[level-membership-for-neurology-category]
Chapter 7 Huntington’s disease
Introduction
Inevitably, a chapter in a physiotherapy textbook will concentrate on the physical aspects of the condition, but for most families and carers a much greater problem is the behavioural change which can lead to irritability, aggression, depression or apathy. Professionals dealing with HD patients need to have an understanding of the neuropsychological and neuro-psychiatric aspects of HD which will help with their management of the patient. Over time the needs of the patient and family change, so professionals need to reassess their management. This chapter will describe the genetic and pathological features before going on to describe the stages of the condition and the associated physiotherapy management. A number of useful texts are available for a wider discussion of the condition including Bates et al. (2002), Quarrell (2009) and Walker (2007).
Prevalence and aspects of natural history
Most UK studies quote a prevalence of 4–10 per 100 000, with similar results from studies from Europe and the US, and low prevalence figures reported from Japan and Finland (Harper, 2002). Whilst HD may be considered a rare disorder it must be remembered that for every affected person there are approximately twice that number who have the mutation and are currently asymptomatic (Conneally, 1984). The median duration of HD is between 16 and 21 years (Foroud et al., 1999; Roos et al., 1993) so this represents a considerable demand on families and carers.
HD can develop at almost any age from under 20 years (and occasionally under 10 years) to over 75 years, but most people develop the condition between the ages of 35 and 55 years, which is after the usual years of reproduction (Harper et al., 1988). One consequence of this is that the 50% risk for an asymptomatic person does not decline appreciably until s/he is going through middle age.
Individuals with an age of onset less than 20 years are described as having the juvenile form of HD. It is difficult to know precisely how many patients have early onset, but 5% is a reasonable estimate. Young people with juvenile HD may develop problems with speech, bradykinesia and dystonic movements earlier in the course in the illness; this topic is reviewed in Quarrell et al. (2009).
Genetics
As HD is a dominant disorder; each cell contains one normal copy and one abnormal copy of the gene. The gene for HD was cloned in 1993 (Huntington’s Disease Collaborative Research Group, 1993) and the protein for which it codes was named huntingtin. The first part of the HD gene has a sequence CAGCAG…CAG which is repeated a number of times. CAG codes for the amino acid glutamine so that the huntingtin protein contains a sequence called a polyglutamine repeat. The mutation causing HD is an expansion of the number of CAG repeats in the gene and therefore of the polyglutamine repeat in the protein. Up to 35 repeats would be considered normal, although there is a possibility that if a person has a repeat size at the upper end of this range it could expand into the pathological range in future generations; a result between 36 and 39 repeats is abnormal, but it is possible that a person may develop the condition late or even live a full life and not develop HD; 40 or more repeats is considered definitely abnormal (ASHG/AMCG statement, 1998). At a mathematical level, there is a correlation between the average age of onset and the CAG repeat size but there is such a wide variation that it is not possible to predict the age of onset for an individual from knowing the CAG repeat size.
An asymptomatic individual with an affected parent has a prior probability of 50% of inheriting the HD gene. Such a person could have a genetic test at this stage. This is called a predictive test. As there is no effective treatment to alter the natural history of HD such tests are used cautiously in conjunction with counselling and according to widely accepted international guidelines drawn up by an ad hoc committee of the International Huntington’s Association/World Federation of Neurology Research Group on Huntington’s chorea (1994). Less than 20% of adults at risk of HD choose to have a predictive test (Tassiker et al., 2009).
