Chapter 40 Imaging of Myocardial Metabolism
METHODS TO IMAGE MYOCARDIAL METABOLISM
Magnetic Resonance Spectroscopy
Magnetic resonance spectroscopy offers the advantages of the ability to measure multiple metabolic pathways simultaneously, the relative ease in performing serial measurements, and the lack of ionizing radiation. Moreover, when combined with MRI, near-simultaneous measurements of myocardial perfusion and mechanical function are possible. A number of biologically important nuclei can be measured, including phosphorous (31P), hydrogen (1H), carbon (13C), sodium (23Na), nitrogen (15N), and fluorine (19F). The fundamental principle of MRS is that the chemical environment of nuclei induces local magnetic fields that shift their resonance frequency. The different frequency shift for different metabolites results in a signal consisting of one or more discrete resonance frequencies. The Fourier transform of the acquired signal produces a spectrum with peaks at distinct frequencies. The MRS spectrum displays the signal intensity as a function of frequency measured in parts per million, relative to the frequency of a reference compound. The signal intensity at a given frequency is proportional to the amount of the respective metabolite and can be used to determine the absolute concentration of the metabolite using appropriate calibrating reference signal.1,2
MRS is limited by low signal-to-noise, concomitant limited spatial resolution, intravoxel signal contamination, and long acquisition times. Compared with nuclear imaging methods, MRS has a much lower sensitivity (detecting millimolar as opposed to nanomolar concentrations). As a consequence, the initial success of imaging of cardiac metabolism using Carbon-13 labeled agents in intact animals has not been translated to the study of humans.3 Of note, cardiac applications for MRS become more limited as one moves from rodent to man as opposed to nuclear methods where the reverse occurs. This appears to be a function of both the higher field strength in the small-bore systems and the use of radiofrequency coils that are in closer proximity to the entire heart used in small-animal imaging. In contrast to rodent hearts, where measurements of the entire left ventricular (LV) myocardium are obtained, measurements in human myocardium are typically limited to the anterior myocardium. Currently, only 31P and 1H have been widely used for in vivo clinical cardiac examinations focusing on myocardial energetics (31P) and lipid accumulation (1H).1,3,4
Single-Photon Emission Computed Tomography (SPECT) (See Chapter 2)
Metabolic processes that can be measured by SPECT include:
Positron Emission Tomography (PET) (See Chapter 3)
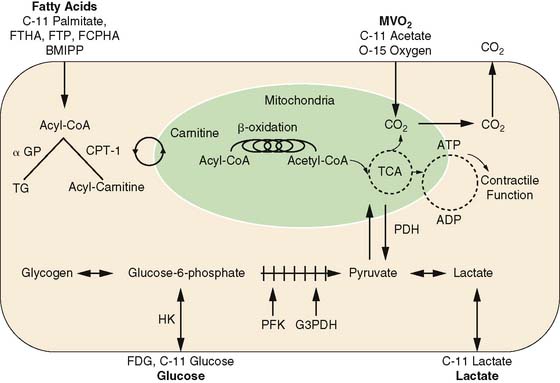
The major advantages of PET are its intrinsic quantitative capability and the use of radiopharmaceuticals labeled with the positron-emitting radionuclides. The PET detection scheme permits accurate quantification of activity in the field of view. The positron-emitting radionuclides of the biologically ubiquitous elements oxygen (15O), carbon (11C), and nitrogen (13N), as well as fluorine (18F) substituting for hydrogen, can be incorporated into a wide variety of substrates or substrate analogs that participate in diverse biochemical pathways without altering the biochemical properties of the substrate of interest (see Fig. 40-1). By combining the knowledge of the metabolic pathways of interest with kinetic models that faithfully describe the fate of the tracer in tissue, an accurate interpretation of the tracer kinetics as they relate to the metabolic process of interest can be achieved. The major disadvantages of PET are its complexity in both radiotracer design and image quantification schemes and expense. Metabolic processes that are typically measured with PET are myocardial oxygen consumption (MVO2), carbohydrate metabolism, and fatty acid metabolism.
Myocardial Oxygen Consumption (MVO2) (See Chapter 38)
Because oxygen is the final electron acceptor in all pathways of aerobic myocardial metabolism, PET with 15O-oxygen has also been used to measure MVO2. The approach provides a measure of myocardial oxygen extraction and measures MVO2 directly. Due to its short physical half-life, 15O-oxygen is readily applicable in studies requiring repetitive assessments, such as those with an acute pharmacologic intervention. Its major disadvantages are the need for a multiple-tracer study (to account for myocardial blood flow and blood volume) and fairly complex compartmental modeling to obtain the measurements.12–14
PET using 11C-acetate is the preferred method of measuring MVO2 noninvasively. After initial extraction, acetate, a two-carbon-chain free fatty acid, is rapidly converted to acetyl-CoA. The primary metabolic fate of acetyl-CoA is metabolism through the tricarboxylic acid (TCA) cycle. Because of the tight coupling of the TCA cycle and oxidative phosphorylation, the myocardial turnover of 11C-acetate reflects overall flux in the TCA cycle and thus overall oxidative metabolism, or MVO2. Either exponential curve fitting or compartmental modeling is used to calculate MVO2. The latter is typically preferable in situations of low cardiac output where marked splaying of the input function and spillover of activity from the lungs to the myocardium can decrease the accuracy of the curve-fitting method.15–19
Carbohydrate Metabolism (See Chapter 38)
Most studies of myocardial glucose metabolism with PET have used FDG. This radiotracer competes with glucose for facilitated transport into the sarcolemma, then for hexokinase-mediated phosphorylation. The resultant FDG-6-phosphate is trapped in the cytosol, and the myocardial uptake of FDG is thought to reflect overall anaerobic and aerobic myocardial glycolytic flux.20–23 The myocardial kinetics of FDG have been well characterized, the acquisition scheme is relatively straightforward, and its production has become routine, owing in part to the rapid growth of its clinical use in oncology. As such, it remains the most widely used tracer for determination of myocardial glucose metabolism. Regional myocardial glucose utilization can be assessed in either relative or absolute terms (i.e., in nmol/g−1/min−1). For quantification, a mathematical correction for the kinetic differences between FDG and glucose called the lumped constant must be used to calculate rates of glucose metabolism. Of note, this value may vary depending upon the prevailing plasma substrate and hormonal conditions, decreasing the accuracy of the measurement.22,24–26 Other disadvantages of FDG include the limited metabolic fate of FDG in tissue, precluding determination of the metabolic fate (i.e., glycogen formation versus glycolysis) of the extracted tracer and glucose, and limitations on the performance of serial measurements of myocardial glucose utilization because of the relatively long physical half-life of 18F.
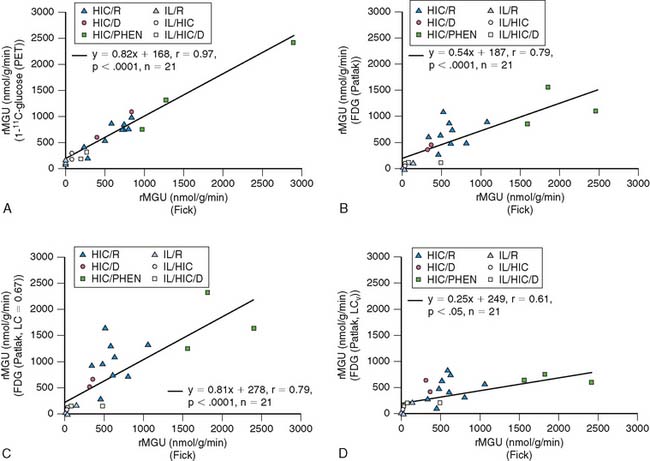
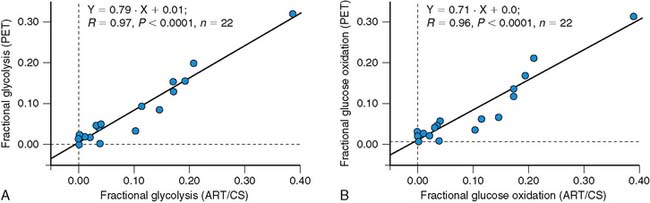
More recently, quantification of myocardial glucose utilization has been performed with PET using glucose radiolabeled in the 1-carbon position with carbon-11 glucose (11C-glucose). Because 11C-glucose is chemically identical to unlabeled glucose, it has the same metabolic fate as glucose, thus obviating the need for the lumped constant correction. It has been demonstrated that measurements of myocardial glucose utilization based on compartmental modeling of tracer kinetics are more accurate with 11C-glucose than with FDG, and can provide estimates of glycogen synthesis, glycolysis, and glucose oxidation (Figs. 40-2 and 40-3).27–29 Disadvantages of this method include compartmental modeling that is more demanding with 11C-glucose than it is with FDG, the need to correct the arterial input function for the production of 11CO2 and 11C-lactate, a fairly complex synthesis of the tracer, and the short physical half-life of 11C, requiring an on-site cyclotron.
