[level-membership-for-internal-medicine-category]Chapter 27
Hypertrophic Cardiomyopathy
1. What is hypertrophic cardiomyopathy?
2. What is the prevalence of HCM?
3. What are the genetic mutations that cause HCM, and how are they transmitted?
4. Who should be screened for HCM?
Patients with known HCM mutations, but without evidence of disease, and first-degree relatives of patients with HCM should be screened. Screening is performed primarily with history, physical examination, 12-lead electrocardiogram (ECG), and two-dimensional echocardiography. Traditionally, screening was performed on a 12- to 18-month basis, usually beginning by age 12 until age 18 to 21. However, it is now recognized that development of the HCM phenotype uncommonly can occur later in adulthood. Therefore, the current recommendation is to extend clinical surveillance into adulthood at about 5-year intervals or to undergo genetic testing (Table 27-1).
TABLE 27-1
CLINICAL SCREENING STRATEGIES FOR DETECTION OF HYPERTROPHIC CARDIOMYOPATHY IN FAMILIES∗
<12 years old
12 to 18-21 years old
>18-21 years old
Probably approximately every 5 years or more frequent intervals with a family history of late-onset hypertrophic cardiomyopathy and/or malignant clinical course
∗In the absence of laboratory-based genetic testing.
From Maron BJ, Seidman JG, Seidman CE: Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. J Am Coll Cardiol 44:2125-2132, 2004.
5. Who should undergo genetic testing?
6. What are the histologic characteristics of HCM?
The histology of HCM is characterized by hypertrophy of cardiac myocytes and myocardial fiber disarray. The abnormal myocytes contain bizarrely shaped nuclei and are arranged in disorganized patterns. The volume of the interstitial collagen matrix is greatly increased, and the arrangement of the matrix components is also disorganized. Myocardial disarray is seen in substantial portions of hypertrophied and nonhypertrophied LV myocardium. Almost all HCM patients have some degree of disarray, and in the majority, at least 5% of the myocardium is involved.
7. What are the common types of HCM?
The distribution and severity of LV hypertrophy in patients with HCM can vary greatly. Even first-degree relatives, with the same genetic mutation, usually show different patterns of hypertrophy. Various patterns of LV hypertrophy have been reported. The most common site of hypertrophy is the anterior interventricular septum (Fig. 27-1), which is seen in more than 80% of HCM patients and is known as asymmetrical septal hypertrophy (ASH). Concentric LV hypertrophy, with maximal thickening at the level of the papillary muscles, is seen in 8% to 10% of patients with HCM. A variant with primary involvement of the apex (apical HCM) is common in Japan and rare in the U.S. (less than 2%) and is characterized by spadelike deformity of the LV.
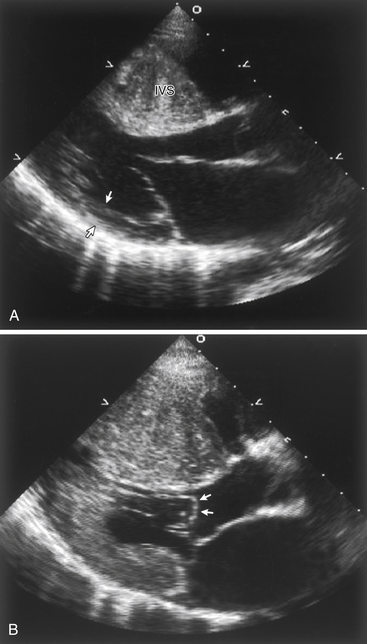
Figure 27-1 A, Echocardiogram of a patient with hypertrophic cardiomyopathy, demonstrating a markedly thickened interventricular septum compared with the normal wall thickness of the posterior wall (arrows). B, During systole, there is systolic anterior motion (SAM) and buckling of the anterior mitral leaflet (arrows), obstructing the left ventricular outflow tract.
8. What are the most common symptoms in patients with HCM?

 Present in more than 90% of symptomatic patients
Present in more than 90% of symptomatic patients

 Also caused by dynamic LV outflow tract (LVOT) obstruction (see Fig. 27-1) leading to elevated intraventricular pressures.
Also caused by dynamic LV outflow tract (LVOT) obstruction (see Fig. 27-1) leading to elevated intraventricular pressures.