Genetics
Pathology
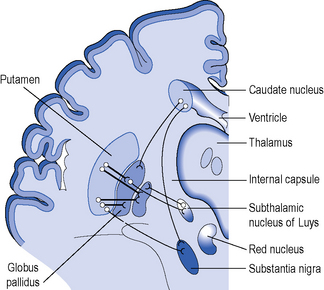
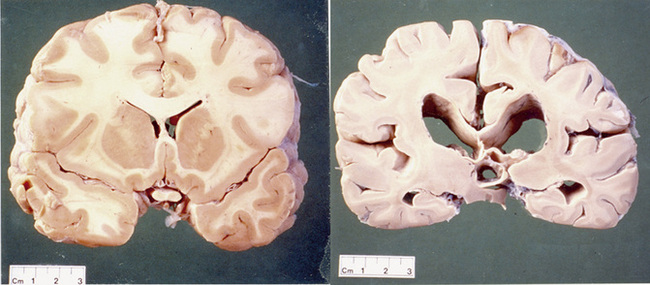
The gene is widely expressed in the cells of the body but some cells within the brain are very sensitive to abnormal huntingtin and there is selective neurodegeneration. An explanation for the selective neurodegeneration characteristic of HD is unclear. The most striking cell loss occurs in the basal ganglia but other areas of the brain including the cortex are also affected. The anatomy of the basal ganglia is shown in Figure 7.1. To emphasize this point it should be noted that the brain of a patient with HD will be smaller and weigh less than that of an age-matched control. The efferent medium spiny neurons within the caudate and putamen nuclei, which are collectively called the striatum, are especially sensitive to the abnormal huntingtin protein (see Figure 7.2).
Implications of pathology for understanding the movement disorder
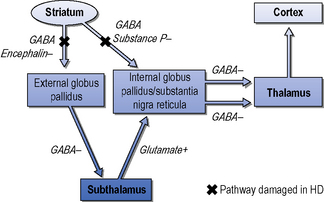
The striatum receives excitatory inputs from the cortex. The efferent medium spiny neurons contain inhibitory neurotransmitters which project to the internal globus pallidus via direct and indirect pathways (Albin et al., 1989). Figure 7.3 shows a simplified diagram of the process. In broad terms, damage to the indirect pathway leads to overstimulation of the thalamo-cortical feedback and chorea (random purposeless movements), whereas loss of the direct pathway results in increased inhibition of the thalamus and less activity of the thalamo-cortical feedback producing bradykinesia and rigidity. Although both pathways degenerate, the balance between them is disturbed. The medium spiny neurons of both the direct and indirect pathways contain dopamine receptors, which are consistently reduced in Huntington’s disease. Conventionally, excitatory D1 receptors are on the direct and inhibitory D2 receptors are on the indirect pathway. This means that whilst it is possible to control the chorea of patients with dopa blocker and dopa depleting drugs, this may worsen the bradykinesia and dystonia.
Understanding pathology at the level of the cell
HD should be easy to explain because one gene is involved with one mutation that affects a single protein. Unfortunately, this is not the case because huntingtin is involved in many cellular processes. Apart from man, no natural animal develops HD. Following the identification of the gene a tremendous research effort involved developing animal models. The first of these, and most studied, was the R6 mice which contain multiple copies of the first part of the HD gene (Mangiarini et al., 1996). These mice die before they develop significant cell death. One feature of the cells is that they contain aggregates of the first part of the huntintin protein (Davies, et al., 1997). These aggregates are now recognized as occurring in human HD brains (DiFiglia et al., 1997). It is unclear as to whether these aggregates are directly related to the cellular pathology or represent a defence mechanism within the cell. There are a number of different animal models; details of their similarities and differences are not relevant to this chapter, but animal models have enabled an understanding that neuronal cells are dysfunctional (‘sick’) before they die which gives hope for interventions to alter the natural history of the disease. This natural history is modelled in Figure 7.4 with neuronal dysfunction preceding the onset of symptoms. In the absence of serendipity, an understanding of the pathology at the cellular level using animal and cellular models is the way forward for developing and subsequently testing new treatments for HD.
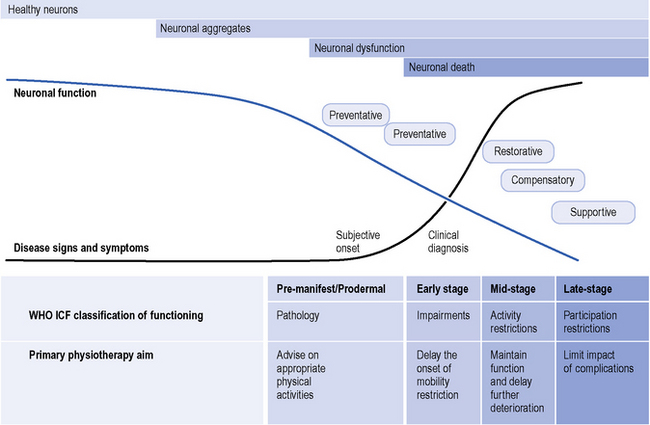
Figure 7.4 Primary physiotherapy aims and the continuum of Huntington’s disease.