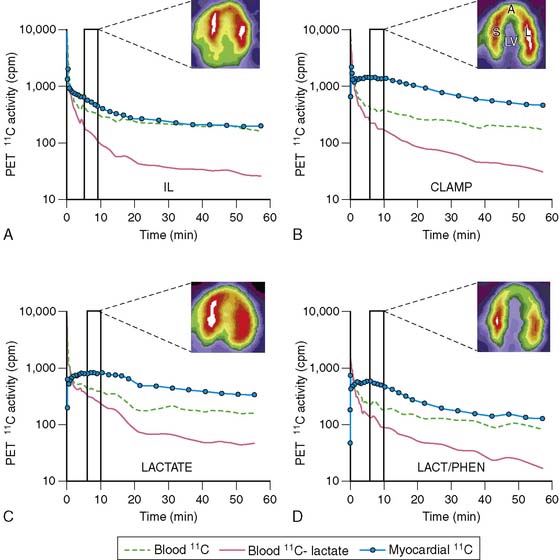
Lactate metabolism in the heart is a key source of energy production, particularly during periods of increased cardiac work. However, to date, the ability to measure myocardial lactate has been limited by the lack of availability of an appropriate radiotracer and analysis scheme. Recently, a multicompartment model was developed for the assessment of myocardial lactate metabolism using PET and L-3-11C-lactate. PET-derived extraction of lactate correlated well with lactate oxidation measured by arterial and coronary sinus sampling over a wide range of conditions (Fig. 40-4).30 This approach may help delineate the clinical role of lactate metabolism in a variety of pathologic conditions such as diabetes mellitus and myocardial ischemia. Moreover, when combined with either FDG or 11C-glucose, it permits a more comprehensive measurement of myocardial carbohydrate metabolism.
Fatty Acid Metabolism
The major advantage of 11C-palmitate is that its myocardial kinetics are identical to labeled palmitate. With appropriate mathematical modeling techniques, its use permits the assessment of various aspects of myocardial fatty acid metabolism such as uptake, oxidation, and storage.31–34 This level of metabolic detail is important because exactly which component of myocardial fatty acid metabolism is the main contributor to a pathologic process is frequently unclear. However, this method does suffer from several disadvantages, including reduced image quality and specificity, a more complex analysis, and the need for an on-site cyclotron and radiopharmaceutical production capability.
Most of the PET tracers in the category of fatty acid tracers that are trapped have been designed to reflect myocardial β-oxidation. One of the first radiotracers developed using this approach was 14-(R,S)-18F-fluoro-6-thiaheptadecanoic acid (FTHA). Initial results were promising, with uptake and retention in the myocardium according with changes in substrate delivery, blood flow, and workload in animal models.35,36 Moreover, PET with FTHA was used to evaluate the effects of various diseases such as coronary artery disease and cardiomyopathy on myocardial fatty acid metabolism.37,38 However, uptake and retention of FTHA has been shown to be insensitive to the inhibition of β-oxidation by hypoxia, reducing enthusiasm for this radiotracer to measure myocardial fatty acid metabolism.39 To circumvent this problem, 16-18F-fluoro-4-thiapalmitate (FTP) has been developed. This modification retains the metabolic trapping function of the radiotracer, which is proportional to fatty acid oxidation under normal oxygenation and hypoxic conditions.39,40 Similar to FDG, quantification of myocardial fatty acid metabolism with FTP requires the use of a lumped constant to correct for kinetic differences between the radiotracer and unlabeled palmitate. However, the stability of the lumped constant under most conditions is unknown. Thus, the ability to quantify fatty acid oxidation with this radiotracer is still unclear. Currently, FTP is undergoing commercialization and entering early phase-1 evaluation. Recently, a new 18F-labeled fatty acid radiotracer, trans-9(RS)-18F-fluoro-3,4(RS,RS) methylene heptadecanoic acid (FCPHA), has been developed.41 This radiotracer is also trapped after undergoing several steps of β-oxidation. Results of initial studies show that uptake of FCPHA into rat myocardium was approximately 1.5% of the injected dose per gram of tissue at 5 minutes, with little change over a period of 60 minutes, and low blood activity over the same period. However, the impact of alterations in plasma substrates, workload, and blood flow on myocardial kinetics is unknown. This radiotracer is also undergoing commercialization.
OVERVIEW OF MYOCARDIAL METABOLISM: NEED FOR FLEXIBILITY IN SUBSTRATE USE
The heart is an omnivore capable of switching between one substrate to another for energy production. This flexibility in substrate permits efficient response to various stimuli such as the pattern of substrate availability, the hormonal milieu, the level of tissue perfusion, and the amount of workload by the heart (Fig. 40-5).42,43 Control of substrate switching can represent either an acute or a chronic adaptation in response to either short or prolonged alterations in the physiologic environment. Inhibitory effects of fatty acid oxidation on glucose oxidation and increasing oxidation of glycogen, lactate, and glucose in response to increasing workload are examples of acute or short-term adaptations. Regulating these rapid changes are a host of enzymes such as pyruvate dehydrogenase complex and enzyme carnitine palmitoyl transferase-1, which is regulated by the concentration of malonyl-CoA.44–48
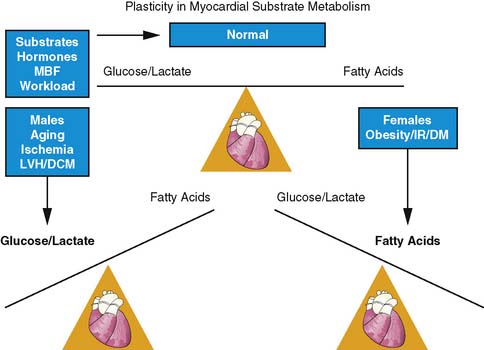
In contrast, chronic metabolic adaptations reflect alterations in gene expression regulating various metabolic pathways. These changes occur primarily at the transcriptional level through the coordinated up-regulation of enzymes and proteins in key metabolic pathways. A prominent example in this case is the nuclear receptor, peroxisome proliferator–activated receptor alpha (PPARα), which is a key regulator of myocardial fatty acid uptake, oxidation, and storage.49 For example, in diabetes mellitus, PPARα activity is increased, leading to an up-regulation in genes controlling fatty acid uptake and oxidation.50 In contrast, pressure-overload hypertrophy PPARα activity is reduced, leading to a down-regulation of genes controlling fat metabolism and in turn leading to an up-regulation of glucose use.51 These chronic adaptations can induce numerous detrimental effects that extend beyond alterations in energy production and may include enhanced oxygen free-radical production, impaired energetics, stimulation of apoptosis, and the induction of LV dysfunction. Discussed in the sections that follow is how metabolic imaging has helped characterize this loss of metabolic flexibility due to these chronic adaptations in various disease processes.
