, and a dense dermal infiltrate of neutrophils in the lesions. In ~10% of the patients, there is an associated malignancy, most commonly acute myelogenous leukemia. Sweet syndrome has also been reported with inflammatory bowel disease, systemic lupus erythematosus, and solid tumors (primarily of the genitourinary tract) as well as drugs (e.g., all-trans-retinoic acid, granulocyte colony-stimulating factor [G-CSF]). The differential diagnosis includes neutrophilic eccrine hidradenitis; bullous forms of pyoderma gangrenosum; and, occasionally, cellulitis. Extracutaneous sites of involvement include joints, muscles, eye, kidney (proteinuria, occasionally glomerulonephritis), and lung (neutrophilic infiltrates). The idiopathic form of Sweet syndrome is seen more often in women, following a respiratory tract infection.
Common causes of erythematous subcutaneous nodules include inflamed epidermoid inclusion cysts, acne cysts, and furuncles. Panniculitis, an inflammation of the fat, also presents as subcutaneous nodules and is frequently a sign of systemic disease. There are several forms of panniculitis, including erythema nodosum, erythema induratum/nodular vasculitis, lupus panniculitis, lipodermatosclerosis, α1-antitrypsin deficiency, factitial, and fat necrosis secondary to pancreatic disease. Except for erythema nodosum, these lesions may break down and ulcerate or heal with a scar. The shin is the most common location for the nodules of erythema nodosum, whereas the calf is the most common location for lesions of erythema induratum. In erythema nodosum, the nodules are initially red but then develop a blue color as they resolve. Patients with erythema nodosum but no underlying systemic illness can still have fever, malaise, leukocytosis, arthralgias, and/or arthritis. However, the possibility of an underlying illness should be excluded, and the most common associations are streptococcal infections, upper respiratory viral infections, sarcoidosis, and inflammatory bowel disease, in addition to drugs (oral contraceptives, sulfonamides, penicillins, bromides, iodides). Less common associations include bacterial gastroenteritis (Yersinia, Salmonella) and coccidioidomycosis followed by tuberculosis, histoplasmosis, brucellosis, and infections with Chlamydophila pneumoniae or Chlamydia trachomatis, Mycoplasma pneumoniae, or hepatitis B virus.
Erythema induratum and nodular vasculitis have overlapping features clinically and histologically, and whether they represent two separate entities or the ends of a single disease spectrum is a point of debate; in general, the latter is usually idiopathic and the former is associated with the presence of Mycobacterium tuberculosis DNA by PCR within skin lesions. The lesions of lupus panniculitis are found primarily on the cheeks, upper arms, and buttocks (sites of abundant fat) and are seen in both the cutaneous and systemic forms of lupus. The overlying skin may be normal, erythematous, or have the changes of discoid lupus. The subcutaneous fat necrosis that is associated with pancreatic disease is presumably secondary to circulating lipases and is seen in patients with pancreatic carcinoma as well as in patients with acute and chronic pancreatitis. In this disorder, there may be an associated arthritis, fever, and inflammation of visceral fat. Histologic examination of deep incisional biopsy specimens will aid in the diagnosis of the particular type of panniculitis.
Subcutaneous erythematous nodules are also seen in cutaneous polyarteritis nodosa and as a manifestation of systemic vasculitis when there is involvement of medium-sized vessels, e.g., systemic polyarteritis nodosa, allergic granulomatosis, or granulomatosis with polyangiitis (Wegener’s) (Chap. 385). Cutaneous polyarteritis nodosa presents with painful subcutaneous nodules and ulcers within a red-purple, netlike pattern of livedo reticularis. The latter is due to slowed blood flow through the superficial horizontal venous plexus. The majority of lesions are found on the lower extremities, and while arthralgias and myalgias may accompany cutaneous polyarteritis nodosa, there is no evidence of systemic involvement. In both the cutaneous and systemic forms of vasculitis, skin biopsy specimens of the associated nodules will show the changes characteristic of a necrotizing vasculitis and/or granulomatous inflammation.
RED-BROWN LESIONS
The cutaneous lesions in sarcoidosis (Chap. 390) are classically red to red-brown in color, and with diascopy (pressure with a glass slide), a yellow-brown residual color is observed that is secondary to the granulomatous infiltrate. The waxy papules and plaques may be found anywhere on the skin, but the face is the most common location. Usually there are no surface changes, but occasionally the lesions will have scale. Biopsy specimens of the papules show “naked” granulomas in the dermis, i.e., granulomas surrounded by a minimal number of lymphocytes. Other cutaneous findings in sarcoidosis include annular lesions with an atrophic or scaly center, papules within scars, hypopigmented papules and patches, alopecia, acquired ichthyosis, erythema nodosum, and lupus pernio (see below).
The differential diagnosis of sarcoidosis includes foreign-body granulomas produced by chemicals such as beryllium and zirconium, late secondary syphilis, and lupus vulgaris. Lupus vulgaris is a form of cutaneous tuberculosis that is seen in previously infected and sensitized individuals. There is often underlying active tuberculosis elsewhere, usually in the lungs or lymph nodes. Lesions occur primarily in the head and neck region and are red-brown plaques with a yellow-brown color on diascopy. Secondary scarring and squamous cell carcinomas can develop within the plaques. Cultures or PCR analysis of the lesions should be performed, along with an interferon γ release assay of peripheral blood, because it is rare for the acid-fast stain to show bacilli within the dermal granulomas.
A generalized distribution of red-brown macules and papules is seen in the form of mastocytosis known as urticaria pigmentosa (Chap. 376). Each lesion represents a collection of mast cells in the dermis, with hyperpigmentation of the overlying epidermis. Stimuli such as rubbing cause these mast cells to degranulate, and this leads to the formation of localized urticaria (Darier’s sign). Additional symptoms can result from mast cell degranulation and include headache, flushing, diarrhea, and pruritus. Mast cells also infiltrate various organs such as the liver, spleen, and gastrointestinal tract, and accumulations of mast cells in the bones may produce either osteosclerotic or osteolytic lesions on radiographs. In the majority of these patients, however, the internal involvement remains indolent. A subtype of chronic cutaneous small-vessel vasculitis, erythema elevatum diutinum (EED), also presents with papules that are red-brown in color. The papules coalesce into plaques on the extensor surfaces of knees, elbows, and the small joints of the hand. Flares of EED have been associated with streptococcal infections.
BLUE LESIONS
Lesions that are blue in color are the result of vascular ectasias, hyperplasias and tumors or melanin pigment within the dermis. Venous lakes (ectasias) are compressible dark-blue lesions that are found commonly in the head and neck region. Venous malformations are also compressible blue papulonodules and plaques that can occur anywhere on the body, including the oral mucosa. When there are multiple rather than single congenital lesions, the patient may have the blue rubber bleb syndrome or Maffucci’s syndrome. Patients with the blue rubber bleb syndrome also have vascular anomalies of the gastrointestinal tract that may bleed, whereas patients with Maffucci’s syndrome have associated osteochondromas. Blue nevi (moles) are seen when there are collections of pigment-producing nevus cells in the dermis. These benign papular lesions are dome-shaped and occur most commonly on the dorsum of the hand or foot or in the head and neck region.
VIOLACEOUS LESIONS
Violaceous papules and plaques are seen in lupus pernio, lymphoma cutis, and cutaneous lupus. Lupus pernio is a particular type of sarcoidosis that involves the tip and alar rim of the nose as well as the earlobes, with lesions that are violaceous in color rather than red-brown. This form of sarcoidosis is associated with involvement of the upper respiratory tract. The plaques of lymphoma cutis and cutaneous lupus may be red or violaceous in color and were discussed above.
PURPLE LESIONS
Purple-colored papules and plaques are seen in vascular tumors, such as Kaposi’s sarcoma (Chap. 226) and angiosarcoma, and when there is extravasation of red blood cells into the skin in association with inflammation, as in palpable purpura (see “Purpura,” below). Patients with congenital or acquired AV fistulas and venous hypertension can develop purple papules on the lower extremities that can resemble Kaposi’s sarcoma clinically and histologically; this condition is referred to as pseudo-Kaposi’s sarcoma (acral angiodermatitis). Angiosarcoma is found most commonly on the scalp and face of elderly patients or within areas of chronic lymphedema and presents as purple papules and plaques. In the head and neck region, the tumor often extends beyond the clinically defined borders and may be accompanied by facial edema.
BROWN AND BLACK LESIONS
Brown- and black-colored papules are reviewed in “Hyperpigmentation,” above.
CUTANEOUS METASTASES
These are discussed last because they can have a wide range of colors. Most commonly, they present as either firm, skin-colored subcutaneous nodules or firm, red to red-brown papulonodules. The lesions of lymphoma cutis range from pink-red to plum in color, whereas metastatic melanoma can be pink, blue, or black in color. Cutaneous metastases develop from hematogenous or lymphatic spread and are most often due to the following primary carcinomas: in men, melanoma, oropharynx, lung, and colon; and in women, breast, melanoma, and ovary. These metastatic lesions may be the initial presentation of the carcinoma, especially when the primary site is the lung.
PURPURA
(Table 72-16) Purpura are seen when there is an extravasation of red blood cells into the dermis and, as a result, the lesions do not blanch with pressure. This is in contrast to those erythematous or violet-colored lesions that are due to localized vasodilatation—they do blanch with pressure. Purpura (≥3 mm) and petechiae (≤2 mm) are divided into two major groups: palpable and nonpalpable. The most frequent causes of nonpalpable petechiae and purpura are primary cutaneous disorders such as trauma, solar (actinic) purpura, and capillaritis. Less common causes are steroid purpura and livedoid vasculopathy (see “Ulcers,” below). Solar purpura are seen primarily on the extensor forearms, whereas steroid purpura secondary to potent topical glucocorticoids or endogenous or exogenous Cushing’s syndrome can be more widespread. In both cases, there is alteration of the supporting connective tissue that surrounds the dermal blood vessels. In contrast, the petechiae that result from capillaritis are found primarily on the lower extremities. In capillaritis, there is an extravasation of erythrocytes as a result of perivascular lymphocytic inflammation. The petechiae are bright red, 1–2 mm in size, and scattered within yellow-brown patches. The yellow-brown color is caused by hemosiderin deposits within the dermis.
|
CAUSES OF PURPURA |
aAlso associated with underlying disorders that lead to hypercoagulability, e.g., factor V Leiden, protein C dysfunction/deficiency. bBacterial (including rickettsial), fungal, or parasitic.
Abbreviation: ITP, idiopathic thrombocytopenic purpura.
Systemic causes of nonpalpable purpura fall into several categories, and those secondary to clotting disturbances and vascular fragility will be discussed first. The former group includes thrombocytopenia (Chap. 140), abnormal platelet function as is seen in uremia, and clotting factor defects. The initial site of presentation for thrombocytopenia-induced petechiae is the distal lower extremity. Capillary fragility leads to nonpalpable purpura in patients with systemic amyloidosis (see “Papulonodular Skin Lesions,” above), disorders of collagen production such as Ehlers-Danlos syndrome, and scurvy. In scurvy, there are flattened corkscrew hairs with surrounding hemorrhage on the lower extremities, in addition to gingivitis. Vitamin C is a cofactor for lysyl hydroxylase, an enzyme involved in the posttranslational modification of procollagen that is necessary for cross-link formation.
In contrast to the previous group of disorders, the purpura (noninflammatory with a retiform outline) seen in the following group of diseases are associated with thrombi formation within vessels. It is important to note that these thrombi are demonstrable in skin biopsy specimens. This group of disorders includes disseminated intravascular coagulation (DIC), monoclonal cryoglobulinemia, thrombocytosis, thrombotic thrombocytopenic purpura, antiphospholipid antibody syndrome, and reactions to warfarin and heparin (heparin-induced thrombocytopenia and thrombosis). DIC is triggered by several types of infection (gram-negative, gram-positive, viral, and rickettsial) as well as by tissue injury and neoplasms. Widespread purpura and hemorrhagic infarcts of the distal extremities are seen. Similar lesions are found in purpura fulminans, which is a form of DIC associated with fever and hypotension that occurs more commonly in children following an infectious illness such as varicella, scarlet fever, or an upper respiratory tract infection. In both disorders, hemorrhagic bullae can develop in involved skin.
Monoclonal cryoglobulinemia is associated with plasma cell dyscrasias, chronic lymphocytic leukemia, and lymphoma. Purpura, primarily of the lower extremities, and hemorrhagic infarcts of the fingers, toes, and ears are seen in these patients. Exacerbations of disease activity can follow cold exposure or an increase in serum viscosity. Biopsy specimens show precipitates of the cryoglobulin within dermal vessels. Similar deposits have been found in the lung, brain, and renal glomeruli. Patients with thrombotic thrombocytopenic purpura can also have hemorrhagic infarcts as a result of intravascular thromboses. Additional signs include microangiopathic hemolytic anemia and fluctuating neurologic abnormalities, especially headaches and confusion.
Administration of warfarin can result in painful areas of erythema that become purpuric and then necrotic with an adherent black eschar; the condition is referred to as warfarin-induced necrosis. This reaction is seen more often in women and in areas with abundant subcutaneous fat—breasts, abdomen, buttocks, thighs, and calves. The erythema and purpura develop between the third and tenth day of therapy, most likely as a result of a transient imbalance in the levels of anticoagulant and procoagulant vitamin K–dependent factors. Continued therapy does not exacerbate preexisting lesions, and patients with an inherited or acquired deficiency of protein C are at increased risk for this particular reaction as well as for purpura fulminans and calciphylaxis.
Purpura secondary to cholesterol emboli are usually seen on the lower extremities of patients with atherosclerotic vascular disease. They often follow anticoagulant therapy or an invasive vascular procedure such as an arteriogram but also occur spontaneously from disintegration of atheromatous plaques. Associated findings include livedo reticularis, gangrene, cyanosis, and ischemic ulcerations. Multiple step sections of the biopsy specimen may be necessary to demonstrate the cholesterol clefts within the vessels. Petechiae are also an important sign of fat embolism and occur primarily on the upper body 2–3 days after a major injury. By using special fixatives, the emboli can be demonstrated in biopsy specimens of the petechiae. Emboli of tumor or thrombus are seen in patients with atrial myxomas and marantic endocarditis.
In the Gardner-Diamond syndrome (autoerythrocyte sensitivity), female patients develop large ecchymoses within areas of painful, warm erythema. Intradermal injections of autologous erythrocytes or phosphatidyl serine derived from the red cell membrane can reproduce the lesions in some patients; however, there are instances where a reaction is seen at an injection site of the forearm but not in the midback region. The latter has led some observers to view Gardner-Diamond syndrome as a cutaneous manifestation of severe emotional stress. More recently, the possibility of platelet dysfunction (as assessed via aggregation studies) has been raised. Waldenström’s hypergammaglobulinemic purpura is a chronic disorder characterized by petechiae on the lower extremities. There are circulating complexes of IgG–anti-IgG molecules, and exacerbations are associated with prolonged standing or walking.
Palpable purpura are further subdivided into vasculitic and embolic. In the group of vasculitic disorders, cutaneous small-vessel vasculitis, also known as leukocytoclastic vasculitis (LCV), is the one most commonly associated with palpable purpura (Chap. 385). Underlying etiologies include drugs (e.g., antibiotics), infections (e.g., hepatitis C virus), and autoimmune connective tissue diseases (e.g., rheumatoid arthritis, Sjögren’s syndrome, lupus). Henoch-Schönlein purpura (HSP) is a subtype of acute LCV that is seen more commonly in children and adolescents following an upper respiratory infection. The majority of lesions are found on the lower extremities and buttocks. Systemic manifestations include fever, arthralgias (primarily of the knees and ankles), abdominal pain, gastrointestinal bleeding, and nephritis. Direct immunofluorescence examination shows deposits of IgA within dermal blood vessel walls. Renal disease is of particular concern in adults with HSP. In polyarteritis nodosa, specific cutaneous lesions result from a vasculitis of arterial vessels (arteritis), or there may be an associated LCV. Arteritis leads to an infarct of the skin, and this explains the irregular outline of the purpura (see below).
Several types of infectious emboli can give rise to palpable purpura. These embolic lesions are usually irregular in outline as opposed to the lesions of LCV, which are circular in outline. The irregular outline is indicative of a cutaneous infarct, and the size corresponds to the area of skin that received its blood supply from that particular arteriole or artery. The palpable purpura in LCV are circular because the erythrocytes simply diffuse out evenly from the postcapillary venules as a result of inflammation. Infectious emboli are most commonly due to gram-negative cocci (meningococcus, gonococcus), gram-negative rods (Enterobacteriaceae), and gram-positive cocci (Staphylococcus). Additional causes include Rickettsia and, in immunocompromised patients, Aspergillus and other opportunistic fungi.
The embolic lesions in acute meningococcemia are found primarily on the trunk, lower extremities, and sites of pressure, and a gunmetal-gray color often develops within them. Their size varies from a few millimeters to several centimeters, and the organisms can be cultured from the lesions. Associated findings include a preceding upper respiratory tract infection; fever; meningitis; DIC; and, in some patients, a deficiency of the terminal components of complement. In disseminated gonococcal infection (arthritis-dermatitis syndrome), a small number of inflammatory papules and vesicopustules, often with central purpura or hemorrhagic necrosis, are found on the distal extremities. Additional symptoms include arthralgias, tenosynovitis, and fever. To establish the diagnosis, a Gram stain of these lesions should be performed. Rocky Mountain spotted fever is a tick-borne disease that is caused by Rickettsia rickettsii. A several-day history of fever, chills, severe headache, and photophobia precedes the onset of the cutaneous eruption. The initial lesions are erythematous macules and papules on the wrists, ankles, palms, and soles. With time, the lesions spread centripetally and become purpuric.
Lesions of ecthyma gangrenosum begin as edematous, erythematous papules or plaques and then develop central purpura and necrosis. Bullae formation also occurs in these lesions, and they are frequently found in the girdle region. The organism that is classically associated with ecthyma gangrenosum is Pseudomonas aeruginosa, but other gram-negative rods such as Klebsiella, Escherichia coli, and Serratia can produce similar lesions. In immunocompromised hosts, the list of potential pathogens is expanded to include Candida and other opportunistic fungi (e.g., Aspergillus, Fusarium).
ULCERS
The approach to the patient with a cutaneous ulcer is outlined in Table 72-17. Peripheral vascular diseases of the extremities are reviewed in Chap. 302, as is Raynaud’s phenomenon.
|
CAUSES OF MUCOCUTANEOUS ULCERS |
aUnderlying atherosclerosis. bAlso associated with underlying disorders that lead to hypercoagulability, e.g., factor V Leiden, protein C dysfunction/deficiency, antiphospholipid antibodies. cReviewed in section on Purpura. dReviewed in section on Papulonodular Skin Lesions. eFavors plantar surface of the foot. fSign of immunosuppression.
Abbreviation: TEN, toxic epidermal necrolysis.
Livedoid vasculopathy (livedoid vasculitis; atrophie blanche) represents a combination of a vasculopathy plus intravascular thrombosis. Purpuric lesions and livedo reticularis are found in association with painful ulcerations of the lower extremities. These ulcers are often slow to heal, but when they do, irregularly shaped white scars form. The majority of cases are secondary to venous hypertension, but possible underlying illnesses include cryofibrinogenemia and disorders of hypercoagulability, e.g., the antiphospholipid syndrome (Chaps. 142 and 379).
In pyoderma gangrenosum, the border of untreated active ulcers has a characteristic appearance consisting of an undermined necrotic violaceous edge and a peripheral erythematous halo. The ulcers often begin as pustules that then expand rather rapidly to a size as large as 20 cm. Although these lesions are most commonly found on the lower extremities, they can arise anywhere on the surface of the body, including sites of trauma (pathergy). An estimated 30–50% of cases are idiopathic, and the most common associated disorders are ulcerative colitis and Crohn’s disease. Less commonly, pyoderma gangrenosum is associated with seropositive rheumatoid arthritis, acute and chronic myelogenous leukemia, hairy cell leukemia, myelofibrosis, or a monoclonal gammopathy, usually IgA. Because the histology of pyoderma gangrenosum may be nonspecific (dermal infiltrate of neutrophils when in untreated state), the diagnosis requires clinicopathologic correlation, in particular, the exclusion of similar-appearing ulcers such as necrotizing vasculitis, Meleney’s ulcer (synergistic infection at a site of trauma or surgery), dimorphic fungi, cutaneous amebiasis, spider bites, and factitial. In the myeloproliferative disorders, the ulcers may be more superficial with a pustulobullous border, and these lesions provide a connection between classic pyoderma gangrenosum and acute febrile neutrophilic dermatosis (Sweet syndrome).
FEVER AND RASH
The major considerations in a patient with a fever and a rash are inflammatory diseases versus infectious diseases. In the hospital setting, the most common scenario is a patient who has a drug rash plus a fever secondary to an underlying infection. However, it should be emphasized that a drug reaction can lead to both a cutaneous eruption and a fever (“drug fever”), especially in the setting of DRESS, AGEP, or serum sickness–like reaction. Additional inflammatory diseases that are often associated with a fever include pustular psoriasis, erythroderma, and Sweet syndrome. Lyme disease, secondary syphilis, and viral and bacterial exanthems (see “Exanthems,” above) are examples of infectious diseases that produce a rash and a fever. Lastly, it is important to determine whether or not the cutaneous lesions represent septic emboli (see “Purpura,” above). Such lesions usually have evidence of ischemia in the form of purpura, necrosis, or impending necrosis (gunmetal-gray color). In the patient with thrombocytopenia, however, purpura can be seen in inflammatory reactions such as morbilliform drug eruptions and infectious lesions.
73 |
Immunologically Mediated Skin Diseases |
A number of immunologically mediated skin diseases and immunologically mediated systemic disorders with cutaneous manifestations are now recognized as distinct entities with consistent clinical, histologic, and immunopathologic findings. Clinically, these disorders are characterized by morbidity (pain, pruritus, disfigurement) and, in some instances, result in death (largely due to loss of epidermal barrier function and/or secondary infection). The major features of the more common immunologically mediated skin diseases are summarized in this chapter (Table 73-1), as are the autoimmune systemic disorders with cutaneous manifestations.
|
IMMUNOLOGICALLY MEDIATED BLISTERING DISEASES |
AUTOIMMUNE CUTANEOUS DISEASES
PEMPHIGUS VULGARIS
Pemphigus refers to a group of autoantibody-mediated intraepidermal blistering diseases characterized by loss of cohesion between epidermal cells (a process termed acantholysis). Manual pressure to the skin of these patients may elicit the separation of the epidermis (Nikolsky’s sign). This finding, while characteristic of pemphigus, is not specific to this group of disorders and is also seen in toxic epidermal necrolysis, Stevens-Johnson syndrome, and a few other skin diseases.
