88 Hypertensive Crisis
Emergency and Urgency
Hypertension is a common problem, and its incidence may be increasing in adults.1 Population data also suggest hypertension is increasing globally; 972 million individuals worldwide now have hypertension,1 and 30% of hypertensive individuals are unaware of their diagnosis.2 Of the 59% of hypertensive individuals being treated for hypertension, only 34% have a blood pressure less than 140/90 mm Hg.2 The exact risk of hypertensive crisis is not clear, but most authors estimate the risk to be less than 1%; it may be increasing.3,4
 Pathophysiology of Hypertensive Crisis
Pathophysiology of Hypertensive Crisis
The precise pathophysiology of hypertensive crisis is unknown. An abrupt increase in blood pressure is one of the initiating events in the transition from simple hypertension or normotension to hypertensive crisis. The product of cardiac output and peripheral vascular resistance determines blood pressure. The initial blood pressure increase is likely secondary to an increase in vascular resistance. Considerable evidence suggests that mechanical stress in the arteriolar wall leads to disruption of endothelial integrity.5 With disruption of vascular integrity, diffuse microvascular lesions develop.6,7 Fibrinoid necrosis of the arterioles is seen in vulnerable organs and is considered the histologic hallmark of hypertensive crisis.6,7 It is unclear whether hypertension alone causes the development to hypertensive crisis or whether other factors are necessary. For example, increases in peripheral vascular resistance result in part from activation of the renin-angiotensin-aldosterone system. Evidence suggests angiotensin II may directly injure the vascular wall by activation of genes for proinflammatory cytokines (interleukin 6) and also of nuclear factor κB.8,9 Other vascular-toxic influences may contribute to increased peripheral vascular resistance, including hyperviscosity, immunologic factors, and other hormones including catecholamines, vasopressin, and endothelin.10–12 The end result of these changes is a significant increase in peripheral vascular resistance, with ischemia of heart, brain, and kidneys.
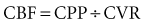
 Diagnosis of Hypertensive Emergencies
Diagnosis of Hypertensive Emergencies
Medical History, Physical Examination, and Laboratory Evaluation
Hypertension from any cause may enter an “emergent” phase. Although hypertensive emergency usually occurs in individuals with a history of essential hypertension, it is also is seen in individuals with secondary hypertension and in individuals with no hypertensive history, as in preeclampsia, pheochromocytoma, drug withdrawal, and acute glomerulonephritis. A medication history, including over-the-counter medications and illegal drug use, should be ascertained from every patient. Malignant hypertension is a unique clinical/pathologic syndrome that is associated with hypertensive crisis. Increases in blood pressure and target-organ damage are caused by changes in the vasculature characterized by fibrinoid necrosis and a proliferative endarteritis. Risk factors associated with the development of malignant hypertension include age between 30 and 50 years,13 male gender,5 African American background,14 and smoking (increases the risk by 2.5- to 5-fold).15
Patients with hypertensive crisis present with a variety of symptoms. The most common is headache. It is either sudden in onset or represents a change from a usual headache pattern and is often worst in the morning. The location is generally occipital or anterior, with a steady quality. Other symptoms include visual complaints (scotoma, diplopia, hemianopsia, blindness), neurologic symptoms (focal deficits, stroke, transient ischemic attacks, confusion, somnolence), ischemic chest pain, renal symptoms (nocturia, polyuria, hematuria), back pain (aortic aneurysm), and gastrointestinal complaints (nausea, vomiting). Weight loss occurs as the high levels of circulating renin and angiotensin induce a diuresis.16 These patients often present with intravascular volume depletion, which has strong implications for treatment.
The blood pressure is measured in both arms and also with the patient lying and standing. In hypertensive emergency, diastolic blood pressures are usually above 120 mm Hg. Pathologic processes that cause stiffening of the vascular wall can prevent vessel compression by external compression with a blood pressure cuff. This results in an artificial increase (at times extreme) in the systolic and diastolic blood pressure, or “pseudohypertension.” Pseudohypertension can occur in atherosclerosis, Monckeberg’s medial calcification, and metastatic calcification, as experienced in end-stage renal disease. Clues to pseudohypertension include a markedly elevated blood pressure in an individual without evidence of end-organ damage. The diagnosis is suggested by a palpable radial artery after proximal compression (Osler’s maneuver).17
A dilated funduscopic examination should be performed on all individuals. Arteriolar thickening reflects chronic hypertension and is manifested by increased light reflex, vascular tortuosity, and arteriovenous nicking where the arterioles cross the venules. These funduscopic findings reflect chronic hypertension and have no prognostic significance with regard to hypertensive crisis. As hypertension increases in severity, there are additional findings caused by the breakdown of the blood-retina barrier, leading to retinal hemorrhage and leakage of lipids, causing hard exudates. Additional findings as the blood pressure continues to increase may include cotton-wool spots as a result of nerve ischemia and swelling of the optic nerve with papilledema.18
The initial laboratory evaluation should include a serum sodium, chloride, potassium, bicarbonate, creatinine and blood urea nitrogen, complete blood count (with a peripheral smear to identify schistocytes), prothrombin time, activated partial thromboplastin time, serum and urine toxicology screen, pregnancy test when appropriate, an electrocardiogram, and a urinalysis. Evidence of intravascular hemolysis is common and may make it difficult to differentiate hypertensive crisis from primary vasculitis with secondary hypertension.19,20 The renin-angiotensin-aldosterone axis is markedly activated, as evidenced by hypokalemia and metabolic alkalosis.3,21 The blood urea nitrogen and creatinine are often elevated. The urinalysis may show small amounts of proteinuria as well as hematuria with occasional erythrocyte casts.5 Marked increases in proteinuria suggest a primary glomerular process such as glomerulonephritis as the etiology of the elevated blood pressure.
If hypertensive encephalopathy is suspected, magnetic resonance imaging (MRI) should be performed. With hypertensive encephalopathy, edema may occur in the posterior regions of the cerebral hemispheres, particularly in the parieto-occipital regions, a finding called posterior leukoencephalopathy on MRI. However, brainstem involvement on MRI has also been reported.22,23 It is important to consider and eliminate other conditions with a similar clinical presentation (Box 88-1). Several important diagnostic considerations help exclude other causes of altered mental status: (1) symptoms of generalized brain dysfunction tend to develop over time (12-24 hours) with hypertensive encephalopathy, as compared to acutely with ischemic stroke or cerebral hemorrhage; (2) focal neurologic findings are unusual with hypertensive encephalopathy unless there is an associated bleed; (3) papilledema is almost always noted with hypertensive encephalopathy and if absent should raise suspicion of another etiology; (4) in comparison to an acute CNS bleed, mental status with hypertensive encephalopathy improves within 24 to 48 hours of treatment.
 Treatment of Hypertensive Emergency
Treatment of Hypertensive Emergency
In most but not all settings, blood pressure can be reduced acutely by 20% to 25% within minutes to hours.3 After the patient is stabilized at this pressure, the blood pressure may be further decreased to 160/100-110 mm Hg over the next 2 to 6 hours.3 If the patient is clinically stable, the blood pressure may then be decreased toward a normal blood pressure over the next 24 to 48 hours.3 With these decreases in blood pressure, CNS blood flow autoregulation is usually maintained. Clinical settings where additional considerations and alternative approaches to reducing blood pressure should be considered include (1) ischemic stroke where immediate reduction of blood pressure is usually not indicated except when the blood pressure is over 220/120 or the patient requires thrombolytic therapy, (2)acute aortic dissection where a rapid blood pressure reduction in 15 to 30 minutes to a systolic blood pressure under 100 mm Hg is clinically warranted if the patient tolerates, and (3) in previously normotensive subjects with abrupt increases in BP.