Gender and Aging
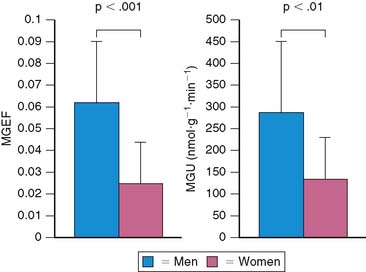
Both gender and aging impact the myocardial metabolic phenotype. Results of studies in animal models show that there are sex differences in myocardial substrate metabolism, with female rats exhibiting less myocardial glucose and more fatty acid metabolism.52,53 Recently, using PET with 11C-glucose and 11C-plamitate, these observations were confirmed in young healthy volunteers.54 Women exhibited lower levels of glucose metabolism compared with men (Fig. 40-6). Although no differences in myocardial fatty acid metabolism were noted, women also exhibited higher MVO2 compared with men, as measured by PET with 11C-acetate. These gender differences in substrate metabolism become more pronounced as one transitions to more pathologic conditions. For example in addition to the changes in glucose metabolism and MVO2, obese women exhibited higher fatty acid uptake and oxidation compared with obese men.55 The differences in myocardial metabolism could not be explained by differences in myocardial blood flow, insulin sensitivity, hemodynamics, myocardial work, or the plasma substrate environment. Differences in hormonal stimulation may play a role in the mechanism of sex-related differences in myocardial substrate metabolism, since results of animal studies have shown that estrogen decreases glucose oxidation, gluconeogenesis, and glycogenolysis and increases fatty acid oxidation in liver and skeletal muscle.56–59 Myocardial glucose utilization may also be decreased by estrogen; it is known to up-regulate nitric oxide synthases, which cause a reduction in GLUT4 translocation to the cell surface, thereby inhibiting myocardial glucose uptake.60–62 Indeed, in a small retrospective study, PET-derived measurements of myocardial fatty acid metabolism were higher in postmenopausal women, compared with either postmenopausal women not receiving hormonal replacement therapy or age-matched men consistent with a decline in glucose uptake.63
In various experimental models of aging, the contribution of fatty acid oxidation to overall myocardial substrate metabolism declines with age.64,65 It appears the cause for the decrease in fatty acid oxidation is multifactorial, ranging from changes in mitochondrial lipid content, oxygen free-radical injury, a decline in carnitine palmitoyltransferase-1 activity, and an age-related decline in myocardial PPARα activity.66–68 Using the PET approaches described above, it has been shown that a similar metabolic shift occurs in healthy older humans.69 However, despite this preference for glucose as an energy substrate, older individuals are unable to increase glucose utilization in response to β-adrenergic stimulation with dobutamine to the same extent as younger individuals. This impaired metabolic response may portend a stress-related energy deprivation state in the aging heart or potentially indicate that the heart is more susceptible to injury during periods of ischemia.70 Recently it has been shown that this impairment in metabolic reserve can be ameliorated by endurance exercise training in older subjects.71 In keeping with our earlier discussion, it appears the myocardial metabolic response to dobutamine following endurance exercise training is gender specific, with men demonstrating an increase in myocardial glucose metabolism and a decline in fatty acid use but women exhibiting increases in both glucose and fatty acid metabolism. Although these gender and age differences in metabolism merit further study, they may provide a partial explanation for the gender- and age-related outcome differences for various cardiovascular diseases where altered myocardial metabolism plays a role.
Ischemia
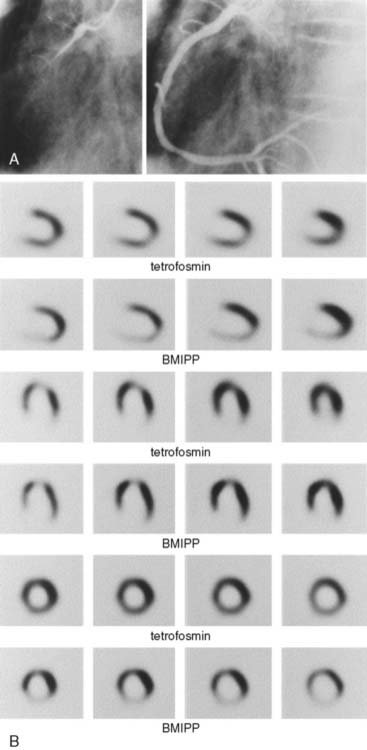
With the induction of myocardial ischemia, fatty acid oxidation ceases and anaerobic metabolism supervenes. Glucose becomes the primary substrate for both increased anaerobic glycolysis and for continued, albeit diminished, oxidative metabolism.72 This metabolic switch is a prerequisite for continued energy production and cell survival. When the ischemic insult is reversed, oxygen availability increases and oxidative metabolism resumes. It appears that these abnormalities in myocardial substrate metabolism may persist well after the resolution of ischemia—so-called ischemic memory. Demonstration of either accelerated myocardial glucose metabolism or reduced fatty acid metabolism using FDG and BMIPP, respectively, has been used to document this phenomenon. Over 20 years ago, it was shown that PET myocardial FDG uptake was increased in patients with unstable angina during pain-free episodes.73 Moreover, in patients with stable angina, increased FDG uptake was demonstrated following exercise-induced ischemia in the absence of either perfusion deficits or ECG abnormalities.74 Similar observations have been made with SPECT using BMIPP. It is apparent in patients with acute chest pain that abnormalities in myocardial BMIPP uptake may persist 24 to 36 hours following the resolution of symptoms (Fig. 40-7).75,76 Moreover, this “metabolic fingerprint” appears superior to perfusion imaging for either identifying coronary artery disease as the cause of the chest pain or assigning prognosis.75 This persistence in the metabolic defect is a significant advantage. It increases the flexibility of radiotracer administration, because it allows for delivery of a unit dose after the patient has already been evaluated. This is in contrast to the use of perfusion radiotracers, which frequently must be available on-site owing to the narrow time window from the resolution of symptoms and normalization of the flow deficit. Based on these observations, BMIPP is currently undergoing phase-3 evaluation for acute chest pain imaging. Of note, the finding of reduced fatty acid metabolism may be a marker of cardiac risk in other patient populations. For example, in patients with end-stage renal disease, cardiac death is significantly associated with highly abnormal BMIPP uptake.77 Metabolic imaging with either FDG or BMIPP has also been used for direct ischemia detection during stress testing—the thought process being that abnormalities in vasodilator reserve with perfusion tracers will underestimate ischemia if oxygen demand and supply remain balanced. Results of initial studies where FDG was injected during exercise appear to support this contention, with a greater detection rate for moderately severe coronary artery stenoses, compared with perfusion imaging.5 Moreover, it appears defects in either glucose or fatty acid metabolism with exercise will persist for approximately 24 hours, providing further credence for the ischemic memory concept.78,79 Despite the promising results with these radiotracers, numerous questions still remain as to the optimal imaging protocols, the impact of alterations in the plasma substrate environment on diagnostic accuracy, whether added diagnostic and prognostic information is provided over perfusion imaging, and whether this information alters clinical management.
Hypertension/Left Ventricular Hypertrophy
There are numerous lines of evidence supporting the close relationship between abnormalities in myocardial substrate metabolism and LV hypertrophy. In animal models of pressure-overload LV hypertrophy there is a reduction in the expression of β-oxidation enzymes, leading to a fall in myocardial fatty acid oxidation and an increase in glucose use.80,81 Moreover, interventions in animals that involve inhibition of mitochondrial fatty acid β-oxidation result in cardiac hypertrophy.81 In humans, variants in genes regulating key aspects of myocardial fatty acid metabolism ranging from PPARα to various key β-oxidative enzymes are associated with LV hypertrophy.82,83
In a rat model of hypertrophy, PET with FDG demonstrated myocardial glucose uptake tracked directly with increasing hypertrophy, confirming this shift in substrate preference in vivo.84 Similar results have been found in man. PET with 11C-palmitate in humans has shown that reduction in myocardial fatty acid oxidation is an independent predictor of LV mass in hypertension.85 Combining measurements of LV myocardial external work (either by echocardiography or MRI) with measurements of MVO2 performed by PET with 11C-acetate or 15O-oxygen, it is possible to estimate cardiac efficiency.13,86 Using this approach in patients with hypertension-induced LV hypertrophy has shown that the decline in myocardial fatty acid metabolism is associated with a decline in efficiency, a condition that may increase the potential for the development of heart failure. An exciting potential application of PET is the phenotyping of patients with genetic variants related to myocyte growth. For example, in patients with hypertrophic cardiomyopathy attributable to a known specific variant in the alpha-tropomyosin gene, it was observed that increased myocardial perfusion, fatty acid metabolism, and efficiency characterize patients with mild hypertrophy, whereas these metabolic alterations decrease as hypertrophy becomes more advanced.87 The results may represent differential penetrance of the gene variant or the effect of modifier gene(s), potentially helping in their identification. This study suggests that metabolic imaging may identify relevant gene variants without waiting for more end-stage manifestations such as LV remodeling and dysfunction to occur, although further study in larger patient populations is warranted.