9. How is HCM differentiated from athlete’s heart?
Long-term athletic training can lead to cardiac hypertrophy, known as athlete’s heart. This clinically benign physiologic condition must be differentiated from HCM, because HCM is the most common cause of sudden death in competitive athletes. Clinical parameters that support the diagnosis of HCM instead of athlete’s heart are asymmetric hypertrophy greater than 16 mm, LV end-diastolic dimension less than 45 mm, enlarged left atrium, impaired LV relaxation on Doppler mitral valve inflow parameters and tissue Doppler echocardiography, absent response to deconditioning (e.g., hypertrophy does not regress with absence of exercise), family history of HCM, and sarcomeric protein mutation identified by genetic testing. These parameters are summarized in Table 27-2.
TABLE 27-2
CLINICAL PARAMETERS USED TO DISTINGUISH HYPERTROPHIC CARDIOMYOPATHY FROM ATHLETE’S HEART
| Parameters | HCM | Athlete’s Heart |
| LV wall thickness | >16 mm | <16 mm |
| Pattern of hypertrophy | Asymmetric, symmetric or apical | Symmetric |
| LV end-diastolic dimension | <45 mm | >55 mm |
| Left atrium size | Enlarged | Normal |
| LV diastolic filling pattern | Impaired relaxation | Normal |
| Response to deconditioning | None | LV wall thickness decreases |
| ECG findings | Very high QRS voltage; Q waves; deep negative T waves | Criteria for LVH but without unusual features |
| Family history of HCM | Present | Absent |
| Sarcomeric protein mutation | Present | Absent |
HCM, Hypertrophic cardiomyopathy; LV, left ventricular; LVH, left ventricular hypertrophy.
Modified from Elliott PM, McKenna WJ: Diagnosis and evaluation of hypertrophic cardiomyopathy. Available at: www.UpToDate.com. Accessed February 2008.
10. Describe the classic murmur of obstructive HCM and bedside maneuvers that differentiate it from other cardiac abnormalities
Maneuvers that increase intracardiac blood volume or decrease contractility typically lead to a decrease in murmur intensity, and maneuvers that decrease intracardiac blood volume or increase contractility lead to an increase in murmur intensity (Table 27-3).

11. How does the carotid pulse in obstructive HCM differ from that in valvular aortic stenosis?
12. What noninvasive studies are helpful in making the diagnosis of HCM?
The ECG is abnormal in the majority of patients with HCM; however, no changes are pathognomonic for HCM. The common abnormalities are ST-segment and T-wave changes, voltage criteria for left ventricular hypertrophy (LVH), prominent Q waves in the inferior (II, III, aVF) or precordial (V2 to V6) leads, left axis deviation, and left atrial enlargement. Apical HCM, seen predominantly in Japanese patients, is characterized by giant negative T waves in the precordial leads.
13. What is systolic anterior motion and what causes it?
 The anterior mitral leaflet is drawn toward the septum by a Venturi effect produced by the lower pressure in the LVOT that occurs as blood is ejected at a high velocity.
The anterior mitral leaflet is drawn toward the septum by a Venturi effect produced by the lower pressure in the LVOT that occurs as blood is ejected at a high velocity.

14. Describe the mechanism of LVOT obstruction in HCM
LVOT obstruction in HCM is produced by SAM of the anterior mitral leaflet and midsystolic contact with the hypertrophic ventricular septum. The magnitude of the subaortic gradient is directly related to the duration of contact between the mitral leaflet and the septum. The subaortic gradient is often dynamic and responds to provocative maneuvers in the same manner as the systolic murmur (see Question 9 and Table 27-3).
15. What are the characteristic hemodynamic findings during cardiac catheterization in obstructive HCM?
Cardiac catheterization is not required for the diagnosis of HCM, and the diagnosis is usually made using noninvasive tests. Cardiac catheterization is generally reserved for assessment of coronary artery disease and evaluation before surgical procedures, such as myectomy. The typical findings during cardiac catheterization are subaortic or midventricular outflow gradient on catheter pullback, spike-and-dome pattern of aortic pressure tracing, elevated left atrial and LV end-diastolic pressures, elevated pulmonary capillary wedge pressure, increased V wave on wedge tracing (as a result of mitral regurgitation), and elevated pulmonary arterial pressure.
16. What is the Brockenbrough-Braunwald sign?
17. What are the risk factors for sudden cardiac death in patients with HCM?
 prior cardiac arrest or sustained ventricular tachycardia (VT)
prior cardiac arrest or sustained ventricular tachycardia (VT)


 hypotensive blood pressure response to exercise
hypotensive blood pressure response to exercise
 nonsustained VT on ambulatory (Holter) monitoring
nonsustained VT on ambulatory (Holter) monitoring


18. What medications should generally be avoided in patients with obstructive HCM?
 Preload reducing agents are diuretics and nitroglycerin. Diuretics may be used cautiously in patients with persistent heart failure symptoms and volume overload.
Preload reducing agents are diuretics and nitroglycerin. Diuretics may be used cautiously in patients with persistent heart failure symptoms and volume overload.