From 40% to 50% of hypertensive crises arise in patients with preexisting hypertension without identifiable secondary causes.24,25 Essential hypertension is the underlying disorder in the majority of African American individuals.26–28 In contrast, from 50% to 60% of white patients with malignant hypertension have an identifiable cause (Box 88-2). Renovascular hypertension secondary to either fibromuscular dysplasia or atherosclerosis is not uncommon. Up to 20% of cases of malignant hypertension occur in patients with underlying chronic glomerulonephritis. Other renal causes include reflex nephropathy (particularly in children) and analgesic nephropathy.3
 Specific Treatment Recommendations for Hypertensive Crisis Based on Etiology
Specific Treatment Recommendations for Hypertensive Crisis Based on Etiology
General Comment on Medication Used to Treat Hypertensive Crisis
The classes of parenteral antihypertensive agents available to treat hypertensive crisis include direct vasodilators (sodium nitroprusside, nitroglycerin), α- and β-adrenergic blockers (labetalol), α-adrenergic blockade (phentolamine), angiotensin-converting enzyme (ACE) inhibitors (enalaprilat), calcium channel blockers (nicardipine), and dopamine agonists (fenoldopam). Some of the advantages and disadvantages of these medications are detailed in Table 88-1. There is no consensus on the most effective antihypertensive medications in the setting of a CNS insult and no large randomized trials demonstrating the superiority of a given agent. Rather, the choice of antihypertensive therapy should be individualized to the patient and clinical setting. However, most authors now caution the use of nitroprusside in the setting of increase in intracranial pressure. Vasodilators increase blood volume and therefore have the potential to increase the intracranial pressure (ICP). Animal and human studies in the setting of a normal ICP show no effect of nitroprusside on ICP.19–21 However, in studies on animals and humans with preexisting increased ICP, nitroprusside increased the ICP, likely reflecting vasodilatation on the background of decreased cranial compliance.29–33 When sodium nitroprusside is contraindicated, other treatment options include labetalol and nicardipine. Fenoldopam, which is an agonist of the vasodilator dopamine-1 receptor, shares with nitroprusside a rapid onset and short duration of action. In addition, fenoldopam, in contrast to nitroprusside, increases renal blood flow, induces natruresis, and produces no toxic metabolites.34–38
TABLE 88-1 Treatment of Hypertensive Crisis: Intravenous Medication
| Drug Name and Mechanism of Action | Indications/Advantages/Dose | Disadvantages/Adverse Effects/Metabolism Cautions |
|---|---|---|
| Sodium nitroprusside: Nitric oxide compound; vasodilation of arteriolar and venous smooth muscle Increases cardiac output by decreasing afterload |
Useful in most hypertensive crisis Onset of action immediate, duration of action 1-2 min Dose: 0.25 µg/kg/min Maximum dose: 8-10 µg/kg/min |
Contraindicated in high-output cardiac failure, congenital optic atrophy. Anemia and liver disease at risk of cyanide toxicity: acidosis, tachycardia, change in mental status, almond smell on breath. Renal disease at risk of thiocyanate toxicity: psychosis, hyperreflexia, seizure, tinnitus. Cautious use with increased intracranial pressure. Do not use maximum dose for more than 10 minutes. Crosses the placenta. |
| Nitroglycerin: Directly interacts with nitrate receptors on vascular smooth muscle Primarily dilates venous bed Decreases preload |
Use with symptoms of cardiac ischemia, perioperative hypertension in cardiac surgery Initial dose: 5 µg/min Maximum dose: 100 µg/min |
Contraindicated in angle-closure glaucoma, increased intracranial pressure. Blood pressure decreased secondary to decreased preload, cardiac output—avoid when cerebral or renal perfusion compromised. Caution with right ventricular infarct. |
| Labetalol: β-Adrenergic blockade and α-adrenergic blockade IV α:β-Blocking ratio is 1 : 7 |
Onset of action 2-5 min Duration 3-6 hours Bolus 20 mg, then 20-80 mg every 10 min for maximum dose 300 mg Infuse at 0.5-2 mg/min |
Avoid in bronchospasm, bradycardia, congestive heart failure, greater than first-degree heart block, second/third trimester pregnancy. Use caution with hepatic dysfunction, inhalational anesthetics (myocardial depression). Enters breast milk. |
| Esmolol: Cardioselective β1-adrenergic blocking agent |
Use with aortic dissection Use during intubation, intraoperative, and postoperative hypertension Onset 60 seconds, duration 10-20 min 200-500 µg/kg/min for 4 min, then infuse 50-300 µg/kg/min |
See labetalol. Not dependent on renal or hepatic function for metabolism (metabolized by hydrolysis in RBC). |
| Fenoldopam: Postsynaptic dopamine-1 agonist; decreases peripheral vascular resistance; 10 times more potent than dopamine as vasodilator |
May be advantageous in kidney disease, increases renal blood flow, increases sodium excretion, no toxic metabolites Initial dose: 0.1 µg/kg/min, with titration every 15 min No bolus |
Contraindicated in glaucoma (may increase intraocular pressure) or allergy to sulfites; hypotension, especially with concurrent beta-blocker. Check serum potassium every 6 hours. Concurrent acetaminophen may significantly increase blood levels. Dose-related tachycardia. |
| Hydralazine: Primarily dilates arteriolar vasculature |
Primarily used in pregnancy/eclampsia Dose: 10 mg every 20-130 min; maximum dose 20 mg Decreases blood pressure in 10-20 min Duration of action 2-4 h |
Reflex tachycardia; give beta-blocker concurrently. May exacerbate angina. Half-life 3 hours, affects blood pressure for 100 hours. Depends on hepatic acetylation for inactivation. |
| Phentolamine: α-Adrenergic blockade |
Used primarily to treat hypertension from excessive catecholamine excess (e.g., pheochromocytoma) Dose: 5-15 mg Onset of action 1-2 min, duration 3-10 min |
β-blockade is generally added to control tachycardia or arrhythmias. As in all catecholamine excess states, beta-blockers should never be given first, as the loss of β-adrenergically mediated vasodilatation will leave α-adrenergically mediated vasoconstriction unopposed and result in increased pressure. |
| Nicardipine: Dihydropyridine calcium channel blocker; inhibits transmembrane influx of calcium ions into cardiac and smooth muscle |
Onset of action 10-20 min, duration 1-4 h Initial dose: 5 mg/h to maximum of 15 mg/h |
Avoid with congestive heart failure, cardiac ischemia. Adverse effects include tachycardia, flushing, HA. |
| Clevidipine: Short-acting dihydropyridine calcium channel hypertension99 |
Initial dose: 1 mg/h; can be increased to 21 mg/h | Reduces blood pressure without affecting cardiac filling pressures or causing reflex tachycardia |
| Enalaprilat: Angiotensin-converting enzyme inhibitor |
Onset of action 15-20 min, duration 12-24 h Dose: 1.25-5 mg every 6 h |
Response not predictable, with high renin states may see acute hypotension. Hyperkalemia in setting of reduced glomerular filtration rate. Avoid in pregnancy. |
| Trimethaphan:Nondepolarizing ganglionic blocking agent; competes with acetylcholine for postsynaptic receptors | Used in aortic dissection Dose: 0.5-5 mg/min |
Does not increase cardiac output. No inotropic cardiac effect. Disadvantages include parasympathetic blockade, resulting in paralytic ileus and bladder atony, and development of tachyphylaxis after 24-96 hours of use. |
Malignant Hypertension
Malignant hypertension is specific syndrome characterized by markedly elevated pressures in conjunction with hypertensive neuroretinopathy. Funduscopic examination often reveals flame-shaped hemorrhages, cotton-wool spots, or papilledema. Malignant hypertension is also associated with nephropathy, encephalopathy, microangiopathic hemolytic anemia, and cardiac ischemia. Untreated malignant hypertension is a rapidly fatal disorder, with a mortality of more than 90 % within 1 year, as reported in a classic series by Kincaid-Smith.6 In this series, deaths were due to renal failure (19%), congestive heart failure (13%), renal failure plus congestive heart failure (48%), stroke (20%), and myocardial infarction (1%).