Nonischemic Dilated Cardiomyopathy
As with LV hypertrophy, alterations in myocardial substrate metabolism have been implicated in the pathogenesis of contractile dysfunction and heart failure. Animal models of heart failure have shown that during the progression from cardiac hypertrophy to ventricular dysfunction, the expression of genes encoding for enzymes regulating β-oxidation is coordinately decreased, resulting in a shift in myocardial substrate metabolism to primarily glucose use, similar to that seen in the fetal heart.80,88,89 These metabolic changes are paralleled by reexpression of fetal isoforms of a variety of contractile and calcium regulatory protein programs. The reactivation of the metabolic fetal gene program may have numerous detrimental consequences on myocardial contractile function, ranging from energy deprivation to the inability to process fatty acids, leading to accumulation of nonoxidized toxic fatty acid derivatives, resulting in lipotoxicity. The importance of this metabolic shift in the pathogenesis of heart failure is underscored by the development of novel therapeutics that target specific aspects of cellular metabolism, such as the partial fatty acid oxidation antagonists and the insulin sensitizer glucagon-like peptide-1.90
The down-regulation in myocardial fatty acid metabolism leading to overdependence on glucose use in heart failure has been well documented using both SPECT and PET techniques. For example, SPECT with BMIPP demonstrated reduced myocardial uptake and increased clearance radiotracer in patients with dilated cardiomyopathy, compared with controls.91 Moreover, the magnitude of the perturbations correlated with other measurements of heart failure severity such as LV size and plasma B-natriuretic peptide levels. PET using 11C-palmitate and 11C-glucose demonstrated that myocardial fatty acid uptake and oxidation are lower in patients with nonischemic dilated cardiomyopathy when compared with age-matched controls. In contrast, myocardial glucose utilization was higher in the cardiomyopathic patients.32 Similarly, PET has also been used to provide mechanistic insights into the myocardial metabolic perturbations associated with heart failure. For example, abrupt lowering of fatty acid delivery with acipimox results in reduced fatty acid uptake, MVO2, and cardiac work, and no change in cardiac efficiency in normal volunteers.92 In contrast, in patients with nonischemic dilated cardiomyopathy, there is a decrease in myocardial fatty acid uptake and cardiac work, no change in MVO2, and a decline in efficiency. Although limited by a small sample size, these results appear to reinforce the central role of loss of flexibility in myocardial substrate metabolism in the pathogenesis of heart failure, with even minor changes in substrate delivery having detrimental consequences on cardiac energy transduction.
Metabolic imaging can also been used to study the mechanisms responsible for the effectiveness of treatment in cardiomyopathy. For example, the efficacy of β-blocker therapy in treatment of heart failure patients is well established. Treatment with the selective β-blocker metoprolol results in a reduction in oxidative metabolism and an improvement in cardiac efficiency, as measured by PET in patients with heart failure.93 Cardiac efficiency has been shown to improve following either exercise training or cardiac resynchronization therapy, implicating improved myocardial energy transduction as a potential mechanism.94,95 Moreover, normalization of the distribution of myocardial glucose metabolism may be partially responsible for the improved energetic state following cardiac resynchronization therapy.96 Partial fatty acid oxidation inhibitors have been proposed for the treatment of heart failure, based on the theory that decreasing myocardial fatty acid oxidation should increase the oxidation of glucose, leading to a more favorable energetic state and improved LV function. This is because much less oxygen is required to oxidize glucose compared with fatty acids. In support of this contention is the finding that the administration of trimetazidine to patients with dilated cardiomyopathy results in a significant improvement in left ventricular ejection fraction.97 However, the improvement in LV function is paralleled by only a mild decrease in myocardial fatty acid oxidation. It appears that with this drug, the improvement in LV function reflects the complex interplay between a mild decrease in myocardial fatty acid oxidation, improved whole-body insulin resistance, and synergistic effects with β-blockade. Metabolic imaging can also be used to predict the response to specific therapies in heart failure patients. For example, in patients with dilated cardiomyopathy, it appears the fraction of myocardial glucose uptake, as measured by PET with FDG, predicts the effectiveness of β-blocker therapy.98 Moreover, in patients with ischemic cardiomyopathy, the extent of viable myocardium as measured with PET and FDG correlated with the hemodynamic response after cardiac resynchronization therapy, suggesting a role for PET in discriminating responders from nonresponders to this therapy.99
Diabetes Mellitus (See Chapters 2 and 29)
Cardiovascular disease is the leading cause of morbidity and mortality in patients with diabetes mellitus.100 The mechanisms responsible for the increased risk are multifactorial and complex, with possibilities including an increased prevalence of hyperlipidemia and hypertension, impaired fibrinolysis, abnormal myocardial endothelial function, and reduced sympathetic neuronal function. However, there is extensive evidence to suggest that abnormalities in myocardial substrate metabolism contribute to the cardiovascular abnormalities observed in diabetic patients.101,102 As mentioned previously, the metabolic phenotype in diabetes is an overdependence on fatty acid metabolism and a decrease in glucose use. Multiple mechanisms contribute to this phenotype. These include increased plasma delivery of fatty acids due to peripheral insulin resistance, decreased insulin signaling, and activation of key transcriptional pathways such as the PPARα/PGC-1 signaling network.103–105 Both insulin-mediated glucose transport and glucose transporter expression decline in diabetes mellitus. However, rates of myocardial glucose uptake are frequently normal owing to the presence of hyperglycemia. The increase in myocardial fatty acid utilization results in increased citrate levels, which inhibit phosphofructokinase. Glucose oxidation is inhibited at the level of pyruvate dehydrogenase complex due to increased mitochondrial acetyl-CoA levels and the phosphorylation of pyruvate dehydrogenase kinase 4 by PPARα activation. Consequently, the maintenance of myocardial glucose uptake but a decrease in its downstream metabolism results in an accumulation of glucose metabolites. Potential detrimental effects associated with this shift in metabolism include: impaired mechanical function as a result of the inability to increase glucose metabolism in response to increased myocardial work, depletion of TCA cycle intermediates due to reduced anaplerosis, electrical instability and apoptosis, greater sensitivity to myocardial ischemia, and myocardial lipid accumulation or lipotoxicity, leading to increased apoptosis.
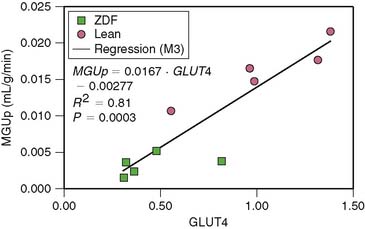
Small-animal imaging has helped clarify the mechanisms responsible for the metabolic alterations that occur in diabetes mellitus. For example, mice with cardiac-restricted overexpression of PPARα demonstrate a metabolic phenotype that is similar to diabetic hearts.106 Small-animal PET studies with 11C-palmitate and FDG in these mice demonstrate an increase in fatty acid uptake and an abnormal suppression of glucose uptake. In contrast, in mice with cardiac-restricted overexpression of PPARβ/σ, small-animal PET measurements demonstrated an increase glucose uptake and reduced fatty acid uptake.107 Taken in sum, these observations demonstrate that PPARα and PPARβ/σ drive different metabolic regulatory programs in the heart and that imaging can help characterize genetic manipulations in mouse heart. However, these studies also demonstrate the challenges in imaging the mouse heart, owing to its small size; only semiquantitative measurements of tracer uptake can currently be performed. However, quantitative measures of myocardial substrate metabolism are now possible with small-animal PET in rat heart. For example, rates of myocardial glucose uptake correlate directly and closely with GLUT4 gene expression in the Zucker diabetic fat (ZDF) rat, a model of type 2 diabetes mellitus (Fig. 40-8).108 Moreover, rates of myocardial glucose uptake and fatty acid uptake and oxidation measured with PET in the same disease model demonstrated the importance of increased fatty acid delivery to defining the metabolic phenotype in diabetes.109