Pemphigus vulgaris (PV) is a mucocutaneous blistering disease that predominantly occurs in patients >40 years of age. PV typically begins on mucosal surfaces and often progresses to involve the skin. This disease is characterized by fragile, flaccid blisters that rupture to produce extensive denudation of mucous membranes and skin (Fig. 73-1). The mouth, scalp, face, neck, axilla, groin, and trunk are typically involved. PV may be associated with severe skin pain; some patients experience pruritus as well. Lesions usually heal without scarring except at sites complicated by secondary infection or mechanically induced dermal wounds. Postinflammatory hyperpigmentation is usually present for some time at sites of healed lesions.
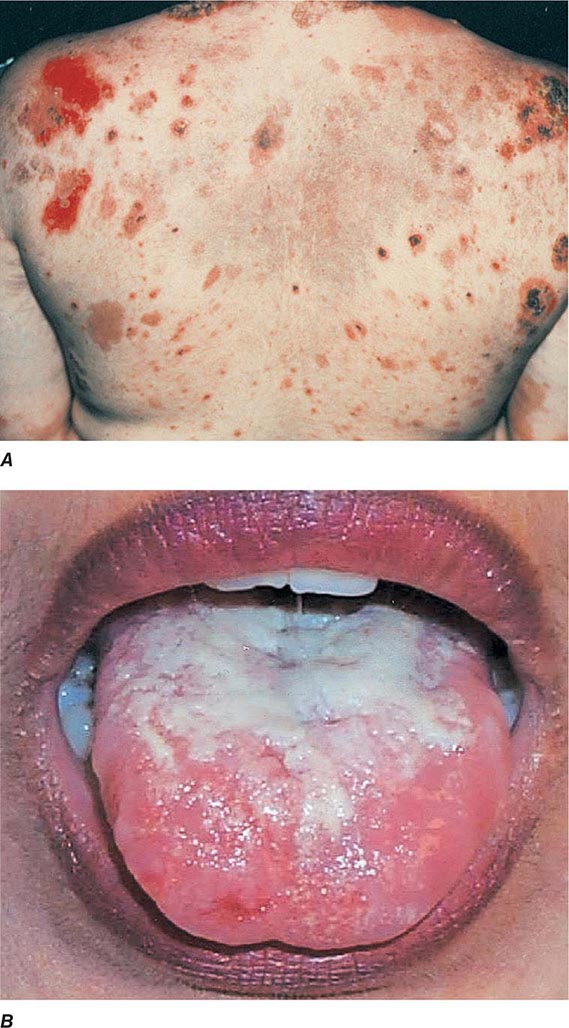
FIGURE 73-1 Pemphigus vulgaris. A. Flaccid bullae are easily ruptured, resulting in multiple erosions and crusted plaques. B. Involvement of the oral mucosa, which is almost invariable, may present with erosions on the gingiva, buccal mucosa, palate, posterior pharynx, or tongue. (B, Courtesy of Robert Swerlick, MD; with permission.)
Biopsies of early lesions demonstrate intraepidermal vesicle formation secondary to loss of cohesion between epidermal cells (i.e., acantholytic blisters). Blister cavities contain acantholytic epidermal cells, which appear as round homogeneous cells containing hyperchromatic nuclei. Basal keratinocytes remain attached to the epidermal basement membrane; hence, blister formation takes place within the suprabasal portion of the epidermis. Lesional skin may contain focal collections of intraepidermal eosinophils within blister cavities; dermal alterations are slight, often limited to an eosinophil-predominant leukocytic infiltrate. Direct immunofluorescence microscopy of lesional or intact patient skin shows deposits of IgG on the surface of keratinocytes; deposits of complement components are typically found in lesional but not in uninvolved skin. Deposits of IgG on keratinocytes are derived from circulating autoantibodies to cell-surface autoantigens. Such circulating autoantibodies can be demonstrated in 80–90% of PV patients by indirect immunofluorescence microscopy; monkey esophagus is the optimal substrate for these studies. Patients with PV have IgG autoantibodies to desmogleins (Dsgs), transmembrane desmosomal glycoproteins that belong to the cadherin family of calcium-dependent adhesion molecules. Such autoantibodies can be precisely quantitated by enzyme-linked immunosorbent assay (ELISA). Patients with early PV (i.e., mucosal disease) have IgG autoantibodies to Dsg3; patients with advanced PV (i.e., mucocutaneous disease) have IgG autoantibodies to both Dsg3 and Dsg1. Experimental studies have shown that autoantibodies from patients with PV are pathogenic (i.e., responsible for blister formation) and that their titer correlates with disease activity. Recent studies have shown that the anti-Dsg autoantibody profile in these patients’ sera as well as the tissue distribution of Dsg3 and Dsg1 determine the site of blister formation in patients with PV. Coexpression of Dsg3 and Dsg1 by epidermal cells protects against pathogenic IgG antibodies to either of these cadherins but not against pathogenic autoantibodies to both.
PV can be life-threatening. Prior to the availability of glucocorticoids, mortality rates ranged from 60% to 90%; the current figure is ~5%. Common causes of morbidity and death are infection and complications of treatment with glucocorticoids. Bad prognostic factors include advanced age, widespread involvement, and the requirement for high doses of glucocorticoids (with or without other immunosuppressive agents) for control of disease. The course of PV in individual patients is variable and difficult to predict. Some patients experience remission, while others may require long-term treatment or succumb to complications of their disease or its treatment. The mainstay of treatment is systemic glucocorticoids. Patients with moderate to severe PV are usually started on prednisone at 1 mg/kg per day. If new lesions continue to appear after 1–2 weeks of treatment, the dose may need to be increased and/or prednisone may need to be combined with other immunosuppressive agents such as azathioprine (2–2.5 mg/kg per day), mycophenolate mofetil (20–35 mg/kg per day), or cyclophosphamide (1–2 mg/kg per day). Patients with severe, treatment-resistant disease may derive benefit from plasmapheresis (six high-volume exchanges [i.e., 2–3 L per exchange] over ~2 weeks), IV immunoglobulin (IVIg) (2 g/kg over 3–5 days every 6–8 weeks), or rituximab (375 mg/m2 per week × 4, or 1000 mg on days 1 and 15). It is important to bring severe or progressive disease under control quickly in order to lessen the severity and/or duration of this disorder. Accordingly, some have suggested that rituximab and daily glucocorticoids should be used early in PV patients to avert the development of treatment-resistant disease.
PEMPHIGUS FOLIACEUS
Pemphigus foliaceus (PF) is distinguished from PV by several features. In PF, acantholytic blisters are located high within the epidermis, usually just beneath the stratum corneum. Hence, PF is a more superficial blistering disease than PV. The distribution of lesions in the two disorders is much the same, except that in PF mucous membranes are almost always spared. Patients with PF rarely have intact blisters but rather exhibit shallow erosions associated with erythema, scale, and crust formation. Mild cases of PF resemble severe seborrheic dermatitis; severe PF may cause extensive exfoliation. Sun exposure (ultraviolet irradiation) may be an aggravating factor.
PF has immunopathologic features in common with PV. Specifically, direct immunofluorescence microscopy of perilesional skin demonstrates IgG on the surface of keratinocytes. Similarly, patients with PF have circulating IgG autoantibodies directed against the surface of keratinocytes. In PF, autoantibodies are directed against Dsg1, a 160-kDa desmosomal cadherin. These autoantibodies can be quantitated by ELISA. As noted for PV, the autoantibody profile in patients with PF (i.e., anti-Dsg1 IgG) and the tissue distribution of this autoantigen (i.e., expression in oral mucosa that is compensated by coexpression of Dsg3) are thought to account for the distribution of lesions in this disease.
Endemic forms of PF are found in south-central rural Brazil, where the disease is known as fogo salvagem (FS), as well as in selected sites in Latin America and Tunisia. Endemic PF, like other forms of this disease, is mediated by IgG autoantibodies to Dsg1. Clusters of FS overlap with those of leishmaniasis, a disease transmitted by bites of the sand fly Lutzomyia longipalis. Recent studies have shown that sand-fly salivary antigens (specifically, the LJM11 salivary protein) are recognized by IgG autoantibodies from FS patients (as well as by monoclonal antibodies to Dsg1 derived from these patients). Moreover, mice immunized with LJM11 produce antibodies to Dsg1. Thus, these findings suggest that insect bites may deliver salivary antigens that initiate a cross-reactive humoral immune response, which may lead to FS in genetically susceptible individuals.
Although pemphigus has been associated with several autoimmune diseases, its association with thymoma and/or myasthenia gravis is particularly notable. To date, >30 cases of thymoma and/or myasthenia gravis have been reported in association with pemphigus, usually with PF. Patients may also develop pemphigus as a consequence of drug exposure; drug-induced pemphigus usually resembles PF rather than PV. Drugs containing a thiol group in their chemical structure (e.g., penicillamine, captopril, enalapril) are most commonly associated with drug-induced pemphigus. Nonthiol drugs linked to pemphigus include penicillins, cephalosporins, and piroxicam. It has been suggested that thiol-containing and non-thiol-containing drugs induce pemphigus via biochemical and immunologic mechanisms, respectively—hence, the better prognosis upon drug withdrawal in cases of pemphigus induced by thiol-containing medications. Some cases of drug-induced pemphigus are durable and require treatment with systemic glucocorticoids and/or immunosuppressive agents.
PF is generally a less severe disease than PV and carries a better prognosis. Localized disease can sometimes be treated with topical or intralesional glucocorticoids; more active cases can usually be controlled with systemic glucocorticoids. Patients with severe, treatment-resistant disease may require more aggressive interventions, as described above for patients with PV.
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus (PNP) is an autoimmune acantholytic mucocutaneous disease associated with an occult or confirmed neoplasm. Patients with PNP typically have painful mucosal erosive lesions in association with papulosquamous and/or lichenoid eruptions that often progress to blisters. Palm and sole involvement are common in these patients and raise the possibility that prior reports of neoplasia-associated erythema multiforme actually may have represented unrecognized cases of PNP. Biopsies of lesional skin from these patients show varying combinations of acantholysis, keratinocyte necrosis, and vacuolar-interface dermatitis. Direct immunofluorescence microscopy of a patient’s skin shows deposits of IgG and complement on the surface of keratinocytes and (variably) similar immunoreactants in the epidermal basement membrane zone. Patients with PNP have IgG autoantibodies to cytoplasmic proteins that are members of the plakin family (e.g., desmoplakins I and II, bullous pemphigoid antigen [BPAG]1, envoplakin, periplakin, and plectin) and to cell-surface proteins that are members of the cadherin family (e.g., Dsg1 and Dsg3). Passive transfer studies have shown that autoantibodies from patients with PNP are pathogenic in animal models.
The predominant neoplasms associated with PNP are non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, thymoma, spindle cell tumors, Waldenström’s macroglobulinemia, and Castleman’s disease; the last-mentioned neoplasm is particularly common among children with PNP. Rare cases of seronegative PNP have been reported in patients with B cell malignancies previously treated with rituximab. In addition to severe skin lesions, many patients with PNP develop life-threatening bronchiolitis obliterans. PNP is generally resistant to conventional therapies (i.e., those used to treat PV); rarely, a patient’s disease may ameliorate or even remit following ablation or removal of underlying neoplasms.
BULLOUS PEMPHIGOID
Bullous pemphigoid (BP) is a polymorphic autoimmune subepidermal blistering disease usually seen in the elderly. Initial lesions may consist of urticarial plaques; most patients eventually display tense blisters on either normal-appearing or erythematous skin (Fig. 73-2). The lesions are usually distributed over the lower abdomen, groin, and flexor surface of the extremities; oral mucosal lesions are found in some patients. Pruritus may be nonexistent or severe. As lesions evolve, tense blisters tend to rupture and be replaced by erosions with or without surmounting crust. Nontraumatized blisters heal without scarring. The major histocompatibility complex class II allele HLA-DQβ1*0301 is prevalent in patients with BP. Despite isolated reports, several studies have shown that patients with BP do not have a higher incidence of malignancy than appropriately age- and gender-matched controls.
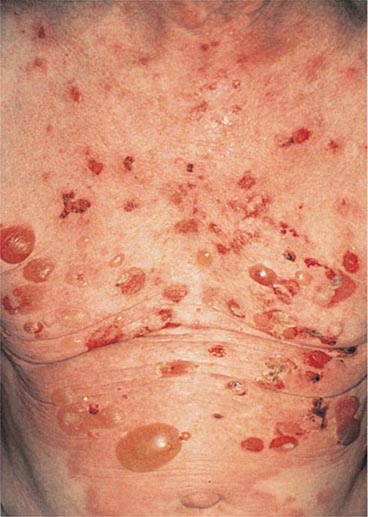
FIGURE 73-2 Bullous pemphigoid with tense vesicles and bullae on erythematous, urticarial bases. (Courtesy of the Yale Resident’s Slide Collection; with permission.)
Biopsies of early lesional skin demonstrate subepidermal blisters and histologic features that roughly correlate with the clinical character of the particular lesion under study. Lesions on normal-appearing skin generally contain a sparse perivascular leukocytic infiltrate with some eosinophils; conversely, biopsies of inflammatory lesions typically show an eosinophil-rich infiltrate at sites of vesicle formation and in perivascular areas. In addition to eosinophils, cell-rich lesions also contain mononuclear cells and neutrophils. It is not possible to distinguish BP from other subepidermal blistering diseases by routine histologic studies alone.
Direct immunofluorescence microscopy of normal-appearing perilesional skin from patients with BP shows linear deposits of IgG and/or C3 in the epidermal basement membrane. The sera of ~70% of these patients contain circulating IgG autoantibodies that bind the epidermal basement membrane of normal human skin in indirect immunofluorescence microscopy. IgG from an even higher percentage of patients reacts with the epidermal side of 1 M NaCl split skin (an alternative immunofluorescence microscopy test substrate used to distinguish circulating IgG autoantibodies to the basement membrane in patients with BP from those in patients with similar, yet different, subepidermal blistering diseases; see below). In BP, circulating autoantibodies recognize 230- and 180-kDa hemidesmosome-associated proteins in basal keratinocytes (i.e., BPAG1 and BPAG2, respectively). Autoantibodies to BPAG2 are thought to deposit in situ, activate complement, produce dermal mast-cell degranulation, and generate granulocyte-rich infiltrates that cause tissue damage and blister formation.
BP may persist for months to years, with exacerbations or remissions. Extensive involvement may result in widespread erosions and compromise cutaneous integrity; elderly and/or debilitated patients may die. The mainstay of treatment is systemic glucocorticoids. Local or minimal disease can sometimes be controlled with topical glucocorticoids alone; more extensive lesions generally respond to systemic glucocorticoids either alone or in combination with immunosuppressive agents. Patients usually respond to prednisone (0.75–1 mg/kg per day). In some instances, azathioprine (2–2.5 mg/kg per day), mycophenolate mofetil (20–35 mg/kg per day), or cyclophosphamide (1–2 mg/kg per day) are necessary adjuncts.
PEMPHIGOID GESTATIONIS
Pemphigoid gestationis (PG), also known as herpes gestationis, is a rare, nonviral, subepidermal blistering disease of pregnancy and the puerperium. PG may begin during any trimester of pregnancy or present shortly after delivery. Lesions are usually distributed over the abdomen, trunk, and extremities; mucous membrane lesions are rare. Skin lesions in these patients may be quite polymorphic and consist of erythematous urticarial papules and plaques, vesiculopapules, and/or frank bullae. Lesions are almost always extremely pruritic. Severe exacerbations of PG frequently follow delivery, typically within 24–48 h. PG tends to recur in subsequent pregnancies, often beginning earlier during such gestations. Brief flare-ups of disease may occur with resumption of menses and may develop in patients later exposed to oral contraceptives. Occasionally, infants of affected mothers have transient skin lesions.
Biopsies of early lesional skin show teardrop-shaped subepidermal vesicles forming in dermal papillae in association with an eosinophil-rich leukocytic infiltrate. Differentiation of PG from other subepidermal bullous diseases by light microscopy is difficult. However, direct immunofluorescence microscopy of perilesional skin from PG patients reveals the immunopathologic hallmark of this disorder: linear deposits of C3 in the epidermal basement membrane. These deposits develop as a consequence of complement activation produced by low-titer IgG anti–basement membrane autoantibodies directed against BPAG2, the same hemidesmosome-associated protein that is targeted by autoantibodies in patients with BP—a subepidermal bullous disease that resembles PG clinically, histologically, and immunopathologically.
The goals of therapy in patients with PG are to prevent the development of new lesions, relieve intense pruritus, and care for erosions at sites of blister formation. Many patients require treatment with moderate doses of daily glucocorticoids (i.e., 20–40 mg of prednisone) at some point in their course. Mild cases (or brief flare-ups) may be controlled by vigorous use of potent topical glucocorticoids. Infants born of mothers with PG appear to be at increased risk of being born slightly premature or “small for dates.” Current evidence suggests that there is no difference in the incidence of uncomplicated live births between PG patients treated with systemic glucocorticoids and those managed more conservatively. If systemic glucocorticoids are administered, newborns are at risk for development of reversible adrenal insufficiency.
DERMATITIS HERPETIFORMIS
Dermatitis herpetiformis (DH) is an intensely pruritic, papulovesicular skin disease characterized by lesions symmetrically distributed over extensor surfaces (i.e., elbows, knees, buttocks, back, scalp, and posterior neck) (see Fig. 70-8). Primary lesions in this disorder consist of papules, papulovesicles, or urticarial plaques. Because pruritus is prominent, patients may present with excoriations and crusted papules but no observable primary lesions. Patients sometimes report that their pruritus has a distinctive burning or stinging component; the onset of such local symptoms reliably heralds the development of distinct clinical lesions 12–24 h later. Almost all DH patients have associated, usually subclinical, gluten-sensitive enteropathy (Chap. 349), and >90% express the HLA-B8/DRw3 and HLA-DQw2 haplotypes. DH may present at any age, including in childhood; onset in the second to fourth decades is most common. The disease is typically chronic.
Biopsy of early lesional skin reveals neutrophil-rich infiltrates within dermal papillae. Neutrophils, fibrin, edema, and microvesicle formation at these sites are characteristic of early disease. Older lesions may demonstrate nonspecific features of a subepidermal bulla or an excoriated papule. Because the clinical and histologic features of this disease can be variable and resemble those of other subepidermal blistering disorders, the diagnosis is confirmed by direct immunofluorescence microscopy of normal-appearing perilesional skin. Such studies demonstrate granular deposits of IgA (with or without complement components) in the papillary dermis and along the epidermal basement membrane zone. IgA deposits in the skin are unaffected by control of disease with medication; however, these immunoreactants diminish in intensity or disappear in patients maintained for long periods on a strict gluten-free diet (see below). Patients with DH have granular deposits of IgA in their epidermal basement membrane zone and should be distinguished from individuals with linear IgA deposits at this site (see below).
Although most DH patients do not report overt gastrointestinal symptoms or have laboratory evidence of malabsorption, biopsies of the small bowel usually reveal blunting of intestinal villi and a lymphocytic infiltrate in the lamina propria. As is true for patients with celiac disease, this gastrointestinal abnormality can be reversed by a gluten-free diet. Moreover, if maintained, this diet alone may control the skin disease and eventuate in clearance of IgA deposits from these patients’ epidermal basement membrane zones. Subsequent gluten exposure in such patients alters the morphology of their small bowel, elicits a flare-up of their skin disease, and is associated with the reappearance of IgA in their epidermal basement membrane zones. As in patients with celiac disease, dietary gluten sensitivity in patients with DH is associated with IgA endomysial autoantibodies that target tissue transglutaminase. Studies indicate that patients with DH also have high-avidity IgA autoantibodies to epidermal transglutaminase 3 and that the latter is co-localized with granular deposits of IgA in the papillary dermis of DH patients. Patients with DH also have an increased incidence of thyroid abnormalities, achlorhydria, atrophic gastritis, and autoantibodies to gastric parietal cells. These associations likely relate to the high frequency of the HLA-B8/DRw3 haplotype in these patients, because this marker is commonly linked to autoimmune disorders. The mainstay of treatment of DH is dapsone, a sulfone. Patients respond rapidly (24–48 h) to dapsone (50–200 mg/d), but require careful pretreatment evaluation and close follow-up to ensure that complications are avoided or controlled. All patients taking dapsone at >100 mg/d will have some hemolysis and methemoglobinemia, which are expected pharmacologic side effects of this agent. Gluten restriction can control DH and lessen dapsone requirements; this diet must rigidly exclude gluten to be of maximal benefit. Many months of dietary restriction may be necessary before a beneficial result is achieved. Good dietary counseling by a trained dietitian is essential.
LINEAR IgA DISEASE
Linear IgA disease, once considered a variant form of DH, is actually a separate and distinct entity. Clinically, patients with linear IgA disease may resemble individuals with DH, BP, or other subepidermal blistering diseases. Lesions typically consist of papulovesicles, bullae, and/or urticarial plaques that develop predominantly on central or flexural sites. Oral mucosal involvement occurs in some patients. Severe pruritus resembles that seen in patients with DH. Patients with linear IgA disease do not have an increased frequency of the HLA-B8/DRw3 haplotype or an associated enteropathy and therefore are not candidates for treatment with a gluten-free diet.
Histologic alterations in early lesions may be virtually indistinguishable from those in DH. However, direct immunofluorescence microscopy of normal-appearing perilesional skin reveals a linear band of IgA (and often C3) in the epidermal basement membrane zone. Most patients with linear IgA disease have circulating IgA basement membrane autoantibodies directed against neoepitopes in the proteolytically processed extracellular domain of BPAG2. These patients generally respond to treatment with dapsone (50–200 mg/d).
EPIDERMOLYSIS BULLOSA ACQUISITA
Epidermolysis bullosa acquisita (EBA) is a rare, noninherited, polymorphic, chronic, subepidermal blistering disease. (The inherited form is discussed in Chap. 427.) Patients with classic or noninflammatory EBA have blisters on noninflamed skin, atrophic scars, milia, nail dystrophy, and oral lesions. Because lesions generally occur at sites exposed to minor trauma, classic EBA is considered a mechanobullous disease. Other patients with EBA have widespread inflammatory scarring and bullous lesions that resemble severe BP. Inflammatory EBA may evolve into the classic, noninflammatory form of this disease. Rarely, patients present with lesions that predominate on mucous membranes. The HLA-DR2 haplotype is found with increased frequency in EBA patients. Studies suggest that EBA is sometimes associated with inflammatory bowel disease (especially Crohn’s disease).
The histology of lesional skin varies with the character of the lesion being studied. Noninflammatory bullae are subepidermal, feature a sparse leukocytic infiltrate, and resemble the lesions in patients with porphyria cutanea tarda. Inflammatory lesions consist of neutrophil-rich subepidermal blisters. EBA patients have continuous deposits of IgG (and frequently C3) in a linear pattern within the epidermal basement membrane zone. Ultrastructurally, these immunoreactants are found in the sublamina densa region in association with anchoring fibrils. Approximately 50% of EBA patients have demonstrable circulating IgG basement membrane autoantibodies directed against type VII collagen—the collagen species that makes up anchoring fibrils. Such IgG autoantibodies bind the dermal side of 1 M NaCl split skin (in contrast to IgG autoantibodies in patients with BP). Studies have shown that passive transfer of experimental or clinical IgG against type VII collagen can produce lesions in mice that clinically, histologically, and immunopathologically resemble those in patients with inflammatory EBA.
