[level-membership-for-dermatology-category]
Hypopigmentation Disorders
Yuin-Chew Chan, Yong-Kwang Tay
Introduction
A diverse group of conditions present with hypopigmentation in neonates and infants. A practical clinical approach would be to categorize them according to the distribution of the hypopigmentation: generalized, mosaic, or localized (Box 23.1). Some may be present at birth, but not noticed until later in infancy because neonates often have a lighter skin at birth than in later life.
Any defect occurring in melanocyte development, melanin synthesis and transport, or distribution of melanosomes to keratinocytes can result in a hypopigmentary disorder. Melanocytes originate from the neural crest and are located in the epidermis, hair bulb, eye (choroid, ciliary body, iris), inner ear (cochlea) and central nervous system (leptomeninges). Melanin is synthesized in melanosomes, which are organelles that share characteristics with lysosomes.
This chapter discusses a variety of clinical conditions causing hypopigmentation in neonates and infants, many of which have a genetic basis.
Generalized hypopigmentation of skin, hair, and eyes
Oculocutaneous albinism
Oculocutaneous albinism (OCA) refers to a group of autosomal recessive disorders involving abnormal melanin synthesis. Affected individuals have absent (type 1A) or reduced (type 1B, 2, 3, and 4) pigmentation in the skin, hair, and eyes from birth. The color varies from white to light brown, depending on ethnicity and the specific type of OCA. Ocular manifestations include nystagmus, photophobia, and decreased visual acuity. These are caused by decreased melanin within the eye or misrouting of optic nerve fibers. The affected infant is typically less pigmented than unaffected siblings.
Historically, OCA was divided into two clinical types based on the presence or absence of tyrosinase, the rate-limiting enzyme in the melanin biosynthetic pathway.1 Advances in molecular genetics have given rise to a more accurate classification and better understanding of pathogenesis.2,3 In OCA, epidermal and follicular melanocytes are present in normal quantity and distribution but do not synthesize melanin adequately (Table 23.1).
TABLE 23.1
Classification of disorders with cutaneous and ocular albinism
| OCA1A Tyrosinase negative | OCA1B Tyrosinase positive | OCA2 Tyrosinase positive | OCA3 Rufous | OCA4 | HPS | CHS | |
| Skin color | White | White at birth, develops some pigmentation in first and second decades | Creamy white to brown at birth. Slight darkening with age | Light brown to red-bronze | Creamy white to brown at birth. Slight darkening with age | White to brown | Creamy white to slate gray |
| Pigmented nevi and freckles | Absent | Present | Present | Absent | Present | Many in exposed areas | Present |
| Hair color | White throughout life; may become light yellow | White to light yellow at birth; turns yellow or blond in first few years | Light yellow, blond to brown at birth; may darken with age | Light brown to red-brown. May darken with age | Silvery white to light yellow at birth. May darken in childhood | White, blond to brown | Blond to light brown; metallic silver-gray sheen |
| Gene (mapping), function | TYR (11q14.3) Encodes tyrosinase |
TYR (11q14.3) Encodes tyrosinase |
OCA2 (15q11.2-q12) Encodes P protein, a melanosomal membrane protein |
TYRP1 (9q23) Encodes dihydroxyindol carboxylic acid oxidase, a melanogenic enzyme |
SLC45A2 (5p13.2) Encodes membrane-associated transporter protein |
HPS1: HPS1 (10q24.2) HPS2: AP3B1 (5q14.1) HPS3: HPS3 (3q24) HPS4: HPS4 (22q12.1) HPS5: HPS5 (11p15.1) HPS6: HPS6 (10q24.32) HPS7: DTNBP1 (6p22.3) HPS8: BLOC1S3 (19q13.32) HPS9: PLDN (15q21.1) Encode proteins involved in biogenesis of lysosomes and lysosome-related organelles, such as melanosomes and platelet-dense granules. Some may play a role in intracellular vesicle trafficking |
LYST (1q42.3) Encodes lysosomal-trafficking regulator, a cytoplasmic protein which may function as an adapter protein that mediates intracellular membrane fusion reactions |
| Mouse model | Albino | Albino | Pink-eye dilution | Brown | Underwhite | HPS1: Pale-ear HPS2: Pearl HPS3: Cocoa HPS4: Light-ear HPS5: Ruby eye 2 HPS6: Ruby eye HPS7: Sandy HPS8: Reduced pigmentation |
Beige |
| Hair bulb melanosomes | Stages I, II | Stages I, II, III | Stages I, II, III | Stages I, II, III, IV | Stages I, II, III | Stages I, II, III | Macromelanosomes and normal to Stage IV |
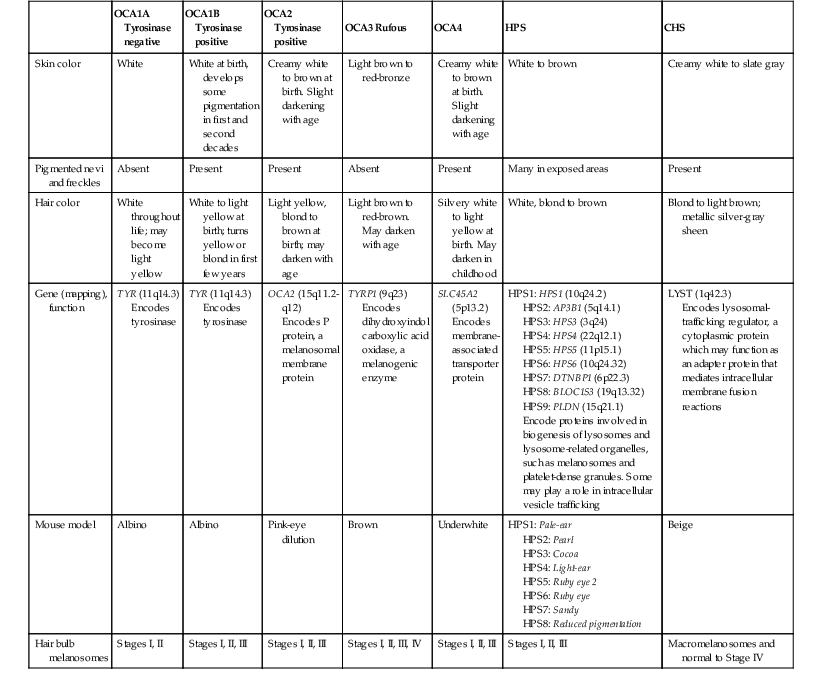
HPS, Hermansky–Pudlak syndrome; CHS, Chediak–Higashi syndrome.
Oculocutaneous albinism type 1
Oculocutaneous albinism type 1 (OCA1) is the second most common OCA worldwide.
Cutaneous findings
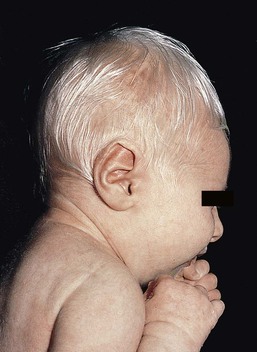
In OCA1A, there is marked generalized hypopigmentation at birth, with white hair and skin (Figs. 23.1, 23.2). As the child matures, the skin remains white. Nevi are not pigmented and sun tanning does not occur. The hair may acquire a slightly yellowish tint as a result of denaturing of hair keratins.
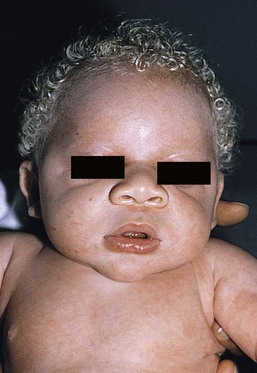
In OCA1B, the variable decrease in tyrosinase activity results in several clinical phenotypes: yellow, minimal pigment, platinum, and temperature-sensitive. Individuals with OCA1B form pheomelanin, which requires less tyrosinase activity, and this results in some pigment production during the first two decades of life.4 Affected individuals have a similar appearance to those with OCA1A at birth, but with time develop some pigment. Hair color can change from white to light blond and even progress to light brown in adolescence. With sun exposure, some individuals with OCA1B may be able to tan, although it is more common to burn without tanning. Pigmented nevi and freckles may develop.5
An interesting subtype of OCA1B is the temperature-sensitive phenotype in which the tyrosinase activity is seen mainly on the extremities. In these patients, the enzyme has no activity at 37°C, but some activity at 35°C. These individuals have white or lightly pigmented hairs on the scalp and trunk (axillae, pubic area) and darkly pigmented hair peripherally (legs, arms). The pattern is similar to that observed in Siamese cats.4
Extracutaneous findings
In OCA1A, the irises are pale blue at birth and throughout life. In bright light, the entire iris can appear pink or red, which is caused by its translucency.5 Severe photophobia results from a lack of retinal pigment. Other ocular abnormalities include decreased visual acuity, nystagmus, and strabismus.
In OCA1B, the irises can progressively darken to light tan or brown. In both subtypes, vision may remain stable or deteriorate with age.
Etiology and pathogenesis
OCA1 is caused by loss-of-function mutations in the tyrosinase gene (TYR), which is mapped to chromosome 11q14.3. Tyrosinase is the key enzyme that catalyzes the first two steps in melanin synthesis, converting tyrosine to L-DOPA (L-3,4-dihyroxyphenylalanine), and L-DOPA to DOPAquinone. The OCA1A subtype is characterized by mutations that result in complete loss of tyrosinase activity, whereas OCA1B is caused by mutations that result in markedly reduced tyrosinase activity (5–10% of the normal level).
Oculocutaneous albinism type 2
Oculocutaneous albinism type 2 (OCA2) is the most common form of OCA. It is most prevalent in people of African descent.6
Cutaneous findings
There is a spectrum of clinical phenotypes, depending on the ethnic background and the dilution of hair and skin pigment, which may be minimal to moderate. Comparison with a first-degree relative may be necessary to distinguish the degree of lightening.4 Most individuals are born with creamy white skin, and light yellow or blond hair. Depending on the individual’s ethnic background, hair may also be reddish blond or brown. Pigmented birthmarks may be present. With maturity, the amount of pigment in the skin and hair tends to increase. In sun-exposed areas, pigmented nevi and freckles can develop.
Extracutaneous findings
The dilution of iris pigment may be mild to moderate. With age, the amount of pigment in the eyes tends to increase. Ocular manifestations are generally not as severe as those seen in OCA1A.2 Visual acuity and nystagmus tend to improve with age.
OCA2 can be found in 1% of individuals with Prader–Willi and Angelman syndromes.6
Etiology and pathogenesis
OCA2 results from loss-of-function mutations of the OCA2 gene (previously called the P gene), which is mapped to chromosome 15q11.2–q13.7 The OCA2 gene encodes the P protein, a melanosomal membrane protein. The specific function of the P protein is currently not known, but is believed to be involved in tyrosine transport within the melanocyte, regulation of melanosome pH and tyrosinase processing and transport.8 The melanocytes of affected individuals are able to synthesize some melanin, but the majority is yellow pheomelanin rather than black-brown eumelanin.
Prader–Willi syndrome involves deletions of the 15q region, including the OCA2 gene, on the paternally inherited copy of chromosome 15, whereas Angelman syndrome involves loss of the maternally inherited allele. Deletion of one copy of the OCA2 gene associated with a mutation in the second copy results in OCA2 in these patients.6
Oculocutaneous albinism type 3
OCA3, previously called ‘rufous OCA’, is most commonly seen in people of African and Puerto Rican descent.9,10
Cutaneous findings
At birth, individuals with this tyrosinase-positive OCA have light brown to red-brown skin and hair. With age, the hair becomes more pigmented. Mild sun tanning is possible.
Extracutaneous findings
At birth, the irises are light brown and become more pigmented with age. Ocular manifestations are present, but less severe. Red reflex on transillumination of the iris and nystagmus are important clues to the diagnosis in dark-skinned people.
Oculocutaneous albinism type 4
OCA4 may be one of the most common types of OCA in Japan, with solute carrier family 45, member 2, SLC45A2 (previously called ‘membrane-associated transporter protein’, MATP) gene mutations found in 24% of 75 unrelated Japanese patients with OCA.13
Extracutaneous findings
Ocular manifestations include nystagmus, decreased iris pigment with iris translucency, reduced retinal pigment, foveal hypoplasia associated with reduction in visual acuity, and strabismus.
Etiology and pathogenesis
OCA4 results from mutations in the SLC45A2 gene, which is mapped to chromosome 5p13.2. The gene encodes a melanosomal membrane protein that is likely to function as a transporter.13 The similar functions of the OCA2 and SLC45A2 genes may explain the phenotypic resemblance of OCA2 and OCA4.
Differential diagnosis
The diagnosis of OCA is usually made clinically and can be confirmed by DNA mutation analysis. Historically, the hair bulb incubator test for tyrosinase activity was used to differentiate between tyrosinase-positive and tyrosinase-negative OCA. In tyrosinase-negative albinism, there is the lack of pigment formation in hair bulbs when incubated with tyrosine, whereas in tyrosinase-positive albinism, pigment is produced. Prenatal diagnosis of OCA using DNA mutation analysis is available.
OCA can be differentiated from other disorders with cutaneous and ocular albinism by the absence of neurological defects, immunodeficiency, and bleeding diathesis.
Treatment and care
No specific treatment is available for OCA. The importance of photoprotection, including sun avoidance, broad-spectrum sunscreen, protective eyewear and clothing, should be stressed to reduce the risk of photodamage and cutaneous malignancies. Early ophthalmologic evaluation and management is important. As squamous cell carcinomas and basal cell carcinomas have been known to develop in all types of OCA, yearly examination by a dermatologist is recommended.
Hermansky–pudlak syndrome
Hermansky–Pudlak syndrome (HPS) comprises nine genetically different autosomal recessive disorders characterized by tyrosinase-positive OCA, a bleeding diathesis, and a lysosomal ceroid storage disease affecting the viscera.16–18 The majority of individuals affected are of Puerto Rican or Dutch descent. HPS type 1 is the most common.
Cutaneous findings
The degree of pigmentary dilution in the skin and hair is highly variable. The color of the skin and hair ranges from white to brown.
