14 Hyperkalemia and Hypokalemia
Hyperkalemia and hypokalemia are the most common electrolyte abnormalities found in hospitalized patients.1 The precise prevalence of potassium abnormalities in critically ill patients is unknown.2 However, owing to comorbid conditions, critically ill patients are likely at a higher risk of developing complications from altered serum potassium levels. Therefore, timely recognition and intervention are essential for minimizing morbidity and mortality.
 Hyperkalemia
Hyperkalemia
Hyperkalemia is defined as a serum potassium concentration (serum [K+]) greater than 5.0 mEq/L. In critically ill patients, hyperkalemia is less frequent than hypokalemia but more likely to cause serious complications. Severe hyperkalemia requires rapid correction to prevent serious cardiovascular complications. The measured value for serum [K+] can be elevated as a result of in vitro phenomena, usually the release of K+ from cells during the clotting process. Pseudohyperkalemia should be recognized and considered in patients with marked elevations of white blood cell or platelet count.3 Simultaneous measurements of plasma (unclotted) and serum (clotted) [K+] should identify this problem. A serum [K+] that is 0.2 to 0.3 mEq/L greater than plasma [K+] is indicative of pseudohyperkalemia. Pseudohyperkalemia also may result from hemolysis of a blood specimen after collection; this event is usually identified in the laboratory and reported.
True hyperkalemia occurs by two mechanisms: (1) impaired K+ excretion and (2) shifts in intracellular and extracellular K+ (Box 14-1). Renal insufficiency is the most common cause of altered K+ excretion. With acute oliguric renal failure, elevated potassium level, if not treated, is life threatening. In most patients with nonoliguric chronic renal failure, mild hyperkalemia is evident.4 With some causes of chronic renal failure, such as diabetes mellitus and tubulointerstitial diseases, hyperkalemia is more pronounced and is probably related to low circulating renin and aldosterone levels.5 Decreased aldosterone production promotes the development of hyperkalemia. Patients with acquired adrenal insufficiency develop hyperkalemia despite normal renal function. Various drugs used in the intensive care unit (ICU) can produce hyperkalemia by impairing K+ excretion.6 Patients with abnormal renal function are more susceptible to drug-induced hyperkalemia, and potassium supplements are the most common cause. Potassium-sparing diuretics (spironolactone, amiloride, and triamterene) inhibit K+ excretion and can produce severe hyperkalemia.7 Spironolactone is the most dangerous of these drugs with respect to impaired K+ excretion, and its effects can be persist even after discontinuation of the drug. Its use has increased significantly after reports of improved mortality in patients with congestive heart failure.8 Angiotensin-converting enzyme (ACE) inhibitors reduce circulating aldosterone levels and are associated with hyperkalemia in patients with renal insufficiency.9 Angiotensin receptor blockers (ARBs) have less impact on circulating aldosterone levels and are less likely to produce hyperkalemia.9 Nonsteroidal antiinflammatory drugs (NSAIDs) and cyclooxygenase-2 (COX-2) inhibitors block prostaglandin synthesis, causing indirect suppression of renin release and aldosterone secretion. NSAIDs and COX-2 inhibitors also reduce renal blood flow and glomerular filtration rate, particularly in patients with prerenal azotemia (due to decreased intravascular volume or heart failure). These compounds may produce hyperkalemia by these mechanisms in patients with or without renal dysfunction.10,11 Heparin inhibits aldosterone synthesis and can cause significant hyperkalemia in patients with altered renal function.12–14 Other drugs that may cause hyperkalemia by decreasing glomerular filtration rate and aldosterone secretion include cyclosporine and tacrolimus.15 Trimethoprim and pentamidine inhibit renal K+ excretion and can cause hyperkalemia in patients with renal insufficiency.15 Hyperkalemia has also been described as one of the manifestations of the propofol infusion syndrome (PRIS), a rare but fatal complication of propofol infusion in critically ill patients.16,17
Box 14-1
Causes of Hyperkalemia
ACE, Angiotensin-converting enzyme; NSAIDs, nonsteroidal antiinflammatory drugs.