(Modified from the Walker et al., Huntington’s Disease. Lancet 2007;369 (9557): 218-28, with permission from Elsevier.)
Pathology
Life history of the disease
If a person has inherited the gene there will be a period of time where she/he is completely asymptomatic. In time, the neurones become dysfunctional. Onset of HD is insidious with non-specific problems such as mood change or being slightly forgetful, which can be attributed easily to other mundane causes. A person in this prodromal stage may become depressed, but a clinical diagnosis cannot be made confidently until motor signs, such as chorea, appear. These may be infrequent, of low amplitude and may not be noticed by the patient, family or clinician with limited experience of the condition. Indeed even at a time when the chorea is quite obvious it may be denied as a problem by the patient (Snowden et al., 1998).
If a patient is seen when there are obvious signs of HD, the CAG repeat size may be measured from a venous blood sample to confirm the clinical diagnosis. It is important to realize that the result of the genetic test will be the same whether the individual is completely asymptomatic, in the prodromal phase, or in the early, middle or later stages of HD. As with any test the interpretationof the test result depends upon the clinical context. This raises an important issue about the genetic test: it is a ‘trait marker’ but says nothing about the state of the illness. There is a significant research effort to determine other laboratory biomarkers (‘state markers’) which can track the development and progression of the disease (Henley et al., 2005). The boundaries between early, middle and late stages are ill defined, but nonetheless they form a useful way of categorizing the impact of HD.
Clinical assessment
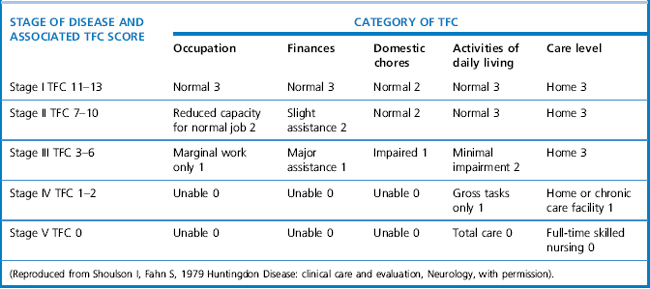
The Unified Huntington’s Disease Rating Scale (UHDRS) (Huntington Study Group, 1996) is a rating system that is used to quantify the severity of Huntington’s disease. It was developed as a clinical rating scale to assess four domains of clinical performance and capacity in individuals with HD: motor function, cognitive function, behavioural abnormalities, and functional capacity. These scores can be calculated by summing the various questions of each section. The UHDRS is useful for tracking changes in the clinical features of HD over time, and appears to be appropriate for repeated administration during clinical studies. Stages in Huntington’s disease are traditionally defined by the Total Functional Capacity scale, developed by Shoulson and Fahn (1979), which classifies patients into one of five stages of disease progression (Marder et al., 2000). The staging evaluates patients based on functioning for ADLs, domestic chores, finances, work and overall care level. Mid-stage is typically defined as levels II-III (see Table 7.1).
Stage I (Early stage)
In the early stages of the disease, the person with HD is able to remain functional and independent, retaining their ability to work and drive (HDSA, 2000). Movement disorders include minor involuntary movements, reduced coordination and linearly progressive motor impersistence or inability to maintain a voluntary muscle contraction at a constant level (Walker, 2007). Even at this stage, it may be possible to detect some slowing of saccadic eye movements (rapidly moving the eye to a new target; this is done by asking the patient to move his/her eye to one corner in response to a cue). Voluntary motor tasks (fine and gross motor skills) may become increasingly difficult as the disease progresses (Aubeeluck & Wilson, 2008). Cognitive tasks also become more difficult with reduced ability to sequence and organize information (HDSA, 2000). Deficits in gait have been identified across the spectrum of HD (Churchyard et al., 2001; Grimbergen et al., 2008; Hausdorff et al., 1998; Koller & Trimble, 1985; Rao et al., 2008). Decreased gait velocity, stride length, and increased time in double support compared to healthy controls may occur (Rao et al., 2008). These impairments appear to worsen with increasing disease severity and can be useful markers of disease progression and treatment efficacy.