Aggressive therapy to prevent progressive ischemic injury in malignant hypertension is critical. Although the autoregulatory range of CNS blood flow is reset upwards in chronic hypertension, the lower limit of the autoregulation remains approximately 25% below the resting mean blood pressure in patients with both normotension and chronic hypertension.39 When the arterial blood pressure falls below this lower limit, cerebral blood flow decreases progressively, and symptoms of low CNS flow including nausea, yawning, hyperventilation, clamminess, and syncope develop. To protect cerebral function, after initial reduction of blood pressure by 20% within the first hour, blood pressure is further reduced over the next 2 to 6 hours to the 160/110 range as long as the patients remains stable. Nitroprusside is one of the most useful intravenous agents for hypertensive emergency. Some patients are highly sensitive to treatment owing to coexisting hypovolemia; therefore, low-dose nitroprusside (0.3 µg/kg/min or less, with titration every 3-5 minutes) is used to reach goal blood pressure. A number of parenteral agents have been used as successful alternatives to nitroprusside, including labetalol, fenoldopam, and nicardipine. Premature discontinuation of parenteral therapy may cause rebound hypertension. Oral therapy is usually started after the pressure has been stabilized on parenteral therapy. Parenteral therapy is then slowly weaned.
Renal failure is common with malignant hypertension. For patients with worsening renal failure due to malignant hypertension, renal failure exacerbates the hypertension. Aggressive treatment can arrest and reverse renal damage. Since the arteriolopathy of malignant hypertension includes fixed anatomic lesions, initial lowering of blood pressure may worsen renal function. Dialysis may be required in patients with a presenting creatinine greater than 4.5 mg/dL.40 In the majority of patients, renal function begins to improve after 2 weeks of therapy. Of the patients who require dialysis, 50% will regain sufficient function to discontinue dialysis.41 Recovery of renal function is predicted when the combined length of both kidneys is 20.2 cm or more, but is felt to be unlikely when the length is 14.2 or less.42 The mean time to recovery is approximately 2 to 3 months, but recovery after up to 26 months has been reported.43 In patients with malignant hypertension secondary to glomerulonephritis, eventual deterioration to end-stage renal disease (ESRD) may occur despite blood pressure control.44 In contrast, renal function tends to remain well preserved in patients without underlying glomerulonephritis if blood pressure is well controlled. Nitroprusside has been one of the preferred agents to treat hypertension and renal failure. The metabolism of nitroprusside results in the production of cyanide, which is taken up by red blood cells and conjugated to thiocyanate in the liver. Cyanide toxicity occurs in patients with anemia or liver disease, whereas thiocyanate toxicity is seen in the setting of renal disease (see Table 88-1). Thiocyanate levels should be monitored and the duration of therapy kept to less than 72 hours whenever possible. Fenoldopam has no toxic metabolites and may protect renal function.34–38
Controversy exists as to the management of the relatively asymptomatic malignant hypertensive patient (i.e., with neuroretinopathy alone).45,46 Although oral medication under close observation has been used successfully,47 we prefer initial parenteral therapy. The progressive breakdown of CNS autoregulation in these patients enhances the sensitivity to ischemia, with abrupt decreases in blood pressure. Of the oral agents, calcium antagonists and minoxidil are effective and safe. ACE inhibitors may cause hyperkalemia in undialyzed patients with significant renal insufficiency.
Hypertensive Encephalopathy
In hypertensive encephalopathy, the MAP exceeds the limits of autoregulation, and brain edema develops from extravasation of plasma proteins. If hypertensive encephalopathy is untreated, coma and death may follow.48 The challenge of hypertensive encephalopathy is appropriate lowering of blood pressure in the setting of CNS ischemia and edema. The hallmark of hypertensive encephalopathy is improvement within 12 to 24 hours of adequate blood pressure reduction. The MAP should be cautiously reduced by no more than 15% over 2 to 3 hours. Neurologic complications have been reported from reductions in MAP of 40% or more.49
Hypertensive encephalopathy is one of the medical conditions believed to cause reversible posterior leukoencephalopathy, a condition that results from reversible vasogenic subcortical edema without infarction.22,23 This syndrome is characterized by headache, decreased alertness, changes in behavior including confusion and diminished speech, seizures, and alterations in visual perceptions and is rapidly reversible with lowering of the blood pressure.22,23 An MRI examination shows characteristic findings including white matter edema in the posterior cerebral hemispheres.22
There is a growing literature supporting a shared pathologic process between hypertensive encephalopathy and eclampsia. Both clinical syndromes share the same clinical features and imaging findings. Eclampsia during pregnancy, as well as postpartum eclampsia, has also been associated with reversible posterior leukoencephalopathy.22,23
Ischemic Cerebral Infarction
Data from animal studies show that in the area surrounding the ischemic infarct, there are “neurons at risk” that rely on collateral circulation to maintain perfusion.55 These neurons are nonfunctional—not dead—and potentially can be “rescued” by reperfusion, a phenomenon referred to as ischemic penumbra.50 The degree to which this occurs in humans is not known. In addition, in acute stroke, autoregulation is impaired, and cerebral blood flow is therefore not preserved in a predictable manner. As a result of these changes, acute reductions in blood pressure could potentially increase the area of infarct, resulting in severe clinical consequences.