Treatment of EBA is generally unsatisfactory. Some patients with inflammatory EBA may respond to systemic glucocorticoids, either alone or in combination with immunosuppressive agents. Other patients (especially those with neutrophil-rich inflammatory lesions) may respond to dapsone. The chronic, noninflammatory form of EBA is largely resistant to treatment, although some patients may respond to cyclosporine, azathioprine, or IVIg.
MUCOUS MEMBRANE PEMPHIGOID
Mucous membrane pemphigoid (MMP) is a rare, acquired, subepithelial immunobullous disease characterized by erosive lesions of mucous membranes and skin that result in scarring of at least some sites of involvement. Common sites include the oral mucosa (especially the gingiva) and conjunctiva; other sites that may be affected include the nasopharyngeal, laryngeal, esophageal, and anogenital mucosa. Skin lesions (present in about one-third of patients) tend to predominate on the scalp, face, and upper trunk and generally consist of a few scattered erosions or tense blisters on an erythematous or urticarial base. MMP is typically a chronic and progressive disorder. Serious complications may arise as a consequence of ocular, laryngeal, esophageal, or anogenital lesions. Erosive conjunctivitis may result in shortened fornices, symblepharon, ankyloblepharon, entropion, corneal opacities, and (in severe cases) blindness. Similarly, erosive lesions of the larynx may cause hoarseness, pain, and tissue loss that, if unrecognized and untreated, may eventuate in complete destruction of the airway. Esophageal lesions may result in stenosis and/or strictures that could place patients at risk for aspiration. Strictures may also complicate anogenital involvement.
Biopsies of lesional tissue generally show subepithelial vesiculobullae and a mononuclear leukocytic infiltrate. Neutrophils and eosinophils may be seen in biopsies of early lesions; older lesions may demonstrate a scant leukocytic infiltrate and fibrosis. Direct immunofluorescence microscopy of perilesional tissue typically reveals deposits of IgG, IgA, and/or C3 in the epidermal basement membrane. Because many patients with MMP exhibit no evidence of circulating basement membrane autoantibodies, testing of perilesional skin is important diagnostically. Although MMP was once thought to be a single nosologic entity, it is now largely regarded as a disease phenotype that may develop as a consequence of an autoimmune reaction to a variety of molecules in the epidermal basement membrane (e.g., BPAG2, laminin-332, type VII collagen, and other antigens yet to be completely defined). Studies suggest that MMP patients with autoantibodies to laminin-332 have an increased relative risk for cancer. Treatment of MMP is largely dependent upon the sites of involvement. Due to potentially severe complications, patients with ocular, laryngeal, esophageal, and/or anogenital involvement require aggressive systemic treatment with dapsone, prednisone, or the latter in combination with another immunosuppressive agent (e.g., azathioprine, mycophenolate mofetil, cyclophosphamide, or rituximab) or IVIg. Less threatening forms of the disease may be managed with topical or intralesional glucocorticoids.
AUTOIMMUNE SYSTEMIC DISEASES WITH PROMINENT CUTANEOUS FEATURES
DERMATOMYOSITIS
The cutaneous manifestations of dermatomyositis (Chap. 388) are often distinctive but at times may resemble those of systemic lupus erythematosus (SLE) (Chap. 378), scleroderma (Chap. 382), or other overlapping connective tissue diseases (Chap. 382). The extent and severity of cutaneous disease may or may not correlate with the extent and severity of the myositis. The cutaneous manifestations of dermatomyositis are similar, whether the disease appears in children or in the elderly, except that calcification of subcutaneous tissue is a common late sequela in childhood dermatomyositis.
The cutaneous signs of dermatomyositis may precede or follow the development of myositis by weeks to years. Cases lacking muscle involvement (i.e., dermatomyositis sine myositis) have also been reported. The most common manifestation is a purple-red discoloration of the upper eyelids, sometimes associated with scaling (“heliotrope” erythema; Fig. 73-3) and periorbital edema. Erythema on the cheeks and nose in a “butterfly” distribution may resemble the malar eruption of SLE. Erythematous or violaceous scaling patches are common on the upper anterior chest, posterior neck, scalp, and the extensor surfaces of the arms, legs, and hands. Erythema and scaling may be particularly prominent over the elbows, knees, and dorsal interphalangeal joints. Approximately one-third of patients have violaceous, flat-topped papules over the dorsal interphalangeal joints that are pathognomonic of dermatomyositis (Gottron’s papules). Thin violaceous papules and plaques on the elbows and knees of patients with dermatomyositis are referred to as Gottron’s sign (Fig. 73-4). These lesions can be contrasted with the erythema and scaling on the dorsum of the fingers that spares the skin over the interphalangeal joints of some SLE patients. Periungual telangiectasia may be prominent in patients with dermatomyositis. Lacy or reticulated erythema may be associated with fine scaling on the extensor and lateral surfaces of the thighs and upper arms. Other patients, particularly those with long-standing disease, develop areas of hypopigmentation, hyperpigmentation, mild atrophy, and telangiectasia known as poikiloderma. Poikiloderma is rare in both SLE and scleroderma and thus can serve as a clinical sign that distinguishes dermatomyositis from these two diseases. Cutaneous changes may be similar in dermatomyositis and various overlap syndromes where thickening and binding down of the skin of the hands (sclerodactyly) as well as Raynaud’s phenomenon can be seen. However, the presence of severe muscle disease, Gottron’s papules, heliotrope erythema, and poikiloderma serves to distinguish patients with dermatomyositis. Skin biopsy of the erythematous, scaling lesions of dermatomyositis may reveal only mild nonspecific inflammation but sometimes may show changes indistinguishable from those found in SLE, including epidermal atrophy, hydropic degeneration of basal keratinocytes, edema of the upper dermis, and a mild mononuclear cell infiltrate. Direct immunofluorescence microscopy of lesional skin is usually negative, although granular deposits of immunoglobulin(s) and complement in the epidermal basement membrane zone have been described in some patients. Treatment should be directed at the systemic disease. Topical glucocorticoids are sometimes useful; patients should avoid exposure to ultraviolet irradiation and aggressively use photoprotective measures, including broad-spectrum sunscreens.
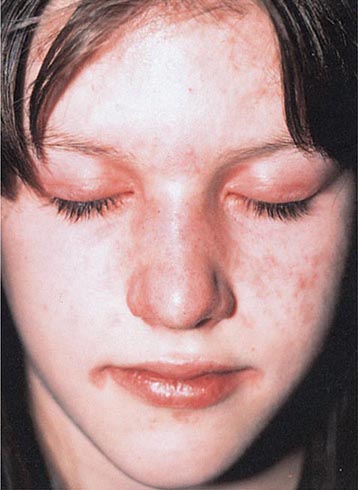
FIGURE 73-3 Dermatomyositis. Periorbital violaceous erythema characterizes the classic heliotrope rash. (Courtesy of James Krell, MD; with permission.)
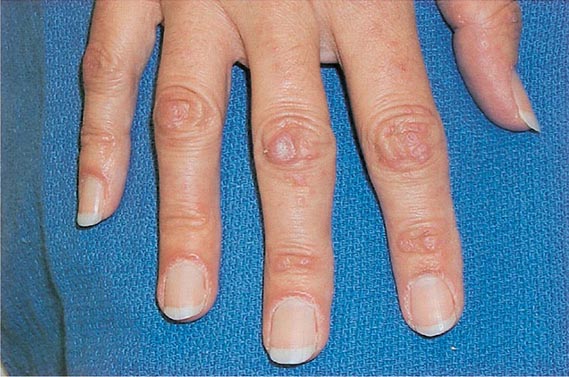
FIGURE 73-4 Gottron’s papules. Dermatomyositis often involves the hands as erythematous flat-topped papules over the knuckles. Periungual telangiectases are also evident.
LUPUS ERYTHEMATOSUS
The cutaneous manifestations of lupus erythematosus (LE) (Chap. 378) can be divided into acute, subacute, and chronic or discoid types. Acute cutaneous LE is characterized by erythema of the nose and malar eminences in a “butterfly” distribution (Fig. 73-5A). The erythema is often sudden in onset, accompanied by edema and fine scale, and correlated with systemic involvement. Patients may have widespread involvement of the face as well as erythema and scaling of the extensor surfaces of the extremities and upper chest (Fig. 73-5B). These acute lesions, while sometimes evanescent, usually last for days and are often associated with exacerbations of systemic disease. Skin biopsy of acute lesions may show only a sparse dermal infiltrate of mononuclear cells and dermal edema. In some instances, cellular infiltrates around blood vessels and hair follicles are notable, as is hydropic degeneration of basal cells of the epidermis. Direct immunofluorescence microscopy of lesional skin frequently reveals deposits of immunoglobulin(s) and complement in the epidermal basement membrane zone. Treatment is aimed at control of systemic disease. Photoprotection is very important in this as well as in other forms of LE.
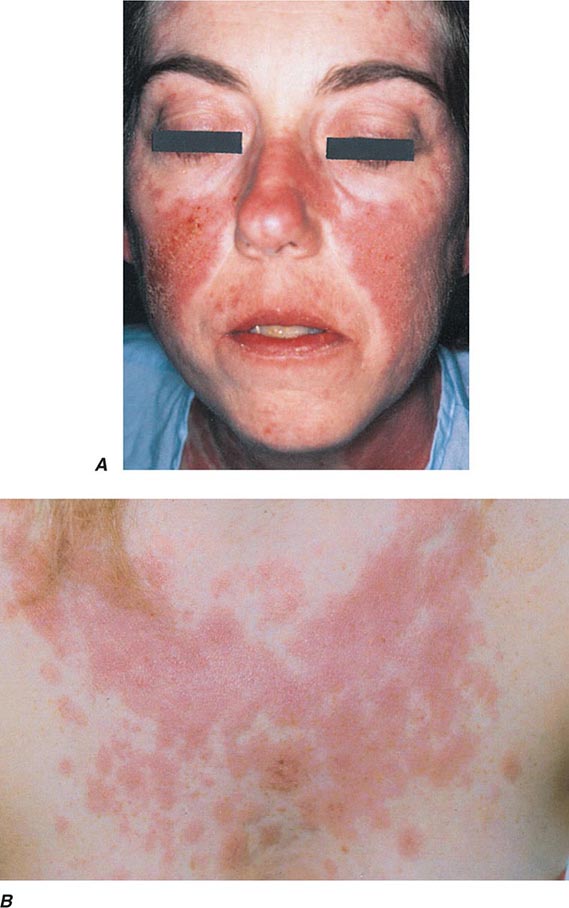
FIGURE 73-5 Acute cutaneous lupus erythematosus (LE). A. Acute cutaneous LE on the face, showing prominent, scaly, malar erythema. Involvement of other sun-exposed sites is also common. B. Acute cutaneous LE on the upper chest, demonstrating brightly erythematous and slightly edematous papules and plaques. (B, Courtesy of Robert Swerlick, MD; with permission.)
Subacute cutaneous lupus erythematosus (SCLE) is characterized by a widespread photosensitive, nonscarring eruption. In most patients, renal and central nervous system involvement is mild or absent. SCLE may present as a papulosquamous eruption that resembles psoriasis or as annular polycyclic lesions that resemble those seen in erythema multiforme. In the papulosquamous form, discrete erythematous papules arise on the back, chest, shoulders, extensor surfaces of the arms, and dorsum of the hands; lesions are uncommon on the central face and the flexor surfaces of the arms as well as below the waist. These slightly scaling papules tend to merge into large plaques, some with a reticulate appearance. The annular form involves the same areas and presents with erythematous papules that evolve into oval, circular, or polycyclic lesions. The lesions of SCLE are more widespread but have less tendency for scarring than lesions of discoid LE. Skin biopsy reveals a dense mononuclear cell infiltrate around hair follicles and blood vessels in the superficial dermis, combined with hydropic degeneration of basal cells in the epidermis. Direct immunofluorescence microscopy of lesional skin reveals deposits of immunoglobulin(s) in the epidermal basement membrane zone in about one-half of these cases. A particulate pattern of IgG deposition throughout the epidermis has been associated with SCLE. Most SCLE patients have anti-Ro autoantibodies. Local therapy alone is usually unsuccessful. Most patients require treatment with aminoquinoline antimalarial drugs. Low-dose therapy with oral glucocorticoids is sometimes necessary. Photoprotective measures against both ultraviolet B and ultraviolet A wavelengths are very important.
Discoid lupus erythematosus (DLE, also called chronic cutaneous LE) is characterized by discrete lesions, most often found on the face, scalp, and/or external ears. The lesions are erythematous papules or plaques with a thick, adherent scale that occludes hair follicles (follicular plugging). When the scale is removed, its underside shows small excrescences that correlate with the openings of hair follicles (so-called “carpet tacking”), a finding relatively specific for DLE. Long-standing lesions develop central atrophy, scarring, and hypopigmentation but frequently have erythematous, sometimes raised borders (Fig. 73-6). These lesions persist for years and tend to expand slowly. Up to 15% of patients with DLE eventually meet the American College of Rheumatology criteria for SLE. However, typical discoid lesions are frequently seen in patients with SLE. Biopsy of DLE lesions shows hyperkeratosis, follicular plugging, atrophy of the epidermis, hydropic degeneration of basal keratinocytes, and a mononuclear cell infiltrate adjacent to epidermal, adnexal, and microvascular basement membranes. Direct immunofluorescence microscopy demonstrates immunoglobulin(s) and complement deposits at the basement membrane zone in ~90% of cases. Treatment is focused on control of local cutaneous disease and consists mainly of photoprotection and topical or intralesional glucocorticoids. If local therapy is ineffective, use of aminoquinoline antimalarial agents may be indicated.
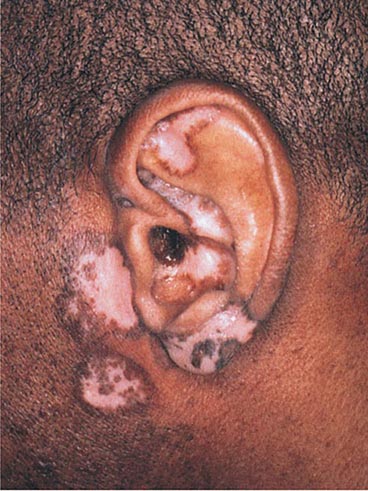
FIGURE 73-6 Discoid (chronic cutaneous) lupus erythematosus. Violaceous, hyperpigmented, atrophic plaques, often with evidence of follicular plugging that may result in scarring, are typical.
SCLERODERMA AND MORPHEA
The skin changes of scleroderma (Chap. 382) usually begin on the hands, feet, and face, with episodes of recurrent nonpitting edema. Sclerosis of the skin commences distally on the fingers (sclerodactyly) and spreads proximally, usually accompanied by resorption of bone of the fingertips, which may have punched out ulcers, stellate scars, or areas of hemorrhage (Fig. 73-7). The fingers may actually shrink and become sausage-shaped, and, because the fingernails are usually unaffected, they may curve over the end of the fingertips. Periungual telangiectases are usually present, but periungual erythema is rare. In advanced cases, the extremities show contractures and calcinosis cutis. Facial involvement includes a smooth, unwrinkled brow, taut skin over the nose, shrinkage of tissue around the mouth, and perioral radial furrowing (Fig. 73-8). Matlike telangiectases are often present, particularly on the face and hands. Involved skin feels indurated, smooth, and bound to underlying structures; hyper- and hypopigmentation are common as well. Raynaud’s phenomenon (i.e., cold-induced blanching, cyanosis, and reactive hyperemia) is documented in almost all patients and can precede development of scleroderma by many years. Linear scleroderma is a limited form of disease that presents in a linear, bandlike distribution and tends to involve deep as well as superficial layers of skin. The combination of calcinosis cutis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia has been termed the CREST syndrome. Centromere antibodies have been reported in a very high percentage of patients with the CREST syndrome but in only a small minority of patients with scleroderma. Skin biopsy reveals thickening of the dermis and homogenization of collagen bundles. Direct immunofluorescence microscopy of lesional skin is usually negative.
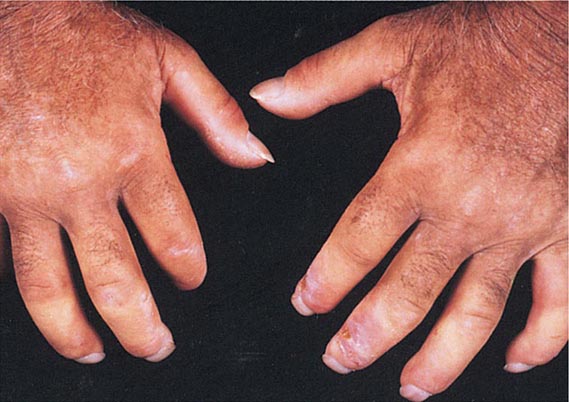
FIGURE 73-7 Scleroderma showing acral sclerosis and focal digital ulcers.
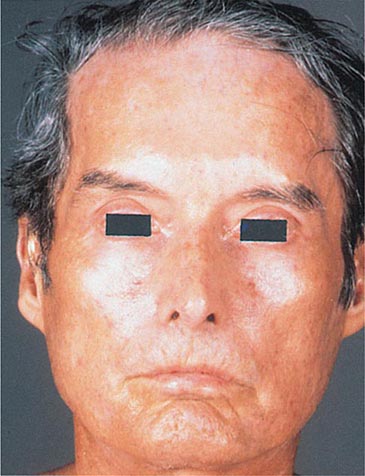
FIGURE 73-8 Scleroderma often eventuates in development of an expressionless, masklike facies.
Morphea is characterized by localized thickening and sclerosis of skin; it dominates on the trunk. This disorder may affect children or adults. Morphea begins as erythematous or flesh-colored plaques that become sclerotic, develop central hypopigmentation, and have an erythematous border. In most cases, patients have one or a few lesions, and the disease is termed localized morphea. In some patients, widespread cutaneous lesions may occur without systemic involvement (generalized morphea). Many adults with generalized morphea have concomitant rheumatic or other autoimmune disorders. Skin biopsy of morphea is indistinguishable from that of scleroderma. Scleroderma and morphea are usually quite resistant to therapy. For this reason, physical therapy to prevent joint contractures and to maintain function is employed and is often helpful. Treatment options for early, rapidly progressive disease include phototherapy (UVA1 or PUVA) or methotrexate (15–20 mg/week) alone or in combination with daily glucocorticoids.
Diffuse fasciitis with eosinophilia is a clinical entity that can sometimes be confused with scleroderma. There is usually a sudden onset of swelling, induration, and erythema of the extremities, frequently following significant physical exertion. The proximal portions of the extremities (upper arms, forearms, thighs, calves) are more often involved than are the hands and feet. While the skin is indurated, it usually displays a woody, dimpled, or “pseudocellulite” appearance rather than being bound down as in scleroderma; contractures may occur early secondary to fascial involvement. The latter may also cause muscle groups to be separated and veins to appear depressed (i.e., the “groove sign”). These skin findings are accompanied by peripheral-blood eosinophilia, increased erythrocyte sedimentation rate, and sometimes hypergammaglobulinemia. Deep biopsy of affected areas of skin reveals inflammation and thickening of the deep fascia overlying muscle. An inflammatory infiltrate composed of eosinophils and mononuclear cells is usually found. Patients with eosinophilic fasciitis appear to be at increased risk for developing bone marrow failure or other hematologic abnormalities. While the ultimate course of eosinophilic fasciitis is uncertain, many patients respond favorably to treatment with prednisone in doses of 40–60 mg/d.
The eosinophilia-myalgia syndrome, a disorder with epidemic numbers of cases reported in 1989 and linked to ingestion of L-tryptophan manufactured by a single company in Japan, is a multisystem disorder characterized by debilitating myalgias and absolute eosinophilia in association with varying combinations of arthralgias, pulmonary symptoms, and peripheral edema. In a later phase (3–6 months after initial symptoms), these patients often develop localized sclerodermatous skin changes, weight loss, and/or neuropathy (Chap. 382). The precise cause of this syndrome, which may resemble other sclerotic skin conditions, is unknown. However, the implicated lots of L-tryptophan contained the contaminant 1,1-ethylidene bis[tryptophan]. This contaminant may be pathogenic or may be a marker for another substance that provokes the disorder.
74 |
Cutaneous Drug Reactions |
Cutaneous reactions are among the most frequent adverse reactions to drugs. Most are benign, but a few can be life threatening. Prompt recognition of severe reactions, drug withdrawal, and appropriate therapeutic interventions can minimize toxicity. This chapter focuses on adverse cutaneous reactions to systemic medications; it covers their incidence, patterns, and pathogenesis and provides some practical guidelines on treatment, assessment of causality, and future use of drugs.
USE OF PRESCRIPTION DRUGS IN THE UNITED STATES
In the United States, more than 3 billion prescriptions for over 60,000 drug products, which include more than 2000 different active agents, are dispensed annually. Hospital inpatients alone annually receive about 120 million courses of drug therapy, and half of adult Americans receive prescription drugs on a regular outpatient basis. Many patients use over-the-counter medicines that may cause adverse cutaneous reactions.
INCIDENCE OF CUTANEOUS REACTIONS
Several large cohort studies established that acute cutaneous reaction to drugs affected about 3% of hospital inpatients. Reactions usually occur a few days to 4 weeks after initiation of therapy.
Many drugs of common use are associated with a 1–2% rate of rashes during premarketing clinical trials. The risk is often higher when medications are used in general, unselected populations. The rate may reach 3–7% for amoxicillin, sulfamethoxazole, many anticonvulsants, and anti-HIV agents.
In addition to acute eruptions, a variety of skin diseases can be induced or exacerbated by prolonged use of drugs (e.g., pruritus, pigmentation, nail or hair disorders, psoriasis, bullous pemphigoid, photosensitivity, and even cutaneous neoplasms). These drug reactions are not frequent, but neither their incidence nor their impact on public health has been evaluated.
In a series of 48,005 inpatients over a 20-year period, morbilliform rash (91%) and urticaria (6%) were the most frequent skin reactions. Severe reactions are actually too rare to be detected in such cohorts. Although rare, severe cutaneous reactions to drugs have an important impact on health because of significant sequelae, including mortality. Adverse drug rashes are responsible for hospitalization, increase the duration of hospital stay, and are life threatening. Some populations are at increased risk of drug reactions, including patients with collagen vascular diseases, bone marrow graft recipients, and those with acute Epstein-Barr virus infection. The pathophysiology underlying this association is unknown, but may be related to immunocompromise or immune dysregulation. Risk of drug allergy, including severe hypersensitivity reactions, is increased with HIV infection; individuals with advanced HIV disease (e.g., CD4 T lymphocyte count <200 cells/μL) have a forty- to fiftyfold increased risk of adverse reactions to sulfamethoxazole (Chap. 226).