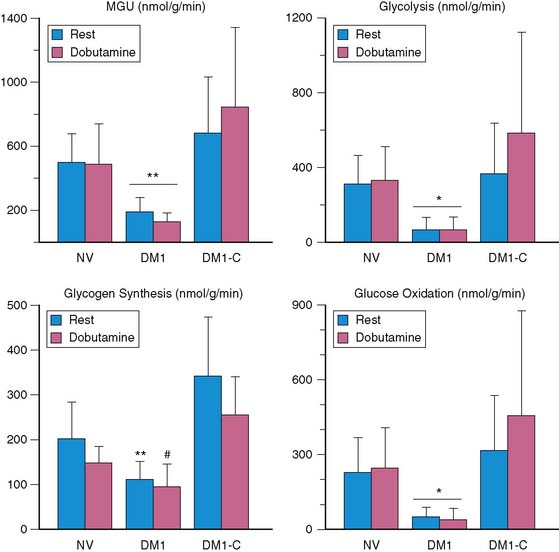
Numerous imaging studies have been performed in humans to assess the impact of diabetes mellitus on myocardial glucose metabolism. These studies have been primarily limited to PET using FDG.110–112 In general, rates of myocardial glucose uptake are reduced in patients with either type 1 or type 2 diabetes mellitus compared with nondiabetics, except under conditions of marked hyperglycemia or supraphysiologic levels in plasma insulin (such as occurs with a hyperinsulinemic-euglycemic clamp). Increased myocardial fatty acid uptake measured by arterial coronary sinus balance studies has been reported in humans with type 1 diabetes mellitus without coronary artery disease.113 Although the impact of plasma levels of free fatty acids on the level of myocardial fatty acid uptake was not determined, a negative correlation between myocardial glucose uptake and plasma fatty acid levels was observed. Recently, studies using PET and 11C-plamitate and 11C-glucose in patients with type 1 diabetes mellitus have helped verify these findings noninvasively. Patients with diabetes exhibited higher levels of fatty acid uptake and oxidation compared with nondiabetics, primarily due to increased plasma fatty acid levels. In contrast, glucose uptake was reduced in these patients, primarily due to decreased glucose transport mechanisms.33 Moreover, the metabolic fate of extracted glucose is impaired in diabetes, with reduced rates of glycolysis and glucose oxidation that become more pronounced with increases in cardiac work induced by dobutamine (Fig. 40-9).114 However, the diabetic myocardium is responsive to changes in plasma insulin and fatty acid levels, but at a cost. Higher insulin levels are needed to achieve the same level of glucose uptake and glucose oxidation compared with nondiabetics, consistent with myocardial insulin resistance. Similarly, in response to higher fatty acid plasma levels, myocardial fatty acid uptake is increased, but at the cost of a greater esterification rate.114,115
The increase in plasma fatty acids is an attractive target to reduce the overdependence of the myocardium on fatty acid metabolism and perhaps improve energetics and function of the left ventricle. For example, the use of the insulin-sensitizing agent troglitazone in ZDF rats results in reduced plasma fatty acid levels, decreased myocardial lipid accumulation, reduced apoptosis, and improved LV function.116 In PET with FDG, studies in patients with type 2 diabetes mellitus—before and 26 weeks after treatment with rosiglitazone—demonstrated nearly a 40% increase in insulin-stimulated myocardial glucose uptake, implying reduced fatty acid uptake, which was attributed in large part to suppression in plasma fatty acid levels.117 Of note, similar metabolic changes were not seen with the biguanide metformin, whose mechanism of action is designed to reduce hepatic glucose production. The metabolic response could not be predicted by changes in the plasma glucose or hemoglobin A1c levels. Thus, metabolic imaging can be used to follow the effects of therapies designed to alter myocardial substrate metabolism in patients with diseases such as diabetes mellitus, where more readily available clinical parameters are not predictive of a therapeutic response.
Obesity and Insulin Resistance
It is now apparent that a significant increase in body mass index (BMI) induces marked increases in myocardial fatty acid metabolism. For example, in either dietary-induced or transgenic models of obesity, myocardial fatty acid uptake and oxidation are significantly increased.116,118 This increase at least initially reflects the increase in fatty acid delivery to the heart secondary to increased lipolysis from both visceral/abdominal and subcutaneous fat stores due to insulin resistance. Similar to diabetes mellitus, the increased delivery of fatty acids likely initiates a cascade of events that lead to increased fatty acid metabolism. Ultimately, fatty acid uptake may exceed oxidation, leading to extracted fatty acids entering nonoxidative pathways, most likely initially forming triglycerides. As mentioned, the accumulation of neutral fats or triglycerides may ultimately become detrimental.116
Imaging of obese young women with PET and 11C-acetate and 11C-palmitate has demonstrated that an increase in BMI is associated with a shift in myocardial substrate metabolism toward greater fatty acid use. Moreover, this dependence on myocardial fatty acid metabolism increased with worsening insulin resistance.34 Of note, little change in myocardial glucose metabolism was observed. Paralleling the preferential use of fatty acids was an increase in MVO2 and a decrease in energy transduction. These findings suggest that metabolic changes in obesity may play a role in the pathogenesis of cardiac dysfunction. Also of note, the myocardial metabolic response to obesity appears to be gender dependent.55 For example, using similar PET techniques, it has been recently demonstrated that in contrast to obese women, obese men had a greater impairment in myocardial glucose metabolism per level of plasma insulin, suggesting greater myocardial insulin resistance. In addition, obesity had less effect on myocardial fatty acid metabolism in men. In contrast, MVO2 was higher in the obese women compared with obese men. Thus it appears there may be a complex interplay between gender and obesity in influencing myocardial substrate metabolism.
BEYOND THE MYOCARDIUM: VASCULAR IMAGING (See Chapter 44)
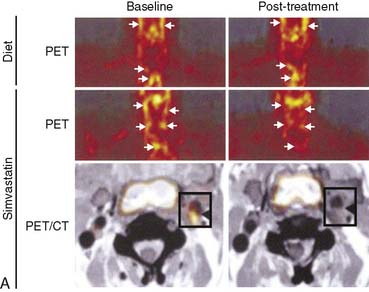
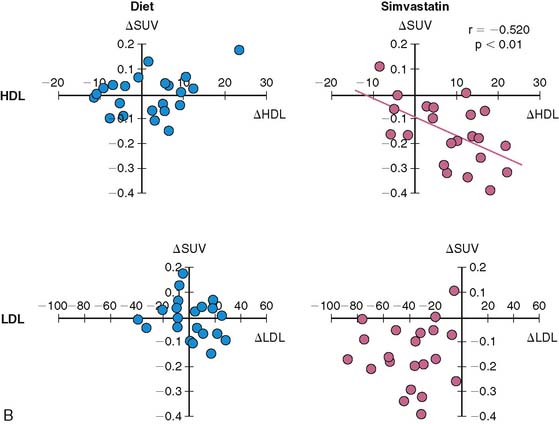
Atherosclerosis is a dynamic immune inflammatory process characterized by cycles of intense activity and progression followed by intervals of stabilization. A common result of this process is coronary artery luminal stenosis that compromises myocardial blood flow and induces ischemia during stress. However, the most devastating event is acute plaque rupture with thrombosis, leading to myocardial infarction and frequently sudden cardiac death. More concerning is that for many patients, acute plaque rupture is the initial clinical manifestation of coronary atherosclerosis. Although many imaging tools are currently available to detect and characterize the extent and severity of coronary atherosclerosis, none of them identifies patients with active disease and at risk of plaque rupture. To this end, FDG is being evaluated for the detection of “biologically active” atherosclerosis, based on the premise that the tracer accumulates in activated macrophages, which are a key component of atherosclerotic plaque. Several groups have established that inflamed arterial vessels have increased uptake of FDG, as measured by PET. The increased uptake has been noted in animal models of atherosclerosis and verified in humans with atherosclerosis of the carotid artery and aorta.119–122 Moreover, a significant correlation between FDG uptake and CD68 cell staining has recently been established. Of interest, a decrease in carotid artery FDG uptake is correlative with an increase in plasma high-density lipoprotein levels following statin therapy (Fig. 40-10).121 However, many questions remain, such as the site of localization of radiotracer (e.g., plaque or smooth muscle), the suitability of the method for evaluating the coronary arteries, and whether the information provides more refined risk stratification compared with other more widely applicable methods or if it alters therapy.
1. Szczepaniak L.S., Babcock E.E., Schick F., et al. Measurement of intracellular triglyceride stores by H spectroscopy: validation in vivo. Am J Physiol. 1999;276(5 Pt 1):E977-E989.
2. Yoshida T. The rate of phosphocreatine hydrolysis and resynthesis in exercising muscle in humans using 31P-MRS. J Physiol Anthropol Appl Human Sci. 2002;21(5):247-255.
3. Lewandowski E.D. Cardiac carbon 13 magnetic resonance spectroscopy: on the horizon or over the rainbow? J Nucl Cardiol. 2002;9(4):419-428.
4. Bottomley P.A., Weiss R.G. Non-invasive magnetic-resonance detection of creatine depletion in non-viable infarcted myocardium. Lancet. 1998;351(9104):714-718.
5. He Z.X., Shi R.F., Wu Y.J., et al. Direct imaging of exercise-induced myocardial ischemia with fluorine-18-labeled deoxyglucose and Tc-99m-sestamibi in coronary artery disease. Circulation. 2003;108(10):1208-1213.