Comprehensive guidelines for the treatment of stroke were recently updated by the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups and affirmed by the American Academy of Neurology.51 In patients determined to be candidates for administration of intravenous recombinant tissue plasminogen activator (tPA), the blood pressure must be reduced if the systolic blood pressure is > 185 mm Hg or the diastolic blood pressure is > 110 mm Hg, and the patient must be carefully monitored before, during, and after administration of this compound. Recombinant tissue plasminogen activator is contraindicated if the systolic blood pressure is >185 mm Hg or diastolic blood pressure is >110 mm Hg.51
Questions remain as to how to manage individuals with ischemic stroke who are not candidates for intravenous recombinant tissue plasminogen activator. There are no large randomized trials to guide this therapy. If there is no indication for acute lowering of the blood pressure (acute ischemic damage to vital organs such as cardiac ischemia, aortic dissection), the current recommendation is that a systolic blood pressure over 220 mm Hg or a diastolic blood pressure over 120 mm Hg be treated to lower the blood pressure 15% to 25% over the first 24 hours.51
However, a recent prospective observational study analyzed the impact of blood pressure lowering in the setting of ischemic stroke in 1092 patients.52 The data suggest an improved outcome at 3 months with modest reductions in blood pressure between 10 and 27 mm Hg. Interestingly, the authors noted that the benefit of blood pressure reduction waned with age. In patients with more than 27 mm Hg blood pressure reduction, a poorer outcome was multiplied by 6 in patients aged 70 to 76 years, by 10 in patients 76 to 80 years, and by 15 in patients older than 80 years.52 The authors recommend that the treatment regime for patients eligible to receive thrombolytic therapy be applied to all patients suffering from ischemic stroke but that sudden decreases in blood pressure to above 10% from baseline be avoided.52
Subarachnoid Hemorrhage
Approximately 10% of cerebrovascular accidents are due to subarachnoid hemorrhage; ruptured congenital berry aneurysms are the most common cause. Aneurysmal subarachnoid hemorrhage remains a devastating entity, with a mortality rate of 50% to 60% at 30 days despite improvement in care and early surgical intervention. Of those who do survive, 50% remain dependent.53 The level of consciousness when the patient presents, global cerebral edema, the size of the subarachnoid blood collection on computed tomography (CT), and a recurrent hemorrhage have been associated with outcome.53 Worse outcomes have also been associated with age, hyperglycemia, and medical complications.53 Subarachnoid hemorrhage increases ICP and decreases cerebral perfusion, causing global ischemia. Complications include an intracerebral hemorrhage or the development of hydrocephalus. Management of these patients is significantly different from those with ischemic stroke. In contrast to ischemia, intracranial bleed induces intense vasospasm in neighboring vessels 4 to 12 days after the initial bleed, increasing the risk for significant cerebral ischemia. The mental status evaluation may be used to guide therapy, with an intact mental status supporting adequate cerebral perfusion.
Markedly elevated pressures increase the risk of rebleeding. The goal is a 20% to 25% reduction in blood pressure over 6 to 12 hours, but to not less than 160 to 180/100 mm Hg.54 Labetalol is the preferred agent, as there are no significant adverse effects on ICP or CPP.3 Given the potential increase in cerebral blood volume and ICP associated with vasodilators, sodium nitroprusside and nitroglycerin are not usually first-line treatments. There are clinical data to show that treatment with oral nimodipine within 4 days of the acute event decreases vasospasm and cerebral ischemia.55 Nimodipine may also directly protect against ischemic damage to nerve cells by blocking calcium uptake into cells.
Intracerebral Hemorrhage
Intracerebral hemorrhage accounts for 10% to 20 % of all strokes.56 Hypertension is a major risk factor; 75% of affected individuals have preexisting hypertension.57 Although patients with intracerebral hemorrhage may present with nausea, vomiting, change in mental status, hypertension, headache, and a focal neurologic examination, the definitive diagnosis must be made by neuroimaging. Unlike ischemic stroke, where blood pressure generally returns to normal within 24 to 48 hours, in intracerebral hemorrhage, the most rapid decline in blood pressure occurs in the first 24 hours, but the blood pressure may remain elevated for 7 to 10 days.50 The hematoma compresses normal tissue, creating an area of ischemia, increasing ICP and further decreasing CPP. Autoregulation is altered, making cerebral perfusion critically dependent on systemic blood pressure.58 The prognosis overall is not encouraging. A retrospective analysis of 411 patients with intracerebral hemorrhage showed that 30% died before reaching the hospital and an additional 50% died within 28 days.59 Independent risk factors associated with this early death included unconsciousness, lateral shift of midline structures, MAP ≥ 134 mm Hg, hyperglycemia, anticoagulant therapy, and ventricular extrasystoles.59
There is no clear consensus on the appropriate treatment of hypertension in the setting of acute intracranial hemorrhage. The decision to treat or not treat blood pressure should be made based on individual considerations including baseline blood pressure, etiology of hemorrhage, age, and elevated ICP, as well as a careful literature review. The central issue is whether aggressive lowering of blood pressure reduces the risk of intracerebral bleeding without disrupting blood flow to collateral areas. Some argue that decreasing blood pressure decreases risk of hemorrhage extension, edema, and associated systemic complications, particularly when systolic blood pressure exceeds 200 mm Hg, a level associated with hematoma growth in some studies.56–58 A retrospective analysis of 76 patients with intracerebral hemorrhage and hypertension showed that maximum systolic blood pressure was significantly associated with hematoma enlargement.60 Furthermore, this analysis suggested that systolic blood pressure ≥160 mm Hg is associated with enlargement of hematoma when compared to a systolic blood pressure ≤150 mm Hg.60 Others argue that not treating hypertension allows continued perfusion of areas at risk from low blood flow.58 It was previously thought rebleeding was rare in the first 24 hours. More recent data suggest that it is more common than thought, occurring in up to a third of affected individuals.57,61 The greatest risk is in the first few hours after the initial insult.61,62 An increased risk of bleed is associated with an initial large irregular bleed,63 coagulopathy, liver disease,64 and a low platelet count.64 No studies have demonstrated a clear relationship between acute hypertension after an intracerebral bleed and the risk of rebleed.58
A recent65 consensus was that (1) aggressive lowering of blood pressure using intravenous medication and blood pressure monitoring every 5 minutes should be considered when the systolic blood pressure is over 200 mm Hg or the mean arterial blood pressure is over 150 mm Hg; (2) in the setting of suspected intracranial hypertension, in addition to ICP monitoring, aggressive lowering of blood pressure with continuous or intermittent intravenous medication should be considered when the systolic blood pressure is over 180 mm Hg or the MAP is over 130 mm Hg, keeping the CPP above 60 to 80 mm Hg; and (3) if there is no suspected elevation of the ICP and the systolic blood pressure is over 180 or the MAP is over 130, consider lowering the target blood pressure to 160/90 mm Hg or to a MAP of 110 mm Hg.
There is no consensus on the agent of choice. Concern revolves around the impact of different antihypertensives on ICP. Common to all agents is a decrease in MAP and a decrease in CPP. Vasodilating agents may increase cerebral blood flow, and in the setting of decreased cranial compliance may potentially increase ICP, further decreasing CPP.32,58 The combination of decreased cerebral compliance, decreased cerebral blood flow, and altered autoregulation—as occurs in chronic hypertension—makes the administration of any antihypertensive agent potentially dangerous. No large randomized studies are available to guide therapy. Combination α- and β-blockers are recommended when antihypertensive treatment is indicated in intracerebral hemorrhage. Risks of this therapy include worsening of bradycardia associated with the Cushing response. However, in the setting of normal cranial compliance and an increased ICP, vasodilators are probably safe. Because of the very high levels of circulating catecholamines with an intracerebral bleed, β-blockade is added when vasodilator therapy alone is ineffective.
Head Trauma
Head trauma complications include skull fractures, epidural hematomas, subdural hematomas, intracerebral hematomas, and diffuse axonal damage. With trauma, there is often edema. Acute increases in ICP are initially prevented by flow of blood and CSF from the cranial vault. However, with increasing edema, ICP eventually increases. In most trauma centers, ICP monitoring has become the standard of care.66 Anywhere from 31% to 61% of patients with a closed head injury may have defective autoregulation.67 If autoregulation is intact, increasing the MAP will cause vasoconstriction and produce no change in ICP. With altered autoregulation, increasing the MAP may cause vasodilatation, increasing blood volume and causing edema and increased ICP. The goal is to maintain a minimum CPP of 70 mm Hg and a MAP above 90 mm Hg. If an antihypertensive agent is needed, a major consideration is its impact on ICP. A combination alpha- and beta-blocker or nicardipine may be preferred when there is decreased intracranial compliance and increased ICP.68,69 In the absence of intracranial hypertension, vasodilators may be preferred.