Extracutaneous findings
The degree of pigmentary dilution in the eyes is highly variable. Other ocular findings include nystagmus, reduced retinal pigment, and foveal hypoplasia with significant reduction in visual acuity. The nystagmus is most obvious during periods of fatigue or emotional change. The bleeding diathesis is caused by platelet storage pool deficiency and results in epistaxis, gingival, menstrual, colonic or post-surgical bleeding. Platelet numbers, prothrombin time, and partial thromboplastin time are normal but bleeding time is prolonged. The absence of dense bodies on electron microscopy of platelets is pathognomonic of HPS.19 Lysosomal ceroid accumulation can result in interstitial pulmonary fibrosis, granulomatous colitis, cardiomyopathy, and renal failure. These life-threatening complications usually develop in adulthood. Some patients with HPS type 2 have persistent neutropenia and suffer from recurrent bacterial infections.20,21
Etiology and pathogenesis
Most of the HPS-related genes encode proteins involved in the biogenesis of lysosome-related organelles. Ceroid is produced by degradation of lipids and glycoproteins within lysosomes. Ceroid accumulation in HPS suggests a defect in the elimination mechanisms of lysosomes.22
Differential diagnosis
The diagnosis of HPS is made on clinical findings of oculocutaneous albinism, bleeding diathesis, and absence of dense bodies on electron microscopy of platelets. Molecular genetic testing of some HPS types, e.g. HPS1, HPS3, is available on a clinical basis. The differential diagnosis includes the Griscelli, Elejalde and Cross syndromes.
Treatment and care
Photoprotection is important, as patients have a predisposition to develop basal cell carcinoma and squamous cell carcinoma. An examination by a dermatologist should be performed annually. Patients should avoid aspirin and trauma to minimize the chance of a bleeding episode. Platelet transfusions may be considered prior to surgical procedures. Cigarette smoking should be avoided, as this reduces pulmonary function and may hasten progression of pulmonary fibrosis.
Chediak–higashi syndrome
Chediak–Higashi syndrome (CHS) is an autosomal recessive disorder characterized by OCA, immunodeficiency, progressive neurological deterioration, a mild bleeding tendency and abnormal inclusions present in a wide variety of cells.
Cutaneous findings
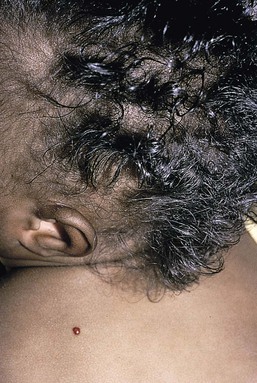
Compared with unaffected family members, the skin and the hair of affected individuals are lighter in color. Cutaneous pigmentation is often slightly to moderately decreased. The hair is blond to light brown, often with a silvery tint (Fig. 23.3). Recurrent skin and systemic pyogenic infections occur in early childhood. Cutaneous involvement usually manifests as a pyoderma, and there are a few reports of deeper involvement resembling pyoderma gangrenosum.23
Extracutaneous findings
Loss of ocular pigmentation results in a translucent iris and pale retina, leading to photophobia and an increased red reflex. Visual acuity is normal, but strabismus and nystagmus are common.
Infections typically involve the skin, lungs, and upper respiratory tract. These intractable infections are often fatal before the age of 10 years. Common culprits include Staphylococcus aureus, Streptococcus pyogenes and S. pneumoniae.
Periodontitis is an important manifestation of the immunologic dysfunction.24
Progressive neurologic deterioration with clumsiness, abnormal gait, paresthesias, and dysesthesias is often apparent later in childhood. Other neurologic abnormalities include peripheral and cranial neuropathies, spinocerebellar degeneration, ataxia, seizures, decreased deep tendon reflexes, cranial nerve palsies, and motor weakness.25–30
Most patients with CHS eventually develop a lymphoproliferative syndrome (‘accelerated phase’) characterized by fever, hepatosplenomegaly, lymphadenopathy, pancytopenia, bleeding, and generalized lymphohistiocytic infiltrates.26 Viral infections, particularly with the Epstein–Barr virus, have been implicated in causing the accelerated phase.31,32
Etiology and pathogenesis
CHS results from mutations in the lysosomal trafficking regulator gene (LYST) gene. The LYST gene (1q 42.1–q42.2) encodes a large cytoplasmic protein that appears to function as an adapter protein to mediate intracellular membrane fusion reactions.33
Natural killer (NK) cell function is drastically decreased. Diminished chemotaxis of granulocytes, monocytes, and lymphocytes has also been reported, as well as decreased antibody-dependent cytotoxicity and reduced suppressor T-cell function.34–36 The resulting susceptibility to infections is caused by the combination of these factors.37
The diagnostic hallmark of CHS is the finding of giant lysosomal granules within leukocytes, melanocytes, platelets, and other cells due to uncontrolled fusion of lysosomes. In bone marrow myeloid cells, the giant granules appear prominent. In melanocytes, giant melanosomes result from uncontrolled fusion of melanosomes, and this failure to disperse melanin to adjacent keratinocytes accounts for the decrease in pigmentation.38 On a cellular level, abnormal intracellular transport to and from the lysosomes has been detected.39 Giant granules within the phagocytic cells cannot discharge their lysosomal and peroxidative enzymes into phagocytic vacuoles.40 Prenatal diagnosis has been successfully performed using light microscopy by examining fetal hair shafts for characteristic clumping of melanosomes.41
Treatment and care
Hematopoietic stem-cell transplantation (HSCT) is the only definitive treatment for this disorder.42 The NK cell defects and immunodeficiencies can be reversed, but the neurological deterioration and pigmentary dilution are not altered.43,44
Without allogenic HSCT, patients who develop an accelerated phase usually die in childhood, usually from pyogenic infections or hemorrhage.26 Patients who do not develop an accelerated phase tend to have fewer or no infections, but usually develop progressively debilitating neurologic manifestations.45,46 Supportive treatments in early infancy include antibiotics for infections, and intravenous γ-globulin. Nonsteroidal anti-inflammatory drugs can exacerbate the bleeding tendency and should be avoided. Ascorbic acid has been shown to partially correct the granulocytic function in some patients.34,35
Cross syndrome
Cross syndrome, or oculocerebral syndrome with hypopigmentation, is an oculocutaneous albinism associated with ocular anomalies, postnatal growth retardation, and neurological defects.47–49 The inheritance is probably autosomal recessive.
Cutaneous findings
The affected neonate has cutaneous generalized hypopigmentation and silvery hair.
Extracutaneous findings
Ocular defects include microphthalmos, a small opaque cornea, and nystagmus. Neurological defects include mental retardation, ataxia, and spasticity.
Treatment and care
Treatment is supportive.
Phenylketonuria (phenylalanine hydroxylase deficiency)
Phenylketonuria (PKU) is an autosomal recessive disorder that results from the impaired conversion of phenylalanine to tyrosine, which is caused by the absence of hepatic phenylalanine hydroxylase (PAH) activity. The absence of this enzyme leads to a build-up of the amino acid phenylalanine and its byproducts in the bloodstream and spinal fluid. The incidence in the USA is estimated at 1 in 10 000 among Caucasians.50 It is most commonly observed in individuals of Scandinavian, Turkish and Irish descent, with males and females equally affected. PKU without treatment results in mental retardation and oculocutaneous pigment dilution. Most affected individuals have blond hair, blue eyes, fair skin, photosensitivity, a musty body odor, and neurologic disturbances.51
Cutaneous findings
At birth, the neonate appears normal but may have a musty odor secondary to urinary and sweat phenylacetic acid or phenylacetaldehyde. Caucasian children with PKU almost invariably have blond hair, blue eyes, fair skin, and photosensitivity. African-American and Asian children tend to be lighter in color than their parents and unaffected siblings. The ability to tan is normal. Endogenous eczema often develops in these patients.
Extracutaneous findings
In affected babies, serum phenylalanine levels begin to rise on the third or fourth day of life. Newborn screening with the Guthrie card bacterial inhibition assay test was implemented in the USA beginning in 1963, testing all newborns for PKU. Prenatal diagnosis is also possible by performing amniocentesis or chorionic villus sampling, with identification of the gene.51 Untreated PKU results in neurologic defects, including mental retardation, seizures, psychosis, hyperreflexia, and growth retardation.
Etiology and pathogenesis
Phenylalanine hydroxylase deficiency is caused by mutations in the PAH gene (mapped to 12q23.2), with more than 400 different mutations identified so far.52,53 Hypotheses to account for the decrease in skin and hair pigmentation include a competitive inhibition of the binding of tyrosine to tyrosinase by excess phenylalanine or a decreased amount of tyrosine.54
Treatment and care
With a low phenylalanine diet, the skin color, photosensitivity, odor, and eczema are reversible. Implementing a diet low in phenylalanine early in infancy can also dramatically reduce the mental retardation.51 Although children with treated PKU typically have a lower IQ than the general population, affected individuals can be expected to have a low-normal to normal intelligence if blood phenylalanine is maintained at a reasonable level in early childhood.55 Supplementation with tyrosine or tryptophan in the diet may be necessary. For women with PAH deficiency who are considering pregnancy, dietary restriction must be started before conception and continued throughout pregnancy.56 Aspartame, an artificial sweetener that contains phenylalanine, should be avoided.
Generalized hypopigmentation involving skin and hair
Griscelli syndrome
Griscelli first described this syndrome in 1978.57 It is a rare autosomal recessive syndrome that results in pigmentary dilution of the skin and hair, the presence of large clumps of pigment in hair shafts, and an accumulation of melanosomes in melanocytes. There are three types:
• Type 1 (GS1): association of hypopigmentation and neurological abnormalities, due to mutations of the MYO5A gene (15q21.2)58,59
• Type 2 (GS2): association of hypopigmentation and immunological abnormalities, due to RAB27A gene (15q21.3) mutation60
• Type 3 (GS3): only hypopigmentation, due to melanophilin (MLPH) gene (2q37.3) mutation.61
Cutaneous findings
In early childhood, individuals with all three types of GS have silvery gray hair, eyebrows, and eyelashes (findings that may also be present in the neonatal period), and skin hypopigmentation.62–65
Histologically, the hair shafts reveal uneven clumps of melanin, mainly in the medulla. Skin biopsy specimens reveal hyperpigmented oval melanocytes and poorly pigmented adjacent keratinocytes. On electron microscopic examination, epidermal melanocytes are found to contain perinuclear stage IV melanosomes. Adjacent keratinocytes contain only sparse melanosomes.66
Prenatal diagnosis of Griscelli syndrome has been accomplished by examination of hair from fetal scalp biopsies performed at 21 weeks’ gestation, with confirmatory postabortion examination of the fetus revealing silvery hair and identical microscopic findings.41
Extracutaneous findings
Neurological defects in GS1 include intracranial hypertension, cerebellar signs, encephalopathy, hemiparesis, peripheral facial palsy, spasticity, hypotonia, seizures, psychomotor retardation, and progressive neurologic deterioration.64–68
Immunological abnormalities in GS2 result in severe pyogenic infections, due to defective release of cytotoxic lysosomal contents from hematopoietic cells, and a hemophagocytic syndrome.66,69 There is combined T- and B-cell immunodeficiency. Frequent pyogenic infections, acute febrile episodes, neutropenia, and thrombocytopenia usually begin between 4 months of age and 4 years.57,64,65,67,70–72 The hemophagocytic syndrome is characterized by acute onset of uncontrolled lymphocyte and macrophage activation, resulting in infiltration and hemophagocytosis in multiple organs and death.
Differential diagnosis
Differentiation from Chediak–Higashi syndrome can be made by pathognomonic light and electron microscopic features. Griscelli syndrome lacks the large cytoplasmic inclusions and granulocyte abnormalities that are characteristic of Chediak–Higashi syndrome. Both diseases, however, are associated with an accelerated phase and carry a poor prognosis without bone marrow transplantation.
Elejalde syndrome
Elejalde syndrome, also called neuroectodermal melanolysosomal disease, is a rare autosomal recessive disorder characterized by silvery hair, hypopigmented skin, severe central nervous system dysfunction, and abnormal intracytoplasmic inclusions in fibroblasts, histiocytes, and lymphocytes.76–78
Cutaneous findings
Neonates have silvery hair and generalized hypopigmentation of the skin, which may develop a bronze color after sun exposure.79
In homozygotes, abnormal melanolysosomes are found in melanocytes and keratinocytes, cultured fibroblasts, and histiocytes of bone marrow.
Extracutaneous findings
Extracutaneous features include mental retardation, hypotonic facies, plagiocephaly, nystagmus, diplopia, micrognathia, crowded teeth, a narrow high palate, pectus excavatum, and cryptorchidism. Neurological abnormalities range from severe hypotonia and the almost complete absence of movements, to seizures and spasticity. The age of onset of neurologic signs ranges from 1 month to 11 years.78
Differential diagnosis
Several authors have suggested that subtypes of Elejalde syndrome and Griscelli syndrome type 1 are the same entity.80–83 However, the absence of immunologic defects allows Elejalde syndrome to be distinguished from the Griscelli syndrome type 1. Chediak–Higashi syndrome should also be considered in the differential diagnosis.
Treatment and care
Treatment is supportive and prognosis is poor.
Menkes disease (and occipital horn syndrome)
Classic Menkes disease is a multisystem disorder that manifests with hypopigmentation, hair abnormalities, failure to thrive, connective tissue changes, seizures, neurological degeneration, and death by the age of 3 years.84 The disorder has a prevalence of 1 in 250 000–350 000 live births.85 Most infants born with Menkes disease appear normal for the first few months of life before showing a rapid decline in growth and neurologic development.
Cutaneous findings
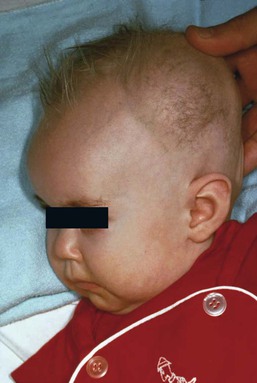
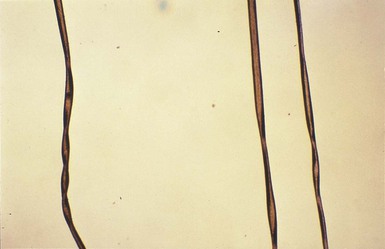
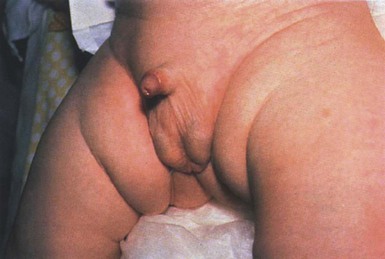
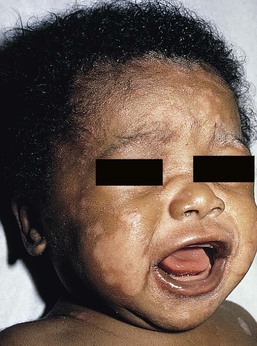
The cutaneous manifestations include alterations in hair, pigmentation, and elasticity of the skin.86 The scalp hair may appear normal at birth, but by about 3 months of age, it becomes sparse, light colored, lusterless, with a ‘steel wool’ quality. The hair is fragile and fractures easily, resulting in generalized alopecia (Fig. 23.4). Pili torti is the most common hair shaft abnormality (Fig. 23.5), demonstrating a flattened appearance under light microscopy with multiple twists of 180° around the long axis of the shaft.87 The twisted hairs result from excessive free sulfhydryl groups and a decrease in copper-dependent disulfide bonds. Cutaneous hypopigmentation is common, may be generalized or localized to the skin folds, and is caused by decreased tyrosinase, a copper-containing enzyme. Scaly dermatitis in a seborrheic distribution occurs frequently88 and transient neonatal erythroderma has been reported as an initial manifestation of Menkes disease.89 The skin is lax, and this doughy laxity is most prominent over the posterior neck, eyebrows, and leg folds (Fig. 23.6).86 Generalized puffiness of the cheeks and feet has been noted.