Stage II and III (Middle stage)
During the middle stages of HD, involuntary movements such as chorea increase. In the early stages of HD, this is typically seen in the fingers, hands and face muscles, but as the disease progresses, it can be seen throughout thebody, including in all four extremities and the trunk. Chorea does not usually result in a direct impairment of motor function, however voluntary motor impairments may have a functional impact (Walker, 2007).
Dystonic postures (abnormal, sustained posturing) in sitting or standing (Louis et al., 1999) and in movements such as shoulder elevation, foot inversion and supination, and trunk extension might be present. People may experience frequent loss of balance and falls, which interfere with their ability to walk and maintain an upright position (Busse et al., 2009). Common tasks during which patients demonstrate balance problems include those where the base of support is decreased; tandem standing and walking, dual tasks, eyes closed and in response to external perturbations (Bilney et al., 2003b; Quinn & Rao, 2002). Falls are frequent in people with HD; 60% of people with early to mid-stage HD in two separate studies (n=45 & n=24) reported two or more falls in the previous 12 months (Busse et al., 2009; Grimbergen et al., 2008). Falls can have physical and psychological consequences (Myers et al., 1996). The fear of future falls may be a more pervasive problem than falls per se, as fear of falls may lead to people restricting their physical activities. Self-imposed restriction of daily activities leads to immobility and consequently osteoporosis, reduced fitness and social isolation (Grimbergen et al., 2004). Individuals with HD have also been found to have lower-limb muscular weakness compared to age-matched controls (Busse et al., 2008a). Strength can be measured clinically by manual muscle testing or by functional observation of strength during task performance (e.g. stair climbing or evidence of gait deviation during walking).
Stage IV and V (Late stage)
In the advanced stages of the disease motor symptoms continue to progress, severely limiting mobility. Choreic and dystonic movement may further increase (see Figure 7.4), but involuntary movements are often overshadowed by Parkinsonian symptoms (Aubeeluck & Wilson, 2008; HDSA, 2000) such as bradykinesia or slowness of movement. People with HD can also experience pain, particularly in the later stage of the disease. The source of this pain can be unknown; however, dystonia or muscle imbalances can often cause musculoskeletal pain. Excessive chorea can cause pain if people injure themselves by hitting their arm or leg into an object or hard surface.
Weight loss can occur in part due to impaired swallowing function (Aubeeluck & Wilson, 2008; HDSA, 2000). People may no longer be able to work or drive and will need assistance when performing some activities of daily living (ADL) (HDSA, 2000).
Even though people with HD appear to be in constant movement, their underlying volitional movements during task performance have been found to be slower than healthy controls for reaching (Quinn et al., 2001) and ambulation (Delval et al., 2006; Delval et al., 2007). Bradykinesia can typically be evaluated by measuring time to complete a task (movement time). Akinesia or delayed initiation of movement is also seen. In research studies, akinesia is typically measured by reaction time. In the clinical setting, it is often difficult to quantitatively evaluate akinesia, but delays in onset of movement for various tasks can be noted. Impaired speech results in difficulties communicating, and cognitive and psychiatric deterioration may continue, but it is thought that patients retain some comprehension (HDSA, 2000). At this stage most people will require assistance in all aspects of daily living, relying fully on nursing care (HDSA, 2000). Despite the severity of the neurological disorder, the primary causes of death in HD are aspiration pneumonia, cardiovascular disease and complications from falls (Sorensen & Fenger, 1992; Walker, 2007).
Physiotherapy assessment
The physiotherapy aims should be both anticipatory and responsive to the disease stage (see Figure 7.4) with interventions to assist HD patients to maintain independence, functional capacity and participation in society (Bilney et al., 2003a).