Aortic Dissection
Aortic dissection begins with a tear in the intima of the aorta that is propagated by the aortic pulse wave (dP/dt). Myocardial contractility, heart rate, and blood pressure contribute to the aortic pulse wave. There are two types of aortic dissection, type A and type B. Type A dissections are often associated with a tear in the intima of the proximal aorta next to a coronary artery and may extend to the aortic arch.70 Type B dissections occur in the descending aortic arch and usually begin with an intimal tear next to the subclavian artery.71 Risk factors for dissection include advanced atherosclerosis, Marfan syndrome, Ehlers-Danlos syndrome, and coarctation of the aorta.72 Symptoms occur as the expanding hematoma causes pressure on the vasculature. This may cause myocardial infarction, stroke, spinal cord/bowel infarction, and acute renal failure. Ischemic kidney may develop, leading to refractory hypertension.73 Dissection to the aortic root can precipitate acute aortic insufficiency.74 Rupture of the ascending aorta leads to hemopericardium and tamponade.74
Both types of dissection may present with severe, often tearing pain in the chest, back, or abdomen, accompanied by diaphoresis, nausea, or vomiting. They are often but not always associated with hypertension.75 Discrepancies in peripheral pulses may be observed. The chest was present in only one half of individuals with type B dissection.76 The diagnosis may be confirmed with CT or MRI. Multiplane transesophageal echocardiography is also used. Type A dissections usually require surgery to prevent the catastrophic consequences of great-vessel occlusion, aortic insufficiency, or tamponade. Type B dissections may usually be treated medically77,78 unless there is rupture, in which case open repair or endovascular repair is indicated. A recent meta-analysis suggested that endovascular repair may be preferred.77
An alternative regimen, preferred by some because of a more potent reduction in the steepness of the pulse wave contour, involves use of the ganglionic blocking agent, trimethaphan.76 This agent prevents increases in cardiac output and left ventricular ejection rate.73,76 The rapid onset (1-2 minutes) and short duration (10 minutes) of action of this drug allows precise pressure control. Any mild reflex increase in heart rate may be treated with subsequent β-blockade. Hydralazine is avoided because it causes unwanted reflex cardiac stimulation.
Pulmonary Edema
Many patients who present with pulmonary edema have long-standing antecedent hypertension with concentric left ventricular hypertrophy and well-preserved systolic contraction.79,80 They develop acute diastolic dysfunction in response to abrupt increases in cardiac afterload due to increased systemic blood pressure.81 With poor diastolic relaxation, the left ventricle requires markedly elevated filling pressures, leading to pulmonary venous hypertension and edema. The therapeutic goal is to decrease afterload, improve diastolic relaxation, and decrease pulmonary pressure. Vasodilators are the agents of choice, as they improve diastolic relaxation and lower pulmonary venous pressure.82 A beta-blocker may also be used. Nitroprusside is often used because it reduces preload and afterload, improving left ventricular function and reducing myocardial oxygen demand. Modest decreases in pressure improve symptoms markedly. In less emergent settings, ACE inhibitors or calcium channel antagonists have been shown to improve diastolic function and cause regression of concentric ventricular hypertrophy.83
In the setting of acute myocardial infarction, acute catecholamine release and sympathetic outflow contribute to hypertension. The hypertension usually resolves in a few hours with sedation and pain control alone. Diastolic blood pressures over 100 mm Hg, should be treated with nitroglycerin. The pressure is rapidly, but cautiously, reduced to near-normotensive levels; overshoot hypotension can worsen coronary perfusion. Therapy can usually be stopped within 24 hours. There is considerable evidence that the early use of β-blocking agents may reduce ultimate infarct size independent of blood pressure control.84
Perioperative Hypertension
Perioperative hypertension is a major risk factor for the development of postoperative hypertension.85 Whenever possible, it is preferred to postpone elective surgery until the blood pressure has been well controlled over days to weeks. However, when waiting is not an option, lowering the blood pressure to below 180/110 prior to noncardiac surgery is recommended.85 In patients with chronic hypertension on adequate treatment, oral medications should be taken the morning of surgery.
Catecholamine-Associated Hypertension
Pheochromocytoma is a very rare cause of hypertension.86 Excess catecholamine secretion by the tumor results in a sustained elevation of blood pressure in the majority of cases, while peripheral catecholamine uptake and storage leads to paroxysmal symptoms when the catecholamines are released in response to stimuli. Symptoms of pheochromocytoma include headache, palpitations, hypertension, anxiety, abdominal pain, and diaphoresis. Patients may present with orthostatic changes in blood pressure, a clue to the diagnosis.87 For the patient with hypertensive emergency, the treatment of choice is the short-acting parenteral α-antagonist, phentolamine. Following blood pressure reduction, β-blockade is generally added to control tachycardia or arrhythmias. As in all catecholamine excess states, beta-blockers should not be used as initial therapy. Loss of β-adrenergically mediated vasodilatation leaves α-adrenergically mediated vasoconstriction unopposed and results in increased pressure. An oral regimen of the nonselective α-antagonist, phenoxybenzamine, can be used in less critical situations. Labetalol has been effective in treating hypotension related to pheochromocytoma in selected patients. However, as its β-blockade exceeds its α-blocking effect, severe hypertension has been reported.88
In patients on MAO-inhibitor therapy, ingestion of foods containing tyramine or sympathomimetic amines can result in hypertension (Table 88-2). Tyramine is metabolized by an alternative pathway to octopamine, which releases catecholamines from peripheral sites by acting as a false neurotransmitter. Nitroprusside or phentolamine is used, with the addition of β-blockade as needed for tachycardia. The episodes are self-limited and last 6 hours or less.
| Chianti wine | Chicken liver |
| Soy sauce | Yeast |
| Avocados | Fermented sausage |
| Bananas | Canned figs |
| Coffee | Certain beers |
| Chocolate | Unpasteurized cheese |
| Pickled herring |
Gestational Hypertension/Preeclampsia/Eclampsia
Gestational hypertension is defined as a systolic blood pressure of at least 140 mm Hg and a diastolic blood pressure of at least 90 mm Hg on two separate blood pressure measurements done 6 hours apart. It occurs after 20 weeks of pregnancy in patients known to previously be normotensive.89 Up to 50% of these women develop preeclampsia if gestational hypertension develops before 30 weeks of gestation. Preeclampsia is defined as gestational hypertension with 300 mg of protein on a 24-hour urine (urine dipstick 1+). A 24-hour urine is necessary because dipstick urine protein correlates poorly with 24-hour urine protein in gestational hypertension.90 Preeclampsia should also be suspected in patients with hypertension developing after 20 weeks’ gestation and associated with nausea, vomiting, cerebral symptoms, abnormal liver function tests, and thrombocytopenia, even in the absence of proteinuria. Preeclampsia develops in 5% of all pregnancies and occurs twice as often in primigravid versus multigravid women.91 In women with a history of multiple pregnancies but with a new partner,91 preeclampsia also appears. In the setting of molar pregnancy, it is seen in up to 70% of individuals.92 During normal pregnancy, blood pressure is initially decreased and then slowly rises toward the normal range during the third trimester. In preeclampsia, intravascular volume is low despite peripheral edema, and the renin-angiotensin system is activated. Progression to seizures defines eclampsia and may occur with diastolic pressures of as low as 100 mm Hg. Clinical treatment includes bed rest and parenteral magnesium.