Alterations in the relationship between intracellular and extracellular [K+] may lead to severe hyperkalemia in critically ill patients, either by increased release of intracellular K+ or by inhibition of extracellular-to-intracellular K+ movement. The effects of acidosis on serum [K+] are complicated and not fully understood. The traditional teaching that acidosis produces a shift of K+ from the intracellular to the extracellular space, thus causing hyperkalemia, was based on observations of hyperkalemia in patients with diabetic ketoacidosis and renal failure.18 This relationship has since been disproved, and changes in serum [K+] in relation to acid-base disorders are more complex than initially thought. Most forms of acute acidosis do not present with hyperkalemia. The most common forms of acute metabolic acidosis in critically ill patients, diabetic ketoacidosis and lactic acidosis, are not associated with shift K+ out of cells.19 Hyperkalemia seen with diabetic ketoacidosis is most likely caused by increased release of intracellular K+ due to the breakdown of muscle cells.20 Hypertonicity of the extracellular fluid causes water to exit cells, and K+ follows. Unless renal function is adequate to eliminate the excess K+, hyperkalemia develops. This situation may occur in patients with uncontrolled diabetes and can lead to severe hyperkalemia in the presence of renal failure and hypoaldosteronism.20 Massive tissue breakdown can occur with trauma, burns, and rhabdomyolysis, leading to release of K+ into the extracellular space. If renal mechanisms for K+ excretion are impaired, severe hyperkalemia may develop. Drugs can affect the transmembrane balance of K+. β-Adrenergic blockers inhibit the entry of K+ into cells and, in combination with renal failure, can promote development of hyperkalemia.21 Succinylcholine blocks normal reentry of K+ into cells after depolarization and causes a transitory increase in serum [K+].22 In patients with severe burns or extensive trauma, the transient hyperkalemia induced by succinylcholine can be more prolonged and severe.23 Digoxin impairs K+ entry into cells by inhibiting the cell membrane Na+/K+-ATPase.24 It does not produce hyperkalemia in therapeutic doses, but may cause hyperkalemia with toxic levels.24,25
Clinical Effects
Most of the clinical consequences of potassium abnormalities are related to the effect on the transmembrane resting cell potential. Cardiac and neuromuscular cells are particularly sensitive to changes in serum [K+]. Most often, hyperkalemia is asymptomatic. However, it affects the cardiac conduction system, as evidenced by characteristic changes in the electrocardiogram (ECG) that serve as indicators of potential life-threatening arrhythmias (Table 14-1). The first sign of increased serum [K+] is tenting of the T wave. Changes associated with progressive increases in serum [K+] include widening of the QRS complex, progressive development of atrioventricular conduction blocks, slow idioventricular rhythm, an ECG tracing that looks like a sine wave, ventricular fibrillation, and finally asystole.26 ECG changes are not always sensitive to changes in serum [K+] levels. There is no absolute level of serum [K+] associated with a particular ECG abnormality, but rapid rises seem to be more dangerous, particularly in patients without a history of chronic renal insufficiency.27,28 However, normal ECGs have been described with extreme hyperkalemia, and in some cases the first manifestation of cardiac compromise from hyperkalemia may be ventricular fibrillation.29,30 Hyperkalemia can cause paresthesias and weakness in the arms and legs, followed by a symmetrical flaccid paralysis of the extremities that ascends toward the trunk, finally involving the respiratory muscles. The cranial nerves are usually not affected by hyperkalemia.