Extracutaneous findings
Progressive neurodegeneration begins at about 2 months of age as a result of gliosis and demyelination of the cerebrum and cerebellum.84 Patients present with seizures, hypothermia, developmental retardation, spontaneous subdural hematomas, muscle hypotonia, and feeding difficulties.90 Urogenital problems include undescended testes, hydronephrosis, hydro-ureter, recurrent urinary tract infections, diverticula of the ureters and bladder, and rupture of the bladder. Chronic intractable diarrhea resulting in malnutrition88,91 and increased frequency of congenital heart defects are seen.92 Skeletal abnormalities are manifested as wormian bones of the skull and spurring of long bone metaphyses. Connective tissue changes are evidenced by loose joints and tortuous blood vessels, such as the carotid and cerebellar arteries, which may cause intracranial hemorrhages. This increased tortuosity is secondary to fragmentation of the internal elastic lamina of the arteries. Low copper and ceruloplasmin levels in the serum and high copper levels in cultured fibroblasts are useful in the diagnosis.93 Menkes disease can be considered a disorder of copper maldistribution.
Etiology and pathogenesis
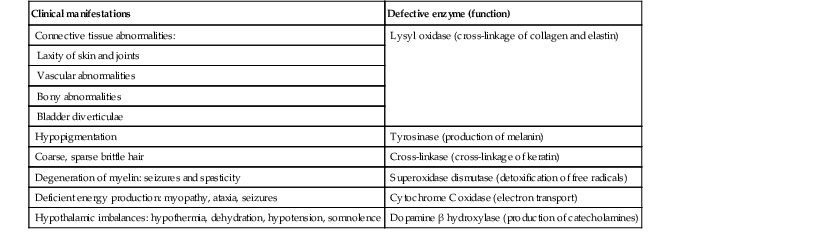
Menkes disease and occipital horn syndrome are rare, allelic, X-linked recessive copper deficiency disorders caused by mutations in the ATP7A gene (mapped to Xq21.1), which encodes a copper-transporting P-type ATPase involved in transport of copper to copper-requiring proteins.94,95 ATPase is localized in the trans-Golgi membrane of cells. The clinical features are due to malfunction of one or more copper-requiring enzymes, such as lysyl oxidase, tyrosinase, cytochrome C oxidase, and dopamine beta-hydroxylase, caused by the deficiency of the ATP7A protein (Table 23.2).86 Most patients are males, although a few female patients have also been reported. Most of the female patients have an X; autosome translocation, where the normal X-chromosome is preferentially inactivated.96
TABLE 23.2
Effects of defective copper-dependent enzymes in Menkes disease
| Clinical manifestations | Defective enzyme (function) |
| Connective tissue abnormalities: | Lysyl oxidase (cross-linkage of collagen and elastin) |
| Laxity of skin and joints | |
| Vascular abnormalities | |
| Bony abnormalities | |
| Bladder diverticulae | |
| Hypopigmentation | Tyrosinase (production of melanin) |
| Coarse, sparse brittle hair | Cross-linkase (cross-linkage of keratin) |
| Degeneration of myelin: seizures and spasticity | Superoxidase dismutase (detoxification of free radicals) |
| Deficient energy production: myopathy, ataxia, seizures | Cytochrome C oxidase (electron transport) |
| Hypothalamic imbalances: hypothermia, dehydration, hypotension, somnolence | Dopamine β hydroxylase (production of catecholamines) |
Occipital horn syndrome, formerly classified as Ehlers–Danlos syndrome type IX or X-linked cutis laxa, is now recognized as a milder form of Menkes disease and is caused by mutations in the same gene. Occipital horn syndrome is characterized primarily by connective tissue abnormalities, including skin laxity, hyperextensible joints, urinary tract diverticuli, hernias, and bony changes such as osteoporosis, arthrosis, and exostoses, such as the presence of a spike of ossification within the occipital insertion of the paraspinal muscles trapezius and sternocleidomastoid muscles at their attachments to the occipital bone (occipital horns), which gives the syndrome its name.97 Intelligence is normal or borderline, and patients can survive into adulthood. The milder phenotype results from the presence of low levels of functional ATP7A, unlike Menkes disease, in which no normal ATP7A activity exists.85,98
Treatment and care
Daily subcutaneous administration of copper-histidine has been shown to be helpful in preventing the severe neurodegenerative problems in some patients with Menkes disease when the treatment is initiated early in life before the onset of significant neurological symptoms.99,100 This treatment, however, does not prevent the development of connective tissue problems, and cannot be regarded as a cure for Menkes disease.100–102
Mosaic hypopigmentation
Mosaicism refers to the presence of two or more genetically distinct cell lines within an individual. These cell lines may be due to X-inactivation, as is normal in all human females, or to postzygotic somatic mutation. When mosaicism affects the skin, the affected skin may show patchy hypopigmentation or hyperpigmentation in a linear or segmental distribution (Figs. 23.7, 23.8). (Pigmentary mosaicism associated with hyperpigmented disorders is discussed in Chapter 24, and other mosaic conditions in Chapter 29.) Segmental hypopigmented lesions may be seen as an isolated cutaneous skin condition or as part of a genetic syndrome. The presence of mosaicism can sometimes be documented by the karyotyping of lymphocytes from peripheral blood or by genomic evaluation of both involved and uninvolved skin.
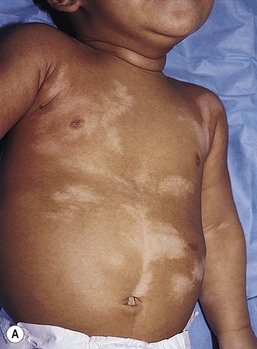
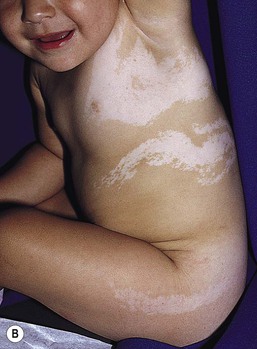
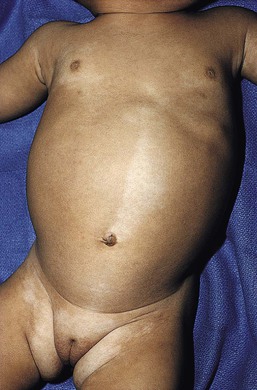
In 1901, Blaschko characterized the distribution of segmental and linear skin abnormalities by examining patients with linear lesions and formulating a patterned composite diagram. He described these patterns as V-shaped or fountain-like over the spine, S-shaped or whorled on the anterior and lateral aspects of the trunk, and linear over the extremities (see Chapter 3). These lines should not be confused with dermatomes, which are the segments of skin that correspond to sensory innervation.103 Hypopigmentation that follows the lines of Blaschko and segmental patterns is thought to reflect cellular migration during embryogenesis affecting pigmentation.104
Nevoid hypopigmentation
In 1952, a Japanese dermatologist named Ito described a 21-year-old woman with hypopigmented cutaneous whorls and streaks.105 As the distribution of the hypopigmentation was analogous to that of the hyperpigmented streaks observed in incontinentia pigmenti, he called the disorder ‘incontinentia pigmenti achromians’. To avoid confusion of these two unrelated entities, the preferred terminology later became ‘hypomelanosis of Ito’ (HI).106
HI is a descriptive term, rather than a diagnosis.107 It has been used for a phenotype with unilateral or bilateral hypopigmented streaks and whorls that follow the lines of Blaschko and which are present at birth or become apparent within the first 2 years of life (Figs 23.7, 23.8).108 There may be associated systemic findings.
Recently, the terms ‘nevoid hypopigmentation,’ ‘mosaic hypopigmentation’ or ‘segmental pigmentary disorder’ with or without systemic anomalies, were adopted to better reflect the heterogeneous nature of this group of disorders.
Cutaneous findings
Hypopigmented whorls and streaks are distributed along the lines of Blaschko. They tend to be stable, although there are reported cases in which the pigmentary changes become more or less pronounced over time.109 In some cases, both hypopigmented and hyperpigmented streaks are evident. Wood’s lamp examination may help to determine the extent of the lesions in fair-skinned patients.
Extracutaneous findings
Extracutaneous findings are variable, and include central nervous, musculoskeletal, and/or ocular abnormalities.110 Defects of teeth, hair, nails, and sweat glands, as well as aplasia cutis, fibromas, and generalized or focal hypertrichosis, have been reported.109,111,112 Additional abnormalities reported include limb-length discrepancies, facial hemiatrophy, scoliosis, sternal abnormalities, dysmorphic facies, genitourinary and cardiac anomalies. Nearly all of the defects are detectable by a thorough physical examination and regular follow-up. Infants should be observed for evidence of CNS involvement, reflected by developmental delay or seizures.109 Most children with CNS involvement manifest with neurological abnormalities before 2 years of age.
Etiology and pathogenesis
Embryonic somatic mutations are the likely pathogenesis, with distribution and pattern of lesions determined by the type of progenitor cell affected and the timing of mutation during embryogenesis. Multiple chromosomal abnormalities have been associated with HI, and most cases are sporadic and have negligible risk of recurrence.106,111,113 On histologic examination, the hypopigmented areas have either normal or reduced numbers of melanocytes, and those melanocytes that are present demonstrate a reduction in the number of melanosomes.114
Differential diagnosis
This includes nevus depigmentosus (also known as nevus achromicus) and Goltz syndrome. Patients with Goltz syndrome have both hyper- and hypopigmentation, as well as depressed areas of depigmentation following Blaschko’s lines.
Treatment and care
Cosmetic cover-up products can be used to conceal the hypopigmented areas but are usually not needed. The use of sunscreens can prevent or lessen the accentuation of pigmentary differences.109
Nevus depigmentosus
Cutaneous findings
Nevus depigmentosus (nevus achromicus) is an uncommon birthmark occurring in 0.4% of newborns.115 It is a well-circumscribed area of hypopigmentation with an off-white color that may occur as a small isolated (circular or rectangular) patch, or develop in a unilateral segmental distribution or follow the lines of Blaschko.116 Hair within a nevus depigmentosus may also be hypopigmented, and the margins may be irregular or serrated. The isolated form is the most common (Fig. 23.9) and the lesions do not usually cross the midline.117 The term depigmentosus is a misnomer because the lesions are actually hypopigmented, not completely depigmented, and become more prominent under Wood’s lamp. They are usually present at birth or become evident shortly thereafter, and remain stable in size and shape. Increase in size is in proportion to the growth of the child. Some lesions may appear during the first 3 years of life,117–119 with the trunk being the most commonly affected site.117,119,120 Occasionally, lesions may appear after the age of 3 years.117,120 In particular, the back and buttocks are most commonly affected, followed by the chest and abdomen.117 Males and females are affected equally and there is no distinct pattern of inheritance.116 The hypothesis is that during embryogenesis a clone of cells with a reduced melanogenic potential arises via a post-zygotic somatic mutation.103
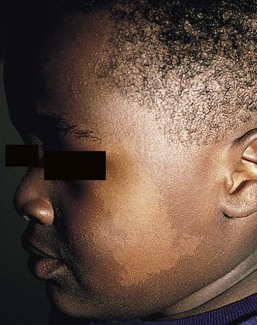
Extracutaneous findings
Systemic manifestations are rare in patients with nevus depigmentosus. Neurologic abnormalities such as seizures and mental retardation have been reported,121 as well as ipsilateral hypertrophy of the extremities.122 In a survey of 50 patients with nevus depigmentosus, none had any extracutaneous features on examination.123 In two studies involving 29 patients with nevus depigmentosus, extracutaneous abnormalities were present in about 10% of the children.118,124 Occasionally, lentigines can develop within the achromic nevi, and this could be explained by the reversion of a mutation in one of the genes involved in pigmentation.125,126 There is a report of an infant developing multiple primary milia within a nevus depigmentosus.127
Etiology and pathogenesis
With dopa-staining, normal melanocytes are seen in nevus depigmentosus, and electron microscopic studies suggest a reduced synthesis of melanosomes and also a defect in their transfer to the keratinocytes, which could account for the hypopigmentation.128 Transfer of melanosomes from melanocytes to keratinocytes is essential for normal pigmentation.
Differential diagnosis
Other entities with which nevus depigmentosus is sometimes confused include nevus anemicus, segmental vitiligo, hypopigmented lesions of tuberous sclerosis, and hypomelanosis of Ito. The distinction between nevus depigmentosus and hypomelanosis of Ito may be artificial, as many patients with segmental hypopigmented macules also have linear pigmentary anomalies, similar to those seen in hypomelanosis of Ito, and underlying mosaicism is the common factor.109 Patients currently diagnosed with either of these conditions might simply be categorized as having nevoid hypopigmentation with or without extracutaneous anomalies. Genetic analysis may be considered in all patients with segmental or linear pigmentary abnormalities, and those with extracutaneous abnormalities to investigate cytogenic anomalies.109
Treatment and care
There has been a report of spontaneous resolution of nevus depigmentosus after a 6-year period.129 One patient with a nevus depigmentosus had partial repigmentation following autologous melanocyte grafting.130 The use of noncultured epidermal cellular grafting has been reported to be successful in the treatment of nevus depigmentosus and a mottled repigmentation of 78% was observed.131 Repigmentation however, may not be permanent and recurrence of a successfully repigmented nevus depigmentosus 6 years after blister roof grafting has been noted.132 Twice weekly narrow-band UVB for 32 sessions was helpful in repigmenting a nevus depigmentosus lesion,133 as was the 308 nm excimer laser.134 Cosmetic camouflage may be helpful.
Localized hypopigmented disorders
Piebaldism
Piebaldism is an autosomal dominant condition caused by defective cell proliferation and migration of melanocytes during embryogenesis.