With regard to hypertensive treatment in pregnancy, the optimal blood pressure has not been defined. The goal is to prolong the pregnancy until the fetus can be delivered. In the case of mild preeclampsia, there are no large studies to guide therapy.91 With more severe preeclampsia, treatment is initiated to prevent cerebral hemorrhage. The recommendation is to initiate antihypertensive therapy when the systolic blood pressure is above 160 mm Hg or the diastolic blood pressure is above 110 mm Hg. Usually, preferred medications include hydralazine and labetalol administered to keep the systolic blood pressure 140 to 155 mm Hg and the diastolic blood pressure 90 to 105 mm Hg.91 Nitroprusside should be avoided owing to the risk of cyanide toxicity in the fetus. ACE inhibitors should also be avoided because of their potential impact on the fetus’s kidney.
Other Hypertensive Situations
The renal crisis of scleroderma is an aggressive form of malignant hypertension in which proliferative endarteritis precedes hypertension. Ischemic-induced activation of the renin-angiotensin system causes the hypertension. The incidence of this condition among patients with scleroderma ranges from 8% to 13%, and it is more common among blacks.93 Progression to ESRD occurs in 1 to 2 months without treatment. Aggressive pressure control with ACE inhibitors leads to a long-term survival of about 50% to 70%.94
Hypertension is a feature of both primary and secondary antiphospholipid antibody syndromes, occurring in up to 93% of patients.95 Malignant hypertension occurs in this syndrome secondary to both microvasculopathy and emboli to the renal artery. Antihypertensive treatment is similar to malignant hypertension. Successful treatment outcomes have been reported with anticoagulation.95
Patients with transverse spinal cord lesions at the T6 level or higher, including patients with Guillain-Barré syndrome, have dysreflexia in which noxious stimulus in dermatomes below the level of lesion trigger a massive sympathetic discharge. This leads to severe hypertension, bradycardia, diaphoresis, and headache. In 90% of patients, distention of the bladder or bowel causes the dysreflexia, and prompt decompression leads to resolution of hypertension.96 Drugs that have been used successfully in treating this condition include nitroprusside, phentolamine, and labetalol.
Hypertension in the renal transplant recipient may be caused by acute rejection, vascular anastomotic stenosis, obstructive uropathy, corticosteroid use, cyclosporine, and native-kidney renin release.97 Oral calcium channel antagonists are effective and well tolerated in these patients. Other rare causes of hypertension include erythropoietin-associated hypertension. This is treated with phlebotomy and dose reduction in conjunction with antihypertensive drugs.98 Diabetics on beta-blockers can experience severe hypertension with hypoglycemic episodes, presumably due to catecholamine release.
 Hypertensive Urgency
Hypertensive Urgency
Hypertensive urgency refers to patients in whom blood pressure is severely elevated, but based on detailed history, physical examination, and laboratory evaluation, there is no evidence of acute end-organ damage. This clinical situation is quite different from that of patients with severe hypertension and chronic stable complications such as those with stable chronic renal failure or stable angina. The decision to treat the latter group in the inpatient or outpatient setting often depends on the associated end-organ involvement (Box 88-3) and reliability of patient follow-up.
The third (and most common) treatment category, termed severe uncomplicated hypertension (see Box 88-3), is used to describe patients with severe blood pressure elevation but no end-organ involvement. Despite markedly elevated pressures (e.g., diastolic of pressures 140 mm Hg at times), these patients are at low risk of immediate complications. Hypertension-related morbidity tends to occur over months to years. The treatment of choice is gradual pressure reduction over a few days in the outpatient setting. The major risk of therapy is rapid pressure reduction. The choice of antihypertensive agent is based on ease of administration and side-effect profile rather than on rapid blood pressure reductions. Frequently, restarting a previously effective regimen is all that is necessary. It is critically important to follow these patients over the next 24 to 48 hours to ensure the blood pressure is appropriately reduced. While medicolegal issues may pressure physicians into loading these patients with medication to observe on-the-spot control of their blood pressure, this practice has recently been questioned as having no clear rational scientific basis.
Adams HPJr, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups. Stroke. 2007;5:1655-1711.
Recent guidelines on total management (including BP) after ischemic CVA.
Freedman BI, Sedor JR. Hypertension-associated kidney disease: perhaps no more. J Am Soc Nephrol. 2008;19:2047-2051.
Fleisher LA, Beckman JA, Brown KA, Calkins H, Chaikof E, Fleischmann KE, et al. ACC/AHA 2007 guidelines on perioperative cardiovascular evaluation and care for noncardiac surgery: executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation. 2007;116:1971-1996.
Consensus statement on management of hypertension (and other CV issues) in the perioperative period.
Wadei HM, Textor SC. Hypertension in the kidney transplant recipient. Transplant Rev. 2010;24:105-120.
Careful recent review of diagnosis and management of hypertension after kidney transplantation.
Kincaid-Smith P, McMichael J, Murphy EA. The clinical course and pathology of hypertension with papilledema (malignant hypertension). Q J Med. 1958;27:117.
Classical description of clinical events in malignant hypertension.
Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of a reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;2:205-210.
Ohwaki K, Yano EM, Nagashima H, Hirata M, Nakagomi T, Tamura A. Blood pressure management in acute intracerebral hemorrhage: relationship between elevated blood pressure and hematoma enlargement. Stroke. 2004;35:1364-1367.
Trivedi M, Coles JP. Blood pressure management in acute head injury. J Intensive Care Med. 2009;24:96-107.
1 Hajjar I, Kotchen JM, Kotchen TA. Hypertension: trends in prevalence, incidence, and control. Annu Rev Public Health. 2006;27:465-490.
2 Chobanian AV, Bakris GL, Black HR, et al. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. JAMA. 2003;289:2560-2572.
3 Nolan CR, Linas SL. Accelerated and malignant hypertension. In: Schrier RW, Gottschalk CW, editors. Diseases of the Kidney. Boston: Little, Brown and Company; 1988:1703.
4 Calhoun D, Oparil S. Treatment of Hypertensive Crisis. N Eng J Med. 1990;323:1177.
5 Beilin LJ, Goldby FS, Mohring J. High arterial pressure versus humoral factors in the pathogensis of the vascular lesions of malignant hypertension. Clin Sci Mol Med. 1977;52:111.
6 Kincaid-Smith P, McMichael J, Murphy EA. The clinical course and pathology of hypertension with papilledema (malignant hypertension). Q J Med. 1958;27:117.
7 Jones DB. Arterial and glomerular lesions associated with severe hypertension. Light and electron microscopic studies. Lab Invest. 1974;31:303.
8 Funakoshi Y, Ichiki T, Ito K, Takeshita A. Induction if interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension. 1999;34:118-125.
9 Muller DN, Dechend R, Mervaala EM, et al. NF-kB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension. 2000;35:193-201.
10 Gudbrandsson T, Hansson L, Herlitz H, et al. Immunological changes in patients with previous malignant essential hypertension. Am J Physiol. 1977;232:F260.