6. DeGrado T.R., Holden J.E., Ng C.K., Raffel D.M., Gatley S.J. Quantitative analysis of myocardial kinetics of 15-p-[iodine-125] iodophenylpentadecanoic acid. J Nucl Med. 1989;30(7):1211-1218.
7. Dormehl I.C., Hugo N., Rossouw D., White A., Feinendegen L.E. Planar myocardial imaging in the baboon model with iodine-123–15-(iodophenyl)pentadecanoic acid (IPPA) and iodine-123–15-(P-iodophenyl)-3-R,S-methylpentadecanoic acid (BMIPP), using time-activity curves for evaluation of metabolism. Nucl Med Biol. 1995;22(7):837-847.
8. Eckelman W.C., Babich J.W. Synthesis and validation of fatty acid analogs radiolabeled by nonisotopic substitution. J Nucl Cardiol. 2007;14(Suppl 3):S100-S109.
9. Reske S.N., Sauer W., Machulla H.J., Knust J., Winkler C. Metabolism of 15 (p 123I iodophenyl-)pentadecanoic acid in heart muscle and noncardiac tissues. Eur J Nucl Med. 1985;10(5–6):228-234.
10. Ambrose K.R., Owen B.A., Goodman M.M., Knapp F.F.Jr. Evaluation of the metabolism in rat hearts of two new radioiodinated 3-methyl-branched fatty acid myocardial imaging agents. Eur J Nucl Med. 1987;12(10):486-491.
11. Goodman M.M., Kirsch G., Knapp F.F.Jr. Synthesis and evaluation of radioiodinated terminal p-iodophenyl-substituted alpha- and beta-methyl-branched fatty acids. J Med Chem. 1984;27(3):390-397.
12. Iida H., Rhodes C.G., Araujo L.I., et al. Noninvasive quantification of regional myocardial metabolic rate for oxygen by use of 15O2 inhalation and positron emission tomography. Theory, error analysis, and application in humans. Circulation. 1996;94(4):792-807.
13. Laine H., Katoh C., Luotolahti M., et al. Myocardial oxygen consumption is unchanged but efficiency is reduced in patients with essential hypertension and left ventricular hypertrophy. Circulation. 1999;100(24):2425-2430.
14. Yamamoto Y., de Silva R., Rhodes C.G., et al. Noninvasive quantification of regional myocardial metabolic rate of oxygen by 15O2 inhalation and positron emission tomography. Experimental validation. Circulation. 1996;94(4):808-816.
15. Armbrecht J.J., Buxton D.B., Schelbert H.R. Validation of [1–11C]acetate as a tracer for noninvasive assessment of oxidative metabolism with positron emission tomography in normal, ischemic, postischemic, and hyperemic canine myocardium. Circulation. 1990;81(5):1594-1605.
16. Brown M., Marshall D.R., Sobel B.E., Bergmann S.R. Delineation of myocardial oxygen utilization with carbon-11-labeled acetate. Circulation. 1987;76(3):687-696.
17. Brown M.A., Myears D.W., Bergmann S.R. Noninvasive assessment of canine myocardial oxidative metabolism with carbon-11 acetate and positron emission tomography. J Am Coll Cardiol. 1988;12(4):1054-1063.
18. Buck A., Wolpers H.G., Hutchins G.D., et al. Effect of carbon-11-acetate recirculation on estimates of myocardial oxygen consumption by PET. J Nucl Med. 1991;32(10):1950-1957.
19. Sun K.T., Yeatman L.A., Buxton D.B., et al. Simultaneous measurement of myocardial oxygen consumption and blood flow using [1-carbon-11]acetate. J Nucl Med. 1998;39(2):272-280.
20. Choi Y., Hawkins R.A., Huang S.C., et al. Parametric images of myocardial metabolic rate of glucose generated from dynamic cardiac PET and 2-[18F]fluoro-2-deoxy-d-glucose studies. J Nucl Med. 1991;32(4):733-738.
21. Gambert S., Vergely C., Filomenko R., et al. Adverse effects of free fatty acid associated with increased oxidative stress in postischemic isolated rat hearts. Mol Cell Biochem. 2006;283(1–2):147-152.
22. Iozzo P., Chareonthaitawee P., Di Terlizzi M., Betteridge D.J., Ferrannini E., Camici P.G. Regional myocardial blood flow and glucose utilization during fasting and physiological hyperinsulinemia in humans. Am J Physiol Endocrinol Metab. 2002;282(5):E1163-E1171.
23. Krivokapich J., Huang S.C., Selin C.E., Phelps M.E. Fluorodeoxyglucose rate constants, lumped constant, and glucose metabolic rate in rabbit heart. Am J Physiol. 1987;252(4 Pt 2):H777-H787.
24. Botker H.E., Bottcher M., Schmitz O., et al. Glucose uptake and lumped constant variability in normal human hearts determined with [18F]fluorodeoxyglucose. J Nucl Cardiol. 1997;4(2 Pt 1):125-132.
25. Hariharan R., Bray M., Ganim R., Doenst T., Goodwin G.W., Taegtmeyer H. Fundamental limitations of [18F]2-deoxy-2-fluoro-D-glucose for assessing myocardial glucose uptake. Circulation. 1995;91(9):2435-2444.
26. Hashimoto K., Nishimura T., Imahashi K.I., Yamaguchi H., Hori M., Kusuoka H. Lumped constant for deoxyglucose is decreased when myocardial glucose uptake is enhanced. Am J Physiol. 1999;276(1 Pt 2):H129-133.
27. Herrero P., Sharp T.L., Dence C., Haraden B.M., Gropler R.J. Comparison of 1-(11)C-glucose and (18)F-FDG for quantifying myocardial glucose use with PET. J Nucl Med. 2002;43(11):1530-1541.
28. Herrero P., Weinheimer C.J., Dence C., Oellerich W.F., Gropler R.J. Quantification of myocardial glucose utilization by PET and 1-carbon-11-glucose. J Nucl Cardiol. 2002;9(1):5-14.
29. Herrero P., Kisrieva-Ware Z., Dence C.S., et al. PET measurements of myocardial glucose metabolism with 1–11C-glucose and kinetic modeling. J Nucl Med. 2007;48(6):955-964.
30. Herrero P., Dence C.S., Coggan A.R., Kisrieva-Ware Z., Eisenbeis P., Gropler R.J. L-3–11C-lactate as a PET tracer of myocardial lactate metabolism: a feasibility study. J Nucl Med. 2007;48(12):2046-2055.
31. Bergmann S.R., Weinheimer C.J., Markham J., Herrero P. Quantitation of myocardial fatty acid metabolism using PET. J Nucl Med. 1996;37(10):1723-1730.
32. Davila-Roman V.G., Vedala G., Herrero P., et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40(2):271-277.
33. Herrero P., Peterson L.R., McGill J.B., et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2006;47(3):598-604.
34. Peterson L.R., Herrero P., Schechtman K.B., et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation. 2004;109(18):2191-2196.
35. DeGrado T.R. Synthesis of 14(R,S)-[18F]fluoro-6-thia-heptadecanoic acid (FTHA). J Label Comp Radiopharm. 1991;29:989-995.
36. DeGrado T.R., Coenen H.H., Stocklin G. 14(R,S)-[18F]fluoro-6-thia-heptadecanoic acid (FTHA): evaluation in mouse of a new probe of myocardial utilization of long chain fatty acids. J Nucl Med. 1991;32(10):1888-1896.
37. Schulz G., von Dahl J., Kaiser H.J., et al. Imaging of beta-oxidation by static PET with 14(R,S)-[18F]-fluoro-6-thiaheptadecanoic acid (FTHA) in patients with advanced coronary heart disease: a comparison with 18FDG-PET and 99Tcm-MIBI SPET. Nucl Med Commun. 1996;17(12):1057-1064.
38. Taylor M., Wallhaus T.R., Degrado T.R., et al. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro-6-thia-heptadecanoic acid and [18F]FDG in Patients with Congestive Heart Failure. J Nucl Med. 2001;42(1):55-62.
39. DeGrado T.R., Wang S., Holden J.E., Nickles R.J., Taylor M., Stone C.K. Synthesis and preliminary evaluation of (18)F-labeled 4-thia palmitate as a PET tracer of myocardial fatty acid oxidation. Nucl Med Biol. 2000;27(3):221-231.