Cutaneous findings
Piebaldism is characterized by congenital depigmented white patches of skin and hair on the forehead, central chest and abdomen, upper arms and lower legs, with normally pigmented skin on the hands and feet (Fig. 23.10). A white forelock, which consists of a tuft of white hair over the midfrontal scalp is present in 80–90% of patients and is associated with depigmentation of the underlying scalp.135 Additional findings include poliosis of the eyebrows and eyelashes. The presence of islands of normally pigmented and hyperpigmented macules within the depigmented patches is typical and aids in the clinical diagnosis.136 The lesions are generally stable in size and increase in proportion to the growth of the child, although in some cases spontaneous contraction or expansion with the appearance of new hyperpigmented macules has been reported.137,138 Spontaneous repigmentation of leukoderma in sun-exposed areas such as the forehead or knees has been reported.139
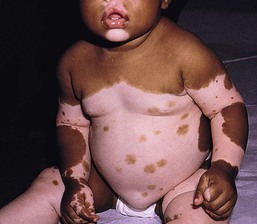
Light and electron microscopy studies show a complete absence of melanin and melanocytes in the epidermis and hair bulbs in the areas of leukoderma and poliosis.128 The hypermelanotic macules contain a normal number of melanocytes, but an abundance of abnormal melanosomes that are granular and spherical in shape.
Extracutaneous findings140–144
Piebaldism is not typically associated with abnormalities of other organs, although associated mental retardation has been reported.140 This may represent contiguous gene deletion syndromes, with inclusion of the KIT gene responsible for piebaldism as well as nearby genes whose absence result in neurologic deficits. There have been at least five reports of piebaldism associated with neurofibromatosis type 1(NF1).141–144 Whether the simultaneous occurrence of these two dominantly inherited diseases is more than chance remains to be established. It has been suggested that café-au-lait macules and intertriginous freckling are occasional features of piebaldism itself and that the diagnosis of NF1 must be based on non-pigmentary diagnostic criteria such as neurofibromas or the presence of an NF1 gene mutation by DNA testing.145,146
Etiology and pathogenesis
Piebaldism results from mutations of the KIT gene on chromosome 4q11–q12.147 This codes for the tyrosine kinase transmembrane cellular receptor for mast/stem cell growth factor, a critical factor for melanoblast migration, proliferation, differentiation, and survival. The KIT proto-oncogenes consist of an extracellular ligand-binding domain, a transmembrane domain, and a cytoplasmic tyrosine kinase domain.148 The severity of the clinical phenotype in piebaldism correlates with the site of the mutation within the KIT gene.149,150 The most severe mutations tend to be dominant negative missense mutations involving the intracellular tyrosine kinase domain. Mutations causing an intermediate severity phenotype have largely been located at the transmembrane region, and the mildest phenotypes are those that occur in the amino terminal extracellular ligand-binding domain. It has been shown that KIT activation induces the expression of a zinc-finger neural crest transcription factor SLUG, and that SLUG is necessary for the normal development of melanocytes, hematopoietic stem cells, and germ cells.151 It has also been shown that deletions in the SLUG (SNA12) gene on chromosome 8q11 are responsible for some cases of piebaldism that lacked mutations in KIT.152
Differential diagnosis
Disorders with similar clinical presentations are vitiligo and Waardenburg syndrome. Vitiligo is acquired later in life, tends to progress, and has a different distribution. Waardenburg syndrome is the major entity in the differential diagnosis of piebaldism, and the patient should be examined for evidence of facial dysmorphism, heterochromia of the irides, and congenital sensorineural hearing loss.
Treatment and care
Photoprotection of the depigmented patches is important, beginning early in life to protect the amelanotic areas from burning with sun exposure and to avoid skin cancers later on. Cosmetic camouflage or the use of a pigmenting tanning product such as dihydroxyacetone to camouflage the depigmented lesions are useful, although temporary.153 PUVA therapy is generally disappointing,154 but a combination of dermabrasion and split-thickness skin grafting followed by minigrafting155 or the use of autologous cultured epidermal grafts156 may be worthwhile in selected patients. Recently, the use of noncultured epidermal cellular grafting131 and melanocyte transplant techniques using noncultured melanocytes (minigrafting)157 has been shown to be helpful with high repigmentation rates.
Waardenburg syndrome
Waardenburg syndrome is a rare autosomal dominant disorder characterized by depigmented patches of the skin and hair, heterochromia iridis, congenital nerve deafness, and craniofacial anomalies. It is caused by the absence of melanocytes in the skin, hair, eyes, and stria vascularis of the cochlea, and is classified as a disorder of neural crest development.158
The estimated incidence of Waardenburg syndrome is 1 in 42 000 in the Netherlands159 and 1 in 20 000 in Kenya.160 It accounts for between 2% and 5% of cases of congenital deafness.161 Both sexes and all races are equally affected.162 Four types of Waardenburg syndrome have been described on clinical and genetic grounds (Box 23.2).
Cutaneous and extracutaneous findings
Affected persons have a depigmented patch, often V-shaped, on the central forehead in association with a white forelock.159 Premature graying of the hair may occur. Depigmented patches with irregular borders containing hyperpigmented macules, resembling piebaldism, as well as hyperpigmented macules on normal skin, have been described.162 Histology reveals a reduced number or complete absence of melanocytes within the depigmented areas.163 Synophrys, or fusion of the medial eyebrows, is typical in Waardenburg syndrome. Heterochromia irides, or differently colored irises, may be present, as well as sectorial areas of diminished pigment in a single iris and bilateral isohypochromia iridis (pale blue eyes) (Fig. 23.11).164 Type 1 Waardenburg syndrome is characterized by the presence of dystopia canthorum, or an increase in the inner canthal distance, without change in the interpupillary distance. If the inner canthal distance divided by the interpupillary distance exceeds 0.6, dystopia canthorum is present.164 In type 2 Waardenburg syndrome, dystopia canthorum is absent.165 Additional facial features include a broad nasal root, hypoplastic alar cartilage, a thin upper lip and a protuberant lower lip.159
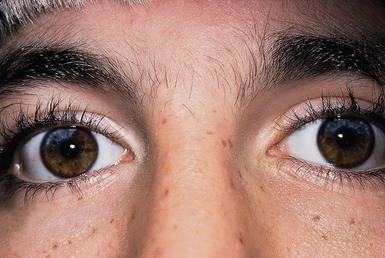
Congenital sensorineural hearing loss is a hallmark of all forms of Waardenburg syndrome and may be unilateral or bilateral.159,165 Histopathologic examination of the inner ears has shown absent organs of Corti, atrophy of the spinal ganglion, and reduction of nerve fibers.166 Hearing loss and heterochromia irides are more common in type 2 than in type 1 Waardenburg syndrome.167 Rarely cleft lip and palate168 and neural tube defects such as spina bifida169 have been reported in association with Waardenburg syndrome.
Waardenburg syndrome type III (Klein–Waardenburg syndrome) is similar to type 1, but is also accompanied by musculoskeletal anomalies of upper limbs and pectoral areas.170 Type IV Waardenburg syndrome (Shah–Waardenburg syndrome) is the association of Waardenburg syndrome with Hirschsprung disease (congenital aganglionic megacolon).171
Etiology and pathogenesis
Waardenburg syndrome types 1 and 3 are allelic variants and due to mutations in the PAX3 gene on chromosome 2q.172 PAX3 is one of a family of paired box genes that control neural crest differentiation by regulating the transcription of a number of other genes involved in embryological development. The neural crest gives rise not only to melanocytes but also to the bony and cartilaginous structures of the central face, accounting for the dysmorphic features associated with Waardenburg syndrome. Most mutations in PAX3 result in Waardenburg syndrome type 1. Simple loss of function of one allele will result in the type 1 phenotype due to haploinsufficiency for the PAX3 gene product (heterozygotes). A small fraction of PAX3 mutations result in the more severe type 3 phenotype, and some of these patients are homozygous for the PAX3 mutation, suggesting a gene dosage effect.173
Waardenburg syndrome type 2 is caused by mutations in the gene encoding the microphthalmia association transcription factor (MITF), mapped to chromosome 3p.174 The MITF gene product is a dimeric transcription factor of the basic–helix–loop–helix–leucine zipper class that is expressed in skin, hair follicles, retina and otic vesicles, and is involved in melanocyte differentiation. Type 2 Waardenburg syndrome is a heterogeneous group and some patients are heterozygous for mutations in the MITF gene, resulting in haploinsufficiency of the MITF protein due to loss of function mutations in one of the alleles of the MITF gene.175 MITF is downstream of PAX3, and this hierarchy of effect explains the lesser facial abnormalities in Waardenburg syndrome type 2 compared to type 1. Homozygous deletions of the SNA12 (SLUG) gene, encoding snail homolog 2, a zinc-finger transcription factor, have been described in two patients with Waardenburg syndrome type 2.176 Recently, SOX10 (SRY-sex determining region Y-box 10) mutations have also been reported to account for about 15–30% of cases of Waardenburg syndrome type 2.177,178 SOX10 is located on chromosome 22q13.1 and is a key transcription factor of neural crest development. It is crucial for the survival and maintenance of pluripotency of migrating neural crest progenitors.179
Waardenburg syndrome type 4 is a rare autosomal recessive condition caused by mutations in the genes for endothelin-3 (EDN3), or one of its receptors, endothelin β receptor (EDNRB) and to the SOX10 gene.180,181 All of these genes are functionally interrelated and contribute to the formation of the nervous system. EDNRB mutations are dosage sensitive: heterozygosity predisposes to isolated Hirschsprung disease, whereas homozygosity results in more complex neurocristopathies associating Hirschsprung disease and Waardenburg syndrome.182 About 20–30% of cases are due to mutations within the EDN3 or the EDNRB genes and about 45–55% result from mutations within the SOX10 gene.183 Mutations in the SOX10 gene are also responsible for an extended syndrome called PCWH (peripheral demyelinating neuropathy, central dysmyelinating leucodystrophy, Waardenburg syndrome, Hirschsprung disease), which is caused mostly by mutations in the last coding exon of SOX10.184
Differential diagnosis
Piebaldism should be considered in patients with dominantly inherited patchy depigmentation. The pigmentary disturbances are very similar in both Waardenburg syndrome and piebaldism, but auditory and facial developmental anomalies are absent in piebaldism.162 Fisch syndrome should be considered in cases of premature graying of the hair and congenital deafness.185 The association of deafness and vitiligo with an autosomal recessive mode of inheritance is known as Rozycki syndrome.186
Treatment and care
Physical findings suggestive of Waardenburg syndrome in neonates warrant a hearing evaluation, as detection of the associated deafness allows for early intervention with hearing aids and specialized education. The use of multichannel cochlear implants in Waardenburg syndrome children with profound deafness was found to be useful for the development and improvement of speech perception and production.187 Spontaneous pigmentation and contraction of the leukodermic patches in Waardenburg syndrome has been reported.188 Treatment options for the leukoderma are the same as those for piebaldism.
Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) is an autosomal dominant condition with variable penetrance, characterized by cutaneous and neurologic abnormalities such as mental retardation and seizures as well as visceral hamartomas. Spontaneous mutations account for 66–86% of cases.189,190 Estimates of prevalence range from 1 : 6000 to 1 : 10 000.191,192
Criteria for diagnosis are shown in Box 23.3.193
Cutaneous findings
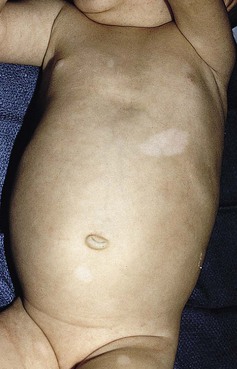
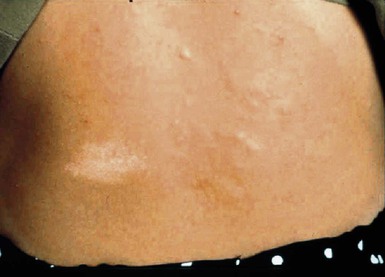
Hypomelanotic macules or patches are the earliest sign of tuberous sclerosis complex and are present in up to 90% of patients.190 The lesions are almost always present from birth, rarely developing later in infancy or childhood. They have a partial rather than complete loss of pigmentation, and perifollicular pigmentation may be observed in some of them.116 Multiple hypopigmented macules are of concern for TSC, although three or fewer may be a variant of normal and occur in otherwise healthy individuals.194 Examination under Wood’s lamp may be necessary to detect subtle lesions in fair-skinned individuals.195 The hypopigmented macules can be polygonal (thumbprint), lance-ovate (ash leaf spot) or guttate (confetti-like) (Figs. 23.12, 23.13).196 Confetti-like lesions appear as multiple small areas of stippled hypopigmentation, typically on the extremities. Poliosis of scalp hair, eyebrows, or eyelashes may be seen in patients with TSC,197 as well as circumscribed hypopigmentation of the iris or fundus.198 Electron microscopy of the hypopigmented macules reveals smaller organelles and a reduction in the size and number of the melanosomes, which exist mainly in the unmelanized stages.128
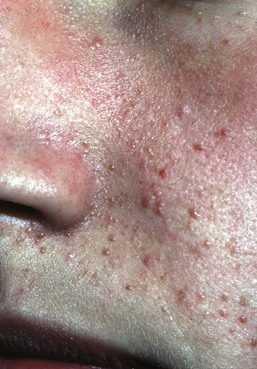
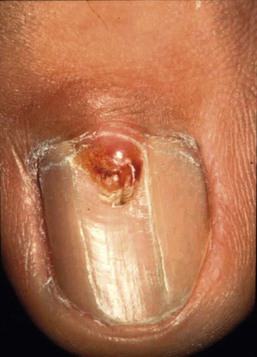
Facial angiofibromas (adenoma sebaceum) are made up of vascular and connective tissue elements and are present in three-quarters of patients representing the most frequent lesion in TSC.199 They are first noticed between the ages of 2 and 8 years as a few small red papules on the malar area, and gradually become larger and more numerous, sometimes extending down the nasolabial folds and onto the chin (Fig. 23.14). A fibrous plaque may be seen on the forehead or scalp, is typically present from birth or early infancy, and has a similar histologic appear to an angiofibroma.193 The shagreen patches, which are connective tissue nevi, are found in 20–30% of TSC patients, typically on the back or flank.190 They appear as slightly raised areas with dimpling at areas of follicular openings, giving the appearance of ‘orange peel’ or ‘gooseflesh’. Ungual and periungual fibromas are nodular or fleshy lesions that arise adjacent to or from underneath the nails (Fig. 23.15). Ungual fibromas usually occur in adolescents or adults, and are seen in 15–20% of TSC patients.190 Although they are highly suggestive of TSC, these lesions may occasionally develop spontaneously200 or after trauma. Examination of the mouth can reveal gingival fibromas and small dental enamel pits. Dental pits are more common in older patients with TSC than in unaffected individuals, and particularly affect the permanent teeth.201
Extracutaneous findings
These include retinal hamartomas, seen in up to 87% of patients, which appear as classic mulberry lesions adjacent to the optic disc.202 Most retinal lesions are clinically insignificant, although occasional patients may have visual impairment due to a large macular lesion, hamartoma enlargement, retinal detachment, or vitreous hemorrhage.
