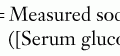72 Hypoglycemia
Etiology and Pathogenesis
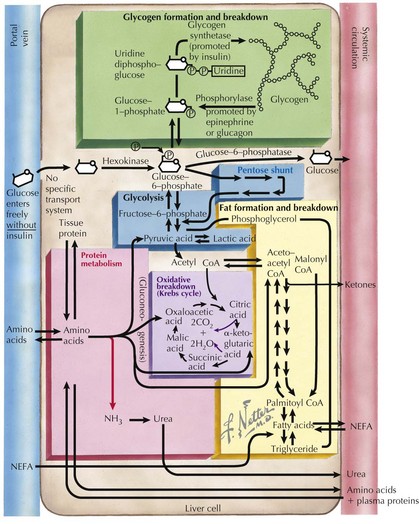
To recognize the different causes of hypoglycemia, it is helpful to understand the fundamentals of energy homeostasis (Figure 72-1). In the fed state, as glucose levels increase, so do insulin levels. Insulin stimulates glucose uptake into cells for use as a source of energy and metabolic intermediate or for storage (see Chapter 4, Figure 4-1). Insulin has other effects that influence energy metabolism; in the liver, insulin inhibits glycogenolysis, gluconeogenesis, and ketogenesis. In the fasted state, insulin secretion is suppressed, allowing glycogenolysis and gluconeogenesis to commence, followed by fatty-acid oxidation, which leads to ketogenesis. In the absence of glucose, the brain uses ketones (e.g., β-hydroxybutyrate and acetoacetate) as energy sources. Insulin secretion from pancreatic β-islet cells is suppressed in the fasted state, and there is increased secretion of counterregulatory hormones that maintain blood glucose levels by stimulating glycogenolysis and gluconeogenesis, such as glucagon, cortisol, growth hormone, and epinephrine. Disorders that impair insulin regulation and counterregulatory hormone secretion, as well as storage or production of glucose, can result in hypoglycemia.
Clinical Presentation
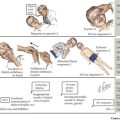
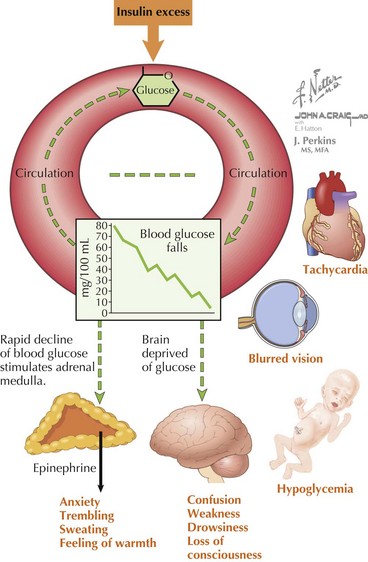
The symptoms of hypoglycemia can result from two different mechanisms. An adrenergic response (e.g., fight or flight) typically is triggered in response to a rapid decrease in blood glucose, as occurs with postprandial hypoglycemia (PPH). By contrast, the slower decline in blood glucose that occurs with fasting hypoglycemia may not trigger an obvious adrenergic response but can manifest as loss of consciousness caused by neuroglycopenia (Figure 72-2). In general, hypoglycemia in infants and children is fasting hypoglycemia. Hypoglycemia in neonates can present with irritability, shakiness, difficulty feeding, hypothermia, pallor, hypotonia, and seizures. In children, symptoms include sweatiness, unsteadiness, headache, hunger, nausea, weakness, tachycardia, change in mentation, and seizures. If symptoms suggestive of hypoglycemia are present, it is imperative, if possible, to send a blood specimen for a glucose level to the laboratory to confirm that hypoglycemia is at the root of the symptoms.
Differential Diagnosis
The differential diagnosis of hypoglycemia is expansive and includes not only disorders of carbohydrate metabolism but also disorders of fat oxidation, hormone deficiencies, and medication-induced hypoglycemia (Box 72-1). It is perhaps most useful to separate these disorders into those associated with hyperinsulinism and those associated with appropriately suppressed levels of insulin. Not included in this classification is transient neonatal hypoglycemia, typically seen within the first 6 hours of life, caused by immaturity of fasting mechanisms and poor glucose stores in premature infants and breastfed infants. In these cases, the hypoglycemia improves with feedings and typically resolves within the first day of life.
Box 72-1 Differential Diagnosis of Hypoglycemia in Infants and Children
Hypoglycemia Secondary to Excessive and Inappropriate Insulin
Other Causes of Hyperinsulinism
Adrenal Insufficiency (Addison’s Disease)
Patients with primary adrenal insufficiency resulting in the loss of glucocorticoid production may present with hypoglycemia (see Chapter 70). Additionally, these patients can present with hyponatremia, hyperkalemia, and dehydration caused by a loss of mineralocorticoid production. Patients may also appear hyperpigmented secondary to the effects of excess ACTH. The diagnosis can be made by checking 8 AM cortisol and ACTH levels or by performing a high-dose ACTH stimulation test.
Evaluation and Management
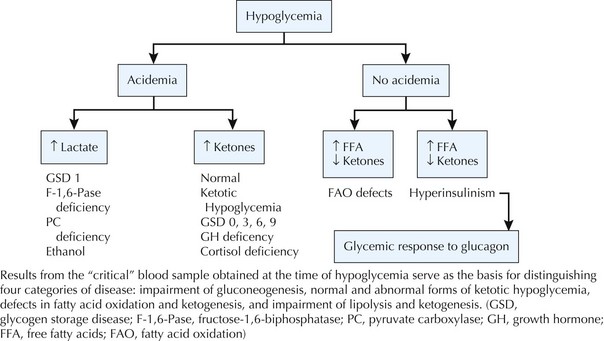
In severely symptomatic or clinically unstable patients with hypoglycemia, the first step should be administration of a 2 mL/kg bolus of 10% dextrose in water IV followed by a continuous infusion of 10% dextrose in an age-appropriate saline concentration at a glucose infusion rate of 6 to 8 mg/kg/min (Box 72-2). Whenever hypoglycemia is suspected in a clinically stable patient, the first step should be to send a “critical” blood sample to the laboratory to confirm hypoglycemia and to obtain the diagnostic tests necessary to establish the underlying cause of hypoglycemia (Box 72-3). If the patient is stable after the critical sample is sent, a glucagon stimulation test will help to refine the differential diagnosis (Figure 72-3).
Box 72-2 Treatment of Hypoglycemia
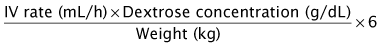
Box 72-3 The Critical Sample and Glucagon Stimulation Test
De León DD, Stanley CA. Mechanisms of disease: advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3:57-68.
De León DD, Stanley CA, Sperling MA. Hypoglycemia in neonates and infants. In Sperling MA, editor: Pediatric Endocrinology, ed 3, Philadelphia: Saunders Elsevier, 2008.
Langdon DR, Stanley CA, Sperling MA. Hypoglycemia in the infant and child. In Sperling MA, editor: Pediatric Endocrinology, ed 3, Philadelphia: Saunders Elsevier, 2008.
Palladino AA, Bennett MJ, Stanley CA. Hyperinsulinism in infancy and childhood: when an insulin level is not always enough. Clin Chem. 2008;54:256-263.
Palladino AA, Stanley CA. A specialized team approach to diagnosis and medical versus surgical treatment of infants with congenital hyperinsulinism. Semin Pediatric Surg. 2011;20:32-37.
Stanley CA. Hypoglycemia in the neonate. Pediatr Endocrinol Rev. 2006;4(suppl):76-81.