40. DeGrado T.R., Kitapci M.T., Wang S., Ying J., Lopaschuk G.D. Validation of 18F-fluoro-4-thia-palmitate as a PET probe for myocardial fatty acid oxidation: effects of hypoxia and composition of exogenous fatty acids. J Nucl Med. 2006;47(1):173-181.
41. Shoup T.M., Elmaleh D.R., Bonab A.A., Fischman A.J. Evaluation of trans-9–18F-fluoro-3,4-Methyleneheptadecanoic acid as a PET tracer for myocardial fatty acid imaging. J Nucl Med. 2005;46(2):297-304.
42. Bing R.J. The metabolism of the heart. Harvey Lect. 1955;50:27-70.
43. Neely J.R., Morgan H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Ann Rev Physiol. 1974;36:413-459.
44. Goodwin G.W., Taegtmeyer H. Regulation of fatty acid oxidation of the heart by MCD and ACC during contractile stimulation. Am J Physiol. 1999;277(4 Pt 1):E772-E777.
45. McGarry J.D., Brown N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur J Biochem. 1997;244(1):1-14.
46. McGarry J.D., Mannaerts G.P., Foster D.W. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest. 1977;60(1):265-270.
47. Sugden M.C., Holness M.J. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. Am J Physiol Endocrinol Metab. 2003;284(5):E855-E862.
48. Young M.E., Goodwin G.W., Ying J., et al. Regulation of cardiac and skeletal muscle malonyl-CoA decarboxylase by fatty acids. Am J Physiol Endocrinol Metab. 2001;280(3):E471-E479.
49. Kelly D.P. PPARs of the heart: three is a crowd. Circ Res. 2003;92(5):482-484.
50. Finck B.N., Han X., Courtois M., et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A. 2003;100(3):1226-1231.
51. Depre C., Shipley G.L., Chen W., et al. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nat Med. 1998;4(11):1269-1275.
52. Desrois M., Sidell R.J., Gauguier D., Davey C.L., Radda G.K., Clarke K. Gender differences in hypertrophy, insulin resistance and ischemic injury in the aging type 2 diabetic rat heart. J Mol Cell Cardiol. 2004;37(2):547-555.
53. Dyck J.R., Lopaschuk G.D. Glucose metabolism, H+ production and Na+/H+-exchanger mRNA levels in ischemic hearts from diabetic rats. Mol Cell Biochem. 1998;180(1–2):85-93.
54. Peterson L.R., Soto P.F., Herrero P., Schechtman K.B., Dence C., Gropler R.J. Sex differences in myocardial oxygen and glucose metabolism. J Nucl Cardiol. 2007;14(4):573-581.
55. Peterson L.R., Soto P.M., Herrero P., et al. Impact of gender on the myocardial metabolic response to Obesity. J Am Coll Cardiol Imaging. 2008;1:424-433.
56. Hatta H., Atomi Y., Shinohara S., Yamamoto Y., Yamada S. The effects of ovarian hormones on glucose and fatty acid oxidation during exercise in female ovariectomized rats. Horm Metab Res. 1988;20(10):609-611.
57. Campbell S.E., Febbraio M.A. Effect of ovarian hormones on mitochondrial enzyme activity in the fat oxidation pathway of skeletal muscle. Am J Physiol Endocrinol Metab. 2001;281(4):E803-E808.
58. Matute M.L., Kalkhoff R.K. Sex steroid influence on hepatic gluconeogenesis and glucogen formation. Endocrinology. 1973;92(3):762-768.
59. Kendrick Z.V., Ellis G.S. Effect of estradiol on tissue glycogen metabolism and lipid availability in exercised male rats. J Appl Physiol. 1991;71(5):1694-1699.
60. Lei B., Matsuo K., Labinskyy V., et al. Exogenous nitric oxide reduces glucose transporters translocation and lactate production in ischemic myocardium in vivo. Proc Natl Acad Sci U S A. 2005;102(19):6966-6971.
61. Weiner C.P., Lizasoain I., Baylis S.A., Knowles R.G., Charles I.G., Moncada S. Induction of calcium-dependent nitric oxide synthases by sex hormones. Proc Natl Acad Sci U S A. 1994;91(11):5212-5216.
62. Recchia F.A., McConnell P.I., Loke K.E., Xu X., Ochoa M., Hintze T.H. Nitric oxide controls cardiac substrate utilization in the conscious dog. Cardiovasc Res. 1999;44(2):325-332.
63. Herrero P., Soto P.F., Dence C.S., et al. Impact of hormone replacement on myocardial fatty acid metabolism: potential role of estrogen. J Nucl Cardiol. 2005;12(5):574-581.
64. Abu-Erreish G.M., Neely J.R., Whitmer J.T., Whitman V., Sanadi D.R. Fatty acid oxidation by isolated perfused working hearts of aged rats. Am J Physiol. 1977;232(3):E258-E262.
65. McMillin J.B., Taffet G.E., Taegtmeyer H., Hudson E.K., Tate C.A. Mitochondrial metabolism and substrate competition in the aging Fischer rat heart. Cardiovasc Res. 1993;27(12):2222-2228.
66. Iemitsu M., Miyauchi T., Maeda S., et al. Aging-induced decrease in the PPAR-alpha level in hearts is improved by exercise training. Am J Physiol Heart Circ Physiol. 2002;283(5):H1750-H1760.
67. Odiet J.A., Boerrigter M.E., Wei J.Y. Carnitine palmitoyl transferase-I activity in the aging mouse heart. Mech Ageing Dev. 1995;79(2–3):127-136.
68. Paradies G., Ruggiero F.M., Gadaleta M.N., Quagliariello E. The effect of aging and acetyl-L-carnitine on the activity of the phosphate carrier and on the phospholipid composition in rat heart mitochondria. Biochim Biophys Acta. 1992;1103(2):324-326.
69. Kates A.M., Herrero P., Dence C., et al. Impact of aging on substrate metabolism by the human heart. J Am Coll Cardiol. 2003;41:293-299.
70. Soto P.F., Herrero P., Kates A.M., et al. Impact of aging on myocardial metabolic response to dobutamine. Am J Physiol Heart Circ Physiol. 2003;285:2158-2164.
71. Soto P.F., Herrero P., Schechtman K.B., et al. Exercise training impacts the myocardial metabolism of older individuals in a gender-specific manner. Am J Physiol Heart Circ Physiol. 2008;295(2):H842-H850.
72. Lopaschuk G. Regulation of carbohydrate metabolism in ischemia and reperfusion. Am Heart J. 2000;139(2 Pt 3):S115-S119.
73. Araujo L.I., Camici P., Spinks T.J., Jones T., Maseri A. Abnormalities in myocardial metabolism in patients with unstable angina as assessed by positron emission tomography. Cardiovasc Drugs Ther. 1988;2(1):41-46.
74. Camici P., Araujo L.I., Spinks T., et al. Increased uptake of 18F-fluorodeoxyglucose in postischemic myocardium of patients with exercise-induced angina. Circulation. 1986;74(1):81-88.
75. Kawai Y., Tsukamoto E., Nozaki Y., Morita K., Sakurai M., Tamaki N. Significance of reduced uptake of iodinated fatty acid analogue for the evaluation of patients with acute chest pain. J Am Coll Cardiol. 2001;38(7):1888-1894.
76. Tamaki N. Role of BMIPP imaging for risk stratification in patients with coronary artery disease. J Nucl Cardiol. 2005;12(2):148-150.
77. Nishimura M., Tokoro T., Nishida M., et al. Myocardial fatty acid imaging identifies a group of hemodialysis patients at high risk for cardiac death after coronary revascularization. Kidney Int. 2008;74(4):513-520.
78. Dilsizian V., Bateman T.M., Bergmann S.R., et al. Metabolic imaging with beta-methyl-p-[(123)I]-iodophenyl-pentadecanoic acid identifies ischemic memory after demand ischemia. Circulation. 2005;112(14):2169-2174.
79. Dou K.F., Yang M.F., Yang Y.J., Jain D., He Z.X. Myocardial 18F-FDG uptake after exercise-induced myocardial ischemia in patients with coronary artery disease. J Nucl Med. 2008;49(12):1986-1991.
80. Barger P.M., Kelly D.P. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci. 1999;318(1):36-42.
81. Rupp H., Jacob R. Metabolically-modulated growth and phenotype of the rat heart. Eur Heart J. 1992;13(Suppl D):56-61.