19. What are the pharmacologic therapies for patients with HCM?
 Beta-adrenergic blocking agents (β-blockers) are generally considered first-line therapy for HCM. The beneficial effects of β-blockers are mediated by their negative chronotropic property, which increases ventricular diastolic filling time, and by the negative inotropic property.
Beta-adrenergic blocking agents (β-blockers) are generally considered first-line therapy for HCM. The beneficial effects of β-blockers are mediated by their negative chronotropic property, which increases ventricular diastolic filling time, and by the negative inotropic property.


20. What nonpharmacologic treatments are available to patients with HCM?
 Septal myectomy surgery has been the gold standard for more than 45 years for patients with severe symptoms that are refractory to medical therapy. Septal myectomy, known as the Morrow procedure, uses a transaortic approach to resect a small amount of muscle from the proximal septum. It is associated with persistent and long-lasting improvement in symptoms, exercise capacity, and possibly survival.
Septal myectomy surgery has been the gold standard for more than 45 years for patients with severe symptoms that are refractory to medical therapy. Septal myectomy, known as the Morrow procedure, uses a transaortic approach to resect a small amount of muscle from the proximal septum. It is associated with persistent and long-lasting improvement in symptoms, exercise capacity, and possibly survival.


21. What are the indications for an implantable cardioverter-defibrillator (ICD) in HCM?
 ICD can be used for secondary prevention in survivors of cardiac arrest or sustained VT.
ICD can be used for secondary prevention in survivors of cardiac arrest or sustained VT.
 ICD is indicated for high-risk patients, defined as having two or more of the major risk factors for SCD (see Question 17).
ICD is indicated for high-risk patients, defined as having two or more of the major risk factors for SCD (see Question 17).


Bibliography, Suggested Readings, and Websites
1. Arnold, J.M.O. Hypertrophic cardiomyopathy. Available at http://www.merckmanuals.com/professional/cardiovascular_disorders/cardiomyopathies/hypertrophic_cardiomyopathy.html. Accessed May 1, 2012
2. Elliot, P.M., McKenna, W.J. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy. Available at www.UpToDate.com. Accessed February 2008
3. Fatkin, D., Seidman, J.G., Seidman, C.E. Hypertrophic cardiomyopathy. In Willerson J.T., Cohn J.N., Wellens H.J.J., Holmes D.R., eds.: Cardiovascular medicine, ed 3, New York: Springer, 2007. pp 1261–1284
4. Gersh, B.J., Maron, B.J., Bonow, R.O., et al. ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:e783–e831.
5. Kimmelstiel, C.D., Maron, B.J. Role of percutaneous septal ablation in hypertrophic obstructive cardiomyopathy. Circulation. 2004;109:452–456.
6. Maron, B.J. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320.
7. Maron, B.J. Hypertrophic cardiomyopathy. In Libby P., Bonow R.O., Mann D.L., Zipes D.P., eds.: Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine, ed 8, Philadelphia: Saunders, 2007.
8. Maron, B.J., Dearani, J.A., Ommen, S.R., et al. The case for surgery in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:2044–2053.
9. Maron, B.J., Pelliccia, A. The heart of trained athletes: cardiac remodeling and the risks of sports, including sudden death. Circulation. 2006;114:1633–1644.
10. Maron, B.J., Seidman, J.G., Seidman, C.E. Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:2125–2132.
11. NHLBI Program for Genomic Applications, Harvard Medical School. Genomics of Cardiovascular Development, Adaptation, and Remodeling: Sarcomere Protein Gene Mutation Database. Available at: http://genepath.med.harvard.edu/~seidman/cg3. Accessed May 1, 2012.
12. Ommen, S.R., Maron, B.J., Olivotto, I., et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46:470–476.
13. Richard, P., Charron, P., Carrier, L., et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232.
14. Sherrid, M.V., Barac, I., McKenna, W.J., et al. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;45:1251–1258.
15. Shah, S.N. Hypertrophic cardiomyopathy. Available at http://www.emedicine.com. Accessed May 1, 2012
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]Chapter 27
Hypertrophic Cardiomyopathy
1. What is hypertrophic cardiomyopathy?
2. What is the prevalence of HCM?
3. What are the genetic mutations that cause HCM, and how are they transmitted?
4. Who should be screened for HCM?
Patients with known HCM mutations, but without evidence of disease, and first-degree relatives of patients with HCM should be screened. Screening is performed primarily with history, physical examination, 12-lead electrocardiogram (ECG), and two-dimensional echocardiography. Traditionally, screening was performed on a 12- to 18-month basis, usually beginning by age 12 until age 18 to 21. However, it is now recognized that development of the HCM phenotype uncommonly can occur later in adulthood. Therefore, the current recommendation is to extend clinical surveillance into adulthood at about 5-year intervals or to undergo genetic testing (Table 27-1).
TABLE 27-1
CLINICAL SCREENING STRATEGIES FOR DETECTION OF HYPERTROPHIC CARDIOMYOPATHY IN FAMILIES∗
<12 years old
12 to 18-21 years old
>18-21 years old
Probably approximately every 5 years or more frequent intervals with a family history of late-onset hypertrophic cardiomyopathy and/or malignant clinical course
∗In the absence of laboratory-based genetic testing.
From Maron BJ, Seidman JG, Seidman CE: Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. J Am Coll Cardiol 44:2125-2132, 2004.
5. Who should undergo genetic testing?
6. What are the histologic characteristics of HCM?
The histology of HCM is characterized by hypertrophy of cardiac myocytes and myocardial fiber disarray. The abnormal myocytes contain bizarrely shaped nuclei and are arranged in disorganized patterns. The volume of the interstitial collagen matrix is greatly increased, and the arrangement of the matrix components is also disorganized. Myocardial disarray is seen in substantial portions of hypertrophied and nonhypertrophied LV myocardium. Almost all HCM patients have some degree of disarray, and in the majority, at least 5% of the myocardium is involved.
7. What are the common types of HCM?
The distribution and severity of LV hypertrophy in patients with HCM can vary greatly. Even first-degree relatives, with the same genetic mutation, usually show different patterns of hypertrophy. Various patterns of LV hypertrophy have been reported. The most common site of hypertrophy is the anterior interventricular septum (Fig. 27-1), which is seen in more than 80% of HCM patients and is known as asymmetrical septal hypertrophy (ASH). Concentric LV hypertrophy, with maximal thickening at the level of the papillary muscles, is seen in 8% to 10% of patients with HCM. A variant with primary involvement of the apex (apical HCM) is common in Japan and rare in the U.S. (less than 2%) and is characterized by spadelike deformity of the LV.
Figure 27-1 A, Echocardiogram of a patient with hypertrophic cardiomyopathy, demonstrating a markedly thickened interventricular septum compared with the normal wall thickness of the posterior wall (arrows). B, During systole, there is systolic anterior motion (SAM) and buckling of the anterior mitral leaflet (arrows), obstructing the left ventricular outflow tract.
8. What are the most common symptoms in patients with HCM?
Present in more than 90% of symptomatic patients
Also caused by dynamic LV outflow tract (LVOT) obstruction (see Fig. 27-1) leading to elevated intraventricular pressures.
9. How is HCM differentiated from athlete’s heart?
Long-term athletic training can lead to cardiac hypertrophy, known as athlete’s heart. This clinically benign physiologic condition must be differentiated from HCM, because HCM is the most common cause of sudden death in competitive athletes. Clinical parameters that support the diagnosis of HCM instead of athlete’s heart are asymmetric hypertrophy greater than 16 mm, LV end-diastolic dimension less than 45 mm, enlarged left atrium, impaired LV relaxation on Doppler mitral valve inflow parameters and tissue Doppler echocardiography, absent response to deconditioning (e.g., hypertrophy does not regress with absence of exercise), family history of HCM, and sarcomeric protein mutation identified by genetic testing. These parameters are summarized in Table 27-2.
TABLE 27-2
[/not-level-membership-for-internal-medicine-category]