Up to two-thirds of infants with TSC will have cardiac rhabdomyomas, detected by echocardiography pre- or postnatally.190 The lesions tend to be multiple, and there is evidence that the majority regress in early childhood.203 Complications include congestive heart failure, cardiac arrhythmia, or cerebral thromboembolism.204
Renal involvement includes angiomyolipomas, which are benign tumors composed of varying amounts of vascular tissue, fat or smooth muscle, and occur in about two-thirds of TSC patients.205 The prevalence of renal tumors increases throughout childhood, and larger lesions are more likely to become symptomatic than smaller tumors. Single or multiple renal cysts tend to appear earlier than the angiomyolipomas, and the combination of the two is characteristic of tuberous sclerosis complex.190 The proximity of the TSC2 gene and the polycystic kidney disease gene may play a synergistic role in cyst development.206 Both angiomyolipomas and renal cysts are often asymptomatic and require no treatment. Hemorrhage is the most common complication of angiomyolipomas, causing hematuria and pain. Renal failure results from obstructive uropathy, or when cysts or tumors replace much of the normal renal parenchyma.
Pulmonary changes are rare, seldom cause symptoms, are five times more common in females, and tend to become clinically manifest in the second decade.207 The most common lesion is pulmonary lymphangiomyomatosis (LAM), which is characterized by a proliferation of abnormal smooth muscle-like LAM cells and leads to pulmonary cysts and pneumothorax.208 Exacerbation during pregnancy and estrogen supplementation has been noted.209 These lesions express estrogen and progesterone receptors, which may explain their gender predilection and sensitivity to hormonal changes. Recurrent spontaneous pneumothorax, dyspnea, cough, hemoptysis, and pulmonary failure are manifestations of pulmonary LAM.
Neurologic manifestations of TSC include mental retardation, seizures, and behavioral problems such as autism,210 hyperkinesis, aggressiveness, and psychosis.211 Infantile spasms are common, occurring in about 70% of affected babies. Neurologic lesions result from impaired cellular interaction, resulting in disrupted neuronal migration along radial glial fibers and abnormal proliferation of glial elements.190 Neuropathologic lesions include subependymal nodules, cortical hamartomas, focal cortical hypoplasia, and heterotopic gray matter. The number of cortical tubers as observed by magnetic resonance imaging (MRI) has been shown to correlate with the severity of the cerebral dysfunction, with increased numbers of cortical tubers seen in patients with more severe cerebral disease.212
Etiology and pathogenesis
Inactivating mutations in either of two tumor-suppressor genes – TSC1 and TSC2 – are the cause of this syndrome, with TSC2 mutations accounting for 80–90% of all mutations.213 TSC1 contains 21 coding exons on chromosome 9q34.13 and encodes hamartin of 1164 amino acids; TSC2 contains 41 exons on 16p13.3, which encodes tuberin of 1807 amino acids.214,215 Hamartin and tuberin function as a complex that suppresses a major pathway for the stimulation of cell growth. The hamartin/tuberin complex inactivates GTPase Rheb, which is an activator of the growth-promoting protein kinases rapamycin (mTOR) and P70 S6 kinase (S6K).216 TSC1 and TSC2 are tumor suppressors that inhibit the mammalian target of rapamycin (mTOR), and if mutated, results in mTOR activation, leading to an increase in protein translation and formation of hamartomas in TSC. Moreover, proto-oncogenes and tumor suppressors, such as Ras and PTEN, were found to regulate growth via the hamartin/tuberin complex.217 Patients with TSC1 mutations generally have milder disease than patients with TSC2 mutations, and patients in whom no mutation was found also had milder disease than patients in whom mutations were found.218
Differential diagnosis
Lesions that should be distinguished from the hypopigmented macules of TSC include nevus depigmentosus, nevus anemicus, post-inflammatory hypopigmentation, pityriasis alba, tinea versicolor, and vitiligo.194 Nevus depigmentosus is most easily confused with the hypopigmented macules of TSC.116 A nevus depigmentosus can only be differentiated from a single ash leaf spot by the absence of other signs or symptoms of TSC.
Treatment and care
Management is multidisciplinary. Special education may be required if mental retardation is present. Facial angiofibromas may be treated with the pulsed dye vascular laser. The more nodular lesions may be treated with shave excision and electrodesiccation or with carbon dioxide laser, but slowly recur.219
Rapamycin (sirolimus) and the related MTOR inhibitor, everolimus, are immunosuppressive agents traditionally used in transplant recipients. Rapamycin has antineoplastic effects which may be mediated by decreasing the production of vascular endothelial growth factor and it corrects aberrant signaling pathways involved in cell growth and apoptosis.220 Oral rapamycin at standard immunosuppressive doses has been used as a targeted therapy with promising results for the renal, lung and neurological manifestations of TSC.221,222 It can induce regression of visceral angiomyolipomas, brain astrocytomas and improve lung function in patients with lymphangioleiomyomatosis.
However, regrowth may occur with renal angiomyolipomas after stopping oral rapamycin therapy.221 Topical rapamycin twice daily has been shown to be an effective and safe treatment for facial angiofibromas.223–228 Improvement was noted within a week of starting treatment223,228 and resolution of lesions in a month to 6 weeks.224,227 Topical rapamycin is especially effective in younger children with flatter lesions.228 Recently, topical rapamycin twice a day for 12 weeks was noted to improve the hypomelanotic macules of TSC.226 Rapamycin has been shown to increase the transcription of microphthalmia transcription factor (MITF), which is involved in melanogenic gene expression.229 The main side-effect of topical rapamycin is cutaneous irritation with undetectable serum rapamycin levels during treatment.225,228 Both angiofibromas and hypomelanotic macules may recur a few months after stopping treatment.226 Rapamycin and its analogues may represent a major advance in therapy.
Examination of the parents and other family members of seemingly sporadic patients with TSC is indicated to exclude the possibility of a mild phenotype of which parents are not aware. Gonadal mosaicism is possible in around 2% of families where parents show no signs of TSC after full clinical evaluation.230
Nevus anemicus
Cutaneous findings
Nevus anemicus is a congenital vascular anomaly characterized by single or multiple hypopigmented patches with a normal texture and irregular margins, sometimes surrounded by satellite macules (Fig. 23.16). It appears at birth or in early childhood, and is more common in females.231,232 Although it occurs most commonly on the trunk, nevus anemicus has also been reported on the extremities and the head and neck.232,233 It persists unchanged throughout life.
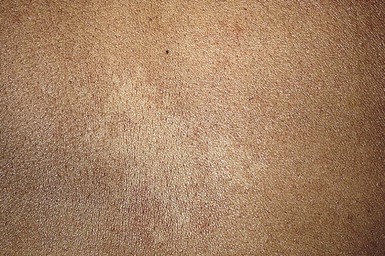
Extracutaneous findings
Nevus anemicus may be seen in close association with port wine stains.232 These twin anomalies may be a result of somatic recombination of allelic mutations for vasoconstriction and vasodilatation.234 Phakomatosis pigmentovascularis, where vascular and pigmented nevi occur in association with nevus anemicus, has been used to support this concept.235 Lesions of nevus anemicus occur with increased frequency in patients with neurofibromatosis.231
Etiology and pathogenesis
Examination by light and electron microscopy reveals no abnormality, and the nevus is best characterized as a pharmacological abnormality, rather than an anatomical one.233 The pallor is due to an increased local vascular reactivity to catecholamines. Donor dominance was demonstrated by grafting lesional skin from nevus anemicus to normal skin, which retained its pale appearance, emphasizing that nevus anemicus is due to increased sensitivity of the blood vessels to catecholamines rather than to increased sympathetic stimulation.236
Differential diagnosis
Under diascopic pressure with a glass microscope slide, the lesion becomes indistinguishable from the blanched surrounding skin.237 Wood’s lamp examination does not accentuate the lesion, and rubbing or temperature change causes erythema in the surrounding area but not within the lesion itself. These maneuvers help to distinguish nevus anemicus from vitiligo, nevus depigmentosus, tuberous sclerosis macules, tinea versicolor, and leprosy.238Treatment is cosmetic, and if desired the discoloration can be concealed with camouflage make-up.
Vitiligo
Vitiligo is an acquired disease characterized by well-demarcated depigmented patches caused by destruction of epidermal and follicular melanocytes. Vitiligo affects up to 2% of pediatric populations.239 It rarely occurs in infancy.
Cutaneous findings
There are several methods of clinical classification. A useful method that provides an indication of the prognosis and treatment is to categorize vitiligo into non-segmental and segmental types.
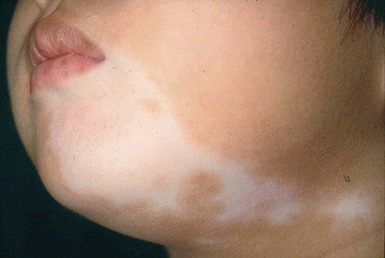
Segmental vitiligo describes unilateral depigmented patches arranged in a segmental, non-dermatomal pattern (Fig. 23.17). It is seen more commonly in children as compared to adults.240 The face is most commonly affected. It progresses rapidly but tends to be confined within the affected segment and remains stable thereafter.241
Non-segmental vitiligo may be generalized, where depigmented patches are distributed symmetrically in more than one region of the body, or localized (focal, acrofacial, acral or mucosal). Children with non-segmental disease are more likely to have more extensive involvement, more frequent progression of disease and thyroid abnormalities on laboratory investigations than children with segmental disease.242
Extracutaneous findings
Autoimmune diseases like thyroiditis, alopecia areata, diabetes mellitus, pernicious anemia and Addison’s disease have been reported in children with non-segmental vitiligo. As the frequency of occurrence of these autoimmune diseases is low, thyroid function tests, full blood count and fasting blood glucose can be performed selectively, for example, in a child who fails to thrive.
Etiology and pathogenesis
There is destruction of functional melanocytes by mechanisms that are not fully understood. The autoimmune theory suggests that melanocytes are destroyed by humoral and cell-mediated immunity.243 Other theories suggest that melanocytes are intrinsically defective, injured by oxidative stress or nerve cells.
Genetic studies provide support for the autoimmune pathogenesis theory in non-segmental vitiligo. NLRP1 and XBP1 are vitiligo susceptibility genes that control key aspects of immune regulation. Non-segmental generalized vitiligo has been epidemiologically and genetically associated with some of the other autoimmune diseases, for example, type 1 diabetes mellitus and Addison disease.244
Differential diagnosis
The differential diagnoses of vitiligo include pityriasis alba, tinea versicolor, post-inflammatory hypopigmentation, piebaldism and nevus depigmentosus.245 Pityriasis alba and tinea versicolor, in contrast to vitiligo, are hypopigmented rather than depigmented and often have scaling and indistinct borders. In post-inflammatory hypopigmentation, irregular mottling of both hyperpigmented and hypopigmented areas is often seen. Piebaldism is an autosomal dominant condition presenting at birth with anterior midline depigmentation and a white forelock (poliosis). Nevus depigmentosus is a segmental hypopigmentation detectable at birth or within the first year of life and grows in proportion to the child’s growth. With a Wood’s lamp, the contrast between lesional and normal skin is less marked than in vitiligo.246
Treatment and care
All children with vitiligo should have photoprotection with a broad-spectrum sunscreen of SPF-30+ during daytime outdoor activity so that sunburn and subsequent koebnerization does not occur.
Cosmetic camouflage such as make-up and self-tanning agents help with the psychosocial burden of vitiligo, especially when extensive and with lesions on exposed skin.247 Self-tanning agents containing dihydroxyacetone (DHA) may be used in older children. It causes the skin to turn brown by polymerizing the amino acids in the skin and is useful to camouflage the depigmented lesions. The desired results usually appear after about 6 h and the resulting pigmentation remains for about 3–4 days.248
In children, mid-potency topical steroids are commonly used as a first-line therapy for localized vitiligo.249 Corticosteroids modulate the immune response by decreasing melanocyte destruction and induces melanocyte repopulation in vitiligo.250 Higher response rates occur in children compared to adults and head and neck lesions respond better.251 Complete repigmentation rates have been reported to be as high as 49.3%.252 Frequent monitoring is required to avoid local side effects like atrophy, telangiectasia, striae and steroid folliculitis.
As monotherapy, calcipotriene is inferior to topical corticosteroids, but when combined with topical steroids, repigmentation rates increase with a faster onset of repigmentation along with greater stability of repigmentation compared with either as monotherapy.253 Eyelid and facial skin respond best to this combination therapy.254 Off-label use of the fixed stable combined preparation of calcipotriene 0.005%-betamethasone dipropionate 0.064% ointment once daily, is a simple treatment option.255 Calcipotriol has been shown to downregulate the production of IL-8, which is believed to enhance the inflammatory destruction of melanocytes.255
Topical calcineurin inhibitors (TCI), tacrolimus and pimecrolimus, are advantageous in treating vitiligo without the side-effect profile of topical steroids. Tacrolimus inhibits T-cell activation, blocking the production and release of pro-inflammatory cytokines like TNF-α.256 A comparative study revealed 0.1% tacrolimus ointment to be almost as effective as 0.05% clobetasol propionate cream, both used twice a day for the treatment of childhood vitiligo, and tacrolimus may be useful for sensitive areas like the eyelids.252 Repigmentation occurs after 4–6 weeks with 0.1% tacrolimus ointment twice daily and pigmentation is maintained for at least 6–9 months after discontinuation of treatment.257 Pimecrolimus 1% cream twice daily has been shown to be effective in repigmentation of vitiligo in the head and neck region after 6 months of treatment.258 This repigmentation effect of topical pimecrolimus on the face is enhanced by the addition of narrow-band ultraviolet B (NB-UVB) thrice weekly.259 Tacrolimus 0.1% ointment has also been shown to enhance the effect of NB-UVB for the treatment of vitiligo.260
Topical psoralen-ultraviolet A (PUVA) can be used in older children with limited body surface area involvement.261 Narrow-band ultraviolet B phototherapy twice or thrice weekly is an effective and safe modality for the treatment of generalized childhood vitiligo with >20% body surface involvement.262–265 Better results are seen in children with recent onset disease (<1 year) with best repigmentation over the face and neck and moderate response on the trunk and proximal extremities.264 If no response is seen after 6 months of NB-UVB phototherapy, treatment should be stopped.265 The monochromatic excimer laser (308 nm) allows for the targeted treatment of specific lesions of vitiligo, avoiding the risk of photoaging of the surrounding skin. Cho and colleagues treated 30 children with localized vitiligo with the 308 nm excimer laser and noted a repigmentation rate of 50–75%, especially over the face, neck and trunk.266
Surgical methods may be chosen for older children with stable vitiligo lesions unresponsive to other treatment modalities. Various surgical techniques have been used such as suction blister epidermal grafting267 and autologous non-cultural epidermal cellular grafting. Better results are seen in segmental vitiligo compared to generalized vitiligo.