PATHOGENESIS OF DRUG REACTIONS
Adverse cutaneous responses to drugs can arise as a result of immunologic or nonimmunologic mechanisms.
NONIMMUNOLOGIC DRUG REACTIONS
Examples of responses that arise from nonimmunologic mechanisms are pigmentary changes related to dermal accumulation of medications or their metabolites; alteration of hair follicles by antimetabolites and signaling inhibitors; and lipodystrophy associated with metabolic effects of anti-HIV medications. These side effects are mostly toxic, predictable, and sometimes can be avoided in part by simple preventive measures.
IMMUNOLOGIC DRUG REACTIONS
Evidence suggests an immunologic basis for most acute drug eruptions. Drug reactions may result from immediate release of preformed mediators (e.g., urticaria, anaphylaxis), antibody-mediated reactions, immune complex deposition, and antigen-specific responses. Drug-specific T cell clones can be derived from the blood or from skin lesions of patients with a variety of drug allergies, strongly suggesting that these T cells play a role in drug allergy in an antigen-specific manner. Specific clones were obtained with penicillin G, amoxicillin, cephalosporins, sulfamethoxazole, phenobarbital, carbamazepine, and lamotrigine (medications that are frequently a cause of drug eruptions). Both CD4 and CD8 clones have been obtained; however, their specific roles in the manifestations of allergy have not been elucidated. Drug presentation to T cells was major histocompatibility complex (MHC)-restricted and may involve drug-peptide complex recognition by specific T cell receptors (TCRs).
Once a drug has induced an immune response, the final phenotype of the reaction probably depends on the nature of effectors: cytotoxic (CD8+) T cells in blistering and certain hypersensitivity reactions, chemokines for reactions mediated by neutrophils or eosinophils, and collaboration with B cells for production of specific antibodies for urticarial reaction. Immunologic reactions have recently been classified into further subtypes that provide a useful framework for designating adverse drug reactions based on involvement of specific immune pathways (Table 74-1).
|
CLASSIFICATION OF ADVERSE DRUG REACTIONS BASED ON IMMUNE PATHWAY |
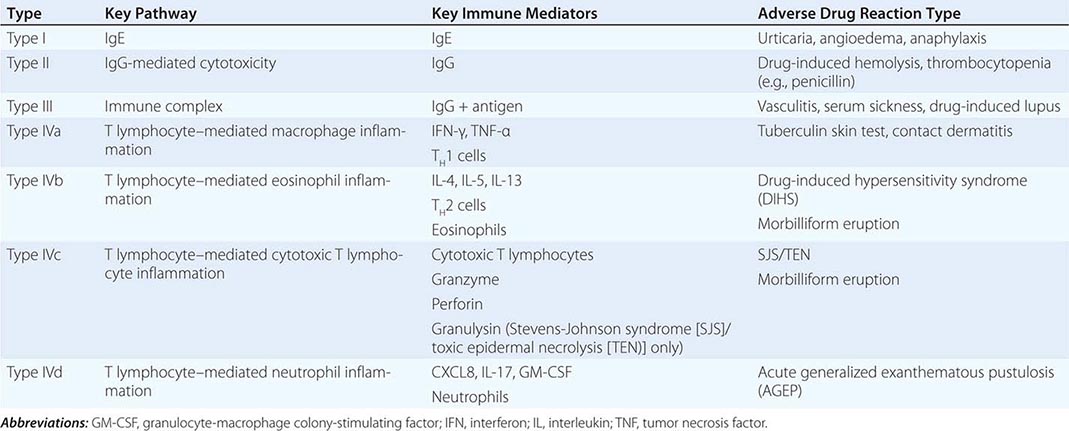
Immediate Reactions Immediate reactions depend on the release of mediators of inflammation by tissue mast cells or circulating basophils. These mediators include histamine, leukotrienes, prostaglandins, bradykinins, platelet-activating factor, enzymes, and proteoglycans. Drugs can trigger mediator release either directly (“anaphylactoid” reaction) or through IgE-specific antibodies. These reactions usually manifest in the skin and gastrointestinal, respiratory, and cardiovascular systems (Chap. 376). Primary symptoms and signs include pruritus, urticaria, nausea, vomiting, abdominal cramps, bronchospasm, laryngeal edema, and, occasionally, anaphylactic shock with hypotension and death. They occur within minutes of drug exposure. Nonsteroidal anti-inflammatory drugs (NSAIDs), including aspirin, and radiocontrast media are frequent causes of direct mast cell degranulation or anaphylactoid reactions, which can occur on first exposure. Penicillins and muscle relaxants used in general anesthesia are the most frequent causes of IgE-dependent reactions to drugs, which require prior sensitization. Release of mediators is triggered when polyvalent drug protein conjugates cross-link IgE molecules fixed to sensitized cells. Certain routes of administration favor different clinical patterns (e.g., gastrointestinal effects from oral route, circulatory effects from intravenous route).
Immune Complex–Dependent Reactions Serum sickness is produced by tissue deposition of circulating immune complexes with consumption of complement. It is characterized by fever, arthritis, nephritis, neuritis, edema, and a urticarial, papular, or purpuric rash (Chap. 385). First described following administration of nonhuman sera, it currently occurs in the setting of monoclonal antibodies and other similar medications. In classic serum sickness, symptoms develop 6 days or more after exposure to a drug, the latent period representing the time needed to synthesize antibody. Cutaneous or systemic vasculitis, a relatively rare complication of drugs, may also be a result of immune complex deposition (Chap. 385). Cephalosporin and other medications, including monoclonal antibodies such as infliximab, rituximab, and omalizumab, may be associated with clinically similar “serum sickness–like” reactions. The mechanism of this reaction is unknown but is unrelated to complement activation and immune complex formation.
Delayed Hypersensitivity While not completely understood, delayed hypersensitivity directed by drug-specific T cells is an important mechanism underlying the most common drug eruptions, i.e., morbilliform eruptions, and also rare and severe forms such as drug-induced hypersensitivity syndrome (DIHS) (also known as drug rash with eosinophilia and systemic symptoms [DRESS]), acute generalized exanthematous pustulosis (AGEP), Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis (TEN) (Table 74-1). Drug-specific T cells have been detected in these types of drug eruptions. For example, drug-specific cytotoxic T cells have been detected in the skin lesions of fixed drug eruptions and of TEN. In TEN, skin lesions contain T lymphocytes reactive to autologous lymphocytes and keratinocytes in a drug-specific, HLA-restricted, and perforin/granzyme-mediated pathway.
The mechanism(s) by which medications result in T cell activation is unknown. Two hypotheses prevail: first, that the antigens driving these reactions may be the native drug itself or components of the drug covalently complexed with endogenous proteins, presented in association with HLA molecules to T cells through the classical antigen presentation pathway, or alternatively, through direct interaction of the drug/metabolite with the T cell receptor or peptide-loaded HLA (e.g., the pharmacologic interaction of drugs with immune receptors, or p-i hypothesis). Recent x-ray crystallography data characterizing binding between specific HLA molecules to particular drugs known to cause hypersensitivity reactions demonstrates unique alterations to the MHC peptide-binding groove, suggesting a molecular basis for T cell activation and the development of hypersensitivity reactions.
GENETIC FACTORS AND CUTANEOUS DRUG REACTIONS
![]() Genetic determinants may predispose individuals to severe drug reactions by affecting either drug metabolism or immune responses to drugs. Polymorphisms in cytochrome P450 enzymes, drug acetylation, methylation (such as thiopurine methyltransferase activity and azathioprine), and other forms of metabolism (such as glucose-6-phosphate dehydrogenase) may increase susceptibility to drug toxicity or underdosing, highlighting a role for differential pharmacokinetic or pharmacodynamic effects.
Genetic determinants may predispose individuals to severe drug reactions by affecting either drug metabolism or immune responses to drugs. Polymorphisms in cytochrome P450 enzymes, drug acetylation, methylation (such as thiopurine methyltransferase activity and azathioprine), and other forms of metabolism (such as glucose-6-phosphate dehydrogenase) may increase susceptibility to drug toxicity or underdosing, highlighting a role for differential pharmacokinetic or pharmacodynamic effects.
Associations between drug hypersensitivities and HLA haplotypes also suggest a key role for immune mechanisms. Hypersensitivity to the anti-HIV medication abacavir is strongly associated with HLA-B*57:01 (Chap. 226). In Taiwan, within a homogeneous Han Chinese population, a 100% association was observed between SJS/TEN (but not DIHS) related to carbamazepine and HLA-B*15:02. In the same population, another 100% association was found between SJS, TEN, or DIHS related to allopurinol and HLA-B*58:01. These associations are drug and phenotype specific; that is, HLA-specific T cell stimulation by medications leads to distinct reactions and may explain why the reaction patterns are so clinically diverse. However, the strong associations found in Taiwan have not been observed in other countries with more heterogeneous populations.
GLOBAL CONSIDERATIONS
 Recognition of the HLA associations with drug hypersensitivity has been acknowledged by recommendations to screen high-risk populations. Genetic screening for HLA-B*57:01 to prevent abacavir hypersensitivity, which carries a 100% negative predictive value when patch test confirmed and 55% positive predictive value generalizable across races, is becoming the clinical standard of care worldwide (number needed to treat = 13). The U.S. Food and Drug Administration recently mandated new labeling of carbamazepine recommending HLA-B*15:02 screening of Asian individuals prior to receiving a new prescription of the medication. The American College of Rheumatology has recommended HLA-B*58:01 screening of Han Chinese patients prescribed allopurinol. To date, screening for a single HLA (but not multiple HLA haplotypes) in specific populations has been determined to be cost-effective.
Recognition of the HLA associations with drug hypersensitivity has been acknowledged by recommendations to screen high-risk populations. Genetic screening for HLA-B*57:01 to prevent abacavir hypersensitivity, which carries a 100% negative predictive value when patch test confirmed and 55% positive predictive value generalizable across races, is becoming the clinical standard of care worldwide (number needed to treat = 13). The U.S. Food and Drug Administration recently mandated new labeling of carbamazepine recommending HLA-B*15:02 screening of Asian individuals prior to receiving a new prescription of the medication. The American College of Rheumatology has recommended HLA-B*58:01 screening of Han Chinese patients prescribed allopurinol. To date, screening for a single HLA (but not multiple HLA haplotypes) in specific populations has been determined to be cost-effective.
Several investigators have proposed that specific HLA haplotypes associated with drug hypersensitivity indeed play a pathogenic role; stimulation of carbamazepine-specific cytotoxic T lymphocytes (CTL) in the context of HLA-B*15:02 results in production of a putative mediator of keratinocyte necrosis in TEN. Other studies have identified CTLs reactive to carbamazepine that use highly restricted V-alpha and V-beta TCR repertoires in patients with carbamazepine hypersensitivity that are not found in carbamazepine-tolerant individuals. Although not yet clinically available, some investigators have suggested combined genetic testing for specific HLA haplotypes and functional screening for TCR repertoire to best identify patients at risk.
CLINICAL PRESENTATION OF CUTANEOUS DRUG REACTIONS
NONIMMUNE CUTANEOUS REACTIONS
Exacerbation or Induction of Dermatologic Diseases A variety of drugs can exacerbate preexisting diseases or sometimes induce a disease that may or may not disappear after withdrawal of the inducing medication. For example, NSAIDs, lithium, beta blockers, tumor necrosis factor (TNF) α cytokine antagonists, interferon (IFN) α, and angiotensin-converting enzyme (ACE) inhibitors can exacerbate plaque psoriasis, whereas antimalarials and withdrawal of systemic glucocorticoids can worsen pustular psoriasis. The situation of TNF-α inhibitors is unusual, as this class of medications is used to treat psoriasis; however, in other cases, they may induce psoriasis (especially palmar-plantar) in patients being treated for other conditions. Acne may be induced by glucocorticoids, androgens, lithium, and antidepressants. Follicular papular or pustular eruptions of the face and trunk, sometimes mimicking acne, frequently occur with epidermal growth factor (EGF) receptor antagonists. In the case of EGF-receptor antagonists, the severity of the eruption correlates with a better anticancer effect. It may be secondarily impetiginized and often spares areas of prior or active radiation. Tetracycline antibiotics, topical corticosteroids, and topical anti-acne treatments (such as benzoyl peroxide and clindamycin lotion) are helpful.
Several medications induce or exacerbate autoimmune disease. Interleukin (IL) 2, IFN-α, and anti-TNF-α are associated with new-onset systemic lupus erythematosus (SLE). Drug-induced lupus is classically marked by antinuclear and antihistone antibodies and, in some cases, anti-double-stranded DNA (D-penicillamine, anti-TNF-α) or p-ANCA (minocycline) antibodies. Minocycline and thiazide diuretics may exacerbate subacute SLE; pemphigus can be induced by D-penicillamine and ACE inhibitors. Furosemide is associated with drug-induced bullous pemphigoid. Vancomycin is associated with linear IgA bullous dermatitis, a transient blistering disorder.
Other medications may cause highly selective cutaneous reactions. Gadolinium contrast has been associated with nephrogenic systemic fibrosis, a condition of sclerosing skin with rare internal organ involvement; advanced renal compromise may be an important risk factor. Granulocyte colony-stimulating factor may induce various neutrophilic dermatoses, including Sweet syndrome and pyoderma gangrenosum. Both systemic and topical glucocorticoids cause a variety of atrophic skin changes, including atrophy and striae, and, in sufficiently high doses, can impede wound healing.
The hypothesis that a drug may be responsible should always be considered, especially in cases with atypical clinical presentation. Resolution of the cutaneous reaction may be delayed upon discontinuation of the medication (e.g., lichenoid drug eruptions may take years to resolve).
Photosensitivity Eruptions Photosensitivity eruptions are usually most marked in sun-exposed areas but may extend to sun-protected areas. The mechanism is almost always phototoxicity. Phototoxic reactions resemble sunburn and can occur with first exposure to a drug. Blistering may occur in drug-related pseudoporphyria, most commonly with NSAIDs (Fig. 74-1). The severity of the reactions depends on the tissue level of the drug, its efficiency as a photosensitizer, and the extent of exposure to the activating wavelengths of ultraviolet (UV) light (Chap. 75).
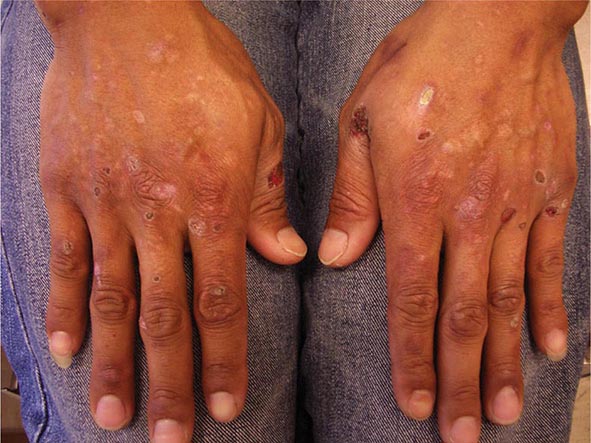
FIGURE 74-1 Pseudoporphyria due to nonsteroidal anti-inflammatory drugs.
Common orally administered photosensitizing drugs include fluoroquinolones and tetracycline antibiotics. Other drugs less frequently encountered are chlorpromazine, thiazides, and NSAIDs. Voriconazole may result in severe photosensitivity, accelerated photo-induced aging, and cutaneous carcinogenesis in organ transplant recipients.
Because UV-A and visible light, which trigger these reactions, are not easily absorbed by nonopaque sunscreens and are transmitted through window glass, photosensitivity reactions may be difficult to block. Photosensitivity reactions abate with removal of either the drug or UV radiation, use of sunscreens that block UV-A light, and treating the reaction as one would a sunburn. Rarely, individuals develop persistent reactivity to light, necessitating long-term avoidance of sun exposure.
Pigmentation Changes Drugs, either systemic or topical, may cause a variety of pigmentary changes in the skin. Oral contraceptives may induce melasma. Long-term minocycline, pefloxacin, and amiodarone may cause blue-gray pigmentation. Phenothiazine, gold, and bismuth result in gray-brown pigmentation of sun-exposed areas. Numerous cancer chemotherapeutic agents may be associated with characteristic patterns of pigmentation (e.g., bleomycin, busulfan, daunorubicin, cyclophosphamide, hydroxyurea, and methotrexate). Clofazimine causes a drug-induced lipofuscinosis with characteristic red-brown coloration. Hyperpigmentation of the face, mucous membranes, and pretibial and subungual areas occurs with antimalarials. Quinacrine causes generalized, cutaneous yellow discoloration. Pigmentation changes may also occur in mucous membranes (busulfan, bismuth), conjunctiva (chlorpromazine, thioridazine, imipramine, clomipramine), nails (zidovudine, doxorubicin, cyclophosphamide, bleomycin, fluorouracil, hydroxyurea), hair, and teeth (tetracyclines).
Warfarin Necrosis of Skin This rare reaction (0.01–0.1%) usually occurs between the third and tenth days of therapy with warfarin, usually in women. Common sites are breasts, thighs, and buttocks (Fig. 74-2). Lesions are sharply demarcated, indurated, and erythematous or purpuric and may progress to form large, hemorrhagic bullae with eventual necrosis and slow-healing eschar formation. These lesions can be life threatening.
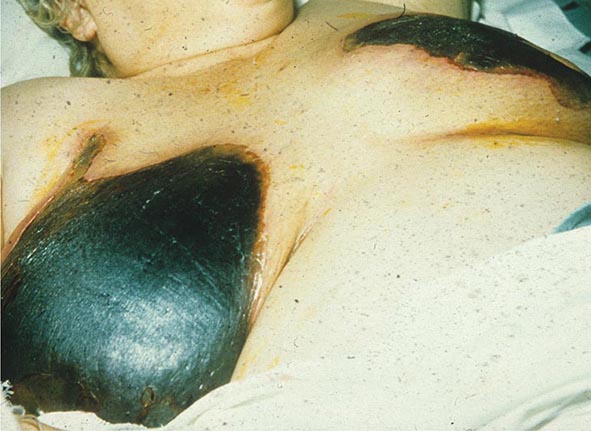
FIGURE 74-2 Warfarin necrosis.
Development of the syndrome is unrelated to drug dose, and the course is not altered by discontinuation of the drug after onset of the eruption. Warfarin anticoagulation in heterozygous protein C deficiency causes a precipitous fall in circulating levels of protein C, permitting hypercoagulability and thrombosis in the cutaneous microvasculature, with consequent areas of necrosis. Heparin-induced necrosis may have clinically similar features but is probably due to heparin-induced platelet aggregation with subsequent occlusion of blood vessels; it can affect areas adjacent to the injection site or more distant sites if infused.
Warfarin-induced cutaneous necrosis is treated with vitamin K, heparin, surgical debridement, and intensive wound care. Treatment with protein C concentrates may also be helpful. Newer agents such as dabigatran etexilate may avoid warfarin necrosis in high-risk patients.
Drug-Induced Hair Disorders • DRUG-INDUCED HAIR LOSS Medications may affect hair follicles at two different phases of their growth cycle: anagen (growth) or telogen (resting). Anagen effluvium occurs within days of drug administration, especially with antimetabolite or other chemotherapeutic drugs. In contrast, in telogen effluvium, the delay is 2 to 4 months following initiation of a new medication. Both present as diffuse nonscarring alopecia most often reversible after discontinuation of the responsible agent. The prevalence and severity of alopecia depend on the drug as well as on an individual’s predisposition.
A considerable number of drugs have been reported to induce hair loss. These include antineoplastic agents (alkylating agents, bleomycin, vinca alkaloids, platinum compounds), anticonvulsants (carbamazepine, valproate), antihypertensive drugs (beta blockers), antidepressants, antithyroid drugs, IFNs (especially IFN-α), oral contraceptives, and cholesterol-lowering agents.
DRUG-INDUCED HAIR GROWTH Medications may also cause hair growth. Hirsutism is an excessive growth of terminal hair with masculine hair growth pattern in a female, most often on the face and trunk, due to androgenic stimulation of hormone-sensitive hair follicles (anabolic steroids, oral contraceptives, testosterone, corticotropin). Hypertrichosis is a distinct pattern of hair growth, not in a masculine pattern, typically located on the forehead and temporal regions of the face. Drugs responsible for hypertrichosis include anti-inflammatory drugs, glucocorticoids, vasodilators (diazoxide, minoxidil), diuretics (acetazolamide), anticonvulsants (phenytoin), immunosuppressive agents (cyclosporine A), psoralens, and zidovudine.
Changes in hair color or structure are uncommon adverse effects from medications. Hair discoloration may occur with chloroquine, IFN-α, chemotherapeutic agents, and tyrosine kinase inhibitors. Changes in hair structure have been observed in patients given epidermal growth factor receptor (EGFR) inhibitors, tyrosine kinase inhibitors (Fig. 74-3), and acitretin.
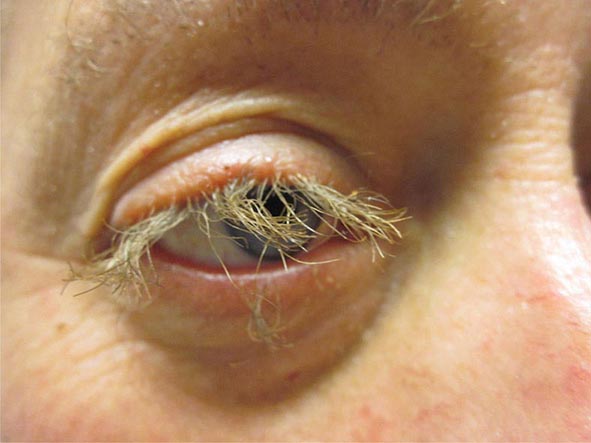
FIGURE 74-3 Dysmorphic eyelashes in association with erlotinib.
Drug-Induced Nail Disorders Drug-related nail disorders usually involve all 20 nails and need months to resolve after withdrawal of the offending agent. The pathogenesis is most often toxic. Drug-induced nail changes include Beau’s line (transverse depression of the nail plate), onycholysis (detachment of the distal part of the nail plate), onychomadesis (detachment of the proximal part of the nail plate), pigmentation, and paronychia (inflammation of periungual skin).
ONYCHOLYSIS Onycholysis occurs with tetracyclines, fluoroquinolones, phenothiazines, and psoralens, as well as in persons taking NSAIDs, captopril, retinoids, sodium valproate, and many chemotherapeutic agents such as anthracyclines or taxanes including paclitaxel and docetaxel. The risk of onycholysis in patients receiving cytotoxic drugs, tetracyclines, quinolones, phenothiazines, and psoralens can be increased by exposure to sunlight.