The World Health Organization International Classification of Functioning, Disability and Health (ICF) (WHO, 2001) is useful as an aid in structuring assessment of a person’s functioning and participation (see Figure 7.5), and in allowing consideration of the triad of motor, cognitive and psychiatric symptoms that are often seen in HD. The ICF can be used for functional status assessment, goal setting and treatment planning, and focuses on aspects of a person’s health and health-related wellbeing in terms of activities and participation, i.e. the description of the tasks (activities) and/or life situations (participation) in which the person wishes to engage, and the impact that impaired body function or structure is having on these aspects. The ICF further takes into consideration the contextual factors (both environmental and personal), which may impact on a person’s life, and the relationships with body function/structure, activities and participation. The specific impairments and associated activities and participations that may need consideration when assessing a person with HD are listed in Table 7.2. Documentation of the impact of the noted impairments on functional abilities, activities and participation is important to help highlight specific factors that contribute to a patient’s functional problems and inform the clinical reasoning process in terms of maximizing functional abilities. Qualifiers of the degree of impairment, and the performance and capacity of activities and participation enable essential documentation of disability and health status.
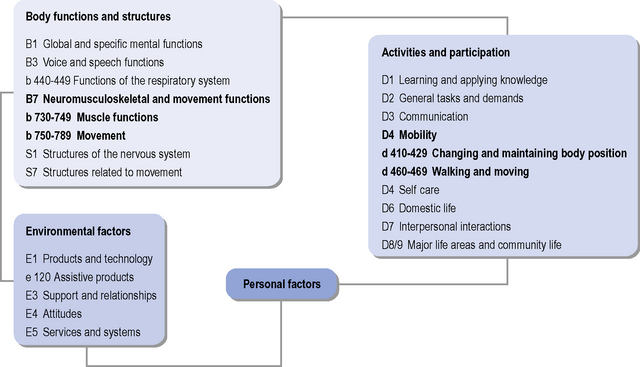
Table 7.2 Specific impairments and associated activities and participations that may need consideration when assessing a person with Huntington’s disease
| Impairments in Body Structure and Function | |
|---|---|
| B1 Global and specific mental functions B3 Voice and speech functions b 440-449 Functions of the respiratory system B7 Neuromusculoskeletal and movement functions b 730-749 Muscle functions b 750-789 Movement functions S1 Structures of the nervous system S7 Structures related to movement |
Cognitive-perceptual: Memory Executive functions Spatial abilities Neuromuscular: Chorea Dystonia Strength Flexibility |
| Functional Abilities | |
|---|---|
| Activities and participation | |
| D1 Learning and applying knowledge D2 General tasks and demands D3 Communication D4 Mobility d 410-429 Changing and maintaining body position d 460-469 Walking and moving D5 Self care D6 Domestic life D7 Interpersonal interactions D8/9 Major life areas and community life For each functional activity, therapists should evaluate and document the level of assistance or caregiver burden needed to complete the task, and also determine the patient’s skill level (e.g. time to complete task, consistency of task performance, etc). This section can include any of the following skills, as appropriate: self-care and home management including activities of daily living (bed mobility, dressing, self care, toileting, bathing, eating, cooking, preparing meals and instrumental activities of daily living) Listing of specific skills assessed (e.g. ambulation, sitting ability, standing ability, sit to stand, wheelchair skills, stair climbing, feeding, bed mobility). Therapists should evaluate and document the functional performance of skills that are pertinent to the person with HD’s independence. |
Speech Feeding, drinking (swallowing, sucking) Bathing, toileting, dressing Household chores Cooking, cleaning Walking (distance, speed, shoes) |
| Environmental Factors | |
|---|---|
| E1 Products and technology e 120 assistive products E3 Support and relationships E4 Attitudes E5 Services and systems For each functional activity, therapists should evaluate and document the level of assistance or caregiver burden needed to complete the task, and also determine the patient’s skill level (e.g. time to complete task, consistency of task performance, etc.). This section can include any of the following skills, as appropriate: Assistive and adaptive devices Environmental, home and work (job/school) barriers |
Equipment: Assistive devices, feeding drinking, dressing equipment Wheelchair, brace, walker |
Specific coding for the domains are shown.