Whatever treatment is used, certain clinical features are poor prognostic markers for repigmentation. These include acral vitiligo, presence of leukotrichia over the vitiligo, and lesions over bony prominences like the elbows, knees and ankles.249
Post-inflammatory hypopigmentation
Cutaneous findings
Post-inflammatory hypopigmentation refers to the partial loss of melanin in previously inflamed areas of skin. The hypochromic macules and patches usually appear in a mottled pattern in the distribution and configuration of the inflammatory process, and are more obvious in dark skin (Fig. 23.18).
Etiology and pathogenesis
Post-inflammatory hypopigmentation can appear in the newborn period after inflammatory conditions such as atopic dermatitis, seborrheic dermatitis, diaper dermatitis, psoriasis, pityriasis lichenoides, and infectious conditions.
Differential diagnosis
The differential diagnosis of post-inflammatory hypopigmentation includes pityriasis alba, pityriasis versicolor and vitiligo. Pityriasis alba is characterized by ill-defined, hypopigmented, minimally scaly macules and patches, commonly seen on the face without erythema or pruritus. It is common, being present in 25–40% of dark-skinned children,268,269 and occasionally seen in the neonatal period. The histology of post-inflammatory hypopigmentation shows nonspecific findings, such as decreased epidermal melanin, superficial lymphohistiocytic infiltration and presence of melanophages in the upper dermis.270 Wood’s lamp will enhance the hypopigmented areas in light skin and helps to distinguish depigmentation from hypopigmentation. Wood’s lamp may help to exclude pityriasis versicolor which displays a coppery-orange fluorescence and a potassium hydroxide wet-mount preparation will show the presence of yeast (‘spaghetti and meatballs’ appearance).
Treatment and care
Treatment of the underlying cause of inflammation will improve the discoloration. Twice-daily application of a mid-strength topical steroid in combination with coal tar has been used to treat post-inflammatory hypopigmentation with some success, although the mechanisms behind this are not well understood.270 Twice-daily application of 1% pimecrolimus cream for 16 weeks has been shown to be beneficial for the treatment of seborrheic dermatitis with associated post-inflammatory hypopigmentation in dark-skinned patients.271 Other treatment options for post-inflammatory hypopigmentation include topical psoralen UVA (PUVA), narrowband UVB phototherapy and the 308 nm excimer laser, which had a response rate of 60–70% after 9 bi-weekly treatments.272 In pityriasis alba, repigmentation is achieved with avoidance of excessive sunlight and topical steroids applied daily for several weeks.
Congenital halo nevi
Cutaneous findings
A halo nevus is a benign melanocytic nevus (usually a compound nevus) surrounded by a ring of depigmentation. Depigmented zones around nevi have also been reported with congenital nevi,273,274 Spitz nevi, blue nevi, neurofibroma, and primary or metastatic malignant melanoma.275
Halo nevi are often multiple, usually occur on the trunk, and appear most commonly in young people. Vitiligo is often associated and develops at distant sites.274 Children with vitiligo tend to have more multiple halo nevi and a personal and/or family history of autoimmune thyroiditis compared with those with only halo nevi.276 Furthermore, patients with halo nevi and nonsegmental vitiligo tend to have a younger age at onset (<18 years), phototypes I–III, truncal involvement and premature graying of the hair compared to patients with vitiligo alone.277 The condition is usually not inherited, although familial cases have been described.278 The nevus tends to flatten and may eventually involute over a period of months to years, leaving an area of depigmentation that persists for up to a decade or longer, but which eventually may repigment.279
Etiology and pathogenesis
The nevus cells appear to be destroyed by cytotoxic CD8+ T-lymphocytes recognizing class 1 HLA antigens on their surfaces.280 This theory of an immunologic mechanism is supported by the fact that a lymphocytic infiltrate is seen around the nevus cells and the nevus cells show cytotoxic changes.281 Unlike acquired halo nevi, congenital halo nevi may have an absence of inflammation on histology and may not involute.273,274
Treatment and care
Usually no treatment is required. Education about the prolonged natural history of halo nevi reassures patients and avoids unnecessary excision.
Miscellaneous hypopigmentation
Alezzandrini syndrome
Cutaneous and extracutaneous findings
The syndrome is characterized by unilateral facial vitiligo, ipsilateral tapetoretinal degeneration, poliosis, and perceptive (sensorineural) deafness.282,283 Associated insulin-dependent diabetes mellitus and unilateral retinal detachment have been reported,284 as well as bilateral retinal detachment283,285 in patients with Alezzandrini syndrome.
Symptoms of visual loss begin gradually in one eye between the ages of 12 and 30 years. Vitiligo and poliosis of the scalp ipsilateral to the retinal lesions tend to occur 3–13 years after the visual decline.284 Ipsilateral café-au-lait patches have been reported in combination with the above features.284 Alezzandrini syndrome has only been reported in a small number of cases.286
Etiology and pathogenesis
The etiology of Alezzandrini syndrome is unknown. Theories involving viral or autoimmune processes have been postulated.287
Differential diagnosis
A possible variant of this syndrome is the Vogt–Koyanagi–Harada syndrome characterized by vitiligo, poliosis, alopecia areata, chronic uveitis, deafness and meningoencephalitis.
Treatment and care
Medical care includes follow-up fundus examinations, visual acuity tests and audiometry.285 Topical steroids, tacrolimus and use of a sunscreen may be tried on localized areas of vitiligo.
Ziprkowski–margolis syndrome
Cutaneous and extracutaneous findings
Ziprkowski–Margolis syndrome (albinism–deafness syndrome) is characterized by diffuse hypomelanosis of the skin and hair, except for the buttocks and genital region.288,289 With time, multiple symmetrical macules of hyperpigmentation appear on the trunk and extremities, giving the skin a leopard-like appearance. Other features are heterochromic irides, congenital nerve deafness, and mutism. The syndrome was first reported in a Sephardi Jewish family of Moroccan origin.
Etiology and pathogenesis
The mode of inheritance is X-linked recessive and the ADFN gene has been mapped to Xq24-q26.290,291 The mutation affects the migration of neural crest-derived precursors of the melanocytes. Female carriers of the diseases can demonstrate sensorineural hearing impairment.292
Treatment and care
Cochlear implantation for hereditary deafness has been reported to improve the speech perception abilities in patients with the albinism–deafness syndrome.293
Access the full reference list at ExpertConsult.com 
[/level-membership-for-dermatology-category][not-level-membership-for-dermatology-category]
Hypopigmentation Disorders
Yuin-Chew Chan, Yong-Kwang Tay
Introduction
A diverse group of conditions present with hypopigmentation in neonates and infants. A practical clinical approach would be to categorize them according to the distribution of the hypopigmentation: generalized, mosaic, or localized (Box 23.1). Some may be present at birth, but not noticed until later in infancy because neonates often have a lighter skin at birth than in later life.
Any defect occurring in melanocyte development, melanin synthesis and transport, or distribution of melanosomes to keratinocytes can result in a hypopigmentary disorder. Melanocytes originate from the neural crest and are located in the epidermis, hair bulb, eye (choroid, ciliary body, iris), inner ear (cochlea) and central nervous system (leptomeninges). Melanin is synthesized in melanosomes, which are organelles that share characteristics with lysosomes.
This chapter discusses a variety of clinical conditions causing hypopigmentation in neonates and infants, many of which have a genetic basis.
Generalized hypopigmentation of skin, hair, and eyes
Oculocutaneous albinism
Oculocutaneous albinism (OCA) refers to a group of autosomal recessive disorders involving abnormal melanin synthesis. Affected individuals have absent (type 1A) or reduced (type 1B, 2, 3, and 4) pigmentation in the skin, hair, and eyes from birth. The color varies from white to light brown, depending on ethnicity and the specific type of OCA. Ocular manifestations include nystagmus, photophobia, and decreased visual acuity. These are caused by decreased melanin within the eye or misrouting of optic nerve fibers. The affected infant is typically less pigmented than unaffected siblings.
Historically, OCA was divided into two clinical types based on the presence or absence of tyrosinase, the rate-limiting enzyme in the melanin biosynthetic pathway.1 Advances in molecular genetics have given rise to a more accurate classification and better understanding of pathogenesis.2,3 In OCA, epidermal and follicular melanocytes are present in normal quantity and distribution but do not synthesize melanin adequately (Table 23.1).
TABLE 23.1
Classification of disorders with cutaneous and ocular albinism
| OCA1A Tyrosinase negative | OCA1B Tyrosinase positive | OCA2 Tyrosinase positive | OCA3 Rufous | OCA4 | HPS | CHS | |
| Skin color | White | White at birth, develops some pigmentation in first and second decades | Creamy white to brown at birth. Slight darkening with age | Light brown to red-bronze | Creamy white to brown at birth. Slight darkening with age | White to brown | Creamy white to slate gray |
| Pigmented nevi and freckles | Absent | Present | Present | Absent | Present | Many in exposed areas | Present |
| Hair color | White throughout life; may become light yellow | White to light yellow at birth; turns yellow or blond in first few years | Light yellow, blond to brown at birth; may darken with age | Light brown to red-brown. May darken with age | Silvery white to light yellow at birth. May darken in childhood | White, blond to brown | Blond to light brown; metallic silver-gray sheen |
| Gene (mapping), function | TYR (11q14.3) Encodes tyrosinase |
TYR (11q14.3) Encodes tyrosinase |
OCA2 (15q11.2-q12) Encodes P protein, a melanosomal membrane protein |
TYRP1 (9q23) Encodes dihydroxyindol carboxylic acid oxidase, a melanogenic enzyme |
SLC45A2 (5p13.2) Encodes membrane-associated transporter protein |
HPS1: HPS1 (10q24.2) HPS2: AP3B1 (5q14.1) HPS3: HPS3 (3q24) HPS4: HPS4 (22q12.1) HPS5: HPS5 (11p15.1) HPS6: HPS6 (10q24.32) HPS7: DTNBP1 (6p22.3) HPS8: BLOC1S3 (19q13.32) HPS9: PLDN (15q21.1) Encode proteins involved in biogenesis of lysosomes and lysosome-related organelles, such as melanosomes and platelet-dense granules. Some may play a role in intracellular vesicle trafficking |
LYST (1q42.3) Encodes lysosomal-trafficking regulator, a cytoplasmic protein which may function as an adapter protein that mediates intracellular membrane fusion reactions |
| Mouse model | Albino | Albino | Pink-eye dilution | Brown | Underwhite | HPS1: Pale-ear HPS2: Pearl HPS3: Cocoa HPS4: Light-ear HPS5: Ruby eye 2 HPS6: Ruby eye HPS7: Sandy HPS8: Reduced pigmentation |
Beige |
| Hair bulb melanosomes | Stages I, II | Stages I, II, III | Stages I, II, III | Stages I, II, III, IV | Stages I, II, III | Stages I, II, III | Macromelanosomes and normal to Stage IV |
HPS, Hermansky–Pudlak syndrome; CHS, Chediak–Higashi syndrome.
Oculocutaneous albinism type 1
Oculocutaneous albinism type 1 (OCA1) is the second most common OCA worldwide.
Cutaneous findings
In OCA1A, there is marked generalized hypopigmentation at birth, with white hair and skin (Figs. 23.1, 23.2). As the child matures, the skin remains white. Nevi are not pigmented and sun tanning does not occur. The hair may acquire a slightly yellowish tint as a result of denaturing of hair keratins.
In OCA1B, the variable decrease in tyrosinase activity results in several clinical phenotypes: yellow, minimal pigment, platinum, and temperature-sensitive. Individuals with OCA1B form pheomelanin, which requires less tyrosinase activity, and this results in some pigment production during the first two decades of life.4 Affected individuals have a similar appearance to those with OCA1A at birth, but with time develop some pigment. Hair color can change from white to light blond and even progress to light brown in adolescence. With sun exposure, some individuals with OCA1B may be able to tan, although it is more common to burn without tanning. Pigmented nevi and freckles may develop.5
An interesting subtype of OCA1B is the temperature-sensitive phenotype in which the tyrosinase activity is seen mainly on the extremities. In these patients, the enzyme has no activity at 37°C, but some activity at 35°C. These individuals have white or lightly pigmented hairs on the scalp and trunk (axillae, pubic area) and darkly pigmented hair peripherally (legs, arms). The pattern is similar to that observed in Siamese cats.4
Extracutaneous findings
In OCA1A, the irises are pale blue at birth and throughout life. In bright light, the entire iris can appear pink or red, which is caused by its translucency.5 Severe photophobia results from a lack of retinal pigment. Other ocular abnormalities include decreased visual acuity, nystagmus, and strabismus.
In OCA1B, the irises can progressively darken to light tan or brown. In both subtypes, vision may remain stable or deteriorate with age.
Etiology and pathogenesis
OCA1 is caused by loss-of-function mutations in the tyrosinase gene (TYR), which is mapped to chromosome 11q14.3. Tyrosinase is the key enzyme that catalyzes the first two steps in melanin synthesis, converting tyrosine to L-DOPA (L-3,4-dihyroxyphenylalanine), and L-DOPA to DOPAquinone. The OCA1A subtype is characterized by mutations that result in complete loss of tyrosinase activity, whereas OCA1B is caused by mutations that result in markedly reduced tyrosinase activity (5–10% of the normal level).
Oculocutaneous albinism type 2
Oculocutaneous albinism type 2 (OCA2) is the most common form of OCA. It is most prevalent in people of African descent.6
Cutaneous findings
There is a spectrum of clinical phenotypes, depending on the ethnic background and the dilution of hair and skin pigment, which may be minimal to moderate. Comparison with a first-degree relative may be necessary to distinguish the degree of lightening.4 Most individuals are born with creamy white skin, and light yellow or blond hair. Depending on the individual’s ethnic background, hair may also be reddish blond or brown. Pigmented birthmarks may be present. With maturity, the amount of pigment in the skin and hair tends to increase. In sun-exposed areas, pigmented nevi and freckles can develop.
Extracutaneous findings
The dilution of iris pigment may be mild to moderate. With age, the amount of pigment in the eyes tends to increase. Ocular manifestations are generally not as severe as those seen in OCA1A.2 Visual acuity and nystagmus tend to improve with age.
OCA2 can be found in 1% of individuals with Prader–Willi and Angelman syndromes.6
Etiology and pathogenesis
OCA2 results from loss-of-function mutations of the OCA2 gene (previously called the P gene), which is mapped to chromosome 15q11.2–q13.7 The OCA2 gene encodes the P protein, a melanosomal membrane protein. The specific function of the P protein is currently not known, but is believed to be involved in tyrosine transport within the melanocyte, regulation of melanosome pH and tyrosinase processing and transport.8 The melanocytes of affected individuals are able to synthesize some melanin, but the majority is yellow pheomelanin rather than black-brown eumelanin.
Prader–Willi syndrome involves deletions of the 15q region, including the OCA2 gene, on the paternally inherited copy of chromosome 15, whereas Angelman syndrome involves loss of the maternally inherited allele. Deletion of one copy of the OCA2 gene associated with a mutation in the second copy results in OCA2 in these patients.6
Oculocutaneous albinism type 3
OCA3, previously called ‘rufous OCA’, is most commonly seen in people of African and Puerto Rican descent.9,10
Cutaneous findings
At birth, individuals with this tyrosinase-positive OCA have light brown to red-brown skin and hair. With age, the hair becomes more pigmented. Mild sun tanning is possible.
Extracutaneous findings
At birth, the irises are light brown and become more pigmented with age. Ocular manifestations are present, but less severe. Red reflex on transillumination of the iris and nystagmus are important clues to the diagnosis in dark-skinned people.
Oculocutaneous albinism type 4
OCA4 may be one of the most common types of OCA in Japan, with solute carrier family 45, member 2, SLC45A2 (previously called ‘membrane-associated transporter protein’, MATP) gene mutations found in 24% of 75 unrelated Japanese patients with OCA.13
Extracutaneous findings
Ocular manifestations include nystagmus, decreased iris pigment with iris translucency, reduced retinal pigment, foveal hypoplasia associated with reduction in visual acuity, and strabismus.
Etiology and pathogenesis
OCA4 results from mutations in the SLC45A2 gene, which is mapped to chromosome 5p13.2. The gene encodes a melanosomal membrane protein that is likely to function as a transporter.13 The similar functions of the OCA2 and SLC45A2 genes may explain the phenotypic resemblance of OCA2 and OCA4.
Differential diagnosis
The diagnosis of OCA is usually made clinically and can be confirmed by DNA mutation analysis. Historically, the hair bulb incubator test for tyrosinase activity was used to differentiate between tyrosinase-positive and tyrosinase-negative OCA. In tyrosinase-negative albinism, there is the lack of pigment formation in hair bulbs when incubated with tyrosine, whereas in tyrosinase-positive albinism, pigment is produced. Prenatal diagnosis of OCA using DNA mutation analysis is available.
OCA can be differentiated from other disorders with cutaneous and ocular albinism by the absence of neurological defects, immunodeficiency, and bleeding diathesis.
Treatment and care
No specific treatment is available for OCA. The importance of photoprotection, including sun avoidance, broad-spectrum sunscreen, protective eyewear and clothing, should be stressed to reduce the risk of photodamage and cutaneous malignancies. Early ophthalmologic evaluation and management is important. As squamous cell carcinomas and basal cell carcinomas have been known to develop in all types of OCA, yearly examination by a dermatologist is recommended.
Hermansky–pudlak syndrome
Hermansky–Pudlak syndrome (HPS) comprises nine genetically different autosomal recessive disorders characterized by tyrosinase-positive OCA, a bleeding diathesis, and a lysosomal ceroid storage disease affecting the viscera.16–18 The majority of individuals affected are of Puerto Rican or Dutch descent. HPS type 1 is the most common.
Cutaneous findings
The degree of pigmentary dilution in the skin and hair is highly variable. The color of the skin and hair ranges from white to brown.
Extracutaneous findings
The degree of pigmentary dilution in the eyes is highly variable. Other ocular findings include nystagmus, reduced retinal pigment, and foveal hypoplasia with significant reduction in visual acuity. The nystagmus is most obvious during periods of fatigue or emotional change. The bleeding diathesis is caused by platelet storage pool deficiency and results in epistaxis, gingival, menstrual, colonic or post-surgical bleeding. Platelet numbers, prothrombin time, and partial thromboplastin time are normal but bleeding time is prolonged. The absence of dense bodies on electron microscopy of platelets is pathognomonic of HPS.19 Lysosomal ceroid accumulation can result in interstitial pulmonary fibrosis, granulomatous colitis, cardiomyopathy, and renal failure. These life-threatening complications usually develop in adulthood. Some patients with HPS type 2 have persistent neutropenia and suffer from recurrent bacterial infections.20,21
Etiology and pathogenesis
Most of the HPS-related genes encode proteins involved in the biogenesis of lysosome-related organelles. Ceroid is produced by degradation of lipids and glycoproteins within lysosomes. Ceroid accumulation in HPS suggests a defect in the elimination mechanisms of lysosomes.22
Differential diagnosis
The diagnosis of HPS is made on clinical findings of oculocutaneous albinism, bleeding diathesis, and absence of dense bodies on electron microscopy of platelets. Molecular genetic testing of some HPS types, e.g. HPS1, HPS3, is available on a clinical basis. The differential diagnosis includes the Griscelli, Elejalde and Cross syndromes.
Treatment and care
Photoprotection is important, as patients have a predisposition to develop basal cell carcinoma and squamous cell carcinoma. An examination by a dermatologist should be performed annually. Patients should avoid aspirin and trauma to minimize the chance of a bleeding episode. Platelet transfusions may be considered prior to surgical procedures. Cigarette smoking should be avoided, as this reduces pulmonary function and may hasten progression of pulmonary fibrosis.
Chediak–higashi syndrome
Chediak–Higashi syndrome (CHS) is an autosomal recessive disorder characterized by OCA, immunodeficiency, progressive neurological deterioration, a mild bleeding tendency and abnormal inclusions present in a wide variety of cells.
Cutaneous findings
Compared with unaffected family members, the skin and the hair of affected individuals are lighter in color. Cutaneous pigmentation is often slightly to moderately decreased. The hair is blond to light brown, often with a silvery tint (Fig. 23.3). Recurrent skin and systemic pyogenic infections occur in early childhood. Cutaneous involvement usually manifests as a pyoderma, and there are a few reports of deeper involvement resembling pyoderma gangrenosum.23
Extracutaneous findings
Loss of ocular pigmentation results in a translucent iris and pale retina, leading to photophobia and an increased red reflex. Visual acuity is normal, but strabismus and nystagmus are common.
Infections typically involve the skin, lungs, and upper respiratory tract. These intractable infections are often fatal before the age of 10 years. Common culprits include Staphylococcus aureus, Streptococcus pyogenes and S. pneumoniae.
Periodontitis is an important manifestation of the immunologic dysfunction.24
Progressive neurologic deterioration with clumsiness, abnormal gait, paresthesias, and dysesthesias is often apparent later in childhood. Other neurologic abnormalities include peripheral and cranial neuropathies, spinocerebellar degeneration, ataxia, seizures, decreased deep tendon reflexes, cranial nerve palsies, and motor weakness.25–30
Most patients with CHS eventually develop a lymphoproliferative syndrome (‘accelerated phase’) characterized by fever, hepatosplenomegaly, lymphadenopathy, pancytopenia, bleeding, and generalized lymphohistiocytic infiltrates.26 Viral infections, particularly with the Epstein–Barr virus, have been implicated in causing the accelerated phase.31,32
Etiology and pathogenesis
CHS results from mutations in the lysosomal trafficking regulator gene (LYST) gene. The LYST gene (1q 42.1–q42.2) encodes a large cytoplasmic protein that appears to function as an adapter protein to mediate intracellular membrane fusion reactions.33
Natural killer (NK) cell function is drastically decreased. Diminished chemotaxis of granulocytes, monocytes, and lymphocytes has also been reported, as well as decreased antibody-dependent cytotoxicity and reduced suppressor T-cell function.34–36 The resulting susceptibility to infections is caused by the combination of these factors.37
The diagnostic hallmark of CHS is the finding of giant lysosomal granules within leukocytes, melanocytes, platelets, and other cells due to uncontrolled fusion of lysosomes. In bone marrow myeloid cells, the giant granules appear prominent. In melanocytes, giant melanosomes result from uncontrolled fusion of melanosomes, and this failure to disperse melanin to adjacent keratinocytes accounts for the decrease in pigmentation.38 On a cellular level, abnormal intracellular transport to and from the lysosomes has been detected.39 Giant granules within the phagocytic cells cannot discharge their lysosomal and peroxidative enzymes into phagocytic vacuoles.40 Prenatal diagnosis has been successfully performed using light microscopy by examining fetal hair shafts for characteristic clumping of melanosomes.41
Treatment and care
Hematopoietic stem-cell transplantation (HSCT) is the only definitive treatment for this disorder.42 The NK cell defects and immunodeficiencies can be reversed, but the neurological deterioration and pigmentary dilution are not altered.43,44
Without allogenic HSCT, patients who develop an accelerated phase usually die in childhood, usually from pyogenic infections or hemorrhage.26 Patients who do not develop an accelerated phase tend to have fewer or no infections, but usually develop progressively debilitating neurologic manifestations.45,46 Supportive treatments in early infancy include antibiotics for infections, and intravenous γ-globulin. Nonsteroidal anti-inflammatory drugs can exacerbate the bleeding tendency and should be avoided. Ascorbic acid has been shown to partially correct the granulocytic function in some patients.34,35
Cross syndrome
Cross syndrome, or oculocerebral syndrome with hypopigmentation, is an oculocutaneous albinism associated with ocular anomalies, postnatal growth retardation, and neurological defects.47–49 The inheritance is probably autosomal recessive.
Cutaneous findings
The affected neonate has cutaneous generalized hypopigmentation and silvery hair.
Extracutaneous findings
Ocular defects include microphthalmos, a small opaque cornea, and nystagmus. Neurological defects include mental retardation, ataxia, and spasticity.
Treatment and care
Treatment is supportive.
Phenylketonuria (phenylalanine hydroxylase deficiency)
Phenylketonuria (PKU) is an autosomal recessive disorder that results from the impaired conversion of phenylalanine to tyrosine, which is caused by the absence of hepatic phenylalanine hydroxylase (PAH) activity. The absence of this enzyme leads to a build-up of the amino acid phenylalanine and its byproducts in the bloodstream and spinal fluid. The incidence in the USA is estimated at 1 in 10 000 among Caucasians.50 It is most commonly observed in individuals of Scandinavian, Turkish and Irish descent, with males and females equally affected. PKU without treatment results in mental retardation and oculocutaneous pigment dilution. Most affected individuals have blond hair, blue eyes, fair skin, photosensitivity, a musty body odor, and neurologic disturbances.51
Cutaneous findings
At birth, the neonate appears normal but may have a musty odor secondary to urinary and sweat phenylacetic acid or phenylacetaldehyde. Caucasian children with PKU almost invariably have blond hair, blue eyes, fair skin, and photosensitivity. African-American and Asian children tend to be lighter in color than their parents and unaffected siblings. The ability to tan is normal. Endogenous eczema often develops in these patients.
Extracutaneous findings
In affected babies, serum phenylalanine levels begin to rise on the third or fourth day of life. Newborn screening with the Guthrie card bacterial inhibition assay test was implemented in the USA beginning in 1963, testing all newborns for PKU. Prenatal diagnosis is also possible by performing amniocentesis or chorionic villus sampling, with identification of the gene.51 Untreated PKU results in neurologic defects, including mental retardation, seizures, psychosis, hyperreflexia, and growth retardation.
Etiology and pathogenesis
Phenylalanine hydroxylase deficiency is caused by mutations in the PAH gene (mapped to 12q23.2), with more than 400 different mutations identified so far.52,53 Hypotheses to account for the decrease in skin and hair pigmentation include a competitive inhibition of the binding of tyrosine to tyrosinase by excess phenylalanine or a decreased amount of tyrosine.54
Treatment and care
With a low phenylalanine diet, the skin color, photosensitivity, odor, and eczema are reversible. Implementing a diet low in phenylalanine early in infancy can also dramatically reduce the mental retardation.51 Although children with treated PKU typically have a lower IQ than the general population, affected individuals can be expected to have a low-normal to normal intelligence if blood phenylalanine is maintained at a reasonable level in early childhood.55 Supplementation with tyrosine or tryptophan in the diet may be necessary. For women with PAH deficiency who are considering pregnancy, dietary restriction must be started before conception and continued throughout pregnancy.56 Aspartame, an artificial sweetener that contains phenylalanine, should be avoided.
Generalized hypopigmentation involving skin and hair
Griscelli syndrome
Griscelli first described this syndrome in 1978.57 It is a rare autosomal recessive syndrome that results in pigmentary dilution of the skin and hair, the presence of large clumps of pigment in hair shafts, and an accumulation of melanosomes in melanocytes. There are three types:
• Type 1 (GS1): association of hypopigmentation and neurological abnormalities, due to mutations of the MYO5A gene (15q21.2)58,59
• Type 2 (GS2): association of hypopigmentation and immunological abnormalities, due to RAB27A gene (15q21.3) mutation60
• Type 3 (GS3): only hypopigmentation, due to melanophilin (MLPH) gene (2q37.3) mutation.61
Cutaneous findings
In early childhood, individuals with all three types of GS have silvery gray hair, eyebrows, and eyelashes (findings that may also be present in the neonatal period), and skin hypopigmentation.62–65
Histologically, the hair shafts reveal uneven clumps of melanin, mainly in the medulla. Skin biopsy specimens reveal hyperpigmented oval melanocytes and poorly pigmented adjacent keratinocytes. On electron microscopic examination, epidermal melanocytes are found to contain perinuclear stage IV melanosomes. Adjacent keratinocytes contain only sparse melanosomes.66
Prenatal diagnosis of Griscelli syndrome has been accomplished by examination of hair from fetal scalp biopsies performed at 21 weeks’ gestation, with confirmatory postabortion examination of the fetus revealing silvery hair and identical microscopic findings.41
Extracutaneous findings
Neurological defects in GS1 include intracranial hypertension, cerebellar signs, encephalopathy, hemiparesis, peripheral facial palsy, spasticity, hypotonia, seizures, psychomotor retardation, and progressive neurologic deterioration.64–68
Immunological abnormalities in GS2 result in severe pyogenic infections, due to defective release of cytotoxic lysosomal contents from hematopoietic cells, and a hemophagocytic syndrome.66,69 There is combined T- and B-cell immunodeficiency. Frequent pyogenic infections, acute febrile episodes, neutropenia, and thrombocytopenia usually begin between 4 months of age and 4 years.57,64,65,67,70–72 The hemophagocytic syndrome is characterized by acute onset of uncontrolled lymphocyte and macrophage activation, resulting in infiltration and hemophagocytosis in multiple organs and death.