ONYCHOMADESIS Onychomadesis is caused by temporary arrest of nail matrix mitotic activity. Common drugs reported to induce onychomadesis include carbamazepine, lithium, retinoids, and chemotherapeutic agents such as cyclophosphamide and vincristine.
PARONYCHIA Paronychia and multiple pyogenic granuloma (Fig. 74-4) with progressive and painful periungual abscess of fingers and toes are side effects of systemic retinoids, lamivudine, indinavir, and anti-EGFR monoclonal antibodies (cetuximab, gefitinib).
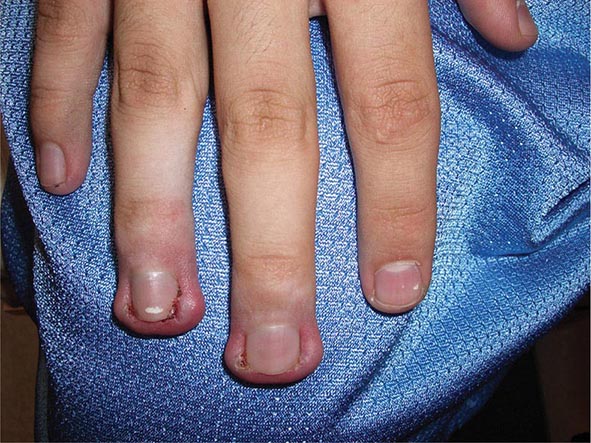
FIGURE 74-4 Pyogenic granuloma in association with isotretinoin.
NAIL DISCOLORATION Some drugs—including anthracyclines, taxanes, fluorouracil, psoralens, and zidovudine—may induce nail bed hyperpigmentation through melanocyte stimulation. It appears to be reversible and dose-dependent.
Toxic Erythema of Chemotherapy and Other Chemotherapy Reactions Because many agents used in cancer chemotherapy inhibit cell division, rapidly proliferating elements of the skin, including hair, mucous membranes, and appendages, are sensitive to their effects. A broad spectrum of chemotherapy-related skin toxicities have been reported, including neutrophilic eccrine hidradenitis, sterile cellulitis, exfoliative dermatitis, and flexural erythema; although previously designated as distinct skin eruptions, recent nomenclature classifies these under the unifying diagnosis of toxic erythema of chemotherapy (TEC). Acral erythema, marked by dysesthesia and an erythematous, edematous eruption of the palms and soles, is caused by cytarabine, doxorubicin, methotrexate, hydroxyurea, and fluorouracil and may be alleviated by pyridoxine supplementation.
The recent introduction of many new monoclonal antibody and small molecular signaling inhibitors for the treatment of cancer has been accompanied by numerous reports of skin and hair toxicity; only the most common of these are mentioned here. Cetuximab and other EGFR antagonists induce follicular eruptions and nail toxicity after a mean interval of 10 days in a majority of patients. Xerosis, eczematous eruptions, acneiform eruptions, and pruritus are common. Erlotinib is associated with marked hair textural changes (Fig. 74-3). Sorafenib, a tyrosine kinase inhibitor, may result in follicular eruptions and bullous palmoplantar eruptions with dysesthesia (Fig. 74-5). BRAF inhibitors are associated with photosensitivity, dyskeratotic (Grover’s-like) rash, hyperkeratotic benign cutaneous neoplasms, and keratoacanthoma-like squamous cell carcinomas. Rash, pruritus, and vitiliginous depigmentation have been reported in association with ipilimumab (anti-CTLA4) treatment.
FIGURE 74-5 Sorafenib-associated hand-foot syndrome.
IMMUNE CUTANEOUS REACTIONS: COMMON
Maculopapular Eruptions Morbilliform or maculopapular eruptions (Fig. 74-6) are the most common of all drug-induced reactions, often start on the trunk or intertriginous areas, and consist of erythematous macules and papules that are symmetric and confluent. Involvement of mucous membranes is unusual; the eruption may be associated with moderate to severe pruritus and fever. Diagnosis is rarely assisted by laboratory testing. Skin biopsy often shows nonspecific inflammatory changes. A viral exanthem is the principal differential diagnostic consideration, especially in children, and graft-versus-host disease is also a consideration in the proper clinical setting. Absence of enanthems; absence of ear, nose, throat, and upper respiratory tract symptoms; and polymorphism of the skin lesions support a drug rather than a viral eruption. Certain medications carry very high rates of morbilliform eruption, including nevirapine and lamotrigine, even in the absence of hypersensitivity reactions. Lamotrigine morbilliform rash is associated with higher starting doses, rapid dose escalation, concomitant use of valproate (which increases lamotrigine levels and half-life) and use in children, especially with seizure disorder.
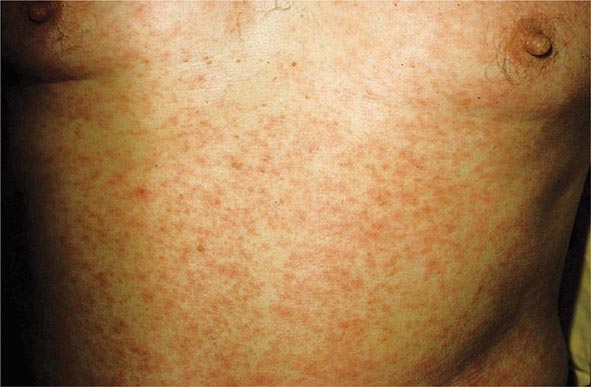
FIGURE 74-6 Morbilliform drug eruption.
Maculopapular reactions usually develop within 1 week of initiation of therapy and last less than 2 weeks. Occasionally, these eruptions resolve despite continued use of the responsible drug. Because the eruption may also worsen, the suspect drug should be discontinued unless it is essential; it is important to note that the rash may continue to progress for a few days to up to one week following medication discontinuation. Oral antihistamines and emollients may help relieve pruritus. Short courses of potent topical glucocorticoids can reduce inflammation and symptoms. Systemic glucocorticoid treatment is rarely indicated.
Pruritus Pruritus is associated with almost all drug eruptions and, in some cases, may represent the only symptom of the adverse cutaneous reaction. It is usually alleviated by antihistamines such as hydroxyzine or diphenhydramine. Pruritus stemming from specific medications may require distinct treatment; opiate-related pruritus may require selective opiate antagonists to gain relief. Pruritus is a common complication of antimalarial therapy, occurring in up to 50% of black patients receiving chloroquine, and may be severe enough to lead to discontinuation of treatment. It is much rarer in Caucasians taking chloroquine. Intense pruritus, sometimes accompanied by an eczematous rash, may occur in 20% of patients receiving IFN and ribavirin for hepatitis C; addition of the protease inhibitor telaprevir may increase this occurrence to 50% of treated patients.
Urticaria/Angioedema/Anaphylaxis Urticaria, the second most frequent type of cutaneous reaction to drugs, is characterized by pruritic, red wheals of varying size rarely lasting more than 24 h. It has been observed in association with nearly all drugs, most frequently ACE inhibitors, aspirin, NSAIDs, penicillin, and blood products. However, medications account for no more than 10–20% of acute urticaria cases. Deep edema within dermal and subcutaneous tissues is known as angioedema and may involve respiratory and gastrointestinal mucous membranes as well. Urticaria and angioedema may be part of a life-threatening anaphylactic reaction.
Drug-induced urticaria may be caused by three mechanisms: an IgE-dependent mechanism, circulating immune complexes (serum sickness), and nonimmunologic activation of effector pathways. IgE-dependent urticarial reactions usually occur within 36 h of drug exposure but can occur within minutes. Immune complex–induced urticaria associated with serum sickness–like reactions usually occurs 6–12 days after first exposure. In this syndrome, the urticarial eruption (typically polycyclic plaques) may be accompanied by fever, hematuria, arthralgias, hepatic dysfunction, and neurologic symptoms. Certain drugs, such as NSAIDs, ACE inhibitors, angiotensin II antagonists, radiographic dye, and opiates, may induce urticarial reactions, angioedema, and anaphylaxis in the absence of drug-specific antibody through direct mast-cell degranulation.
Radiocontrast agents are a common cause of urticaria and, in rare cases, can cause anaphylaxis. High-osmolality radiocontrast media were about five times more likely to induce urticaria (1%) or anaphylaxis than were newer low-osmolality media. About one-third of those with mild reactions to previous exposure react on reexposure. Pretreatment with prednisone and diphenhydramine reduces reaction rates. Persons with a reaction to a high-osmolality contrast media may be given low-osmolality media if later contrast studies are required.
The treatment of urticaria or angioedema depends on the severity of the reaction. In severe cases with respiratory or cardiovascular compromise, epinephrine is the mainstay of therapy, but its effect is reduced in patients using beta blockers. Treatment with intravenous systemic glucocorticoids is helpful. For patients with urticaria without symptoms of angioedema or anaphylaxis, drug withdrawal and oral antihistamines are usually sufficient. Future drug avoidance is recommended; rechallenge, especially in individuals with severe reactions, should only occur in an intensive care setting.
Anaphylactoid Reactions Vancomycin is associated with red man syndrome, a histamine-related anaphylactoid reaction characterized by flushing, diffuse maculopapular eruption, and hypotension. In rare cases, cardiac arrest may be associated with rapid IV infusion of the medication.
Irritant/Allergic Contact Dermatitis Patients using topical medications may develop an irritant or allergic contact dermatitis to the medication itself or to a preservative or other component of the formulation. Reactions to chlorhexidine, neomycin sulfate, and polymyxin B are common. Allergic contact dermatitis to topical glucocorticoids may also occur and is paradoxically partially masked by the anti-inflammatory nature of the medication itself; typically this allergy is selective for one of the four classes of glucocorticoid types, as subdivided by allergenic properties. Patch testing can be useful to determine whether a patient is steroid allergic. Desoximetasone is rarely allergenic.
Fixed Drug Eruptions These less common reactions are characterized by one or more sharply demarcated, dull red to brown lesions, sometimes with central bulla (Fig. 74-7). Hyperpigmentation often results after resolution of the acute inflammation. With rechallenge, the lesion recurs in the same (e.g., fixed) location. Lesions often involve the lips, hands, legs, face, genitalia, and oral mucosa and cause a burning sensation. Most patients have multiple lesions. Fixed drug eruptions have been associated with pseudoephedrine (frequently a nonpigmented reaction), phenolphthalein (in laxatives), sulfonamides, tetracyclines, NSAIDs, and barbiturates.
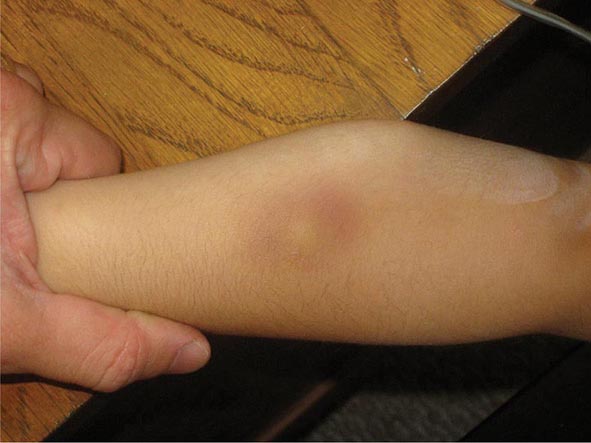
FIGURE 74-7 Fixed drug eruption.
IMMUNE CUTANEOUS REACTIONS: RARE AND SEVERE
Vasculitis Cutaneous small-vessel vasculitis often presents as palpable purpuric lesions that may be generalized or limited to the lower extremities or other dependent areas (Chap. 385). Pustular lesions and hemorrhagic blisters also occur. Vasculitis may involve other organs, including the liver, kidney, brain, and joints. Drugs are implicated as a cause of 10–15% of all cases of small-vessel vasculitides. Infection, malignancy, and collagen vascular disease are responsible for the majority of non-drug-related cases.
Propylthiouracil induces a cutaneous vasculitis that is accompanied by leukopenia and splenomegaly. Direct immunofluorescent changes in these lesions suggest immune-complex deposition. Common drugs implicated in vasculitis include allopurinol, thiazides, sulfonamides, antimicrobials, and NSAIDs. The presence of eosinophils in the perivascular infiltrate of skin biopsy suggests a drug etiology.
Pustular Eruptions AGEP is a rare reaction pattern (3–5 cases per million per year) that is often associated with exposure to drugs (Fig. 74-8). Usually beginning on the face or intertriginous areas, small nonfollicular pustules overlying erythematous and edematous skin may coalesce and lead to superficial erosion. Differentiating this eruption from TEN in its initial stages may be difficult. A skin biopsy is important and shows neutrophil collections and sparse necrotic keratinocytes in the upper part of the epidermis instead of the full-thickness epidermal necrosis that characterizes TEN. Fever and leukocytosis are common, and eosinophilia occurs in one-third of cases. Acute pustular psoriasis is the principal differential diagnostic consideration. DIHS with pustular features must also be clinically considered, although the timing for the onset of DIHS is distinct (much later onset). AGEP often begins within a few days of initiating drug treatment, most notably antibiotics, but may occur as late as 7–14 days after initiation of treatment. A broad range of drug classes (anticonvulsants, mercury, radiocontrast dye) and infections (viral, Mycoplasma) are also associated with AGEP. Patch testing with the responsible drug results in a localized pustular eruption.
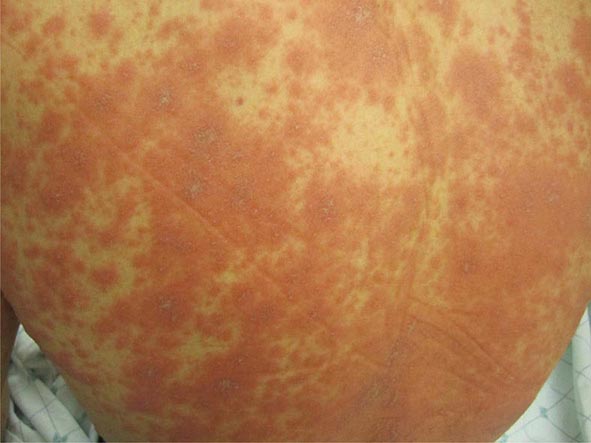
FIGURE 74-8 Acute generalized exanthematous pustulosis.
Drug-induced Hypersensitivity Syndrome Drug-induced hypersensitivity syndrome (DIHS) is a multiorgan drug reaction previously known as DRESS (drug reaction with eosinophilia and systemic symptoms); since eosinophilia is not always present, the term DIHS is now preferred. Allopurinol is the most common cause. Although less frequently prescribed, abacavir has been reported to cause DIHS with an incidence as high as 4–8%. It presents as a widespread erythematous eruption that may become purpuric, pustular, or lichenoid and is accompanied by many of the following features: fever, facial edema, lymphadenopathy, leukocytosis (often with atypical lymphocytes and eosinophilia), hepatitis, myositis (including myocarditis), and sometimes nephritis (with proteinuria) or pneumonitis. Distinct patterns of timing of onset and organ involvement may exist; e.g., allopurinol classically induces DIHS with renal involvement. Cardiac and lung involvement is more common with minocycline; gastrointestinal involvement is almost exclusively seen with abacavir, and some medications typically lack eosinophilia (abacavir, dapsone, lamotrigine). The cutaneous reaction usually begins 2–8 weeks after the drug is started and lasts longer than mild eruptions after drug cessation. Signs and symptoms may persist for several weeks, especially those associated with hepatitis. The eruption recurs with rechallenge, and cross-reactions among aromatic anticonvulsants, including phenytoin, carbamazepine, and barbiturates, are frequent. Other drugs causing this syndrome include sulfonamides and other antibiotics. Hypersensitivity to reactive drug metabolites, hydroxylamine for sulfamethoxazole, and arene oxide for aromatic anticonvulsants may be involved in the pathogenesis of DIHS. Reactivation of herpes viruses, especially herpesvirus 6 and Epstein-Barr virus (EBV), has been frequently reported in this syndrome, although the causal role of viral infection has been debated. Recent research suggests that inciting drugs may reactivate quiescent herpes viruses, resulting in expansion of viral-specific CD8+ T lymphocytes with subsequent end-organ damage. Viral reactivation may be associated with a worse clinical prognosis. Mortality rates as high as 10% have been reported; mortality is highest in association with hepatitis. Systemic glucocorticoids (prednisone, 1–2 mg/kg daily) should be started with slow taper over 8–12 weeks. A steroid-sparing agent, such as mycophenolate mofetil, may be indicated in cases of rapid recurrence upon steroid taper. In all cases, rapid withdrawal of the suspected drug is required. Given the severe long-term complications of myocarditis, patients should undergo cardiac evaluation if heart involvement is suspected by hypotension or arrhythmia. Patients should be closely monitored for resolution of organ dysfunction and for development of late-onset autoimmune thyroiditis (up to 6 months).
Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis SJS and TEN are characterized by blisters and mucosal/epidermal detachment resulting from full-thickness epidermal necrosis in the absence of substantial dermal inflammation (Fig. 74-9). The term Stevens-Johnson syndrome describes cases with blisters developing on target lesions, dusky or purpuric macules in which mucosal involvement is significant, and total body surface area blistering and eventual detachment in <10% of cases. The term Stevens-Johnson syndrome/toxic epidermal necrolysis overlap is used to describe cases with 10–30% detachment, and TEN is used to describe cases with >30% detachment.
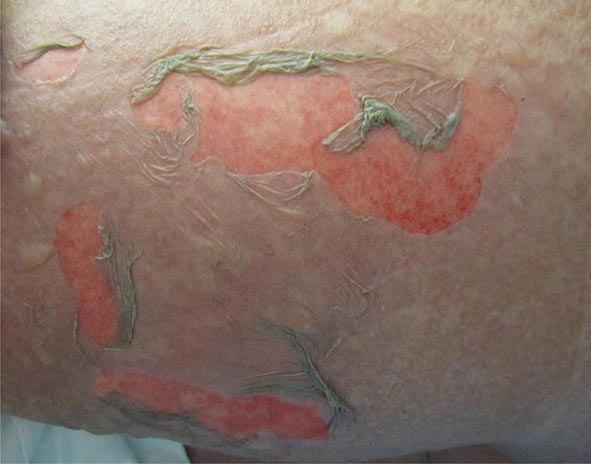
FIGURE 74-9 Toxic epidermal necrolysis. (Photo credit: Lindy Peta Fox, MD, and Jubin Ryu, MD, PhD.)
Other blistering eruptions with mucositis associated with infections may be confused with SJS/TEN. Erythema multiforme (EM) associated with herpes simplex virus is characterized by mucosal involvement and target lesions often more acrally distributed and with limited skin detachment. Mycoplasma infection in children causes a clinically distinct presentation with prominent mucositis and limited blistering lesions; some believe that this clinical entity is the syndrome originally described by Stevens and Johnson.
Patients with SJS, SJS/TEN, or TEN initially present with acute onset of painful skin lesions, fever >39°C (102.2°F), sore throat, and conjunctivitis resulting from mucosal lesions. Intestinal and pulmonary involvement is associated with a poor prognosis, as are a greater extent of epidermal detachment and older age. About 10% and 30% of SJS- and TEN-affected persons die from their disease, respectively. Drugs that most commonly cause SJS or TEN are sulfonamides, nevirapine (1 in 1000 risk of SJS or TEN), allopurinol, lamotrigine, aromatic anticonvulsants, and NSAIDs, specifically oxicam. Frozen-section skin biopsy may aid in rapid diagnosis. At this time, SJS and TEN have no proven effective treatment. The best results come from early diagnosis, immediate discontinuation of any suspected drug, supportive therapy, and paying close attention to ocular complications and infection. Systemic glucocorticoid therapy (prednisone 1–2 mg/kg) may be useful early in the evolution of the disease, but long-term systemic glucocorticoid use has been associated with higher mortality. Cyclosporine may be a possible therapy for SJS/TEN. After initial enthusiasm for the use of intravenous immunoglobulin (IVIG) in the treatment of SJS/TEN, some recent data questions whether IVIG benefits these patients. Randomized studies to more definitively assess the potential benefit of systemic glucocorticoids and IVIG are lacking and difficult to perform but are necessary.
Overlap Hypersensitivity Syndromes An important emerging concept in the clinical approach to severe drug eruptions is the presence of overlap syndromes, most notably DIHS with TEN-like features, DIHS with pustular eruption (AGEP-like), and AGEP with TEN-like features. In several case series of AGEP, 50% of cases had TEN-like or DRESS-like features, and 20% of cases had mucosal involvement resembling SJS/TEN. In one study, up to 20% of all severe drug eruptions had overlap features, suggesting that AGEP, DIHS, and SJS/TEN represent a clinical spectrum with common pathophysiologic mechanisms. Designation of a single diagnosis based on cutaneous and extracutaneous involvement may not always be possible in cases of hypersensitivity.
MANAGEMENT OF A PATIENT WITH A DRUG ERUPTION
There are four main questions to answer regarding an eruption:
1. Is it a drug reaction?
2. Is it a severe eruption or the onset of a form that may become severe?
3. Which drug(s) is (are) suspected, and which drug(s) should be withdrawn?
4. What is recommended for future use of drugs?
EARLY DIAGNOSIS OF SEVERE ERUPTIONS
Rapid recognition of adverse drug reactions that may become serious or life threatening is paramount. Table 74-2 lists clinical and laboratory features that, if present, suggest that the reaction may be serious. Table 74-3 provides key features of the most serious adverse cutaneous reactions. Intensity of symptoms and rapid progression of signs should raise the suspicion of a severe eruption. Any doubt should lead to prompt consultation with a dermatologist and/or referral of the patient to a specialized center.
|
CLINICAL AND LABORATORY FINDINGS ASSOCIATED WITH MORE SERIOUS DRUG-INDUCED CUTANEOUS CLINICAL FINDINGS |
Source: Adapted from JC Roujeau, RS Stern: N Engl J Med 331:1272, 1994.
|
CLINICAL FEATURES OF SEVERE CUTANEOUS DRUG REACTIONS |
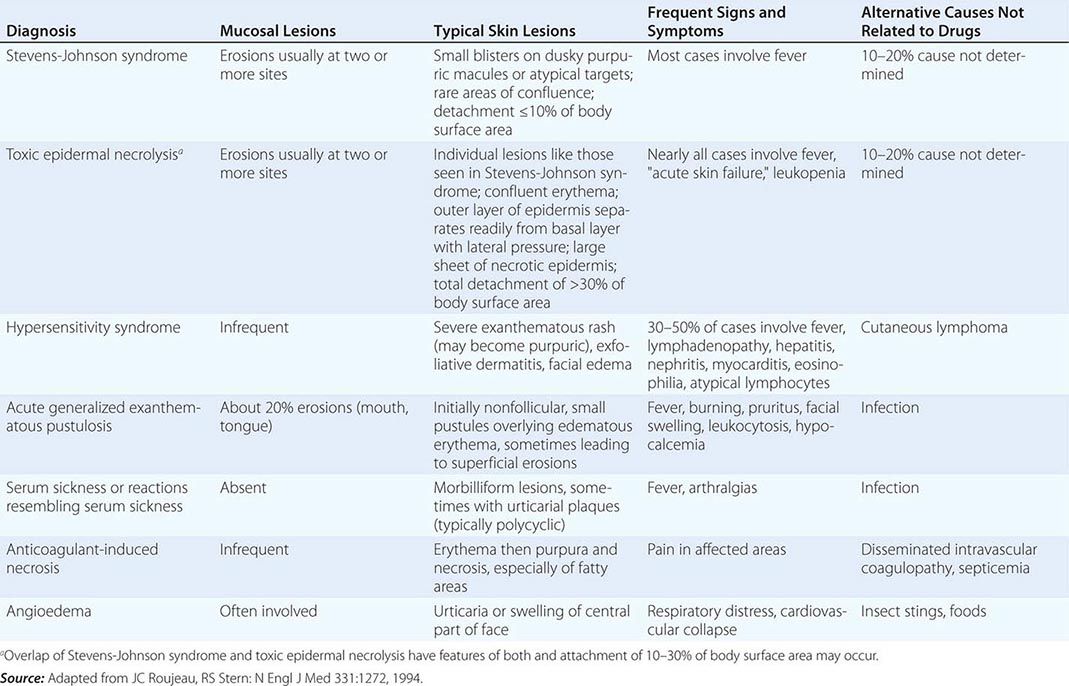
CONFIRMATION OF DRUG REACTION
The probability of drug etiology varies with the pattern of the reaction. Only fixed drug eruptions are always drug-induced. Morbilliform eruptions are usually viral in children and drug-induced in adults. Among severe reactions, drugs account for 10–20% of anaphylaxis and vasculitis and between 70–90% of AGEP, DIHS, SJS, or TEN. Skin biopsy helps in characterizing the reaction but does not indicate drug causality. Blood counts and liver and renal function tests are important for evaluating organ involvement. The association of mild elevation of liver enzymes and high eosinophil count is frequent but not specific for a drug reaction. Blood tests that could identify an alternative cause, antihistone antibody tests (to rule out drug-induced lupus), and serology or polymerase chain reaction for infections may be of great importance to determine a cause.
