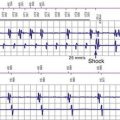
17 ICD Therapy in Channelopathies
Cardiac channelopathies, also called ion channel disorders, include long QT syndrome, short QT syndrome, Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia. This group of genetically determined diseases has low prevalence in the general population, usually less than 5 per 10,000.1 Affected individuals have a structurally normal heart but an increased risk of syncope and sudden cardiac death (SCD) caused by ventricular tachycardia (VT) or ventricular fibrillation (VF). The genes affected by causative mutations encode proteins that form cardiac ion channels, regulate their function, or participate in ionic-cellular imbalance, regulating the electrical function of the heart and resulting in primary electrical diseases. The penetrance of channelopathies is variable, and individuals who show the most severe phenotype are at higher risk of presenting with life-threatening episodes of ventricular arrhythmias.
Symptoms usually begin early in life, and risk stratification is paramount because patients might present with SCD, in some cases as the first symptom. Although genetic information is entering clinical practice and being integrated in the risk stratification schemes,2,3 family history of SCD has not been identified as a reliable marker of high risk in patients with channelopathies.
There are no large, randomized trials of treatment for these diseases, and probably never will be. Current indications for risk stratification and treatment are based on information from large registries and retrospective analysis. Thus, the level of evidence for all indications is low. Patients diagnosed with channelopathies are advised to restrict exercise, even if exercise is not a recognized trigger for their arrhythmic events.4 This chapter reviews risk stratification and implantable cardioverter-defibrillator (ICD) indications for the channelopathies, as well as benefits and drawbacks of ICD implantation in these patients.
 Long QT Syndrome
Long QT Syndrome
Described for the first time by Jervell and Lange-Nielsen5 in 1957 (autosomal recessive pattern of inheritance; associated genes: KCNQ1 and KCNE1)6 and by Romano et al. in 1963 and Ward et al.7 in 1964 (autosomal dominant inheritance; associated genes: KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, and SNTA1),6 long QT syndrome (LQTS) is the most studied cardiac channelopathy. Established in 1979, the International LQTS Registry includes more than 3000 patients and provides information about causative mutations, clinical course, risk factors, and response to treatment.8
The typical features of LQTS are prolonged corrected QT (QTc) interval in the 12-lead electrocardiogram (ECG), T-wave abnormalities, and symptoms that vary from syncope to SCD. Symptoms are secondary to torsades de pointes (TdP), a rapid polymorphic VT that may be self-limited or may degenerate into VF (mean age of onset, 12 years).9 The arrhythmic episodes are often triggered by adrenergic stimuli (e.g., exercise, emotion) but may also occur at rest.10 TdP may be pause dependent or adrenergic dependent.11,12 Pause-dependent TdP mainly follows post-extrasystolic pauses but may also appear after pauses during sinus arrhythmia or sinus arrest. These pauses enhance early afterdepolarizations (EADs), manifesting on ECG as more prolonged QT interval and U-wave augmentation in the beat immediately after the pause, and may trigger TdP. Adrenergic-dependent TdP follows sinus tachycardia during physical or emotional stress. In this case, tachycardia and high sympathetic tone increase the dispersion of repolarization by shortening it in different degrees across the myocardial layers. Also during sinus tachycardia, however, ventricular extrasystoles and the following post-extrasystolic pause are able to trigger TdP.13
Risk Stratification
Factors in risk stratification of LQTS patients include ECG, age, gender, genotype, and symptoms.
Electrocardiogram
Many studies have confirmed a direct relationship between prolonged QTc interval and risk of arrhythmia-related symptoms in patients with LQTS.3,14–19 A QTc interval of 500 msec or higher implies high risk for arrhythmic events.3 The strength of QTc interval to predict arrhythmic risk varies when dividing patients into subgroups according to age or gender, but it always maintains a strong value as an independent risk factor. Serial QTc measurements may have a role in risk stratification, because maximum QTc interval during adolescence was the strongest predictor of cardiac events.20
Age and Gender
Observation of the natural history of LQTS reveals how gender and age interact as risk factors.8,21,22 Several analyses of the probability of arrhythmic events during different decades of life showed that risk is significantly higher in boys than girls (5% vs. 1%).17 Also, the gender-related risk reverses in the final stage of adolescence, about age 17, with respect to any cardiac events, and about age 23 with SCD and sudden cardiac arrest (SCA).15–18
Genotype
Genotype testing identifies a causative mutation in up to 68% of probands of LQTS; 90% of patients are LQT1, LQT2, or LQT3.23,24 Genotype is useful for risk stratification. Patients with LQT1 and LQT2 have more cardiac events (mainly syncope) than patients with LQT3, who present a higher rate of lethal or potentially lethal events.2 Triggers for the arrhythmic events in LQTS are also associated with the genotype: exercise (especially swimming) in patients with LQT1, sudden loud noise in LQT2, and rest/sleep in LQT3 patients.10 Furthermore, the response to β-blocker therapy for prevention of cardiac events is significantly better in LQT1 than in LQT2 or LQT3 patients.9 A given type of mutation or its location in the affected gene can influence risk.25–27
Symptoms
Syncope is the strongest predictor of cardiac events and SCD in the patient with LQTS. In all age groups, recent or remote syncope is associated with a 2.7- to 18-fold increased risk of SCD.15–18
Unrelated Factors
Electrophysiologic study (EPS)1,28 and family history of SCD29 have proved not to influence risk stratification in LQTS.