TABLE 14-1 Electrocardiogram Changes Caused by Abnormal [K+]
| Hyperkalemia | Hypokalemia |
|---|---|
| Peaked T waves | Broad, flat T waves |
| Loss of P waves | ST depression |
| Widening QRS complexes | U wave |
| Sine wave | QT interval prolongation |
| Ventricular arrhythmias | Ventricular arrhythmias |
| Asystole |
Treatment
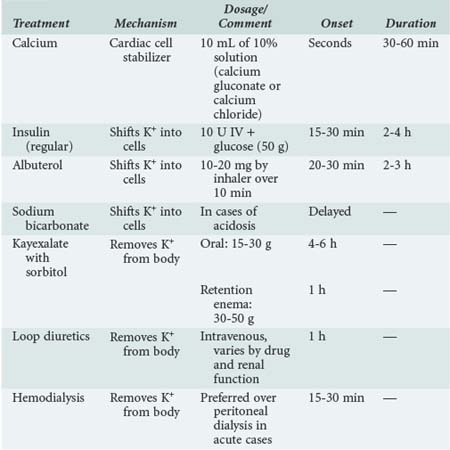
The primary goal of treating hyperkalemia is to prevent adverse cardiac complications. Treatment modalities are aimed at one of three mechanisms to prevent or decrease these complications: (1) direct antagonism of hyperkalemic effect on the cell membrane polarization, (2) movement of extracellular K+ into the intracellular compartment, and (3) removal of K+ from the body. Patients with a serum [K+] greater than 6.5 mEq/L or ECG signs suggestive of hyperkalemia should be treated emergently.31
Direct Antagonism of Hyperkalemic Effect on Cell Membrane Polarization
The intravenous (IV) infusion of calcium gluconate antagonizes the effects of hyperkalemia on the heart. This effect occurs within minutes and lasts 30 to 60 minutes. If a salutary effect is noted, repeat doses may be used. The recommended dose is 10 mL of 10% calcium gluconate or chloride. Extreme caution must be used in patients with hyperkalemia and digitalis toxicity, because the administration of ionized calcium may potentiate the effects of digoxin on the conduction system.32 Calcium should be avoided in the setting of digoxin toxicity. Finally, IV hypertonic saline (3%) has been shown to reverse the ECG changes of hyperkalemia in patients with concomitant hyponatremia.33 This effect is likely due to direct action on the cardiac cells and has not been demonstrated to be effective in patients with normal or elevated serum sodium levels.
Movement of Extracellular K+ Into the Intracellular Compartment
Administration of insulin shifts K+ into cells; this effect occurs in 15 to 30 minutes and lasts approximately 2 to 4 hours.34 The recommended dose is 10 units of regular insulin IV; dextrose (50 g) should be added to avoid hypoglycemia. This dose will decrease serum [K+] by 0.5 to 1.5 mEq/L. Patients without IV access can be treated with inhaled β2-adrenergic agonists such as albuterol. Albuterol drives K+ into cells by increasing Na+/K+-ATPase activity. Albuterol (10 to 20 mg in 4 mL of saline by nasal inhalation over 10 minutes) can lower the serum [K+] by 0.5 to 1.5 mEq/L.35 Sodium bicarbonate is much less effective than either insulin or albuterol but may produce shifting of [K+] into cells.36 The use of sodium bicarbonate should be limited to situations in which it is indicated for the treatment of concurrent acidosis.
Removal of K+ from the Body
Removal of K+ is necessary to prevent a recurrence of hyperkalemia once the effects of the preceding measures have waned. Loop diuretics can be helpful in patients with sufficient renal function (dosing depends on medication and renal function); however, most often, other measures are needed. Sodium polystyrene sulfonate (Kayexalate) binds to K+ secreted in the colon. Each gram of resin removes 0.5 to 1 mEq of K+. The usual dose of Kayexalate is 15 to 30 g orally. Because the resin causes constipation, sorbitol (15 mL of a 70% solution) should be administered to induce osmotic diarrhea. If oral administration is not feasible, Kayexalate can be given as a retention enema consisting of 30 to 50 g of the resin in 70% sorbitol solution. It is important, however, that the enema be retained for at least 30 to 60 minutes to obtain the desired therapeutic effect. The effects of Kayexalate on serum [K+] occur in 4 to 6 hours when the agent is given orally and in 1 to 2 hours when it is given as an enema. Serious side effects of Kayexalate and sorbitol include bowel necrosis and perforation. These complications seem to be more likely in severely immunocompromised patients or shortly after surgery.37,38 Kayexalate should be avoided in these circumstances. Both peritoneal dialysis and hemodialysis are very effective in removing K+ from the body. In acute cases when serum [K+] needs to be corrected rapidly, hemodialysis is preferred. Hemodialysis can quickly remove 50 to 125 mEq of K+ and should be used as definitive treatment when other treatments fail. Peritoneal dialysis is also effective in removing K+ from the body, but its effects are slower than those achieved with hemodialysis or cation exchange resins. In addition to the implementation of rapid treatment, the causes of hyperkalemia should be sought and corrected, and offending drugs should be discontinued when possible. Table 14-2 summarizes the treatment for hyperkalemia.