WHAT DRUG(S) TO SUSPECT AND WITHDRAW
Most cases of drug eruptions occur during the first course of treatment with a new medication. A notable exception is IgE-mediated urticaria and anaphylaxis that need presensitization and develop a few minutes to a few hours after rechallenge. Characteristic times of onset to drug reaction are as follows: 4–14 days for morbilliform eruptions, 2–4 days for AGEP, 5–28 days for SJS/TEN, and 14–48 days for DIHS. A drug chart, compiling information of all current and past medications/supplements and the timing of administration relative to the rash, is a key diagnostic tool to identifying the inciting drug. Medications introduced for the first time in the relevant time frame are prime suspects. Two other important elements to suspect causality at this stage are (1) previous experience with the drug in the population and (2) alternative etiologic candidates.
The decision to continue or discontinue any medication will depend on the severity of the reaction, the severity of the primary disease, the degree of suspicion of causality, and the feasibility of an alternative safer treatment. In any potentially fatal drug reaction, elimination of all possible suspect drugs or unnecessary medications should be attempted. Some rashes may resolve when “treating through” a benign drug-related eruption. The decision to treat through an eruption should, however, remain the exception and withdrawal of every suspect drug the general rule. On the other hand, drugs that are not suspected and are important for the patient (e.g., antihypertensive agents) generally should not be quickly withdrawn. This approach prevents reluctance to future use of these agents.
RECOMMENDATION FOR FUTURE USE OF DRUGS
The aims are (1) to prevent the recurrence of the drug eruption and (2) not to compromise future treatments by contraindicating otherwise useful medications.
Begin with thorough assessment of drug causality. Drug causality is evaluated based on timing of the reaction, evaluation of other possible causes, effect of drug withdrawal or continuation, and knowledge of medications that have been associated with the observed reaction. Combination of these criteria leads to considering the causality as definite, probable, possible, or unlikely. The RegiSCAR group has proposed a useful algorithm called Algorithm of Drug Causality for Epidermal Necrolysis (ALDEN) to determine drug causality in SJS/TEN. A drug with a “definite” or “probable” causality should be contraindicated, a warning card or medical alert tag (e.g., wristband) should be given to the patient, and the drugs should be listed in the patient’s medical chart as an allergy.
A drug with a “possible” causality may be submitted to further investigations depending on the expected need for future treatment.
A drug with “unlikely” causality or that has been continued when the reaction improved or was reintroduced without a reaction can be administered safely.
The usefulness of laboratory tests to determine causality is still debated. Many in vitro immunologic assays have been developed, but the predictive value of these tests has not been validated in any large series of affected patients; these tests exist primarily for research and not clinical purposes.
In some cases, diagnostic rechallenge may be appropriate, even for drugs with high rates of adverse reactions. Desensitization is often successful in HIV-infected patients with morbilliform eruptions to sulfonamides but is not recommended in HIV-infected patients who manifested erythroderma or a bullous reaction in response to their earlier sulfonamide exposure.
In patients with history suggesting immediate IgE-mediated reactions to penicillin, skin-prick testing with penicillins or cephalosporins has proved useful for identifying patients at risk of anaphylactic reactions to these agents. However, skin tests themselves carry a small risk of anaphylaxis. Negative skin tests do not totally rule out IgE-mediated reactivity, but the risk of anaphylaxis in response to penicillin administration in patients with negative skin tests is about 1%. In contrast, two-thirds of patients with a positive skin test experience an allergic response upon rechallenge.
For patients with delayed-type hypersensitivity, the clinical utility of skin tests is more questionable. At least one of a combination of several tests (prick, patch, and intradermal) is positive in 50–70% of patients with a reaction “definitely” attributed to a single medication. This low sensitivity corresponds to the observation that readministration of drugs with negative skin testing resulted in eruptions in 17% of cases.
CROSS-SENSITIVITY
Because of the possibility of cross-sensitivity among chemically related drugs, many physicians recommend avoidance of not only the medication that induced the reaction but also all drugs of the same pharmacologic class.
There are two types of cross-sensitivity. Reactions that depend on a pharmacologic interaction may occur with all drugs that target the same pathway, whether they are structurally similar or not. This is the case with angioedema caused by NSAIDs and ACE inhibitors. In this situation, the risk of recurrence varies from drug to drug in a particular class; however, avoidance of all drugs in the class is usually recommended. Immune recognition of structurally related drugs is the second mechanism by which cross-sensitivity occurs. A classic example is hypersensitivity to aromatic antiepileptics (barbiturates, phenytoin, carbamazepine) with up to 50% reaction to a second drug in patients who reacted to one. For other drugs, in vitro as well as in vivo data have suggested that cross-reactivity existed only between compounds with very similar chemical structures. Sulfamethoxazole-specific lymphocytes may be activated by other antibacterial sulfonamides but not diuretics, antidiabetic drugs, or anti-COX2 NSAIDs with a sulfonamide group. Approximately 10% of patients with penicillin allergies will also develop allergic reactions to cephalosporin class antibiotics.
Recent data suggest that although the risk of a drug eruption to another drug was increased in persons with a prior reaction, “cross-sensitivity” was probably not the explanation. As an example, persons with a history of an allergic-like reaction to penicillin were at higher risk to develop a reaction to antibacterial sulfonamides than to cephalosporins.
These data suggest that the list of drugs to avoid after a drug reaction should be limited to the causative one(s) and to a few very similar medications.
Because of growing evidence that some severe cutaneous reactions to drugs are associated with HLA genes, it is recommended that first-degree family members of patients with severe cutaneous reactions also should avoid these causative medications. This may be most relevant to sulfonamides and antiseizure medications.
Desensitization can be considered in those with a history of reaction to a medication that must be used again. Efficacy of such procedures has been demonstrated in cases of immediate reaction to penicillin and positive skin tests, anaphylactic reactions to platinum chemotherapy, and delayed reactions to sulfonamides in patients with AIDS. Various protocols are available, including oral and parenteral approaches. Oral desensitization appears to have a lower risk of serious anaphylactic reactions. However, desensitization carries the risk of anaphylaxis regardless of how it is performed and should be performed in monitored clinical settings such as an intensive care unit. After desensitization, many patients experience non-life-threatening reactions during therapy with the culprit drug.
REPORTING
Any severe reaction to drugs should be reported to a regulatory agency or to pharmaceutical companies (e.g., MedWatch, http://www.fda.gov/Safety/MedWatch/default.htm). Because severe reactions are too rare to be detected in premarketing clinical trials, spontaneous reports are of critical importance for early detection of unexpected life-threatening events. To be useful, the report should contain enough details to permit ascertainment of severity and drug causality. This enables recognition of similar cases that may be reported from several different sources.
ACKNOWLEDGMENT
We acknowledge the contribution of Dr. Jean-Claude Roujeau to this chapter in the 17th edition.
75 |
Photosensitivity and Other Reactions to Light |
SOLAR RADIATION
Sunlight is the most visible and obvious source of comfort in the environment. The sun provides the beneficial effects of warmth and vitamin D synthesis. However, acute and chronic sun exposure also has pathologic consequences. Few effects of sun exposure beyond those affecting the skin have been identified, but cutaneous exposure to sunlight is the major cause of human skin cancer and can have immunosuppressive effects as well.
The sun’s energy reaching the earth’s surface is limited to components of the ultraviolet (UV) spectrum, the visible spectrum, and portions of the infrared spectrum. The cutoff at the short end of the UV spectrum at ~290 nm is due primarily to stratospheric ozone—formed by highly energetic ionizing radiation—that prevents penetration to the earth’s surface of the shorter, more energetic, potentially more harmful wavelengths of solar radiation. Indeed, concern about destruction of the ozone layer by chlorofluorocarbons released into the atmosphere has led to international agreements to reduce production of those chemicals.
Measurements of solar flux showed a twentyfold regional variation in the amount of energy at 300 nm that reaches the earth’s surface. This variability relates to seasonal effects, the path that sunlight traverses through ozone and air, the altitude (a 4% increase for each 300 m of elevation), the latitude (increasing intensity with decreasing latitude), and the amount of cloud cover, fog, and pollution.
The major components of the photobiologic action spectrum that are capable of affecting human skin include the UV and visible wavelengths between 290 and 700 nm. In addition, the wavelengths beyond 700 nm in the infrared spectrum primarily emit heat and in certain circumstances may exacerbate the pathologic effects of energy in the UV and visible spectra.
The UV spectrum reaching the earth represents <10% of total incident solar energy and is arbitrarily divided into two major segments, UV-B and UV-A, which constitute the wavelengths from 290 to 400 nm. UV-B consists of wavelengths between 290 and 320 nm. This portion of the photobiologic action spectrum is the most efficient in producing redness or erythema in human skin and thus is sometimes known as the “sunburn spectrum.” UV-A includes wavelengths between 320 and 400 nm and is ~1000-fold less efficient in producing skin redness than is UV-B.
The wavelengths between 400 and 700 nm are visible to the human eye. The photon energy in the visible spectrum is not capable of damaging human skin in the absence of a photosensitizing chemical. Without the absorption of energy by a molecule, there can be no photosensitivity. Thus, the absorption spectrum of a molecule is defined as the range of wavelengths it absorbs, whereas the action spectrum for an effect of incident radiation is defined as the range of wavelengths that evoke the response.
Photosensitivity occurs when a photon-absorbing chemical (chromophore) present in the skin absorbs incident energy, becomes excited, and transfers the absorbed energy to various structures or to molecular oxygen.
UV RADIATION (UVR) AND SKIN STRUCTURE AND FUNCTION
Human skin consists of two major compartments: the outer epidermis, which is a stratified squamous epithelium, and the underlying dermis, which is rich in matrix proteins such as collagens and elastin. Both compartments are susceptible to damage from sun exposure. The epidermis and the dermis contain several chromophores capable of absorbing incident solar energy, including nucleic acids, proteins, and lipids. The outermost epidermal layer, the stratum corneum, is a major absorber of UV-B, and <10% of incident UV-B wavelengths penetrate through the epidermis to the dermis. Approximately 3% of radiation below 300 nm, 20% of radiation below 360 nm, and 33% of short visible radiation reach the basal cell layer in untanned human skin. In contrast, UV-A readily penetrates to the dermis and is capable of altering structural and matrix proteins that contribute to photoaging of chronically sun-exposed skin, particularly in individuals of light complexion. Thus, longer wavelengths can penetrate more deeply into the skin.
Molecular Targets for UVR-Induced Skin Effects Epidermal DNA—predominantly in keratinocytes and in Langerhans cells, which are dendritic antigen-presenting cells—absorbs UV-B and undergoes structural changes between adjacent pyrimidine bases (thymine or cytosine), including the formation of cyclobutane dimers and 6,4-photoproducts. These structural changes are potentially mutagenic and are found in most basal cell and squamous cell carcinomas (BCCs and SCCs, respectively). They can be repaired by cellular mechanisms that result in their recognition and excision and the restoration of normal base sequences. The efficient repair of these structural aberrations is crucial, since individuals with defective DNA repair are at high risk for the development of cutaneous cancer. For example, patients with xeroderma pigmentosum, an autosomal recessive disorder, have a variably deficient repair of UV-induced photoproducts. The skin of these patients often shows the dry, leathery appearance of prematurely photoaged skin, and these patients have an increased frequency of skin cancer already in the first two decades of life. Studies in transgenic mice have verified the importance of functional genes that regulate these repair pathways in preventing the development of UV-induced skin cancer. DNA damage in Langerhans cells may also contribute to the known immunosuppressive effects of UV-B (see “Photoimmunology,” below).
In addition to DNA, molecular oxygen is a target for incident solar UVR, leading to the generation of reactive oxygen species (ROS). These ROS can damage skin components, such as epidermal lipids—either free lipids in the stratum corneum or cell membrane lipids. UVR also can target proteins, leading to increased cross-linking and degradation of matrix proteins in the dermis and accumulation of abnormal dermal elastin leading to photoaging changes known as solar elastosis.
Cutaneous Optics and Chromophores Chromophores are endogenous or exogenous chemical components that can absorb physical energy. Endogenous chromophores are of two types: (1) normal components of skin, including nucleic acids, proteins, lipids, and 7-dehydrocholesterol (the precursor of vitamin D); and (2) components that are synthesized elsewhere in the body and that circulate in the bloodstream and diffuse into the skin, such as porphyrins. Normally, only trace amounts of porphyrins are present in the skin, but, in selected diseases known as the porphyrias (Chap. 430), porphyrins are released into the circulation in increased amounts from the bone marrow and the liver and are transported to the skin, where they absorb incident energy both in the Soret band (around 400 nm; short visible) and, to a lesser extent, in the red portion of the visible spectrum (580–660 nm). This energy absorption results in the generation of ROS that can mediate structural damage to the skin, manifested as erythema, edema, urticaria, or blister formation. It is of interest that photoexcited porphyrins are currently used in the treatment of nonmelanoma skin cancers and their precursor lesions, actinic keratoses. Known as photodynamic therapy, this modality generates ROS in the skin, leading to cell death. Topical photosensitizers used in photodynamic therapy are the porphyrin precursors 5-aminolevulinic acid and methyl aminolevulinate, which are converted to porphyrins in the skin. It is believed that photodynamic therapy targets tumor cells for destruction more selectively than it targets adjacent nonneoplastic cells. The efficacy of such therapy requires appropriate timing of the application of methyl aminolevulinate or 5-aminolevulinic acid to the affected skin followed by exposure to artificial sources of visible light. High-intensity blue light has been used successfully for the treatment of thin actinic keratoses. Red light has a longer wavelength, penetrates more deeply into the skin, and is more beneficial in the treatment of superficial BCCs.
Acute Effects of Sun Exposure The acute effects of skin exposure to sunlight include sunburn and vitamin D synthesis.
SUNBURN This painful skin condition is an acute inflammatory response of the skin, predominantly to UV-B. Generally, an individual’s ability to tolerate sunlight is inversely proportional to that individual’s degree of melanin pigmentation. Melanin, a complex polymer of tyrosine derivatives, is synthesized in specialized epidermal dendritic cells known as melanocytes and is packaged into melanosomes that are transferred via dendritic processes into keratinocytes, thereby providing photoprotection and simultaneously darkening the skin. Sun-induced melanogenesis is a consequence of increased tyrosinase activity in melanocytes. Central to the suntan response is the melanocortin-1 receptor (MC1R), and mutations in this gene contribute to the wide variation in human skin and hair color; individuals with red hair and fair skin typically have low MC1R activity. Genetic studies have revealed additional genes that influence skin color variation in humans, such as the gene for tyrosinase (TYR) and the genes APBA2[OCA2], SLC45A2, and SLC24A5. The human MC1R gene encodes a G protein–coupled receptor that binds α-melanocyte-stimulating hormone, which is secreted in the skin mainly by keratinocytes in response to UVR. The UV-induced expression of this hormone is controlled by the tumor suppressor p53, and absence of functional p53 attenuates the tanning response. Activation of the melanocortin receptor leads to increased intracellular cyclic adenosine 5′-monophosphate (cAMP) and protein kinase A activation, resulting in an increased transcription of the microphthalmia-associated transcription factor (MITF), which stimulates melanogenesis. Since the precursor of α-melanocyte-stimulating hormone, proopiomelanocortin, is also the precursor of β-endorphins, UVR may result in not only increased pigmentation but also in increased β-endorphin production, an effect that has been hypothesized to promote sun-seeking behaviors.
The Fitzpatrick classification of human skin phototypes is based on the efficiency of the epidermal-melanin unit, which usually can be ascertained by asking an individual two questions: (1) Do you burn after sun exposure? (2) Do you tan after sun exposure? The answers to these questions permit division of the population into six skin types, varying from type I (always burn, never tan) to type VI (never burn, always tan) (Table 75-1).
|
SKIN TYPE AND SUNBURN SENSITIVITY (FITZPATRICK CLASSIFICATION) |
Sunburn erythema is due to vasodilation of dermal blood vessels. There is a lag time (usually 4–12 h) between skin exposure to sunlight and the development of visible redness. The action spectrum for sunburn erythema includes UV-B and UV-A, although UV-B is much more efficient than UV-A in evoking the response. However, UV-A may contribute to sunburn erythema at midday, when much more UV-A than UV-B is present in the solar spectrum. The erythema that accompanies the inflammatory response induced by UVR results from the orchestrated release of cytokines along with growth factors and the generation of ROS. Furthermore, UV-induced activation of nuclear factor κB–dependent gene transcription can augment release of several proinflammatory cytokines and vasoactive mediators. These cytokines and mediators accumulate locally in sunburned skin, providing chemotactic factors that attract neutrophils, macrophages, and T lymphocytes, which promote the inflammatory response. UVR also stimulates infiltration of inflammatory cells through induced expression of adhesion molecules such as E-selectin and intercellular adhesion molecule 1 on endothelial cells and keratinocytes. UVR also has been shown to activate phospholipase A2, resulting in increases in eicosanoids such as prostaglandin E2, which is known to be a potent inducer of sunburn erythema. The role of eicosanoids in this reaction has been verified by studies showing that nonsteroidal anti-inflammatory drugs (NSAIDs) can reduce it.
Epidermal changes in sunburn include the induction of “sunburn cells,” which are keratinocytes undergoing p53-dependent apoptosis as a defense, with elimination of cells that harbor UV-B-induced structural DNA damage.
VITAMIN D SYNTHESIS AND PHOTOCHEMISTRY Cutaneous exposure to UV-B causes photolysis of epidermal 7-dehydrocholesterol, converting it to pre–vitamin D3, which then undergoes temperature-dependent isomerization to form the stable hormone vitamin D3. This compound diffuses to the dermal vasculature and circulates to the liver and kidney, where it is converted to the dihydroxylated functional hormone 1,25-dihydroxyvitamin D3. Vitamin D metabolites from the circulation and those produced in the skin itself can augment epidermal differentiation signaling and inhibit keratinocyte proliferation. These effects are exploited therapeutically in psoriasis with the topical application of synthetic vitamin D analogues. In addition, vitamin D is increasingly thought to have beneficial effects in several other inflammatory conditions, and some evidence suggests that—besides its classic physiologic effects on calcium metabolism and bone homeostasis—it is associated with a reduced risk of various internal malignancies. There is controversy regarding the risk-to-benefit ratio of sun exposure in vitamin D homeostasis. At present, it is important to emphasize that no clear-cut evidence suggests that the use of sunscreens substantially diminishes vitamin D levels. Since aging also substantially decreases the ability of human skin to photocatalytically produce vitamin D3, the widespread use of sunscreens that filter out UV-B has led to concerns that the elderly might be unduly susceptible to vitamin D deficiency. However, the amount of sunlight needed to produce sufficient vitamin D is small and does not justify the risks of skin cancer and other types of photodamage linked to increased sun exposure or tanning behavior. Nutritional supplementation of vitamin D is a preferable strategy for patients with vitamin D deficiency.
Chronic Effects of Sun Exposure: Nonmalignant The clinical features of photoaging (dermatoheliosis) consist of wrinkling, blotchiness, and telangiectasia as well as a roughened, irregular, “weather-beaten” leathery appearance.
UVR is important in the pathogenesis of photoaging in human skin, and ROS are likely involved. The dermis and its connective tissue matrix are major targets for sun-associated chronic damage that manifests as solar elastosis, a massive increase in thickened irregular masses of abnormal-appearing elastic fibers. Collagen fibers are also abnormally clumped in the deeper dermis of sun-damaged skin. The chromophore(s), the action spectra, and the specific biochemical events orchestrating these changes are only partially understood, although more deeply penetrating UV-A seems to be primarily involved. Chronologically aged sun-protected skin and photoaged skin share important molecular features, including connective tissue damage and elevated levels of matrix metalloproteinases (MMPs). MMPs are enzymes involved in the degradation of the extracellular matrix. UV-A induces expression of some MMPs, including MMP-1 and MMP-3, leading to increased collagen breakdown. In addition, UV-A reduces type I procollagen mRNA expression. Thus, chronic UVR alters the structure and function of dermal collagen. On the basis of these observations, it is not surprising that high-dose UV-A phototherapy may have beneficial effects in some patients with localized fibrotic diseases of the skin, such as localized scleroderma.
Chronic Effects of Sun Exposure: Malignant One of the major known consequences of chronic excessive skin exposure to sunlight is nonmelanoma skin cancer. The two most common types of nonmelanoma skin cancer are BCC and SCC (Chap. 105). A model for skin cancer induction involves three major steps: initiation, promotion, and progression. Exposure of human skin to sunlight results in initiation, a step by which structural (mutagenic) changes in DNA evoke an irreversible alteration in the target cell (keratinocyte) that begins the tumorigenic process. Exposure to a tumor initiator such as UV-B is believed to be a necessary but not a sufficient step in the malignant process, since initiated skin cells not exposed to tumor promoters generally do not develop tumors. The second stage in tumor development is promotion, a multistep process by which chronic exposure to sunlight evokes further changes that culminate in the clonal expansion of initiated cells and cause the development, over many years, of premalignant growths known as actinic keratoses, a minority of which may progress to form SCCs. As a result of extensive studies, it seems clear that UV-B is a complete carcinogen, meaning that it can act as both a tumor initiator and a tumor promoter. The third and final step in the malignant process is malignant conversion of benign precursors into malignant lesions, a process thought to require additional genetic alterations.