The subjective assessment allows the physiotherapist to evaluate the patient’s presenting problem(s), in the contextof their past medical history and how this may affect their ability to live independently and within society. This also provides the opportunity to build a therapeutic, collaborative relationship, and facilitate realistic goal setting. It must be noted that the patient’s caregiver(s) should be involved, particularly if the patient has difficulties communicating as a result of cognitive or physical impairment. The objective assessment should include assessment of neuromuscular, musculoskeletal (posture, range of motion, pain and muscle strength), cardio-respiratory and cardio-vascular impairments. An overview of the HD specific considerations for the physiotherapy assessment is shown in Table 7.3. These are useful to aid clinical reasoning and goal setting but are not necessarily outcomes that are sensitive to change over time. There are numerous outcome measures that have potential utility in the assessment of efficacy of physiotherapy interventions for people with HD. Measurement tools that have been used in previous HD-related studies are presented in Table 7.4 and are classified according to the WHO ICF. The Berg Balance Scale, Timed Up and Go Test and the Functional Reach Test have all been found to be valid and responsive clinical measures in HD and are considered useful to detect those at risk of falls (Busse et al., 2009; Grimbergen et al., 2008; Rao et al., 2009).
Table 7.3 Specific assessment points for consideration in Huntington’s disease
| Subjective Assessment |
|---|
| Patient information and demographics Current condition: concerns that led the patient to seek services of physiotherapist; current and prior therapeutic interventions; current stage of Huntington’s disease (HD) and age of onset Past medical history: prior hospitalizations, surgeries and pre-existing medical and other health-related conditions; family history Medications: list any medications taken for HD symptoms as well as for other medical conditions Home environment, i.e. living environment and community characteristics; family and living situation; family and caregiver resources; assistive devices and equipment Employment/work, i.e. current work situation and requirements Health status, i.e. prior functional status in self care and home management, including activities of daily living (ADLs) and instrumental activities of daily living (IADLs). Community, leisure and social activity participation General health perception/quality of life Physical function (e.g. mobility, sleep patterns, restricted bed days) Psychological function (e.g. memory, reasoning ability, depression, anxiety) Behavioural health risks (e.g. smoking, drug abuse) Level of physical fitness |
| Objective Assessment | Specific Assessment Points |
|---|---|
| Posture: Head/neck/trunk, upper extremities and lower extremities | Standing and sitting |
| Respiratory function: Lung function tests may highlight obstructive or restrictive disorders of the respiratory system. Measurement of vital capacity in supine and upright positions can identify weakness of the diaphragm (American Thoracic Society, 2002). Forced expiratory Volume in 1 second (FEV1), Forced Vital Capacity (FVC), FEV1/FVC ratio, Peak Expiratory Flow Rate (PEFR) should also be considered using standardized spirometry techniques (Miller et al., 2005). |
Ausculatation Breathing patterns Cough Exercise tolerance |
| Skin integrity | Decubiti, abrasions, open wounds |
| Pain | Location, type, pain scale rating |
| Sensation | Location, type |
| Reflexes | Clonus Babinski |
| Range of motion: Head/neck/trunk, upper extremities and lower extremities | Active and passive movements |
| Movement patterns: Chorea, dystonia, bradykinesia, tremor and rigidity | Consider all affected body segments |
| Mobility: Gait assessment |
Base of support Tonal changes Deviations |
| Endurance |
| Additional Considerations | |
|---|---|
| Behaviour | Combative Labile Impulsive |
| Cooperation | Carry over of new skill from session to session; follows simple, one step directions and follows multiple step directions |
| Safety awareness and problem solving | |
Table 7.4 Classification of outcome measures according to the WHO ICF
| Participation | Activities | Impairments |
|---|---|---|
| The Short Form-36 (Quinn & Rao, 2002; Ware & Sherbourne, 1992) | Self paced and fast paced 10 m walk (Busse et al., 2009; Quinn & Rao, 2002; Watson, 2002) | Berg Balance scale (Berg et al., 1992; Busse et al., 2009; Grimbergen et al., 2008; Quinn & Rao, 2002) |
| Barthel Index (Zinzi et al., 2007) | The Timed Up & Go (TUG) Test (Busse et al., 2009; Podsiadlo & