Chapter 565 Hypoparathyroidism
Etiology
Hypocalcemia is common between 12 and 72 hr of life, especially in premature infants, in infants with asphyxia, and in infants of diabetic mothers (early neonatal hypocalcemia) (Chapter 100) (Table 565-1). After the 2nd to 3rd day and during the 1st wk of life, the type of feeding also is a determinant of the level of serum calcium (late neonatal hypocalcemia). The role played by the parathyroid glands in these hypocalcemic infants remains to be clarified, although functional immaturity of the parathyroid glands is invoked as one pathogenetic factor. In a group of infants with transient idiopathic hypocalcemia (1-8 wk of age), serum levels of parathyroid hormone (PTH) are significantly lower than those in normal infants. It is possible that the functional immaturity is a manifestation of a delay in development of the enzymes that convert glandular PTH to secreted PTH; other mechanisms are possible.
Table 565-1 ETIOLOGIC CLASSIFICATION OF HYPOCALCEMIA
PARATHYROID HORMONE DEFICIENCY
PARATHYROID HORMONE RECEPTOR DEFECTS (PSEUDOHYPOPARATHYROIDISM)
MITOCHONDRIAL DNA MUTATIONS
MAGNESIUM DEFICIENCY
EXOGENOUS INORGANIC PHOSPHATE EXCESS
VITAMIN D DEFICIENCY
APECED, autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy; cAMP, cyclic adenosine monophosphate; PTH, parathyroid hormone.
Aplasia or Hypoplasia of the Parathyroid Glands
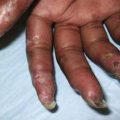
Aplasia or hypoplasia of the parathyroid glands is often associated with the DiGeorge/velocardiofacial syndrome (see Fig. 564-1). This syndrome occurs in 1/4,000 newborns. In 90% of patients, the condition is caused by a deletion of chromosome 22q11.2. Approximately 25% of these patients inherit the chromosomal abnormality from a parent. Neonatal hypocalcemia occurs in 60% of affected patients, but it is transitory in the majority; hypocalcemia can recur or can have its onset later in life. Associated abnormalities of the 3rd and 4th pharyngeal pouches are common; these include conotruncal defects of the heart in 25%, velopharyngeal insufficiency in 32%, cleft palate in 9%, renal anomalies in 35%, and aplasia of the thymus with severe immunodeficiency in 1%. This syndrome has also been reported in a small number of patients with a deletion of chromosome 10p13, in infants of diabetic mothers, and in infants born to mothers treated with retinoic acid for acne early in pregnancy.
Hdr Syndrome
Hypoparathyroidism, sensorineural deafness, and renal anomaly occur owing to mutations of the GATA3 gene. The protein encoded by this gene is essential in the development of the parathyroids, auditory system, and kidneys. The GATA3 gene is located at chromosome 10p14 and is nonoverlapping with the DiGeorge critical region at 10p13 (see Fig. 564-1).
Autosomal Dominant Hypoparathyroidism
Patients with autosomal dominant hypoparathyroidism have an activating (gain-of-function) mutation of the Ca2+-sensing receptor, forcing the receptor to an “on” state with subsequent depression of PTH secretion even during hypocalcemia. The patients have hypercalciuria. The hypocalcemia is usually mild and might not require treatment beyond childhood (see Fig. 564-1).
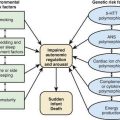
Hypoparathyroidism Associated with Mitochondrial Disorders
Mitochondrial DNA mutations in Kearns-Sayre syndrome and in mitochondrial trifunctional protein have been associated with hypoparathyroidism. A diagnosis of mitochondrial cytopathy should be considered in patients with unexplained symptoms such as ophthalmoplegia, sensorineural hearing loss, cardiac conduction disturbances, and tetany (see Fig. 564-1).
Idiopathic Hypoparathyroidism
Clinical Manifestations
Permanent physical and mental deterioration occur if initiation of treatment is long delayed.
Differential Diagnosis
Poisoning with inorganic phosphate leads to hypocalcemia and tetany. Infants administered large doses of inorganic phosphates, either as laxatives or as sodium phosphate enemas, have had sudden onset of tetany, with serum calcium levels <5 mg/dL and markedly elevated levels of phosphate. Symptoms are quickly relieved by intravenous administration of calcium. The mechanism of the hypocalcemia is not clear (Chapter 52.6).
Alimohammadi M, Björklund P, Hallgren A, et al. Autoimmune polyendocrine syndrome type 1 and NALP5, a parathyroid autoantigen. N Engl J Med. 2008;358:1018-1028.
Bowl MR, Nesbit MA, Harding B, et al. An interstitial deletion-insertion involving chromosome 2p25.3 and Xq27.1, near SOX3, causes X-linked recessive hypoparathyroidism. J Clin Invest. 2005;115(10):2822-2831.
Brown EM. The calcium-sensing receptor (CaR) and its disorders. Hormones. 2002;1:10-21.
Ding C, Buckingham B, Levine M. Familial isolated hypoparathyroidism caused by a mutation in the gene for the transcription factor GCMB. J Clin Invest. 2001;108:1215-1220.
Gavalas NG, Kemp EH, Krohn KJ, et al. The calcium-sensing receptor is a target of autoantibodies in patients with autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2007;92(6):2107-2114.
Glaudemans B, van der Wijst J, Scola RH, et al. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest. 2009;119(4):936-942.
Kahn KT, Uma R, Farag TI, et al. Kenny-Caffey syndrome in six Bedouin sibships: autosomal recessive inheritance is confirmed. Am J Med Genet. 1997;69:126-132.
Mannstadt M, Bertrand G, Muresan M, et al. Dominant-negative GCMB mutations cause an autosomal dominant form of hypoparathyroidism. J Clin Endocrinol Metab. 2008;93(9):3568-3576.
Marx SJ. Hyperparathyroid and hypoparathyroid disorders. N Engl J Med. 2000;343:1863-1875.
Oskarsdottir S, Persson C, Eriksson BO, et al. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005;164(3):146-153.
Parvari R, Hershkovitz F, Grossman N, et al. Mutation of TBCE causes hypoparathyroidism-retardation-dysmorphism and autosomal recessive Kenny-Caffey syndrome. Nat Genet. 2002;32:448-452.
Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab. 2006;91(8):2843-2850.
Tengan CH, Kiyomoto BH, Moraes CT, et al. Mitochondrial encephalomyopathy and hypoparathyroidism associated with a duplication and a deletion of mitochondrial deoxyribonucleic acid. J Clin Endocrinol Metab. 1998;83:125-129.
Thomas BR, Bennett JD. Symptomatic hypocalcemia and hypoparathyroidism in two infants of mothers with hyperparathyroidism and familial benign hypercalcemia. J Perinatol. 1995;15:23-26.
Yagi H, Furutani Y, Hamada H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362(9393):1366-1373.