11 Mohring J, Mohring B, Petri M, Haack D. Vasopressor role of ADH in the pathogenesis of malignant DOC hypertension. Am J Physiol. 1977;232:F260.
12 Robertson AL, Khairallah PA. Effects of angiotension II and some analogues on vascular permeability in the rabbit. Circ Res. 1972;31:923.
13 Milliez P, et al. The natural course of malignant hypertension. In: Bock LD, Cottier P, editors. Essential Hypertension: an international symposium. Berlin: Springer-Verlag; 1960:214.
14 Patel R, Ansari A, Grim CE. Prognosis and predisposing factors for essential malignant hypertension in predominantly black patients. Am J Cardiol. 1990;66:868.
15 Bloxham CA, Beevers DF, Walker JM. Malignant hypertension and cigarette smoking. Br J Med. 1979;1:581.
16 Barraclough MA. Sodium and water depletion with acute malignant hypertension. Am J Med. 1979;1:579.
17 Messerli FH. Osler’s maneuver, pseudohypertension, and true hypertension in the elderly. Am J Med. 1980;80:906-910.
18 DellaCroce JT. Vitale, Albert. Hypertension and the eye. Curr Opin Ophthalmol. 2008;19:493-498.
19 Gavras H, Oliver N, Aitchison J, et al. Abnormalities of Coagulation and the development of malignant phase hypertension. Kidney Int. 1975;suppl 2:S252.
20 Laragh JH, Ulick S, Janusezewicz V, et al. Aldosterone secretion and primary malignant hypertension. J Clin Invest. 1960;39:1091.
21 McAllister RGJr, Van Way CW, Dayani K, et al. Malignant hypertension effect of therapy on renin and aldosterone. Circ Res. 1971;28(suppl2):16.
22 Hinchey J, Chaves C, Appignani B, et al. A Reversible posterior Leukoencephalopathy Syndrome. New Engl J Med. 1996;334:494-500.
23 Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of a reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;2:205-210.
24 Milne FJ, James SH, Veriava Y. Malignant hypertension and its renal complications in black South Africans. S Afr Med J. 1989;76:164.
25 Patel R, Ansari A, Grim CE, Hidaka M. Prognosis and predisposing factors for essential malignant hypertension in predominantly black patients (published erratum appears in Am J Cardiol 67:341, 1991). Am J Cardiol. 1990;66:868.
26 Freedman BI, Sedor JR, et al. Hypertension-Associated Kidney Disease. J Am Soc Nephrol. 2008;19:2047-2051.
27 Holland NH, Kotchen T, Bhathena D. Hypertension in Children with chronic pyelonephritis. Kidney Int. 1975;suppl 8:s243.
28 Kincaid-Smith P. Malignant Hypertension: mechanisms and management. Pharmacol Ther. 1980;9:245.
29 Michenfelder JD, Theye RA. Canine systemic and cerebral effects of hypotension induced by hemorrhage, trimethaphan, halothane, or nitroprusside. Anesthesiology. 1977;46:188-195.
30 Turner JM, Powell D, Gibson RM, et al. Intracranial Pressure exchanges in Neurosurgical patients during hypotension induced sodium nitroprusside or trimethaphan. Br J Anaesth. 1977;49:419.
31 Cottrell JE, Patel K, Turndorf H, et al. Intracranial pressure changes induced by sodium nitroprusside in patients with intracranial mass lesion. J Neurosurg. 1978;48:329.
32 Marsh ML, Shapiro HM, Smith RW, et al. Changes in neurologic status and intracranial pressure associated with sodium nitroprusside administration. Anesthesiology. 1979;51:336.
33 Anile C, Zanghi F, Bracali A, et al. Sodium nitroprusside and intracranial pressure. Acta Neurochir. 1981;58:203-211.
34 Tumlin J, Dunbar LM, Oparil S, et al. Fenoldopam, a Dopamine agonist, for Hypertensive Emergency: Fenoldopam Study Group. A Multicenter Randomized Trial. Acad Emerg Med. 2000;7:653-662.
35 Murphy MB, Murray C, Shorten GD. Fenoldopam: a selective peripheral dopamine-receptor agonist for the treatment of severe hypertension. N Engl J Med. 2001;345:1548-1557.
36 Brogden RN, Markham A. Fenoldopam: a review of its pharmacodynamic and pharmacokinetic properties and intravenous clinical potential in the management of hypertensive urgencies and emergencies. Drugs. 1997;54:634-650.
37 Oparil S, Aronson S, Deeb M. Fenoldopam: A New Parenteral Antihypertensive: Consensus roundtable on the management of perioperative hypertension and hypertensive crisis. Am J Hypertens. 1999;12:653-664.
38 Post JB4th, Frishman WH. Fenoldopam: a new dopamine agonist for the treatment of hypertensive urgencies and emergencies. J Clin Pharm. 1998;38:2-13.
39 Blumenfeld JD, Laragh JH. Management of hypertensive crisis. Am J Hypertens. 2001;14:1154-1167.
40 Lawton WJ. The short-term course of renal function in malignant hypertensives with renal insufficiency. Clin Nephrol. 1982;17:277.
41 Bakir AA, Bazilinski N, Dunea G. Transient and sustained recovery from renal shutdown in accelerated hypertension. Am J Med. 1986;80:172.
42 Nicholson GD. Long-term survival after recovery from malignant nephrosclerosis. Am J Hypertens. 1988;1:73.
43 Mandani BH, Lim VS, Mahurker SD, et al. Recovery of prolonged renal failure in patients with accelerated hypertensive emergencies. N Engl J Med. 1983;291:95.
44 Mroczek WJ, Davidov M, Govrilovich L, et al. The value of aggressive therapy in hypertensive patients with azotemia. Circulation. 1969;40:893.
45 Gifford RWJr. Management of Hypertensive Crisis. JAMA. 1991;266:829.
46 Gifford RWJr. Effect of reducing elevated blood pressure on cerebral circulation. Hypertension. 1983;5:III17.
47 Iisles CG, Hohson AO, Milne FJ. Slow-release nifedipine and atenolol as initial treatments in blacks with malignant hypertension. Br J Clin Pharmacol. 1986;21:377.
48 Reed G, Devous M. Cerebral blood flow autoregulation and hypertension. Am J Med Sci. 1985;289:37.
49 Harden RA, Russell RP. Iatrogenically induced hypertensive encephalopathy. Johns Hopkins Med J. 1979;145:44.
50 Powers WJ. Acute hypertension after stroke: The scientific basis for treatment decisions. Neurology. 1993;43:461-467.
51 Adams HPJr, del Zoppo G, Alberts MJ, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: The American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Stroke. 2007;5:1655-1711.
52 Leira R, Millian M, Diez-Tejedor E, et al. Age Determines the Effects of Blood pressure During the Acute phase of Ischemic Stroke. The TICA Study. Hypertension. 2009;54:769-774.
53 Roux AA, Wallace MC. Outcome and Cost of Aneurysmal Subarachnoid Hemorrhage Aneurysmal Subarachnoid Hemorrhage. Neurosurg Clin North Am. 2010;21:235-246.
54 Gifford RWJr. Management of hypertensive crisis. JAMA. 1989;266:257.
55 Rinkel GJ, Feigin VL, Vermeulen M, et al. Calcium antagonists for aneurysmal subarachnoid bleed. Cochrane Database Sys Rev. 2002;4:CD000277.