On a molecular level, skin carcinogenesis results from the accumulation of gene mutations that cause inactivation of tumor suppressors, activation of oncogenes, or reactivation of cellular signaling pathways that normally are expressed only during epidermal embryologic development. Accumulation of mutations in the tumor-suppressor gene p53 secondary to UV-induced DNA damage occurs in both SCCs and BCCs and is important in promoting skin carcinogenesis. Indeed, the majority of both human and murine UV-induced skin cancers have characteristic p53 mutations (C → T and CC → TT transitions). Studies in mice have shown that sunscreens can substantially reduce the frequency of these signature mutations in p53 and inhibit the induction of tumors.
BCCs also harbor inactivating mutations in the tumor-suppressor gene patched, which result in activation of the sonic hedgehog signaling pathway and increased cell proliferation. Thus, these tumors can manifest mutations in tumor suppressors (p53 and patched) or oncogenes (smoothened). New evidence links alterations in the Wnt/β-catenin signaling pathway, which is known to be critical for hair follicle development, to skin cancer as well. Thus interactions between this pathway and the hedgehog signaling pathway appear to be involved in both skin carcinogenesis and embryologic development of the skin and hair follicles.
Clonal analysis in mouse models of BCC revealed that tumor cells arise from long-term resident progenitor cells of the interfollicular epidermis and the upper infundibulum of the hair follicle. These BCC-initiating cells are reprogrammed to resemble embryonic hair follicle progenitors, whose tumor-initiating ability depends on activation of the Wnt/β-catenin signaling pathway.
SCC initiation occurs both in the interfollicular epidermis and in the hair follicle bulge stem cell populations. In mouse models, the combination of mutant K-Ras and p53 is sufficient to induce invasive SCCs from these cell populations.
The transcription factor Myc is important for stem cell maintenance in the skin, and oncogenic activation of Myc has been implicated in the development of BCCs and SCCs. Thus, nonmelanoma skin cancer involves mutations and alterations in multiple genes and pathways that occur as a result of their chronic accumulation driven by exposure to environmental factors such as UVR.
Epidemiologic studies have linked excessive sun exposure to an increased risk of nonmelanoma cancers and melanoma of the skin; the evidence is far more direct for nonmelanoma skin cancers (BCCs and SCCs) than for melanoma. Approximately 80% of nonmelanoma skin cancers develop on sun-exposed body areas, including the face, neck, and hands. Major risk factors include male sex, childhood sun exposures, older age, fair skin, and residence at latitudes relatively close to the equator. Individuals with darker-pigmented skin have a lower risk of skin cancer than do fair-skinned individuals. More than 2 million individuals in the United States develop nonmelanoma skin cancer annually, and the lifetime risk that a fair-skinned individual will develop such a neoplasm is estimated at ~15%. The incidence of nonmelanoma skin cancer in the population is increasing at a rate of 2–3% per year. One potential explanation is the widespread use of indoor tanning. It is estimated that 30 million people tan indoors in the United States annually, including >2 million adolescents.
The relationship of sun exposure to melanoma development is less direct, but strong evidence supports an association. Clear-cut risk factors include a positive family or personal history of melanoma and multiple dysplastic nevi. Melanomas can occur during adolescence; the implication is that the latent period for tumor growth is shorter than that for nonmelanoma skin cancer. For reasons that are only partially understood, melanomas are among the most rapidly increasing human malignancies (Chap. 105). Epidemiologic studies indicate that indoor tanning is a risk factor for melanoma, which may contribute to the increasing incidence of melanoma formation. Furthermore, epidemiologic studies suggest that life in a sunny climate from birth or early childhood may increase the risk of melanoma development. In general, risk does not correlate with cumulative sun exposure but may be related to the duration and extent of exposure in childhood.
However, in contrast to nonmelanoma skin cancers, melanoma frequently develops in sun-protected skin, and oncogenic mutations in melanoma may also not be UVR-signature mutations; these observations suggest that UVR-independent factors contribute to melanomagenesis. Low MC1R activity leads to production of the red/yellow pheomelanin pigment in individuals with red hair and fair skin, while high MC1R activity results in increased production of the black/brown eumelanin. Experiments in mice suggest that high pheomelanin content in skin (as in individuals with red hair and fair skin) leads to a UVR-independent increase in the risk of melanoma through a mechanism that involves oxidative damage. Thus, both UVR-dependent and UVR-independent factors are likely to contribute to melanoma formation.
Photoimmunology Exposure to solar radiation causes both local immunosuppression (through inhibition of immune responses to antigens applied at the irradiated site) and systemic immunosuppression (through inhibition of immune responses to antigens applied at remote, unirradiated sites). For example, human skin exposure to modest doses of UV-B can deplete the epidermal antigen-presenting cells known as Langerhans cells, thereby reducing the degree of allergic sensitization to application of the potent contact allergen dinitrochlorobenzene at the irradiated skin site.
An example of the systemic immunosuppressive effects of higher doses of UVR is the diminished immunologic response to antigens introduced either epicutaneously or intracutaneously at sites distant from the irradiated site. Various immunomodulatory factors and immune cells have been implicated in UVR-induced systemic immunosuppression, including tumor necrosis factor α, interleukin 4, interleukin 10, cis-urocanic acid, and eicosanoids. Experimental evidence suggests that prostaglandin E2 signaling through prostaglandin E receptor subtype 4 mediates UVR-induced systemic immunosuppression by elevating the number of regulatory T cells, and this effect can be inhibited with NSAIDs.
The major chromophores in the upper epidermis that are known to initiate UV-mediated immunosuppression include DNA, trans-urocanic acid, and membrane components. The action spectrum for UV-induced immunosuppression closely mimics the absorption spectrum of DNA. Pyrimidine dimers in Langerhans cells may inhibit antigen presentation. The absorption spectrum of epidermal urocanic acid closely mimics the action spectrum for UV-B-induced immunosuppression. Urocanic acid is a metabolic product of the essential amino acid histidine and accumulates in the upper epidermis through breakdown of the histidine-rich protein filaggrin due to the absence of its catabolizing enzyme in keratinocytes. Urocanic acid is synthesized as a trans-isomer, and UV-induced trans-cis isomerization of urocanic acid in the stratum corneum drives immunosuppression. Cis-urocanic acid may exert its immunosuppressive effects through a variety of mechanisms, including inhibition of antigen presentation by Langerhans cells.
One important consequence of chronic sun exposure and associated immunosuppression is an enhanced risk of skin cancer. In part, UV-B activates regulatory T cells that suppress antitumor immune responses via interleukin 10 expression, whereas, in the absence of high UV-B exposure, epidermal Langerhans cells present tumor-associated antigens and induce protective immunity, thereby inhibiting skin tumorigenesis. UV-induced DNA damage is a major molecular trigger of this immunosuppressive effect.
Perhaps the most graphic demonstration of the role of immunosuppression in enhancing the risk of nonmelanoma skin cancer comes from studies of organ transplant recipients who require lifelong immunosuppressive/antirejection drug regimens. More than 50% of organ transplant recipients develop BCCs and SCCs, and these cancers are the most common types of malignancies arising in these patients. Rates of BCC and SCC increase with the duration and degree of immunosuppression. These patients ideally should be screened prior to organ transplantation, be monitored closely thereafter, and adhere to rigorous photoprotection measures, including the use of sunscreens and protective clothing as well as sun avoidance. Notably, immunosuppressive drugs that target the mTOR pathway, such as sirolimus and everolimus, may reduce the risk of nonmelanoma skin cancer in organ transplant recipients from that associated with the use of calcineurin inhibitors (cyclosporine and tacrolimus), which may contribute to nonmelanoma skin cancer formation not only through their immunosuppressive effects but also through suppression of p53-dependent cancer cell senescence pathways independent of host immunity.
PHOTOSENSITIVITY DISEASES
The diagnosis of photosensitivity requires elicitation of a careful history in order to define the duration of signs and symptoms, the length of time between exposure to sunlight and the development of subjective symptoms, and visible changes in the skin. The age of onset can also be a helpful diagnostic clue; for example, the acute photosensitivity of erythropoietic protoporphyria almost always begins in childhood, whereas the chronic photosensitivity of porphyria cutanea tarda (PCT) typically begins in the fourth and fifth decades of life. A patient’s history of exposure to topical and systemic drugs and chemicals may provide important diagnostic clues. Many classes of drugs can cause photosensitivity on the basis of either phototoxicity or photoallergy. Fragrances such as musk ambrette that were previously present in numerous cosmetic products are also potent photosensitizers.
Examination of the skin may offer important clues. Anatomic areas that are naturally protected from direct sunlight, such as the hairy scalp, the upper eyelids, the retroauricular areas, and the infranasal and submental regions, may be spared, whereas exposed areas show characteristic features of the pathologic process. These anatomic localization patterns are often helpful, but not infallible, in making the diagnosis. For example, airborne contact sensitizers that are blown onto the skin may produce dermatitis that can be difficult to distinguish from photosensitivity despite the fact that such material may trigger skin reactivity in areas shielded from direct sunlight.
Many dermatologic conditions may be caused or aggravated by sunlight (Table 75-2). The role of light in evoking these responses may be dependent on genetic abnormalities ranging from well-described defects in DNA repair that occur in xeroderma pigmentosum to the inherited abnormalities in heme synthesis that characterize the porphyrias. The chromophore has been identified in certain photosensitivity diseases, but the energy-absorbing agent remains unknown in the majority.
|
CLASSIFICATION OF PHOTOSENSITIVITY DISEASES |
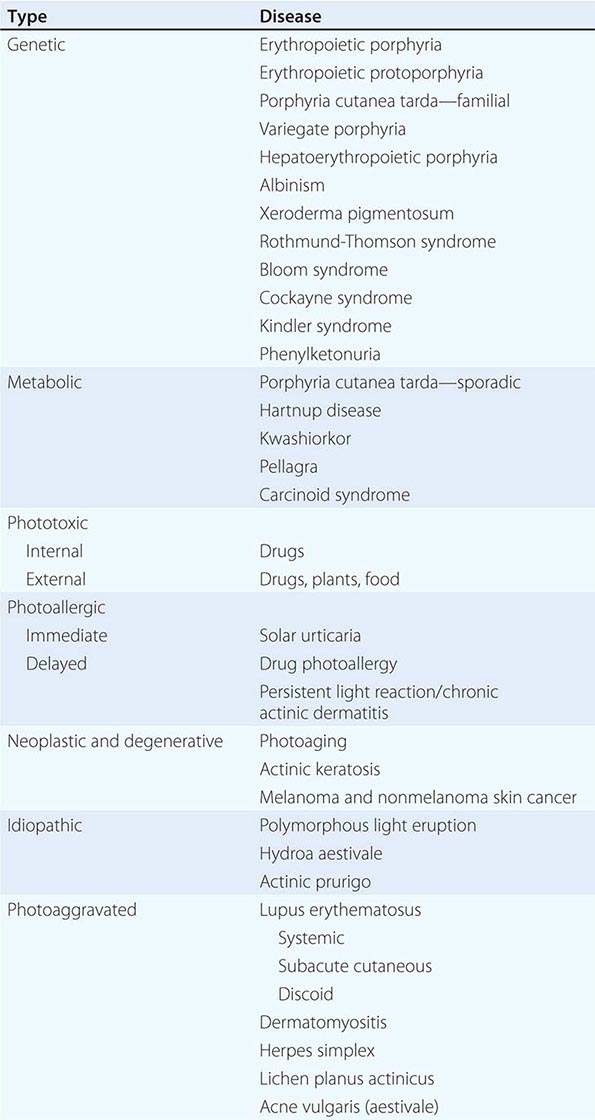
Polymorphous Light Eruption A common type of photosensitivity disease is polymorphous light eruption (PMLE). Many affected individuals never seek medical attention because the condition is often transient, becoming manifest in the spring with initial sun exposure but then subsiding spontaneously with continuing exposure, a phenomenon known as “hardening.” The major manifestations of PMLE include (often intensely) pruritic erythematous papules that may coalesce into plaques in a patchy distribution on exposed areas of the trunk and forearms. The face is usually less seriously involved. Whereas the morphologic skin findings remain similar for each patient with subsequent recurrences, significant interindividual variations in skin findings are characteristic (hence the term “polymorphous”).
A skin biopsy and phototest procedures in which skin is exposed to multiple erythemal doses of UV-A and UV-B may aid in the diagnosis. The action spectrum for PMLE is usually within these portions of the solar spectrum.
Whereas the treatment of an acute flare of PMLE may require topical or systemic glucocorticoids, approaches to preventing PMLE are important and include the use of high-SPF and high UVA-protection broad-spectrum sunscreens as well as the induction of “hardening” by the cautious administration of artificial UV-B (broad-band or narrow-band) and/or UV-A radiation or the use of psoralen plus UV-A (PUVA) photochemotherapy for 2–4 weeks before initial sun exposure. Such prophylactic phototherapy or photochemotherapy at the beginning of spring may prevent the occurrence of PMLE throughout the summer.
Phototoxicity and Photoallergy These photosensitivity disorders are related to the topical or systemic administration of drugs and other chemicals. Both reactions require the absorption of energy by a drug or chemical with consequent production of an excited-state photosensitizer that can transfer its absorbed energy to a bystander molecule or to molecular oxygen, thereby generating tissue-destructive chemical species, including ROS.
Phototoxicity is a nonimmunologic reaction that can be caused by drugs and chemicals, a few of which are listed in Table 75-3. The usual clinical manifestations include erythema resembling a sunburn reaction that quickly desquamates, or “peels,” within several days. In addition, edema, vesicles, and bullae may occur.
|
DRUGS THAT MAY CAUSE A PHOTOTOXIC REACTION |
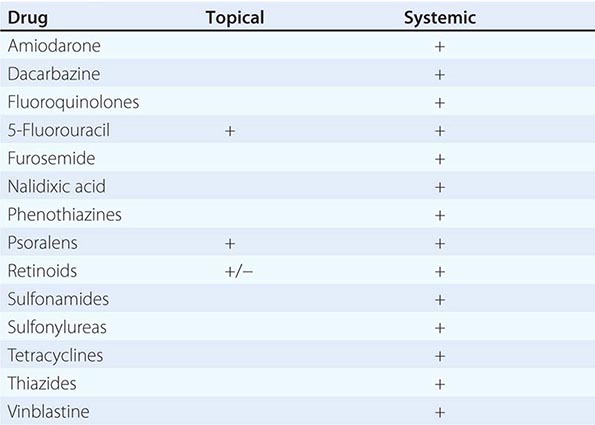
Photoallergy is much less common and is distinct in that it is an immunopathologic process. The excited-state photosensitizer may create highly unstable haptenic free radicals that bind covalently to macromolecules to form a functional antigen capable of evoking a delayed-type hypersensitivity response. Some drugs and chemicals that can produce photoallergy are listed in Table 75-4. The clinical manifestations typically differ from those of phototoxicity in that an intensely pruritic eczematous dermatitis tends to predominate and evolves into lichenified, thickened, “leathery” changes in sun-exposed areas. A small subset (perhaps 5–10%) of patients with photoallergy may develop a persistent exquisite hypersensitivity to light even when the offending drug or chemical is identified and eliminated, a condition known as persistent light reaction.
|
DRUGS THAT MAY CAUSE A PHOTOALLERGIC REACTION |
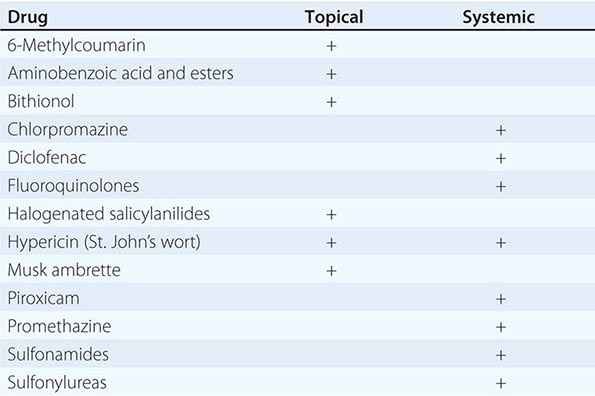
A very uncommon type of persistent photosensitivity is known as chronic actinic dermatitis. The affected patients are typically elderly men with a long history of preexisting allergic contact dermatitis or photosensitivity. These individuals are usually exquisitely sensitive to UV-B, UV-A, and visible wavelengths.
Phototoxicity and photoallergy often can be diagnostically confirmed by phototest procedures. In patients with suspected phototoxicity, determining the minimal erythemal dose (MED) while the patient is exposed to a suspected agent and then repeating the MED after discontinuation of the agent may provide a clue to the causative drug or chemical. Photopatch testing can be performed to confirm the diagnosis of photoallergy. In this simple variant of ordinary patch testing, a series of known photoallergens is applied to the skin in duplicate, and one set is irradiated with a suberythemal dose of UV-A. The development of eczematous changes at sites exposed to sensitizer and light is a positive result. The characteristic abnormality in patients with persistent light reaction is a diminished threshold to erythema evoked by UV-B. Patients with chronic actinic dermatitis usually manifest a broad spectrum of UV hyperresponsiveness and require meticulous photoprotection, including avoidance of sun exposure, use of high-SPF (>30) sunscreens, and, in severe cases, systemic immunosuppression, such as with azathioprine.
The management of drug photosensitivity involves first and foremost the elimination of exposure to the chemical agents responsible for the reaction and the minimization of sun exposure. The acute symptoms of phototoxicity may be ameliorated by cool moist compresses, topical glucocorticoids, and systemically administered NSAIDs. In severely affected individuals, a rapidly tapered course of systemic glucocorticoids may be useful. Judicious use of analgesics may be necessary.
Photoallergic reactions require a similar management approach. Furthermore, patients with persistent light reaction and chronic actinic dermatitis must be meticulously protected against light exposure. In selected patients to whom chronic systemic high-dose glucocorticoids pose unacceptable risks, it may be necessary to employ an immunosuppressive drug such as azathioprine, cyclophosphamide, cyclosporine, or mycophenolate mofetil.
Porphyria The porphyrias (Chap. 430) are a group of diseases that have in common inherited or acquired derangements in the synthesis of heme. Heme is an iron-chelated tetrapyrrole or porphyrin, and the nonmetal chelated porphyrins are potent photosensitizers that absorb light intensely in both the short (400–410 nm) and the long (580–650 nm) portions of the visible spectrum.
Heme cannot be reutilized and must be synthesized continuously. The two body compartments with the largest capacity for its production are the bone marrow and the liver. Accordingly, the porphyrias originate in one or the other of these organs, with an end result of excessive endogenous production of potent photosensitizing porphyrins. The porphyrins circulate in the bloodstream and diffuse into the skin, where they absorb solar energy, become photoexcited, generate ROS, and evoke cutaneous photosensitivity. The mechanism of porphyrin photosensitization is known to be photodynamic, or oxygen-dependent, and is mediated by ROS such as singlet oxygen and superoxide anions.
Porphyria cutanea tarda is the most common type of porphyria and is associated with decreased activity of the enzyme uroporphyrinogen decarboxylase. There are two basic types of PCT: (1) the sporadic or acquired type, generally seen in individuals ingesting ethanol or receiving estrogens; and (2) the inherited type, in which there is autosomal dominant transmission of deficient enzyme activity. Both forms are associated with increased hepatic iron stores.
In both types of PCT, the predominant feature is chronic photosensitivity characterized by increased fragility of sun-exposed skin, particularly areas subject to repeated trauma such as the dorsa of the hands, the forearms, the face, and the ears. The predominant skin lesions are vesicles and bullae that rupture, producing moist erosions (often with a hemorrhagic base) that heal slowly, with crusting and purplish discoloration of the affected skin. Hypertrichosis, mottled pigmentary change, and scleroderma-like induration are associated features. The diagnosis can be confirmed biochemically by measurement of urinary porphyrin excretion, plasma porphyrin assay, and assay of erythrocyte and/or hepatic uroporphyrinogen decarboxylase. Multiple mutations of the uroporphyrinogen decarboxylase gene have been identified in human populations. Some patients with PCT have associated mutations in the HFE gene, which is linked to hemochromatosis; these mutations could contribute to the iron overload seen in PCT, although iron status as measured by serum ferritin, iron levels, and transferrin saturation is no different from that in PCT patients without HFE mutations. Prior hepatitis C virus infection appears to be an independent risk factor for PCT.
Treatment of PCT consists of repeated phlebotomies to diminish the excessive hepatic iron stores and/or intermittent low doses of chloroquine and hydroxychloroquine. Long-term remission of the disease can be achieved if the patient eliminates exposure to porphyrinogenic agents and prolonged exposure to sunlight.
Erythropoietic protoporphyria originates in the bone marrow and is due to a decrease in the mitochondrial enzyme ferrochelatase secondary to numerous gene mutations. The major clinical features include acute photosensitivity characterized by subjective burning and stinging of exposed skin that often develops during or just after sun exposure. There may be associated skin swelling and, after repeated episodes, a waxlike scarring.
The diagnosis is confirmed by demonstration of elevated levels of free erythrocyte protoporphyrin. Detection of increased plasma protoporphyrin helps distinguish erythropoietic protoporphyria from lead poisoning and iron-deficiency anemia, in both of which erythrocyte protoporphyrin levels are elevated in the absence of cutaneous photosensitivity and elevated plasma protoporphyrin levels.
Treatment includes reduction of sun exposure and oral administration of the carotenoid β-carotene, which is an effective scavenger of free radicals. This drug increases tolerance to sun exposure in some affected individuals, although it has no effect on deficient ferrochelatase.
An algorithm for managing patients with photosensitivity is presented in Fig. 75-1.