 Hypokalemia
Hypokalemia
Hypokalemia is more common than hyperkalemia and is defined as serum [K+] less than 3.6 mEq/L. Hypokalemia usually occurs as a consequence of K+ depletion due to either increased excretion or inadequate intake. Shifts in extracellular and intracellular [K+] also can cause hypokalemia (Box 14-2). Low serum [K+] reflects an imbalance of normal K+ homeostasis, with one rare exception. In patients with leukemia and markedly elevated white cell count, K+ can be taken up by the abnormal cells in the test tube and produce pseudohypokalemia.39 However, as noted earlier, in vitro changes in [K+] more commonly produce pseudohyperkalemia.
In critically ill patients, increased losses are more commonly responsible for K+ depletion than is inadequate ingestion. The use of diuretics is the most common cause of hypokalemia in hospitalized patients. Both loop and thiazide diuretics cause increased delivery of Na+ and Cl− to the collecting duct, promoting the secretion of K+ and causing hypokalemia. Diuretics are often used in high doses or administered by continuous infusion in critically ill patients, increasing the risk of hypokalemia. K+ losses can also occur from increased stool output. Because K+ is secreted into the colon, patients with high outputs from ileal or jejunal ostomies do not develop hypokalemia. Causes of upper gastrointestinal (GI) losses, such as vomiting or nasogastric suctioning, usually do not promote depletion of K+ directly. However, upper GI losses are associated with hypochloremia and metabolic alkalosis, both of which may cause increased renal K+ excretion, exacerbating the resultant hypokalemia. Large doses of laxatives or repeated enemas lead to excessive K+ losses and hypokalemia. Magnesium depletion and some forms of renal tubular acidosis (type 1 and some forms of type 2) can cause renal K+ wasting.40 Other drugs also can lead to hypokalemia. For example, fludrocortisone and hydrocortisone increase K+ excretion. Aminoglycosides, amphotericin B, cisplatin, and foscarnet cause magnesium depletion and increased K renal losses.41 Penicillin and its synthetic derivatives, when given IV, cause increased Na+ delivery to the distal nephron, promoting K+ secretion and potentially causing hypokalemia.41 Alkalosis can cause movement of K+ into cells. This effect is seen with both metabolic and respiratory alkalosis and occurs as a consequence of hydrogen ions leaving the cell to minimize changes in extracellular pH, and K+ moving into the cells to maintain electroneutrality. The direct effects of alkalosis on serum [K+] are small, and the hypokalemia seen with metabolic alkalosis is more often caused by chloride losses producing increased delivery of Na+ to the distal nephron, which stimulates K+ losses. A number of β2-adrenergic agonist drugs, including bronchodilators, decongestants, and tocolytics, can cause K+ shifts into cells and transient hypokalemia.42 Theophylline stimulates cell membrane Na+/K+-ATPase and promotes K+ entry into cells; hypokalemia is commonly seen with theophylline toxicity.43 Barium can block the exit of K+ from cells and cause hypokalemia.44 Thyroid hormone can stimulate Na+/K+-ATPase, and hypokalemia is sometimes seen with hyperthyroidism. Increased endogenous β-adrenergic stimulation occurs with delirium tremens, producing intracellular movement of K+ and hypokalemia.45 Familial hypokalemic periodic paralysis, a rare hereditary disease, is associated with a mutation in cell membrane calcium channels and causes episodes of severe hypokalemia triggered by high sodium intake or exercise.46 These patients can present with severe muscle weakness and respiratory failure from hypoventilation.