Aborted Cardiac Arrest
A simplified scheme for risk stratification of aborted cardiac arrest (ACA) or SCD in LQTS patients divides patients in three groups: (1) high risk (history of ACA or documented TdP), with an estimated rate of ACA or SCD of 14% at 5-year follow-up; (2) intermediate risk (QTc >500 msec and/or time-dependent syncopal history), with an estimated rate of ACA or SCD at 5-year follow up of 3%; and (3) low risk (QTc ≤500 msec and/or no history of prior syncope), with 0.5% estimated rate of ACA or SCD at 5-year follow-up.30
Current Therapeutic Recommendations
Current guidelines suggest that all patients diagnosed with LQTS should avoid triggers of arrhythmias (exercise, emotional stress, loud noises) as well as drugs that prolong the QTc interval. Patients should receive β-blockers (class I indication in clinical diagnosis, i.e., prolonged QTc interval; class IIa indication in molecular diagnosis and normal QTc interval). Beta-adrenergic blockers should be given at the highest dose tolerated by the patient, and the dose may be titrated by exercise testing.1
Torsades de pointes in patients with congenital LQTS is often pause dependent (up to 74%),31 and cardiac pacing at lower rate limit (LRL) >70 bpm has proved helpful for preventing TdP development in acquired LQTS,32 in which TdP is almost always pause dependent. Therefore, strategies to avoid “short-long-short” sequences were proposed in these patients as a complement to pharmacologic treatment. However, patients undergoing pacemaker implantation and β-blocker therapy still presented unacceptably high rates of aborted SCD (aSCD) and SCD, leading to further indication for ICDs in high-risk patients with LQTS.33 Even though this is currently the preferred approach, LQTS patients can still obtain additional benefits from their ICD if antibradycardia therapy is correctly programmed, in combination with pharmacologic treatment and antitachycardia settings (see later Antibradycardia Therapy in Long QT Syndrome).
The LQTS patients with aSCD are at high risk of repeating a fatal or near-fatal arrhythmic event and thus are candidates for ICD implantation. Patients who continue to experience syncope or TdP while taking maximally tolerated β-blockers also are candidates. These two groups of LQTS patients are also candidates for left sympathetic cardiac denervation.34 Genotype information may help guide the decision about ICD implantation. However, it is also important to consider other clinical markers of high risk, which have proved very useful and simple to evaluate, such as QTc duration, symptoms, presence of sinus pauses, T-wave alternans, and 2 : 1 atrioventricular (AV) block.35
Several studies support a role for ICDs in preventing or reducing SCD in high-risk patients with LQTS. However, most patients do not belong to this subgroup and remain at a relatively low risk of cardiac events; these patients may be appropriately protected from arrhythmic events by a combination of noninvasive and simple measures. Moss et al.14 reported a 5%/yr incidence of syncope and 0.9%/yr incidence of SCD in probands. In affected family members, incidence of cardiac events was even lower: 0.5%/yr for syncope and 0.2%/yr for SCD.
Some studies may have overestimated the benefits of ICD implantation, comparing ICD patients with non-ICD patients who were not adequately treated with drugs,22 or by making “appropriate ICD therapy” synonymous with SCD. Considering that TdP is often nonsustained, and that the rates of “lifesaving therapies” in several studies were higher than the expected mortality in LQTS, some “appropriate ICD shocks” likely were inappropriate, delivered for ventricular arrhythmia that otherwise would have terminated spontaneously.36–39
 Short QT Syndrome
Short QT Syndrome
Initially described in 2000, short QT syndrome (SQTS) is characterized by an abnormally short QT interval, often followed by morphologic T-wave abnormalities (tall, narrow), and increased susceptibility to atrial fibrillation (AF) and VF.40,41 To date, mutations in five genes have been found in almost 25% of SQTS patients. Mutations in KCNH2, KCNQ1, and KCNJ242–44 result in gain of function in the encoded potassium ion (K+) channels and lead to shortening of the ventricular repolarization phase. Mutations in CACNA1C and CACNB2 cause loss of function in the cardiac L-type calcium ion (Ca2+) channel and produce an overlap between Brugada syndrome and SQTS.45 At present, it is still unclear whether diagnosis of SQTS should be based on QT or QTc interval; sensitivity and specificity of different QT/QTc cutoff values also remain undefined.1
Age of onset of symptoms varies from 4 months to 62 years, and incidence of SQTS is higher among males. Atrial and ventricular effective refractory periods are shortened and AF/VF easily induced in patients undergoing EPS (up to 60%).46 The risk for arrhythmic events is high in SQTS patients. Cardiac arrest is frequently the first manifestation of disease and may occur during the first year of life. Patients also present with syncope and AF (up to 30%) at any age (even intrauterine), making SQTS a differential diagnosis to rule out in young patients with lone AF. The use of quinidine in a group of patients with SQT1 proved useful in making VT noninducible by prolonging ventricular refractory periods.47
Risk Stratification
Up to 2008, only 51 patients with SQTS had been diagnosed worldwide.48 Thus, risk stratification is based on limited data; currently, symptoms are the only tool available to evaluate risk of arrhythmic events. Patients with SQTS and SCA and those with syncope of unknown origin should have an ICD for secondary and primary prevention, respectively, even if no data exist on the usefulness of syncope to predict cardiac events.49 Although ECG helps diagnose the disease, its role in risk stratification is still undefined (e.g., it is unknown if degree of QT shortening identifies patients at higher risk of arrhythmic events). Age, gender, genotype, and inducibility of ventricular tachyarrhythmias during EPS have not yet proved to have a predictive value for cardiac events in this group of patients.
Current Therapeutic Recommendations
Management of patients with SQTS is poorly established. Although limited data show that quinidine may suppress inducibility of ventricular arrhythmias during EPS,50,51 whether it confers long-term protection for malignant arrhythmias remains unknown. Most SQTS patients receive an ICD, with a high rate of complications reported, mostly inappropriate shocks caused by T-wave oversensing.52
 Brugada Syndrome
Brugada Syndrome
The Brugada syndrome (BS) is one of the leading causes of SCD in patients with structurally normal hearts.53 A characteristic electrocardiographic pattern (“type 1 ECG”) in at least two right precordial leads, spontaneously or after pharmacologic challenge with a class I antiarrhythmic agent confirms the diagnosis, in conjunction with at least one clinical diagnostic criterion reflecting documented ventricular arrhythmias, a positive family history of SCD or BS, or arrhythmia-related symptoms.54 The disease has autosomal dominant inheritance; to date, mutations in eight genes have been linked to BS,55–60 identified in up to 30% of patients, most affecting SCN5A.53 Patients are predominantly male, up to 83% in most series.61–67 Diagnosis is made and cardiac events occur mainly in the fourth decade of life,3,53,63–65 although cardiac arrest has been reported in neonates and children.62,63,68 Fever is a predisposing factor for cardiac arrest in the BS patient.68–72
Risk Stratification
There is an ongoing controversy about risk stratification in BS. All groups agree that patients who recover from an episode of SCD are at high risk of repeating a fatal or near-fatal arrhythmic event (17%-62% at follow-up of 24-40 months)62,63,65 and should receive an ICD.1 However, the best management of patients without a history of SCD remains unclear, specifically asymptomatic patients. Brugada et al.63,64 described a high rate of events in patients with previous syncope (19%) and even in asymptomatic patients (8%) after mean follow-up of 24 months, and inducibility during EPS and previous syncope were independent predictors of cardiac events. Giustteto et al.66 emphasized the role of symptoms as predictors of future cardiac events in 136 consecutive BS patients, supporting the role of EPS as a valuable tool for risk stratification. However, Priori et al.,62 Eckardt et al.,65 and more recently Probst et al.67 found that the incidence of cardiac events was much lower, and inducibility in EPS did not prove useful as a predictor of future arrhythmic events.
Current Therapeutic Recommendations
Quinidine has proved useful in BS patients with ICD and frequent shocks.73 It is also effective in episodes of electrical storm,74 a class IIb indication.1 Some studies show that quinidine in BS patients with inducible VF may render the EPS noninducible for sustained ventricular arrhythmias.75–77 Isoproterenol is also indicated in patients with BS and electrical storm74,78 (class IIa indication).1 Other pharmacologic options are currently being investigated.79–82
 Catecholaminergic Polymorphic Ventricular Tachycardia
Catecholaminergic Polymorphic Ventricular Tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is considered one of the most malignant channelopathies. Clinical manifestations include syncope and SCD triggered by adrenergic stimuli (exercise, emotion) from ventricular arrhythmias: bidirectional VT (35% of cases) and polymorphic VT (PVT), which sometimes degenerate into VF. Symptoms often begin early in life (age 7-9 years), and by age 40, up to 80% of patients have developed symptoms.83 Overall mortality without treatment is 30% to 50%.84
To date, mutations in two genes have been linked to CPVT. RyR2 (encoding cardiac receptor of ryanodine)85 and CASQ2 (calsequestrin 2)86 are involved in intracellular management of Ca2+ in the myocardium.87,88 Mutations in both genes result in a cytosolic Ca2+ overload from the sarcoplasmic reticulum in the myocytes, giving rise to delayed afterdepolarizations (DADs). During β-adrenergic stimulation, DADs increase in number and magnitude and may trigger multiple action potentials, resulting in symptomatic ventricular arrhythmias. Mutations in RyR2 account for 50% to 55% of genotyped CPVT patients,89 with autosomal dominant inheritance and mean penetrance of 83%.90 Mutations in CASQ2 represent only 5% to 10% of patients, with autosomal recessive inheritance.
Risk Stratification
Current risk stratification is based on limited data because the number of patients identified with CPVT is low. The largest published series includes 101 patients,83 estimated prevalence of CPVT is 1 : 10,000, and available follow-up periods are short. As in other channelopathies, family history of SCD has not proved a predictive factor for cardiac events in CPVT. The baseline ECG is not useful as a diagnostic or prognostic tool. The natural history does not differ among CPVT patients with or without identified mutations or among patients with RyR2– and CASQ2-related mutations. Silent mutation carriers (asymptomatic, with no arrhythmias at exercise testing or on Holter monitoring) present a similar event rate to symptomatic patients.83 Neither gender seems to have a higher risk of cardiac events or SCD. EPS is not useful to diagnose or evaluate risk of cardiac events, because arrhythmias in CPVT patients are usually not inducible. Young age at diagnosis, history of ACA, and absence of β-blocker therapy have been identified as independent predictors of fatal or near-fatal cardiac events. Syncope before diagnosis did not identify patients at higher risk for cardiac events; however, these results may be biased because most patients with a diagnosis of CPVT and syncope were receiving β-blockers, resulting in a significant reduction of cardiac events and SCD.83
Current Therapeutic Recommendations
Exercise testing is the most useful tool for diagnosis of CPVT because the ventricular arrhythmias are highly reproducible with exercise. It is also of value to titrate the dose of β-blockers. All patients diagnosed with CPVT must be treated with β-blockers; asymptomatic gene carriers show a similar rate of arrhythmic events to symptomatic patients.83 Patients who recovered from an episode of SCD are considered at high risk of cardiac events, and ICD implantation plus β-blockers for secondary prevention are mandatory. Patients with complex ventricular arrhythmias or symptoms despite full doses of β-blockers are also candidates for ICD (class IIa indication). Some reports suggest that the use of Ca2+ antagonists (in combination with β-blockers)91 and left cardiac sympathetic denervation may be useful in patients with recurrent ventricular arrhythmias despite full-dose β-blockers and in those with ICD and electrical storm.92
 General Considerations
General Considerations
Correct risk stratification and treatment in patients with cardiac channelopathies is challenging. Some patients are at risk of fatal or near-fatal arrhythmias at a very young age, and all efforts should be directed to avoid these events. Table 17-1 summarizes current indications for ICD implantation, according to the American College of Cardiology/American Heart Association (ACC/AHA) Task Force and the European Society of Cardiology (ESC) guidelines.1 ICD therapy has proved the most effective tool to prevent arrhythmic SCD,22 and its indication in high-risk patients is clearly justified (Fig. 17-1), especially considering the cumulative increased risk of very young patients who present for primary or secondary prevention. However, it is important to highlight that ICD therapy does have associated complications.