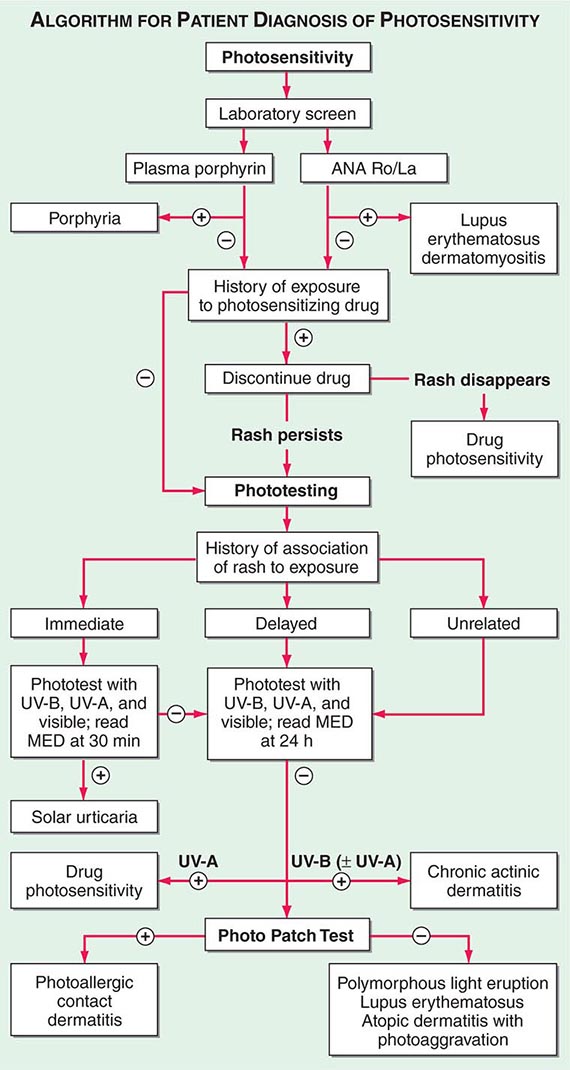
FIGURE 75-1 Algorithm for the diagnosis of a patient with photosensitivity. ANA, antinuclear antibody; MED, minimal erythemal dose; UV-A and UV-B, ultraviolet spectrum segments including wavelengths of 320–400 nm and 290–320 nm, respectively.
PHOTOPROTECTION
Since photosensitivity of the skin results from exposure to sunlight, it follows that absolute avoidance of sunlight will eliminate these disorders. However, contemporary lifestyles make this approach impractical for most individuals. Thus better approaches to photoprotection have been sought.
Natural photoprotection is provided by structural proteins in the epidermis, particularly keratins and melanin. The amount of melanin and its distribution in cells are genetically regulated, and individuals of darker complexion (skin types IV–VI) are at decreased risk for the development of acute sunburn and cutaneous malignancy.
Other forms of photoprotection include clothing and sunscreens. Clothing constructed of tightly woven sun-protective fabrics, irrespective of color, affords substantial protection. Wide-brimmed hats, long sleeves, and trousers all reduce direct exposure. Sunscreens are now considered over-the-counter drugs, and a monograph from the U.S. Food and Drug Administration (FDA) has recognized category I ingredients as safe and effective. Those ingredients are listed in Table 75-5. Sunscreens are rated for their photoprotective effect by their sun protection factor (SPF). The SPF is simply a ratio of the time required to produce sunburn erythema with and without sunscreen application. The SPF of most sunscreens reflects protection from UV-B but not from UV-A. The FDA monograph stipulates that sunscreens must be rated on a scale ranging from minimal (SPF ≥2 and <12) to moderate (SPF ≥12 and <30) to high (SPF ≥30, labeled as 30+).
|
FDA CATEGORY I MONOGRAPHED SUNSCREEN INGREDIENTSa |
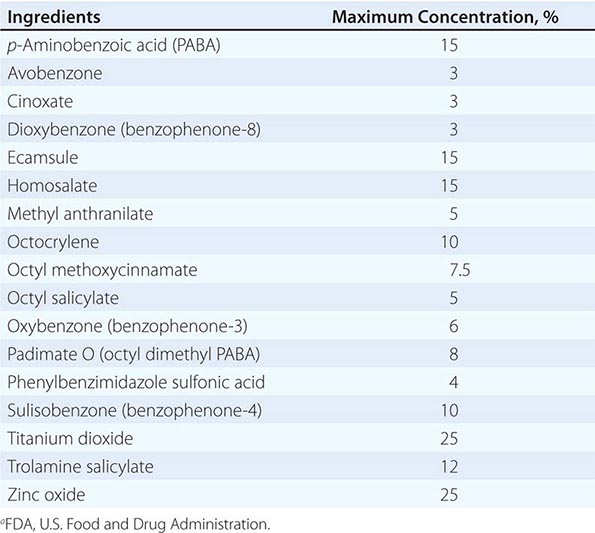
Broad-spectrum sunscreens contain both UV-B-absorbing and UV-A-absorbing chemicals, the latter including avobenzone and ecamsule (terephthalylidene dicamphor sulfonic acid). These chemicals absorb UVR and transfer the absorbed energy to surrounding cells. In contrast, physical UV blockers (zinc oxide and titanium dioxide) scatter or reflect UVR.
In addition to light absorption, a critical determinant of the sustained photoprotective effect of sunscreens is their water resistance. The FDA monograph has defined strict testing criteria for sunscreens that claim to possess a high degree of water resistance.
Some degree of photoprotection can be achieved by limiting the time of sun exposure during the day. Since a large part of an individual’s total lifetime sun exposure may occur by age 18, it is important to educate parents and young children about the hazards of sunlight. Simply eliminating exposure at midday will substantially reduce lifetime UVR exposure.
PHOTOTHERAPY AND PHOTOCHEMOTHERAPY
UVR can be used therapeutically. The administration of UV-B alone or in combination with topically applied agents can induce remissions of many dermatologic diseases, including psoriasis and atopic dermatitis. In particular, narrow-band UV-B treatments (with fluorescent bulbs emitting radiation at ~311 nm) have enhanced efficacy over that obtained with broad-band UV-B in the treatment of psoriasis.
Photochemotherapy in which topically applied or systemically administered psoralens are combined with UV-A (PUVA) is effective in treating psoriasis and the early stages of cutaneous T cell lymphoma and vitiligo. Psoralens are tricyclic furocoumarins that, when intercalated into DNA and exposed to UV-A, form adducts with pyrimidine bases and eventually form DNA cross-links. These structural changes are thought to decrease DNA synthesis and to be related to the amelioration of psoriasis. Why PUVA photochemotherapy is effective in cutaneous T cell lymphoma is only partially understood, but it has been shown to induce apoptosis of atypical T lymphocyte populations in the skin. Consequently, direct treatment of circulating atypical lymphocytes by extracorporeal photochemotherapy (photopheresis) has been used in Sézary syndrome as well as in other severe systemic diseases with circulating atypical lymphocytes, such as graft-versus-host disease.
In addition to its effects on DNA, PUVA photochemotherapy stimulates epidermal thickening and melanin synthesis; the latter property, together with its anti-inflammatory effects, provides the rationale for use of PUVA in the depigmenting disease vitiligo. Oral 8-methoxypsoralen and UV-A appear to be most effective in this regard, but as many as 100 treatments extending over 12–18 months may be required for satisfactory repigmentation.
Not surprisingly, the major side effects of long-term UV-B phototherapy and PUVA photochemotherapy mimic those seen in individuals with chronic sun exposure and include skin dryness, actinic keratoses, and an increased risk of skin cancer. Despite these risks, the therapeutic index of these modalities continues to be excellent. It is important to choose the most appropriate phototherapeutic approach for a specific dermatologic disease. For example, narrow-band UV-B has been reported in several studies to be as effective as PUVA photochemotherapy in the treatment of psoriasis but to pose a lower risk of skin cancer development than PUVA.
76e |
Atlas of Skin Manifestations of Internal Disease |
In the practice of medicine, virtually every clinician encounters patients with skin disease. Physicians of all specialties face the daily task of determining the nature and clinical implication of dermatologic disease. In patients with skin disease, the physician must confront the question of whether the cutaneous process is confined to the skin, representing a purely dermatologic event, or whether it is a manifestation of internal disease related to the patient’s overall medical condition. Evaluation and accurate diagnosis of skin lesions are particularly critical given the marked rise in both melanoma and nonmelanoma skin cancer. Dermatologic conditions can be classified and categorized in many ways. In this atlas, a selected group of inflammatory skin eruptions and neoplastic conditions are grouped in the following manner: (1) common skin diseases and lesions, (2) nonmelanoma skin cancer, (3) melanoma and benign pigmented lesions, (4) infectious disease and the skin, (5) immunologically mediated skin disease, and (6) skin manifestations of internal disease.
COMMON SKIN DISEASES AND LESIONS
(Figs. 76e-1 to 76e-19) While most of these common inflammatory skin diseases and benign neoplastic and reactive lesions usually present as a predominantly dermatologic process, underlying systemic associations may be found in some settings. Atopic dermatitis is often present in patients with an atopic diathesis, including asthma or sinusitis. Psoriasis ranges from limited patches on the elbows and knees to severe erythrodermic and pustular involvement and associated psoriatic arthritis. Some patients with alopecia areata may have an underlying thyroid abnormality requiring screening. Finally, even acne vulgaris, one of the most common inflammatory dermatoses, can be associated with a systemic process such as polycystic ovarian syndrome.
FIGURE 76e-1 Acne vulgaris, with inflammatory papules, pustules, and comedones. (Courtesy of Kalman Watsky, MD; with permission.)
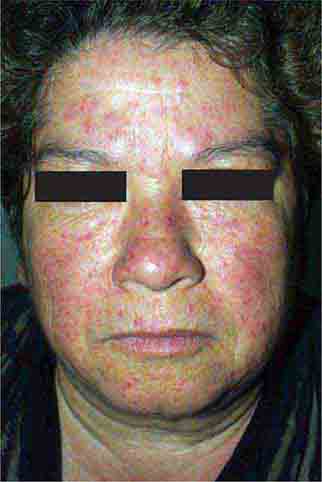
FIGURE 76e-2 Acne rosacea, with prominent facial erythema, telangiectasias, scattered papules, and small pustules. (Courtesy of Robert Swerlick, MD; with permission.)
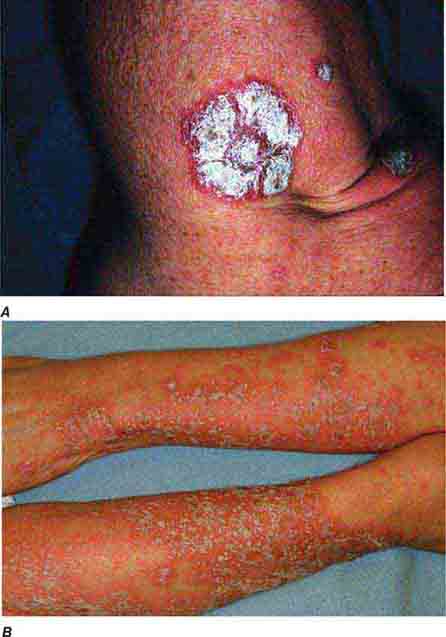
FIGURE 76e-3 Psoriasis. A. Typical psoriasis is characterized by small and large erythematous plaques with adherent silvery scale. B. Acute inflammatory variants of psoriasis may present with widespread superficial pustules.
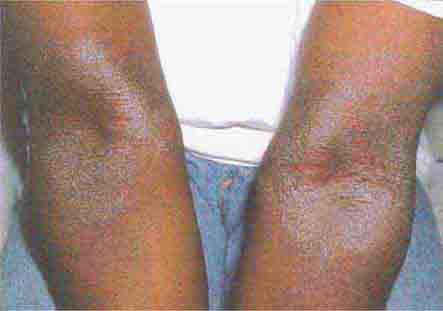
FIGURE 76e-4 Atopic dermatitis, with hyperpigmentation, lichenification, and scaling in the antecubital fossae. (Courtesy of Robert Swerlick, MD; with permission.)
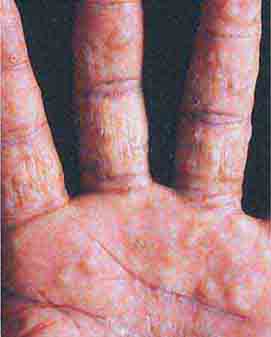
FIGURE 76e-5 Dyshidrotic eczema, characterized by deep-seated vesicles and scaling on palms and lateral fingers, is often associated with an atopic diathesis.
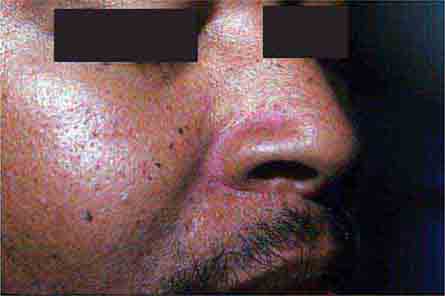
FIGURE 76e-6 Seborrheic dermatitis, with erythema and scale in the nasolabial fold. (Courtesy of Robert A. Swerlick, MD; with permission.)
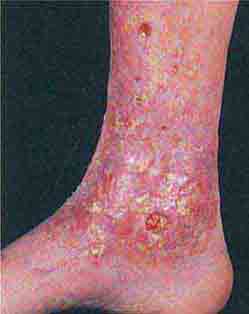
FIGURE 76e-7 Stasis dermatitis, with erythematous, scaly, and oozing patches over the lower leg. Several stasis ulcers are also seen in this patient.
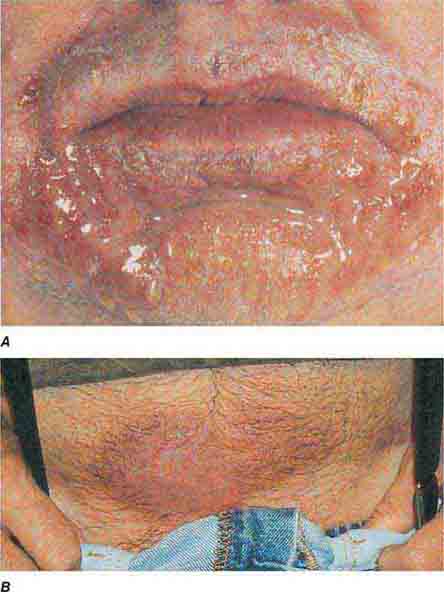
FIGURE 76e-8 Allergic contact dermatitis. A. Acute phase, with sharply demarcated, weeping, eczematous plaques in a perioral distribution. B. Allergic contact reaction to nickel, chronic phase, with an erythematous, lichenified, weeping plaque on skin chronically exposed to a metal snap. (B: Courtesy of Robert Swerlick, MD; with permission.)
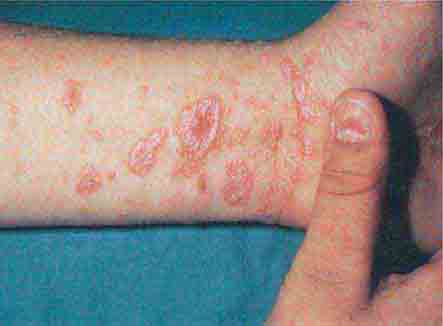
FIGURE 76e-9 Lichen planus, with multiple flat-topped, violaceous papules and plaques. Nail dystrophy, as seen in this patient’s thumbnail, may also be a feature. (Courtesy of Robert Swerlick, MD; with permission.)
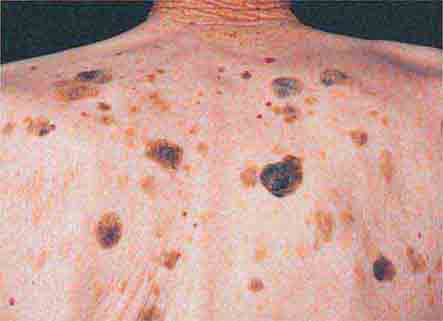
FIGURE 76e-10 Seborrheic keratoses are “stuck on,” waxy, verrucous papules and plaques with a variety of colors ranging from light tan to black.
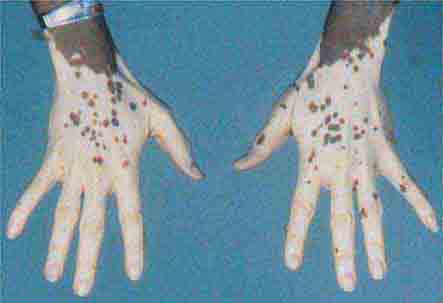
FIGURE 76e-11 Vitiligo in a typical acral distribution, with striking cutaneous depigmentation as a result of melanocyte loss.
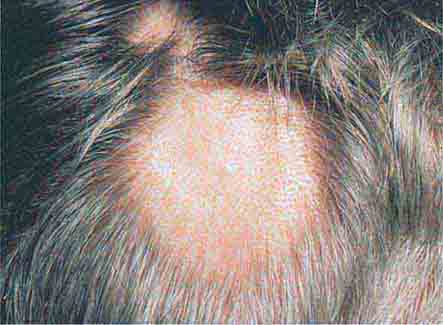
FIGURE 76e-12 Alopecia areata, characterized by a sharply demarcated circular patch of scalp completely devoid of hairs. Preservation of follicular orifices is indicative of nonscarring alopecia. (Courtesy of Robert Swerlick, MD; with permission.)
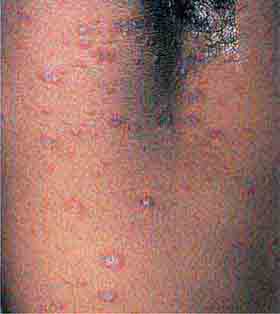
FIGURE 76e-13 Pityriasis rosea. Multiple round or oval erythematous patches with fine central scale are distributed along the skin tension lines on the trunk.
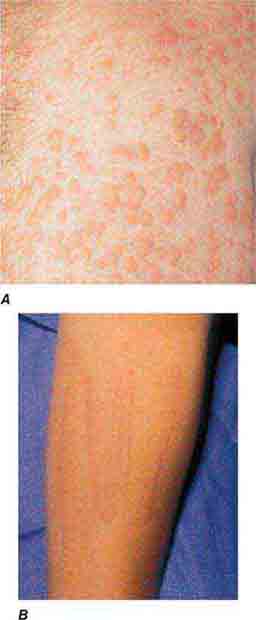
FIGURE 76e-14 A. Urticaria, with characteristic discrete and confluent, edematous, erythematous papules and plaques. B. Dermatographism. Erythema and whealing developed after firm stroking of the skin. (B: Courtesy of Robert Swerlick, MD; with permission.)
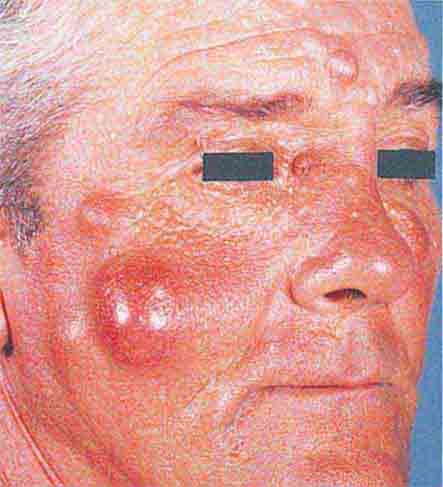
FIGURE 76e-15 Epidermoid cysts. Several inflamed and noninflamed firm cystic nodules are seen in this patient. Often a patulous follicular punctum is observed on the overlying epidermal surface.
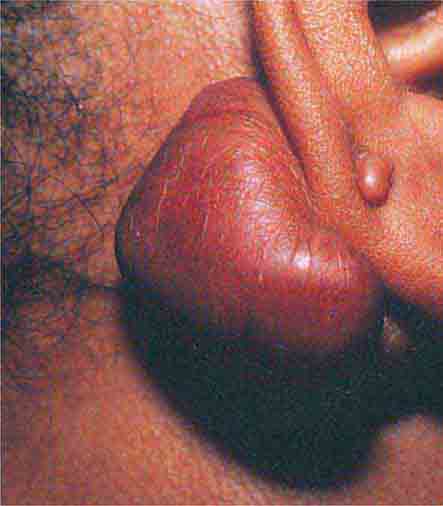
FIGURE 76e-16 Keloids resulting from ear piercing, with firm exophytic flesh-colored to erythematous nodules of scar tissue.
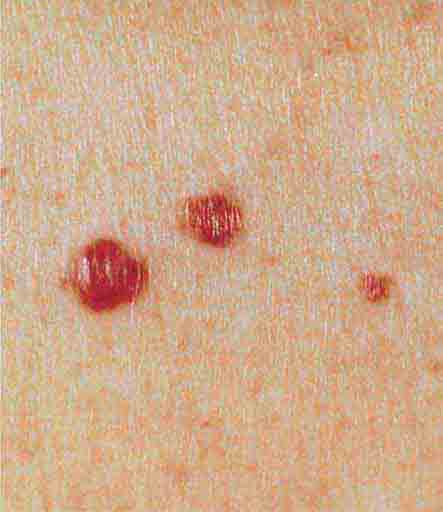
FIGURE 76e-17 Cherry hemangiomas—multiple erythematous to dark-purple papules, usually located on the trunk—are very common and arise in middle-aged to older adults.
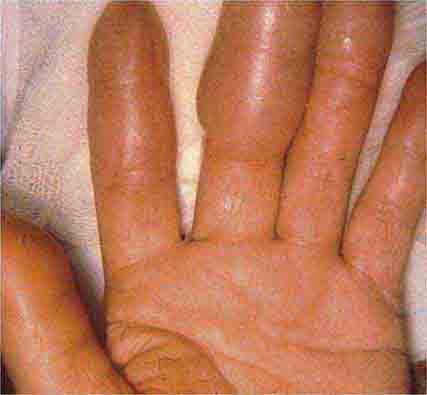
FIGURE 76e-18 Frostbite of the hand, with vesiculation surrounded by edema and erythema. (Courtesy of Daniel F. Danzl, MD; with permission.)
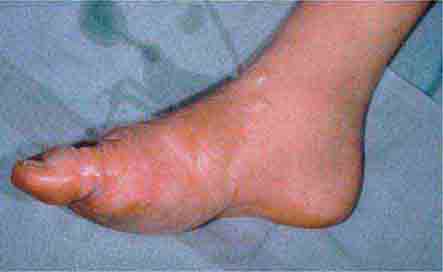
FIGURE 76e-19 Frostbite of the foot, with vesiculation surrounded by edema and erythema. (Courtesy of Daniel F. Danzl, MD; with permission.)
NONMELANOMA SKIN CANCERS
(Figs. 76e-20 to 76e-27) In fair-skinned ethnic populations, rates of nonmelanoma skin cancer are increasing at an alarming rate. Basal cell carcinoma is the most common cancer in humans and is strongly linked to ultraviolet radiation. Squamous cell carcinoma, including keratoacanthoma, is the second most common skin cancer in most ethnic groups and is also most commonly linked to ultraviolet radiation. Less common cutaneous malignancies include cutaneous T cell lymphoma (mycosis fungoides) and carcinoma and lymphoma metastatic to skin.
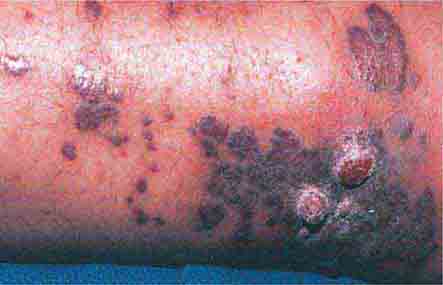
FIGURE 76e-20 Kaposi’s sarcoma in a patient with AIDS. Patch, plaque, and tumor stages are shown.