82. Blair E., Redwood C., Ashrafian H., et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10(11):1215-1220.
83. Jamshidi Y., Montgomery H.E., Hense H.W., et al. Peroxisome proliferator—activated receptor alpha gene regulates left ventricular growth in response to exercise and hypertension. Circulation. 2002;105(8):950-955.
84. Handa N., Magata Y., Mukai T., Nishina T., Konishi J., Komeda M. Quantitative FDG-uptake by positron emission tomography in progressive hypertrophy of rat hearts in vivo. Ann Nucl Med. 2007;21(10):569-576.
85. de las Fuentes L., Herrero P., Peterson L.R., Kelly D.P., Gropler R.J., Davila-Roman V.G. Myocardial fatty acid metabolism: independent predictor of left ventricular mass in hypertensive heart disease. Hypertension. 2003;41(1):83-87.
86. de las Fuentes L., Soto P.F., Cupps B.P., et al. Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. J Nucl Cardiol. 2006;13(3):369-377.
87. Tuunanen H., Kuusisto J., Toikka J., et al. Myocardial perfusion, oxidative metabolism, and free fatty acid uptake in patients with hypertrophic cardiomyopathy attributable to the Asp175Asn mutation in the alpha-tropomyosin gene: a positron emission tomography study. J Nucl Cardiol. 2007;14(3):354-365.
88. Buttrick P.M., Kaplan M., Leinwand L.A., Scheuer J. Alterations in gene expression in the rat heart after chronic pathological and physiological loads. J Mol Cell Cardiol. 1994;26(1):61-67.
89. Sack M.N., Kelly D.P. The energy substrate switch during development of heart failure: gene regulatory mechanisms (Review). Int J Mol Med. 1998;1(1):17-24.
90. Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation. 2004;110(8):894-896.
91. Nakae I., Matsuo S., Koh T., Mitsunami K., Horie M. Iodine-123 BMIPP scintigraphy in the evaluation of patients with heart failure. Acta Radiol. 2006;47(8):810-816.
92. Tuunanen H., Engblom E., Naum A., et al. Decreased myocardial free fatty acid uptake in patients with idiopathic dilated cardiomyopathy: evidence of relationship with insulin resistance and left ventricular dysfunction. J Card Fail. 2006;12(8):644-652.
93. Beanlands R.S.B., Nahmias C., Gordon E., et al. The effects of β1-blockade on oxidative metabolism and the metabolic cost of ventricular work in patients with left ventricular dysfunction: a double-blind, placebo-controlled, positron-emission tomography study. Circulation. 2000;102:2070-2075.
94. Stolen K.Q., Kemppainen J., Ukkonen H., et al. Exercise training improves biventricular oxidative metabolism and left ventricular efficiency in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2003;41(3):460-467.
95. Sundell J., Engblom E., Koistinen J., et al. The effects of cardiac resynchronization therapy on left ventricular function, myocardial energetics, and metabolic reserve in patients with dilated cardiomyopathy and heart failure. J Am Coll Cardiol. 2004;43(6):1027-1033.
96. Nowak B., Sinha A.M., Schaefer W.M., et al. Cardiac resynchronization therapyhomogenizes myocardial glucosemetabolism and perfusion in dilated cardiomyopathy and left bundle branch block. J Am Coll Cardiol. 2003;41(9):1523-1528.
97. Tuunanen H., Engblom E., Naum A., et al. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118(12):1250-1258.
98. Hasegawa S., Kusuoka H., Maruyama K., Nishimura T., Hori M., Hatazawa J. Myocardial positron emission computed tomographic images obtained with fluorine-18 fluoro-2-deoxyglucose predict the response of idiopathic dilated cardiomyopathy patients to beta-blockers. J Am Coll Cardiol. 2004;43(2):224-233.
99. van Campen C.M., Visser F.C., van der Weerdt A.P., et al. FDG PET as a predictor of response to resynchronisation therapy in patients with ischaemic cardiomyopathy. Eur J Nucl Med Mol Imaging. 2007;34(3):309-315.
100. Kannel W.B., Hjortland M., Castelli W.P. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29-34.
101. Stanley W.C., Lopaschuck G.D., McCormack J.G. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res. 1997;34(1):25-33.
102. Rodrigues B., Cam M.C., McNeill J.H. Myocardial substrate metabolism: implications for diabetic cardiomyopathy. J Mol Cell Cardiol. 1995;27:169-179.
103. Stanley W.C., Lopaschuk G.D., McCormack J.G. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res. 1997;34(1):25-33.
104. Taegtmeyer H., McNulty P., Young M.E. Adaptation and maladaptation of the heart in diabetes: Part I: general concepts. Circulation. 2002;105(14):1727-1733.
105. Young M.E., McNulty P., Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation. 2002;105(15):1861-1870.
106. Finck B.N., Lehman J.J., Leone T.C., et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109(1):121-130.
107. Burkart E.M., Sambandam N., Han X., et al. Nuclear receptors PPARbeta/delta and PPARalpha direct distinct metabolic regulatory programs in the mouse heart. J Clin Invest. 2007;117(12):3930-3939.
108. Shoghi K.I., Gropler R.J., Sharp T., et al. Time course of alterations in myocardial glucose utilization in the Zucker diabetic fatty rat with correlation to gene expression of glucose transporters: a small-animal PET investigation. J Nucl Med. 2008;49(8):1320-1327.
109. Welch M.J., Lewis J.S., Kim J., et al. Assessment of myocardial metabolism in diabetic rats using small-animal PET: a feasibility study. J Nucl Med. 2006;47(4):689-697.
110. Monti L.D., Landoni C., Setola E., et al. Myocardial insulin resistance associated with chronic hypertriglyceridemia and increased FFA levels in Type 2 diabetic patients. Am J Physiol Heart Circ Physiol. 2004;287(3):H1225-H1231.
111. Monti L.D., Lucignani G., Landoni C., et al. Myocardial glucose uptake evaluated by positron emission tomography and fluorodeoxyglucose during hyperglycemic clamp in IDDM patients. Role of free fatty acid and insulin levels. Diabetes. 1995;44(5):537-542.
112. vom Dahl J., Herman W.H., Hicks R.J., et al. Myocardial glucose uptake in patients with insulin-dependent diabetes mellitus assessed quantitatively by dynamic positron emission tomography. Circulation. 1993;88(2):395-404.
113. Avogaro A., Nosadini R., Doria A., Fioretto P., Velussi M., Vigorito C., Sacca L., Toffolo G., Cobelli C., Trevisan R., et al. Myocardial metabolism in insulin-deficient diabetic humans without coronary artery disease. Am J Physiol. 1990;258(4 Pt 1):E606-E618.
114. Herrero P., McGill J.B., Lesniak D., et al. Pet dection of the impact of dobutamine on myocardial glucose metabolism in women with type 1 diabetes mellitus. J Nucl Cardiol. 2008;15(6):598-604.
115. Peterson L.R., Herrero P., McGill J., et al. Fatty acids and insulin modulate myocardial substrate metabolism in humans with type 1 diabetes. Diabetes. 2008;57(1):32-40.
116. Zhou Y.T., Grayburn P., Karim A., et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000;97(4):1784-1789.
117. Hallsten K., Virtanen K.A., Lonnqvist F., et al. Enhancement of insulin-stimulated myocardial glucose uptake in patients with Type 2 diabetes treated with rosiglitazone. Diabet Med. 2004;21(12):1280-1287.
118. Commerford S.R., Pagliassotti M.J., Melby C.L., Wei Y., Gayles E.C., Hill J.O. Fat oxidation, lipolysis, and free fatty acid cycling in obesity-prone and obesity-resistant rats. Am J Physiol Endocrinol Metab. 2000;279(4):E875-E885.
119. Ogawa M., Ishino S., Mukai T., et al. (18)F-FDG accumulation in atherosclerotic plaques: immunohistochemical and PET imaging study. J Nucl Med. 2004;45(7):1245-1250.
120. Rudd J.H., Warburton E.A., Fryer T.D., et al. Imaging atherosclerotic plaque inflammation with [18F]-fluorodeoxyglucose positron emission tomography. Circulation. 2002;105(23):2708-2711.
121. Tahara N., Kai H., Ishibashi M., et al. Simvastatin attenuates plaque inflammation: evaluation by fluorodeoxyglucose positron emission tomography. J Am Coll Cardiol. 2006;48(9):1825-1831.
122. Tawakol A., Migrino R.Q., Bashian G.G., et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol. 2006;48(9):1818-1824.