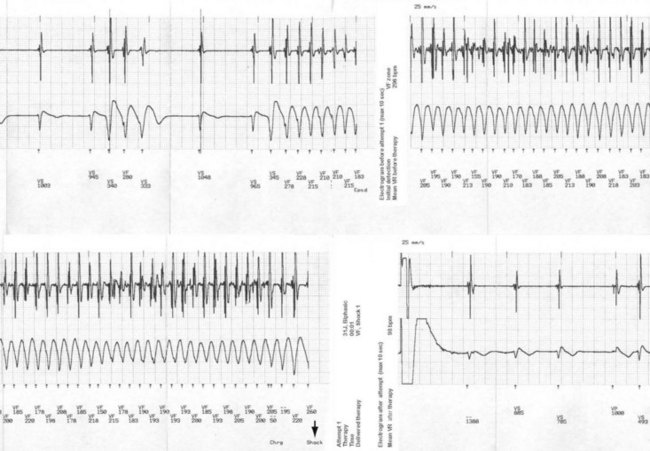
Some series of BS patients with an ICD reported low rates of appropriate shocks (8%-15%; median follow-up, 45 months; annual appropriate discharge rate, 2.6%) and high rates of complications (28%), including inappropriate shocks (20%-36%), which often greatly exceeded (2-2.5 times) the rate of appropriate shocks.93–95 However, some studies of LQTS patients may have promoted ICD use based on incidence of ICD shock delivery, considering “appropriate ICD therapy” equivalent to SCD, with resulting rates of “livesaving therapies” higher than the expected mortality for the disease. In randomized trials for primary and secondary prevention of SCD in LQTS patients, the number of appropriate shocks consistently exceeded the sudden death rate in the control group by 2 : 1. Because most TdP episodes terminate spontaneously, some of those “appropriate ICD shocks” may have been inappropriate, treating a ventricular arrhythmia that would have terminated spontaneously.36–39 Other studies also showed excellent response to β-blocker therapy (especially in LQT1) when patients were fully compliant and avoided QT-prolonging medications.96
 Antibradycardia Therapy in Long QT Syndrome
Antibradycardia Therapy in Long QT Syndrome
Most episodes of TdP in congenital LQTS are preceded by “short-long-short” sequences. Therefore, antibradycardia therapy was proposed in these patients to avoid sudden increases in R-R intervals, which may be arrhythmogenic. The simplest way to avoid pauses when the patient carries a pacemaker or ICD is to increase the lower rate limit. Different groups suggest programming LRL above 70 bpm,32 between 70 and 80 bpm,13 or at 80 bpm or higher97 in adults diagnosed with LQTS, increasing this value for short periods when the patient is known to be at increased risk for developing TdP (e.g., postpartum period, surgical procedures). Maintenance of higher LRL for long periods, however, may lead to tachycardia-induced cardiomyopathy.
To prevent pauses in LQTS patients through pacing, other antibradycardia parameters of the ICD should be correctly programmed, as follows98:
 Complications
Complications
Implantation
The reported overall risk of any early complication after device implant was 6.7%, with 4.9% of patients requiring an invasive treatment; rate of late procedure-related complications was 3.1%.99 Diagnosis of implant complications requires a high index of suspicion. Routine examination after implantation should include physical examination, chest radiograph, and device interrogation.