56 Butcher K, Laidlaw J. Current intracerebral hemorrhage management. J Clin Neuro Sci. 2003;10:158-167.
57 Kazui S, Minematsu H, Yamamato H, et al. Predisposing factors to enlargement of spontaneous intracerebral hematoma. Stroke. 1997;28:2370-2375.
58 Adams RE, Powers WJ. Management of Hypertension in Acute Intracerebral Hemorrhage. Crit Care Clin. 1997;13:131-161.
59 Fogelholm R, Murros K, Rissanen A, Avikainen S. Long-term survival after primary intracerebral haemorrhage: a retrospective population based study. J Neurol Neurosurg Psychiatry. 2005;76:1534-1538.
60 Ohwaki K, Yano E, Nagashima H, et al. Blood pressure management in acute intracerebral hemorrhage: relationship between elevated blood pressure and hematoma enlargement. Stroke. 2004;35:1364-1367.
61 Broderick JP, Brott TG, Tomsick T, et al. Ultra-early evaluation of intracerebral hemorrhage. J Neurosurg. 1990;72:195.
62 Mayer SA, Sacco RL, Shi T, et al. Neurologic deterioration in noncomatose patients with supratentorial intracerebral hemorrhage. Neurology. 1994;44:1379.
63 Fijii Y, Tanaka R, Takeuchi S, et al. Hematoma enlargement in spontaneous intracerebral hemorrhage. J Neurosurg. 1994;80:51.
64 Hayashi M, Kobayashi H, Kawano H, et al. Treatment of systemic hypertension and intracranial hypertension in cases of brain hemorrhage. Stroke. 1988;19:314.
65 Broderick J, Connolly S, Feldmann E, et al. Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Stroke. 2007;6:2001-2023.
66 Marik PE, Varon J, Trask T. Management of Head trauma. Chest. 2002;122:699-711.
67 Kaji S, Akasaka T, Katayama M, et al. Prognosis of Retrograde Dissection from the Descending to the Ascending Aorta. Circulation. September 2003;108(Suppl. 1):300-306.
68 Narotam PK, Puri V, Roberto JM, et al. Management of hypertensive emergencies in acute brain disease; evaluation of the treatment effects of intravenous nicardipine on cerebral oxygenation. J Neurosurg. 2008;109:1065-1074.
69 Trivedi M, Coles JP. Blood Pressure Management in Acute Head Injury. J Intensive Care Med. 2009;24:96-107.
70 Slater EE, DeSanctis RW. The clinical recognition dissecting aortic aneurysm. Am J Med. 1987;60:1060.
71 DeSanctis RE, Doroghazi RM, Austen WG, Buckley MJ. Aortic Dissection. N Engl J Med. 1987;317:1060.
72 Wheat MWJr. Acute dissecting aneurysms of the aorta: diagnosis and treatment. Am Heart J. 1980;99:373.
73 Slater EE, DeSanctis RW. Dissection of the aorta. Med Clin North Am. 1979;63:141.
74 Suzuki T, Mehta RH, Ince H, et al. Clinical Profiles and Outcomes of Acute Type B Aortic Dissection in the Current Era: Lessons from the International Registry of Aortic Dissection (IRAD). Circulation. 2003;109:1312-1317.
75 Macura KJ, Szarf G, Fishman EK, Bluemke DA. Role of Computed tomography and magnetic imaging in assessment of acute aortic syndromes. Semin Ultrasound CT MR. 2003;24:232-252.
76 Abdelwahab W, Frishman W, Landau A. Management of Hypertensive urgencies and emergencies. J Clin Pharmacol. 1995;35:747-762.
77 Jonker FH, et al. Meta-analysis of open endovascular repair for ruptured descending thoracic aortic aneurysm. J Vasc Surg. 2010;51:1026-1032.
78 Hiratzka LF, Bakris MD, et al. 2010/CCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients With Thoracic Aortic Disease. J Am Coll Cardiol. 2010;55:e27-130.
79 Staufer JC, Gaasch WH. Recognition and treatment of left ventricular diastolic dysfunction. Prog Cardiovasc Dis. 1990;32:319.
80 Silva HB, Bortolotto LA, Giorgi DM, et al. Ventricular function by radionucleotide ventriculography in malignant hypertension. Hypertension. 1992;19:11210.
81 Zile MR, Gaasch WH. Mechanical loads and isovolumic and filling indices of left ventricular relaxation. Prog Cardiovasc Dis. 1990;32:333.
82 Marek PE, Varon J. Hypertensive crises: challenges and management. Chest. 2007;131:1949-1962.
83 Weber JR. Left ventricular hypertrophy: its prime importance as a controllable risk factor. Am Heart J. 1988;116:272.
84 First International Study of Infarct Survival Collaborative Group. Randomized trial of intravenous atenolol among 16,027 cases of suspected acute myocardial infarction. Lancet. 1986;2:57.
85 Fleisher LA, Beckman JA, Brown KA, et al. ACC/AHA 2007 Guidelines on Perioperative Cardiovascular Evaluation and Care for Noncardiac Surgery; Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines on Perioperative Cardiovascular Evaluation for Noncardiac Surgery). Circulation. 2007;116:1971-1996.
86 Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma. A review of the literature and report of one institution experience. Medicine. 1991;70:46.
87 Rubenstein EB, Escalante C. Hypertensive Crisis. Crit Care Clin. 1989;5:477.
88 Briggs RS, Birtwell AJ, Pohl JE. Hypertensive response to labetalol in pheochromocytoma (letter). Lancet. 1978;1:1045.
89 Sibai BM. Diagnosis and management of gestational hypertension and preeclampsia. Obstet Gynecol. 2003;102:181-192.
90 Barton JR, O’Brien JM, Bergauer NK, et al. Mild gestational diabetes remote from term: progression and outcome. Am J Obstet Gynecol. 2001;184:979-983.
91 Jim B, Sharma S, Kebeds T, et al. Hypertension in Pregnancy: A Comprehensive Update. Cardiol Rev. 2010;18:178-189.
92 Meyer NL, Mercer BM, Friedman SA, et al. Urinary dipstick protein: a poor predictor of absent or severe proteinuria. Am J Obstet Gynecol. 1994;182:938-942.
93 Traub YM, Shapiro AP, Rodnan GP, et al. Hypertension and renal failure (scleroderma renal crisis) in progressive systemic sclerosis. Review of 25 years experience with 68 cases. Medicine. 1983;62:335.
94 Beckett VL, Donadio JV, Brennan LAJr. Use of captopril as early therapy for renal scleroderma: A prospective study. Mayo Clin Proc. 1985;60:763.
95 Nzerue CM, Hewan-Lowe K, Pierangeli S, et al. Perspective in Renal Medicine: “Black swan in the kidney: Renal involvement in the antiphospholipid antibody syndrome. Kidney Int. 2002;62:733-744.
96 Erickson RP. Autonomic hyperreflexia: pathophysiology and medical management. Arch Phys Med Rehab. 1980;61:431.
97 Wadei HM, Textor SC. Hypertension in the kidney transplant recipient. Transplant Rev. 2010;24:105-120.
98 Roger SD, Macdougall IC, Raine AE. Phlebotomy for erythropoietin-associated malignant hypertension. (letter). Lancet. 1991;337:1606.
99 Kenyon KW, Clevidipine. An Ultra Short-acting Calcium Channel Antagonist for Acute Hypertension. Ann Pharmacol Ther. 2009;43:1258-1265.