Clinical Effects
It is estimated that approximately 20% of hospitalized patients have a serum [K+] less than 3.6 mEq/L; most are asymptomatic. As discussed earlier, the consequences of changes in serum [K+] occur as a result of alterations in the resting membrane potential, making cardiac and neuromuscular cells the most susceptible targets. The most serious and potentially fatal effects of hypokalemia are related to disturbances in cardiac electrical activity that can lead to cardiac arrest. However, cardiac arrest caused by hypokalemia occurs almost exclusively in patients with underlying cardiac disease or patients taking digitalis.47 Hypokalemia is also associated with characteristic ECG changes (see Table 14-1). Progressive decreases in serum [K+] produce broad, flat T waves; ST depression; and the appearance of U waves, QT interval prolongation, and finally ventricular arrhythmias, leading to cardiac arrest.26 When serum [K+] is less than 3.0 mEq/L, generalized weakness can develop. When serum [K+] decreases to less than 2.5 mEq/L, muscle necrosis and rhabdomyolysis can occur. With progression of hypokalemia, an ascending muscle paralysis develops, leading to respiratory failure and arrest.
Treatment
The immediate goal of treatment in hypokalemia is to prevent or correct cardiac electrical disturbances and serious neuromuscular weakness. The long-term goal of treatment is to achieve repletion of total body potassium to normal levels. Supplementation of [K+] is the principal treatment for hypokalemia and is achieved with the administration of potassium chloride or potassium phosphate. In general, plasma [K+] decreases by approximately 0.3 mEq/L for each 100 mEq decrease in total body K+. This relationship is more difficult to estimate when serum [K+] is less than 2 mEq/L.42 K+ replacement should be given orally except when severe hypokalemia is associated with respiratory or cardiac instability, in which case the IV route is recommended. Intravenous administration of K+ should not exceed 20 mEq/h to minimize possible iatrogenic hyperkalemia. For infusion of K+, an infusion pump and continuous cardiac monitoring are mandatory. In the case of life-threatening arrhythmias due to severe hypokalemia, more rapid infusion into a central vein may be appropriate. In these rare circumstances, KCl should be diluted to 10 mEq per 100 mL of infusion fluid. In most cases, oral supplementation of K+ is preferred because this route is safer and produces a more gradual increase in serum [K+]. Because supplementation of K+ is usually not an emergency, it is best accomplished using moderate doses of KCl (20 to 40 mEq once or twice a day) over several days. Potassium phosphate is used when hypophosphatemia is also present (as in diabetic ketoacidosis); occasionally, potassium bicarbonate is used in the setting of metabolic acidosis and hypokalemia. However, for most cases of hypokalemia, KCl is the salt of choice for replacement of K+. Serum [K+] should be followed closely, especially when using IV or higher doses, to prevent the development of hyperkalemia. If magnesium levels are low, they should be corrected because hypomagnesemia promotes renal loss of K+, making correction of hypokalemia more difficult. Finally, prevention of further episodes should be addressed with proper K+ intake and supplementation in patients with a continuous cause for hypokalemia. Nurse-driven protocols for electrolyte (potassium) supplementation have been shown to be effective in preventing hypokalemia in patients admitted to the ICU.48
Key Points
Weisberg LS, Weisberg LS. Management of severe hyperkalemia. Crit Care Med. 2008;36:3246-3251.
Buckley MS, Leblanc JM, Cawley MJ. Electrolyte disturbances associated with commonly prescribed medications in the intensive care unit. Crit Care Med. 2010;38(Suppl):S253-S264.
Adrogue HJ, Madias NE. Changes in plasma potassium concentration during acute acid-base disturbances. Am J Med. 1981;71:456-467.
Montague BT, Ouellette JR, Buller GK. Retrospective review of frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330.
1 Acker CG, Johnson JP, Palevsky PM, Greenberg A. Hyperkalemia in hospitalized patients: Causes, adequacy of treatment, and results of an attempt to improve physician compliance with published therapy guidelines. Arch Intern Med. 1998;158:917-924.
2 Sedlacek M, Schoolwerth AC, Remillard BD. Electrolyte disturbances in the intensive care unit. Semin Dial. 2006;19(6):496-501.
3 Ong YL, Deore R, El-Agnaf M. Pseudohyperkalaemia is a common finding in myeloproliferative disorders that may lead to inappropriate management of patients. Int J Lab Hematol. 2010;32(1 Pt 1):e151-e157.
4 Kupin WL, Narins RG. The hyperkalemia of renal failure: Pathophysiology, diagnosis and therapy. Contrib Nephrol. 1993;102:1-22.
5 DeFronzo RA. Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney Int. 1980;17:118-134.
6 Buckley MS, Leblanc JM, Cawley MJ. Electrolyte disturbances associated with commonly prescribed medications in the intensive care unit. Crit Care Med. 2010;38(Suppl):S253-S264.
7 Rimmer JM, Horn JF, Gennari FJ. Hyperkalemia as a complication of drug therapy. Arch Intern Med. 1987;147:867-869.
8 Effectiveness of spironolactone added to an angiotensin-converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (the Randomized Aldactone Evaluation Study [RALES]). Am J Cardiol. 1996;78:902-907.
9 Bakris GL, Weir MR. Angiotensin-converting enzyme inhibitor–associated elevations in serum creatinine: Is this a cause for concern? Arch Intern Med. 2000;160:685-693.
10 Noroian G, Clive D. Cyclo-oxygenase-2 inhibitors and the kidney: A case for caution. Drug Saf. 2002;25:165-172.
11 Clive DM, Stoff JS. Renal syndromes associated with nonsteroidal anti-inflammatory drugs. N Engl J Med. 1984;310:563-572.
12 Gheno G, Savarino C, Vellar S, Cinetto L. Heparin-induced life-threatening hyperkalemia. Ann Ital Med Int. 2002;17:51-53.
13 Thomas CM, Thomas J, Smeeton F, et al. Heparin-induced hyperkalemia. Diabetes Res Clin Pract. May 2008;80(2):e7-e8.
14 Gonzalez-Martin G, Diaz-Molinas MS, Martinez AM, Ortiz M. Heparin-induced hyperkalemia: A prospective study. Int J Clin Pharmacol Ther Toxicol. 1991;29:446-450.
15 Gennari FJ. Disorders of potassium homeostasis: Hypokalemia and hyperkalemia. Crit Care Clin. 2002;18:273-288.
16 Fudickar A, Berthold B, Tonner PH. Propofol infusion syndrome in anaesthesia and intensive care medicine. Curr Opin Anaesthesiol. 2006;19:404-410.
17 Mali AR, Patil VP, Pramesh CS, Mistry RC. Hyperkalemia during surgery: is it an early warning of propofol infusion syndrome? J Anesth. 2009;23(3):421-423.
18 Burnell JM, Scribner BH, Uyeno BT, Villamil MF. The effect in humans of extracellular pH change on the relationship between serum potassium concentration and intracellular potassium. J Clin Invest. 1956;35:935-939.
19 Adrogue HJ, Madias NE. Changes in plasma potassium concentration during acute acid-base disturbances. Am J Med. 1981;71:456-467.
20 Goldfarb S, Cox M, Singer I, Goldberg M. Acute hyperkalemia induced by hyperglycemia: Hormonal mechanisms. Ann Intern Med. 1976;84:426-432.
21 Arrizabalaga P, Montoliu J, Martinez Vea A, et al. Increase in serum potassium caused by beta-2 adrenergic blockade in terminal renal failure: Absence of mediation by insulin or aldosterone. Proc Eur Dial Transplant Assoc. 1983;20:572-576.
22 Gronert GA, Gronert GA. Succinylcholine-induced hyperkalemia and beyond. 1975. Anesthesiology. Dec 2009;111(6):1372-1377.
23 Martyn JA, Richtsfeld M. Succinylcholine-induced hyperkalemia in acquired pathologic states: etiologic factors and molecular mechanisms. Anesthesiology. Jan 2006;104(1):158-169.
24 Josephson GW. Digoxin intoxication and hyperkalemia. JAMA. 1980;244:1557-1558.
25 Rees SM, Nelson L. Digoxin, hyperkalemia, and kidney failure. Ann Emerg Med. 1997;29:694-695. author reply, 696-697
26 Slovis C, Jenkins R. ABC of clinical electrocardiography: Conditions not primarily affecting the heart. BMJ. 2002;324:1320-1323.
27 Schim van der Loeff HJ, Strack van Schijndel RJ, Thijs LG. Cardiac arrest due to oral potassium intake. Intensive Care Med. 1988;15:58-59.
28 Montague BT, Ouellette JR, Buller GK. Retrospective review of frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324-330.
29 Szerlip HM, Weiss J, Singer I. Profound hyperkalemia without electrocardiographic manifestations. Am J Kidney Dis. 1986;7:461-465.
30 Dodge HT, Grant RP, Seavey PW. The effect of induced hyperkalemia on the normal and abnormal electrocardiogram. Am Heart J. 1953;45:725-740.
31 Weisberg LS, Weisberg LS. Management of severe hyperkalemia. Crit Care Med. 2008;36(12):3246-3251.
32 Bower JOMH. The additive effect of calcium and digitalis: A warning with a report of two deaths. JAMA. 1936;106:1151-1153.
33 Garcia-Palmieri MR. Reversal of hyperkalemic cardiotoxicity with hypertonic saline. AM Heart J. 1962;64:483-488.
34 Allon M, Takeshian A, Shanklin N. Effect of insulin-plus-glucose infusion with or without epinephrine on fasting hyperkalemia. Kidney Int. 1993;43:212-217.
35 Allon M, Dunlay R, Copkney C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426-429.
36 Blumberg A, Weidmann P, Ferrari P. Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure. Kidney Int. 1992;41:369-374.
37 Cheng ES, Stringer KM, Pegg SP. Colonic necrosis and perforation following oral sodium polystyrene sulfonate (Resonium A/Kayexalate in a burn patient). Burns. 2002;28:189-190.
38 Gerstman BB, Kirkman R, Platt R. Intestinal necrosis associated with postoperative orally administered sodium polystyrene sulfonate in sorbitol. Am J Kidney Dis. 1992;20:159-161.
39 Naparstek Y, Gutman A. Case report: Spurious hypokalemia in myeloproliferative disorders. Am J Med Sci. 1984;288:175-177.
40 Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol. 2007;18(10):2649-2652.
41 Ben Salem C, Hmouda H, Bouraoui K. Drug-induced hypokalaemia. Curr Drug Saf. 2009;4(1):55-61.
42 Gennari FJ. Hypokalemia. N Engl J Med. 1998;339:451-458.
43 Shannon M. Hypokalemia, hyperglycemia and plasma catecholamine activity after severe theophylline intoxication. J Toxicol Clin Toxicol. 1994;32:41-47.
44 Sigue G, Gamble L, Pelitere M, et al. From profound hypokalemia to life-threatening hyperkalemia: A case of barium sulfide poisoning. Arch Intern Med. 2000;160:548-551.
45 Tonnesen E. Delirium tremens and hypokalemia. Lancet. 1982;2:97.
46 Sillen A, Sorensen T, Kantola I, et al. Identification of mutations in the CACNL1A3 gene in 13 families of Scandinavian origin having hypokalemic periodic paralysis and evidence of a founder effect in Danish families. Am J Med Genet. 1997;69:102-106.
47 Schulman M, Narins RG. Hypokalemia and cardiovascular disease. Am J Cardiol. 1990;65:4E-9E. discussion, 22E-23E
48 Kanji Z, Jung K. Evaluation of an electrolyte replacement protocol in an adult intensive care unit: a retrospective before and after analysis. Intensive Crit Care Nurs. 2009;25(4):181-189.