Infection
Pocket infection and device infection are some of the most severe complications related to ICD implantation, requiring aggressive and expensive treatment: prolonged in-hospital antibiotic administration and usually surgical explantation of the device. Identified risk factors for device infections in patients with channelopathies include multiple implant procedures (up to fivefold increased risk)100,101 and young age at implantation,102 especially during childhood. Lead and infection complications were more prevalent in children, doubling the incidence of device infection in patients under 40 versus over age 40.103 Evidence also indicates that ICDs become infected more frequently than pacemakers.104,105
Inappropriate Shocks
Inappropriate shocks are defined as shocks delivered by the ICD for a non-VT/VF rhythm. Identified causes of inappropriate shocks are common in patients with channelopathies, including sinus tachycardia (Fig. 17-2); atrial tachyarrhythmias (Fig. 17-3), found in up to 30% of BS patients,53 in 70% of SQTS patients, and in CPVT patients; and T-wave oversensing, especially in SQTS. Young patients may lead more active physical lives and inadvertently damage their leads, causing inappropriate shocks by sensing artifacts (“noise”) or myopotentials. Lead failures, which cause one third to one half of inappropriate shocks in younger patients, are more frequent in children than adults.102,106–108 In adults, the lead failure rate reaches 2% to 3% in short-term follow-up, versus a 10-fold rate among the pediatric population. There have been reports of a high rate of lead failure beyond 10 years after implantation (up to 20% cumulative risk), which is relevant in this population, who have a long survival expectation (especially after ICD implantation).109
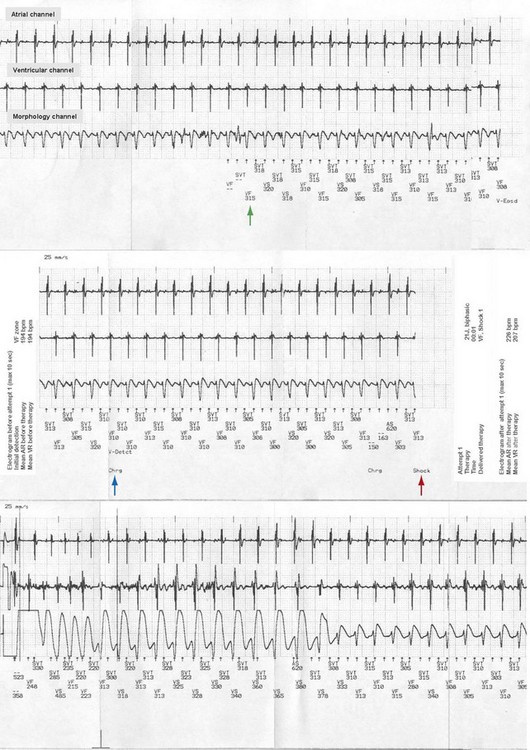
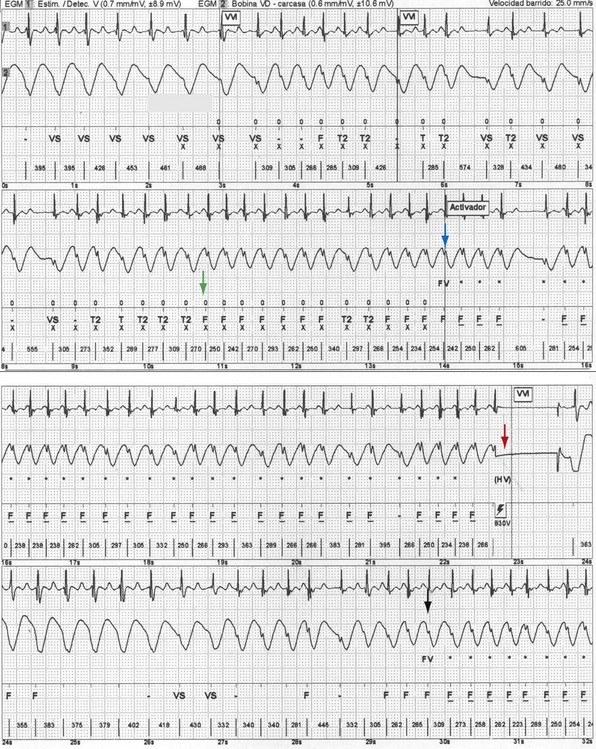
The incidence of inappropriate shocks in young patients is high: 25% after 16-month follow-up.106 Reported rate of inappropriate shocks in BS patients is 20% at 38 months and 36% at 54 months.94 Inappropriate shocks leading to cardiac arrest or SCD by triggering VT or VF are extremely rare. Even if these do not trigger fatal or near-fatal arrhythmias, inappropriate shocks have negative consequences on the quality of life of these patients.95,110 In CPVT patients, an important concern is electrical storms, which may lead to death secondary to exhaustion of therapies by the ICD after shocks (appropriate and inappropriate).111 Inappropriate shocks after sinus tachycardia or supraventricular arrhythmias cause increased release of endogenous catecholamines, leading to polymorphic VT and more shocks.112 The patient also may receive an inadequate shock for nonsustained ventricular tachycardia (NSVT), which is common in patients with channelopathies (Fig. 17-4). Special care must be taken during programming the ICD to avoid treating NSVT (see Strategies to Avoid Complications).
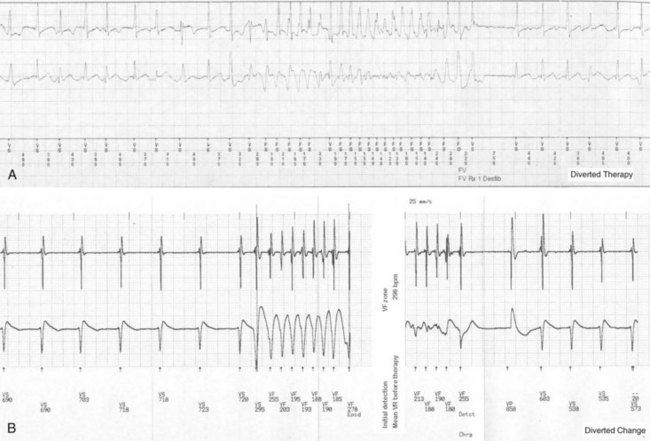
Risk of Never Using Device
In patients with no myocardial disease, the percentage of surviving patients in the “never used device” category may remain high, because death from heart failure is low or completely unexpected.53,94 In patients with BS and LQTS, mortality was near or at 0%, and survival without appropriate discharge was 85% to 93%.39,94 In young patients, this poses a special problem; besides being exposed for decades to the risk of sudden death, they will also be exposed to the risks of ICD implantation, but accrue none of the benefit, which may fade over time.113
Strategies to Avoid Complications
Table 17-2 summarizes suggested strategies to avoid complications with ICD therapy. Other recommendations include the following:
TABLE 17-2 Tips to Avoid Complications with ICD Therapy in Patients with Channelopathies
| Tips at Implantation | Programming Tips |
|---|---|
| 1. IV antibiotic prophylaxis ≥30 min before surgery. | 1. Only one therapy zone (VF). |
ATP, Antitachycardia pacing; IV, intravenous; LQTS; long QT syndrome; NID, number of intervals to detect; NSVT, nonsustained ventricular tachycardia; VF, ventricular fibrillation.