Differential diagnosis
Differentiation from Chediak–Higashi syndrome can be made by pathognomonic light and electron microscopic features. Griscelli syndrome lacks the large cytoplasmic inclusions and granulocyte abnormalities that are characteristic of Chediak–Higashi syndrome. Both diseases, however, are associated with an accelerated phase and carry a poor prognosis without bone marrow transplantation.
Elejalde syndrome
Elejalde syndrome, also called neuroectodermal melanolysosomal disease, is a rare autosomal recessive disorder characterized by silvery hair, hypopigmented skin, severe central nervous system dysfunction, and abnormal intracytoplasmic inclusions in fibroblasts, histiocytes, and lymphocytes.76–78
Cutaneous findings
Neonates have silvery hair and generalized hypopigmentation of the skin, which may develop a bronze color after sun exposure.79
In homozygotes, abnormal melanolysosomes are found in melanocytes and keratinocytes, cultured fibroblasts, and histiocytes of bone marrow.
Extracutaneous findings
Extracutaneous features include mental retardation, hypotonic facies, plagiocephaly, nystagmus, diplopia, micrognathia, crowded teeth, a narrow high palate, pectus excavatum, and cryptorchidism. Neurological abnormalities range from severe hypotonia and the almost complete absence of movements, to seizures and spasticity. The age of onset of neurologic signs ranges from 1 month to 11 years.78
Differential diagnosis
Several authors have suggested that subtypes of Elejalde syndrome and Griscelli syndrome type 1 are the same entity.80–83 However, the absence of immunologic defects allows Elejalde syndrome to be distinguished from the Griscelli syndrome type 1. Chediak–Higashi syndrome should also be considered in the differential diagnosis.
Treatment and care
Treatment is supportive and prognosis is poor.
Menkes disease (and occipital horn syndrome)
Classic Menkes disease is a multisystem disorder that manifests with hypopigmentation, hair abnormalities, failure to thrive, connective tissue changes, seizures, neurological degeneration, and death by the age of 3 years.84 The disorder has a prevalence of 1 in 250 000–350 000 live births.85 Most infants born with Menkes disease appear normal for the first few months of life before showing a rapid decline in growth and neurologic development.
Cutaneous findings
The cutaneous manifestations include alterations in hair, pigmentation, and elasticity of the skin.86 The scalp hair may appear normal at birth, but by about 3 months of age, it becomes sparse, light colored, lusterless, with a ‘steel wool’ quality. The hair is fragile and fractures easily, resulting in generalized alopecia (Fig. 23.4). Pili torti is the most common hair shaft abnormality (Fig. 23.5), demonstrating a flattened appearance under light microscopy with multiple twists of 180° around the long axis of the shaft.87 The twisted hairs result from excessive free sulfhydryl groups and a decrease in copper-dependent disulfide bonds. Cutaneous hypopigmentation is common, may be generalized or localized to the skin folds, and is caused by decreased tyrosinase, a copper-containing enzyme. Scaly dermatitis in a seborrheic distribution occurs frequently88 and transient neonatal erythroderma has been reported as an initial manifestation of Menkes disease.89 The skin is lax, and this doughy laxity is most prominent over the posterior neck, eyebrows, and leg folds (Fig. 23.6).86 Generalized puffiness of the cheeks and feet has been noted.
Extracutaneous findings
Progressive neurodegeneration begins at about 2 months of age as a result of gliosis and demyelination of the cerebrum and cerebellum.84 Patients present with seizures, hypothermia, developmental retardation, spontaneous subdural hematomas, muscle hypotonia, and feeding difficulties.90 Urogenital problems include undescended testes, hydronephrosis, hydro-ureter, recurrent urinary tract infections, diverticula of the ureters and bladder, and rupture of the bladder. Chronic intractable diarrhea resulting in malnutrition88,91 and increased frequency of congenital heart defects are seen.92 Skeletal abnormalities are manifested as wormian bones of the skull and spurring of long bone metaphyses. Connective tissue changes are evidenced by loose joints and tortuous blood vessels, such as the carotid and cerebellar arteries, which may cause intracranial hemorrhages. This increased tortuosity is secondary to fragmentation of the internal elastic lamina of the arteries. Low copper and ceruloplasmin levels in the serum and high copper levels in cultured fibroblasts are useful in the diagnosis.93 Menkes disease can be considered a disorder of copper maldistribution.
Etiology and pathogenesis
Menkes disease and occipital horn syndrome are rare, allelic, X-linked recessive copper deficiency disorders caused by mutations in the ATP7A gene (mapped to Xq21.1), which encodes a copper-transporting P-type ATPase involved in transport of copper to copper-requiring proteins.94,95 ATPase is localized in the trans-Golgi membrane of cells. The clinical features are due to malfunction of one or more copper-requiring enzymes, such as lysyl oxidase, tyrosinase, cytochrome C oxidase, and dopamine beta-hydroxylase, caused by the deficiency of the ATP7A protein (Table 23.2).86 Most patients are males, although a few female patients have also been reported. Most of the female patients have an X; autosome translocation, where the normal X-chromosome is preferentially inactivated.96
TABLE 23.2
Effects of defective copper-dependent enzymes in Menkes disease
| Clinical manifestations | Defective enzyme (function) |
| Connective tissue abnormalities: | Lysyl oxidase (cross-linkage of collagen and elastin) |
| Laxity of skin and joints | |
| Vascular abnormalities | |
| Bony abnormalities | |
| Bladder diverticulae | |
| Hypopigmentation | Tyrosinase (production of melanin) |
| Coarse, sparse brittle hair | Cross-linkase (cross-linkage of keratin) |
| Degeneration of myelin: seizures and spasticity | Superoxidase dismutase (detoxification of free radicals) |
| Deficient energy production: myopathy, ataxia, seizures | Cytochrome C oxidase (electron transport) |
| Hypothalamic imbalances: hypothermia, dehydration, hypotension, somnolence | Dopamine β hydroxylase (production of catecholamines) |
Occipital horn syndrome, formerly classified as Ehlers–Danlos syndrome type IX or X-linked cutis laxa, is now recognized as a milder form of Menkes disease and is caused by mutations in the same gene. Occipital horn syndrome is characterized primarily by connective tissue abnormalities, including skin laxity, hyperextensible joints, urinary tract diverticuli, hernias, and bony changes such as osteoporosis, arthrosis, and exostoses, such as the presence of a spike of ossification within the occipital insertion of the paraspinal muscles trapezius and sternocleidomastoid muscles at their attachments to the occipital bone (occipital horns), which gives the syndrome its name.97 Intelligence is normal or borderline, and patients can survive into adulthood. The milder phenotype results from the presence of low levels of functional ATP7A, unlike Menkes disease, in which no normal ATP7A activity exists.85,98
Treatment and care
Daily subcutaneous administration of copper-histidine has been shown to be helpful in preventing the severe neurodegenerative problems in some patients with Menkes disease when the treatment is initiated early in life before the onset of significant neurological symptoms.99,100 This treatment, however, does not prevent the development of connective tissue problems, and cannot be regarded as a cure for Menkes disease.100–102
Mosaic hypopigmentation
Mosaicism refers to the presence of two or more genetically distinct cell lines within an individual. These cell lines may be due to X-inactivation, as is normal in all human females, or to postzygotic somatic mutation. When mosaicism affects the skin, the affected skin may show patchy hypopigmentation or hyperpigmentation in a linear or segmental distribution (Figs. 23.7, 23.8). (Pigmentary mosaicism associated with hyperpigmented disorders is discussed in Chapter 24, and other mosaic conditions in Chapter 29.) Segmental hypopigmented lesions may be seen as an isolated cutaneous skin condition or as part of a genetic syndrome. The presence of mosaicism can sometimes be documented by the karyotyping of lymphocytes from peripheral blood or by genomic evaluation of both involved and uninvolved skin.
In 1901, Blaschko characterized the distribution of segmental and linear skin abnormalities by examining patients with linear lesions and formulating a patterned composite diagram. He described these patterns as V-shaped or fountain-like over the spine, S-shaped or whorled on the anterior and lateral aspects of the trunk, and linear over the extremities (see Chapter 3). These lines should not be confused with dermatomes, which are the segments of skin that correspond to sensory innervation.103 Hypopigmentation that follows the lines of Blaschko and segmental patterns is thought to reflect cellular migration during embryogenesis affecting pigmentation.104
Nevoid hypopigmentation
In 1952, a Japanese dermatologist named Ito described a 21-year-old woman with hypopigmented cutaneous whorls and streaks.105 As the distribution of the hypopigmentation was analogous to that of the hyperpigmented streaks observed in incontinentia pigmenti, he called the disorder ‘incontinentia pigmenti achromians’. To avoid confusion of these two unrelated entities, the preferred terminology later became ‘hypomelanosis of Ito’ (HI).106
HI is a descriptive term, rather than a diagnosis.107 It has been used for a phenotype with unilateral or bilateral hypopigmented streaks and whorls that follow the lines of Blaschko and which are present at birth or become apparent within the first 2 years of life (Figs 23.7, 23.8).108 There may be associated systemic findings.
Recently, the terms ‘nevoid hypopigmentation,’ ‘mosaic hypopigmentation’ or ‘segmental pigmentary disorder’ with or without systemic anomalies, were adopted to better reflect the heterogeneous nature of this group of disorders.
Cutaneous findings
Hypopigmented whorls and streaks are distributed along the lines of Blaschko. They tend to be stable, although there are reported cases in which the pigmentary changes become more or less pronounced over time.109 In some cases, both hypopigmented and hyperpigmented streaks are evident. Wood’s lamp examination may help to determine the extent of the lesions in fair-skinned patients.
Extracutaneous findings
Extracutaneous findings are variable, and include central nervous, musculoskeletal, and/or ocular abnormalities.110 Defects of teeth, hair, nails, and sweat glands, as well as aplasia cutis, fibromas, and generalized or focal hypertrichosis, have been reported.109,111,112 Additional abnormalities reported include limb-length discrepancies, facial hemiatrophy, scoliosis, sternal abnormalities, dysmorphic facies, genitourinary and cardiac anomalies. Nearly all of the defects are detectable by a thorough physical examination and regular follow-up. Infants should be observed for evidence of CNS involvement, reflected by developmental delay or seizures.109 Most children with CNS involvement manifest with neurological abnormalities before 2 years of age.
Etiology and pathogenesis
Embryonic somatic mutations are the likely pathogenesis, with distribution and pattern of lesions determined by the type of progenitor cell affected and the timing of mutation during embryogenesis. Multiple chromosomal abnormalities have been associated with HI, and most cases are sporadic and have negligible risk of recurrence.106,111,113 On histologic examination, the hypopigmented areas have either normal or reduced numbers of melanocytes, and those melanocytes that are present demonstrate a reduction in the number of melanosomes.114
Differential diagnosis
This includes nevus depigmentosus (also known as nevus achromicus) and Goltz syndrome. Patients with Goltz syndrome have both hyper- and hypopigmentation, as well as depressed areas of depigmentation following Blaschko’s lines.
Treatment and care
Cosmetic cover-up products can be used to conceal the hypopigmented areas but are usually not needed. The use of sunscreens can prevent or lessen the accentuation of pigmentary differences.109
Nevus depigmentosus
Cutaneous findings
Nevus depigmentosus (nevus achromicus) is an uncommon birthmark occurring in 0.4% of newborns.115 It is a well-circumscribed area of hypopigmentation with an off-white color that may occur as a small isolated (circular or rectangular) patch, or develop in a unilateral segmental distribution or follow the lines of Blaschko.116 Hair within a nevus depigmentosus may also be hypopigmented, and the margins may be irregular or serrated. The isolated form is the most common (Fig. 23.9) and the lesions do not usually cross the midline.117 The term depigmentosus is a misnomer because the lesions are actually hypopigmented, not completely depigmented, and become more prominent under Wood’s lamp. They are usually present at birth or become evident shortly thereafter, and remain stable in size and shape. Increase in size is in proportion to the growth of the child. Some lesions may appear during the first 3 years of life,117–119 with the trunk being the most commonly affected site.117,119,120 Occasionally, lesions may appear after the age of 3 years.117,120 In particular, the back and buttocks are most commonly affected, followed by the chest and abdomen.117 Males and females are affected equally and there is no distinct pattern of inheritance.116 The hypothesis is that during embryogenesis a clone of cells with a reduced melanogenic potential arises via a post-zygotic somatic mutation.103
Extracutaneous findings
Systemic manifestations are rare in patients with nevus depigmentosus. Neurologic abnormalities such as seizures and mental retardation have been reported,121 as well as ipsilateral hypertrophy of the extremities.122 In a survey of 50 patients with nevus depigmentosus, none had any extracutaneous features on examination.123 In two studies involving 29 patients with nevus depigmentosus, extracutaneous abnormalities were present in about 10% of the children.118,124 Occasionally, lentigines can develop within the achromic nevi, and this could be explained by the reversion of a mutation in one of the genes involved in pigmentation.125,126 There is a report of an infant developing multiple primary milia within a nevus depigmentosus.127
Etiology and pathogenesis
With dopa-staining, normal melanocytes are seen in nevus depigmentosus, and electron microscopic studies suggest a reduced synthesis of melanosomes and also a defect in their transfer to the keratinocytes, which could account for the hypopigmentation.128 Transfer of melanosomes from melanocytes to keratinocytes is essential for normal pigmentation.
Differential diagnosis
Other entities with which nevus depigmentosus is sometimes confused include nevus anemicus, segmental vitiligo, hypopigmented lesions of tuberous sclerosis, and hypomelanosis of Ito. The distinction between nevus depigmentosus and hypomelanosis of Ito may be artificial, as many patients with segmental hypopigmented macules also have linear pigmentary anomalies, similar to those seen in hypomelanosis of Ito, and underlying mosaicism is the common factor.109 Patients currently diagnosed with either of these conditions might simply be categorized as having nevoid hypopigmentation with or without extracutaneous anomalies. Genetic analysis may be considered in all patients with segmental or linear pigmentary abnormalities, and those with extracutaneous abnormalities to investigate cytogenic anomalies.109
Treatment and care
There has been a report of spontaneous resolution of nevus depigmentosus after a 6-year period.129 One patient with a nevus depigmentosus had partial repigmentation following autologous melanocyte grafting.130 The use of noncultured epidermal cellular grafting has been reported to be successful in the treatment of nevus depigmentosus and a mottled repigmentation of 78% was observed.131 Repigmentation however, may not be permanent and recurrence of a successfully repigmented nevus depigmentosus 6 years after blister roof grafting has been noted.132 Twice weekly narrow-band UVB for 32 sessions was helpful in repigmenting a nevus depigmentosus lesion,133 as was the 308 nm excimer laser.134 Cosmetic camouflage may be helpful.
Localized hypopigmented disorders
[/not-level-membership-for-dermatology-category]