 Conclusion
Conclusion1 Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death), developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e385-e484.
2 Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med. 1998;339:960-965.
3 Priori SG, Schwartz PJ, Napolitano C, et al. Risk stratification in the long QT syndrome. N Engl J Med. 2003;348:1866-1874.
4 Heidbuchel H, Corrado D, Biffi A, et al. Recommendations for participation in leisure-time physical activity and competitive sports of patients with arrhythmias and potentially arrhythmogenic conditions. Part II. Ventricular arrhythmias, channelopathies and implantable defibrillators. Eur J Cardiovasc Prev Rehabil. 2006;13:676-686.
5 Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59-68.
6 Hedley PL, Jorgensen P, Schlamowitz S, et al. The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat. 2009;30:1486-1511.
7 Ward OC. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103-106.
8 Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: a prospective international study. Circulation. 1985;71:17-21.
9 Priori SG, Napolitano C, Schwartz PJ, et al. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341-1344.
10 Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype correlation in the long QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89-95.
11 Jackman WM, Friday KJ, Anderson JL, et al. The long QT syndromes: a critical review, new clinical observations and a unifying hypothesis. Prog Cardiovasc Dis. 1988;31:115-172.
12 Roden DM, Lazzara R, Rosen M, et al. Multiple mechanisms in the long-QT syndrome: current knowledge, gaps, and future directions. The SADS Foundation Task Force on LQTS. Circulation. 1996;94:1996-2012.
13 Moss AJ, Liu JE, Gottlieb S, et al. Efficacy of permanent pacing in the management of high-risk patients with long QT syndrome. Circulation. 1991;84:1524-1529.
14 Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: prospective longitudinal study of 328 families. Circulation. 1991;84:1136-1144.
15 Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249-1254.
16 Sauer AJ, Moss AJ, McNitt S, et al. Long QT syndrome in adults. J Am Coll Cardiol. 2007;49:329-337.
17 Goldenberg I, Moss AJ, Peterson DR, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long QT syndrome. Circulation. 2008;117:2184-2191.
18 Goldenberg I, Moss AJ, Bradley J, et al. Long-QT syndrome after age 40. Circulation. 2008;117:2192-2201.
19 Spazzolini C, Mullally J, Moss AJ, et al. Clinical implications for patients with long QT syndrome who experience a cardiac event during infancy. J Am Coll Cardiol. 2009;54:832-837.
20 Goldenberg I, Mathew J, Moss AJ, et al. Corrected QT variability in serial electrocardiograms in long QT syndrome: the importance of the maximum corrected QT for risk stratification. J Am Coll Cardiol. 2006;48:1047-1052.
21 Locati EH, Zareba W, Moss AJ, et al. Age- and sex-related differences in clinical manifestations in patients with congenital long QT syndrome: findings from the International LQTS Registry. Circulation. 1998;97:2237-2244.
22 Zareba W, Moss AJ, Locati EH, et al. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol. 2003;42:103-109.
23 Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507-517.
24 Splawski I, Shen J, Timothy KW, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178-1185.
25 Moss AJ, Shimizu W, Wilde AA, et al. Clinical aspects of type-1 long QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481-2489.
26 Jons C, Moss AJ, Lopes CM, et al. Mutations in conserved amino acids in the KCNQ1 channel and risk of cardiac events in type-1 long QT syndrome. J Cardiovasc Electrophysiol. 2009;20:859-865.
27 Moss AJ, Zareba W, Kaufman ES, et al. Increased risk of arrhythmic events in long QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794-799.
28 Bhandari AK, Shapiro WA, Morady F, et al. Electrophysiologic testing in patients with the long QT syndrome. Circulation. 1985;71:63-71.
29 Kaufman ES, McNitt S, Moss AJ, et al. Risk of death in the long QT syndrome when a sibling has died. Heart Rhythm. 2008;5:831-836.
30 Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol. 2008;51:2291-2300.
31 Viskin S, Fish R, Zeltser D, et al. Arrhythmias in the congenital long QT syndrome: how often is torsade de pointes pause dependent? Heart. 2000;83:661-666.
32 Pinski SL, Eguia LE, Trohman RG. What is the minimal pacing rate that prevents torsades de pointes? Insights from patients with permanent pacemakers. Pacing Clin Electrophysiol. 2002;25:1612-1615.
33 Dorostkar PC, Eldar M, Belhassen B, Scheinman MM. Long-term follow-up of patients with long-QT syndrome treated with beta-blockers and continuous pacing. Circulation. 1999;100:2431-2436.
34 Schwartz PJ, Priori SG, Cerrone M, et al. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long QT syndrome. Circulation. 2004;109:1826-1833.
35 Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD: true or false? Heart Rhythm. 2009;6:113-120.
36 Groh WJ, Silka MJ, Oliver RP, et al. Use of implantable cardioverter-defibrillators in the congenital long QT syndrome. Am J Cardiol. 1996;78:703-706.
37 Etheridge SP, Sanatani S, Cohen MI, et al. Long QT syndrome in children in the era of implantable defibrillators. J Am Coll Cardiol. 2007;50:1335-1340.
38 Goel AK, Berger S, Pelech A, Dhala A. Implantable cardioverter-defibrillator therapy in children with long QT syndrome. Pediatr Cardiol. 2004;25:370-378.
39 Monnig G, Kobe J, Loher A, et al. Implantable cardioverter-defibrillator therapy in patients with congenital long QT syndrome: a long-term follow-up. Heart Rhythm. 2005;2:497-504.
40 Gussak I, Brugada P, Brugada J, et al. Idiopathic short QT interval: a new clinical syndrome? Cardiology. 2000;94:99-102.
41 Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108:965-970.
42 Brugada R, Hong K, Dumaine R, et al. Sudden death associated with short QT syndrome linked to mutations in HERG. Circulation. 2004;109:30-35.
43 Bellocq C, van Ginneken AC, Bezzina CR, et al. Mutation in the KCNQ1 gene leading to the short QT interval syndrome. Circulation. 2004;109:2394-2397.
44 Priori SG, Pandit SV, Rivolta I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005;96:800-807.
45 Antzelevitch C, Pollevick GD, Cordeiro JM, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442-449.
46 Giustetto C, Di MF, Wolpert C, et al. Short QT syndrome: clinical findings and diagnostic-therapeutic implications. Eur Heart J. 2006;27:2440-2447.
47 Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494-1499.
48 Maury P, Extramiana F, Sbragia P, et al. Short QT syndrome: update on a recent entity. Arch Cardiovasc Dis. 2008;101:779-786.
49 Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: a familial cause of sudden death. Circulation. 2003;108:965-970.
50 Gaita F, Giustetto C, Bianchi F, et al. Short QT syndrome: pharmacological treatment. J Am Coll Cardiol. 2004;43:1494-1499.
51 Wolpert C, Schimpf R, Giustetto C, et al. Further insights into the effect of quinidine in short QT syndrome caused by a mutation in HERG. J Cardiovasc Electrophysiol. 2005;16:54-58.
52 Schimpf R, Wolpert C, Bianchi F, et al. Congenital short QT syndrome and implantable cardioverter-defibrillator treatment: inherent risk for inappropriate shock delivery. J Cardiovasc Electrophysiol. 2003;14:1273-1277.
53 Antzelevitch C, Brugada P, Borggrefe M, et al. Brugada syndrome: report of the Second Consensus Conference, endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659-670.
54 Wilde A, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002;106:2514-2519.
55 Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293-296.
56 London B, Michalec M, Mehdi H, et al. Mutation in glycerol-3-phosphate dehydrogenase 1-Like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260-2268.
57 Watanabe H, Koopmann TT, Le SS, et al. Sodium channel beta-1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest. 2008;118:2260-2268.
58 Delpon E, Cordeiro JM, Nunez L, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol. 2008;1:209-218.
59 Hu D, Barajas-Martinez H, Burashnikov E, et al. A mutation in the beta-3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet. 2009;2:270-278.
60 Medeiros-Domingo A, Tan BH, Crotti L, et al. Gain-of-function mutation, S422L, in the KCNJ8-encoded cardiac K ATP channel Kir6.1 as a pathogenic substrate for J wave syndromes. Heart Rhythm. 2010:••.
61 Benito B, Sarkozy A, Mont L, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol. 2008;52:1567-1573.
62 Priori SG, Napolitano C, Gasparini M, et al. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation. 2002;105:1342-1347.
63 Brugada J, Brugada R, Antzelevitch C, et al. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation. 2002;105:73-78.
64 Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation. 2003;108:3092-3096.
65 Eckardt L, Probst V, Smits JPP, et al. Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada syndrome. Circulation. 2005;111:257-263.
66 Giustetto C, Drago S, Demarchi PG, et al. Risk stratification of the patients with Brugada type electrocardiogram: a community-based prospective study. Europace. 2009;11:507-513.
67 Probst V, Veltmann C, Eckardt L, et al. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation. 2010;121:635-643.
68 Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome—a multicenter report. J Am Coll Cardiol. 1992;20:1391-1396.
69 Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803-809.
70 Mok NS, Priori SG, Napolitano C, et al. A newly characterized SCN5A mutation underlying Brugada syndrome unmasked by hyperthermia. J Cardiovasc Electrophysiol. 2003;14:407-411.
71 Saura D, Garcia-Alberola A, Carrillo P, et al. Brugada-like electrocardiographic pattern induced by fever. Pacing Clin Electrophysiol. 2002;25:856-859.
72 Ortega-Carnicer J, Benezet J, Ceres F. Fever-induced ST-segment elevation and T-wave alternans in a patient with Brugada syndrome. Resuscitation. 2003;57:315-317.
73 Hermida JS, Denjoy I, Clerc J, et al. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol. 2004;43:1853-1860.
74 Maury P, Hocini M, Haissaguerre M. Electrical storms in Brugada syndrome: review of pharmacologic and ablative therapeutic options. Indian Pacing Electrophysiol J. 2005;5:25-34.
75 Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110:1731-1737.
76 Belhassen B, Glick A, Viskin S. Excellent long-term reproducibility of the electrophysiologic efficacy of quinidine in patients with idiopathic ventricular fibrillation and Brugada syndrome. PACE. 2009;32:294-301.
77 Belhassen B, Viskin S, Fish R, et al. Effects of electrophysiologic-guided therapy with class IA antiarrhythmic drugs on the long-term outcome of patients with idiopathic ventricular fibrillation with or without the Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10:1301-1312.
78 Maury P, Couderc P, Delay M, et al. Electrical storm in Brugada syndrome successfully treated using isoprenaline. Europace. 2004;6(2):130-133.
79 Antzelevitch C. Brugada syndrome. PACE. 2006;29:1130-1159.
80 Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660-1666.
81 Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2002;13:698-701.
82 Abud A, Bagattin D, Goyeneche R, Becker C. Failure of cilostazol in the prevention of ventricular fibrillation in a patient with Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17:210-212.
83 Hayashi M, Denjoy I, Extramiana F, et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119:2426-2434.
84 Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca2+ regulation. Heart Rhythm. 2009;6:1652-1659.
85 Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196-200.
86 Lahat H, Eldar M, Levy-Nissenbaum E, et al. Autosomal recessive catecholamine- or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13-21. Circulation. 2001;103:2822-2827.
87 Di Barletta MR, Viatchenko-Karpinski S, Nori A, et al. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114:1012-1019.
88 Marks AR, Priori S, Memmi M, et al. Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia. J Cell Physiol. 2002;190:1-6.
89 Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69-74.
90 Napolitano C, Priori SG, Bloise R. Catecholaminergic polymorphic ventricular tachycardia. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington: Seattle, 1993-2004.
91 Rosso R, Kalman JM, Rogowski O, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149-1154.
92 Wilde AA, Bhuiyan ZA, Crotti L, et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic ventricular tachycardia. N Engl J Med. 2008;358:2024-2029.
93 Sacher F, Probst V, Iesaka Y, et al. Outcome after implantation of a cardioverter-defibrillator in patients with Brugada syndrome: a multicenter study. Circulation. 2006;114:2317-2324.
94 Sarkozy A, Boussy T, Kourgiannides G, et al. Long-term follow-up of primary prophylactic implantable cardioverter-defibrillator therapy in Brugada syndrome. Eur Heart J. 2007;28:334-344.
95 Rosso R, Glick A, Glikson M, et al. Outcome after implantation of cardioverter-defibrillator in patients with Brugada syndrome: a multicenter Israeli study (ISRABRU). Isr Med Assoc J. 2008;10:435-439.
96 Vincent GM, Schwartz PJ, Denjoy I, et al. High efficacy of beta-blockers in long QT syndrome type 1: contribution of noncompliance and QT-prolonging drugs to the occurrence of beta-blocker treatment “failures.”. Circulation. 2009;119:215-221.
97 Viskin S, Alla SR, Barron HV, et al. Mode of onset of torsade de pointes in congenital long QT syndrome. J Am Coll Cardiol. 1996;28:1262-1268.
98 Viskin S. Cardiac pacing in the long QT syndrome: review of available data and practical recommendations. J Cardiovasc Electrophysiol. 2000;11:593-600.
99 Kiviniemi MS, Pirnes MA, Eranen HJ, et al. Complications related to permanent pacemaker therapy. Pacing Clin Electrophysiol. 1999;22:711-720.
100 Harcombe AA, Newell SA, Ludman PF, et al. Late complications following permanent pacemaker implantation or elective unit replacement. Heart. 1998;80:240-244.
101 Catanchin A, Murdock CJ, Athan E. Pacemaker infections: a 10-year experience. Heart Lung Circ. 2007;16:434-439.
102 Link MS, Hill SL, Cliff DL, et al. Comparison of frequency of complications of implantable cardioverter-defibrillators in children versus adults. Am J Cardiol. 1999;83:263-266.
103 Klug D, Vaksmann G, Jarwe M, et al. Pacemaker lead infection in young patients. Pacing Clin Electrophysiol. 2003;26:1489-1493.
104 Voigt A, Shalaby A, Saba S. Rising rates of cardiac rhythm management device infections in the United States: 1996 through 2003. J Am Coll Cardiol. 2006;48:590-591.
105 Uslan DZ, Sohail MR, St Sauver JL, et al. Permanent pacemaker and implantable cardioverter-defibrillator infection: a population-based study. Arch Intern Med. 2007;167:669-675.
106 Alexander ME, Cecchin F, Walsh EP, et al. Implications of implantable cardioverter-defibrillator therapy in congenital heart disease and pediatrics. J Cardiovasc Electrophysiol. 2004;15:72-76.
107 Korte T, Koditz H, Niehaus M, et al. High incidence of appropriate and inappropriate ICD therapies in children and adolescents with implantable cardioverter-defibrillator. Pacing Clin Electrophysiol. 2004;7:924-932.
108 Eicken A, Kolb C, Lange S, et al. Implantable cardioverter-defibrillator (ICD) in children. Int J Cardiol. 2006;107:30-35.
109 Kleemann T, Becker T, Doenges K, et al. Annual rate of transvenous defibrillation lead defects in implantable cardioverter-defibrillators over a period of >10 years. Circulation. 2007;115:2474-2480.
110 DeMaso DR, Lauretti A, Spieth L, et al. Psychosocial factors and quality of life in children and adolescents with implantable cardioverter-defibrillators. Am J Cardiol. 2004;93:582-587.
111 Mohamed U, Gollob MH, Gow RM, Krahn AD. Sudden cardiac death despite an implantable cardioverter-defibrillator in a young female with catecholaminergic ventricular tachycardia. Heart Rhythm. 2006;3:1486-1489.
112 Pizzale S, Gollob MH, Gow R, Birnie DH. Sudden death in a young man with catecholaminergic polymorphic ventricular tachycardia and paroxysmal atrial fibrillation. J Cardiovasc Electrophysiol. 2008;19:1319-1321.
113 Sherrid MV, Daubert JP. Risks and challenges of implantable cardioverter-defibrillators in young adults. Prog Cardiovasc Dis. 2008;51:237-263.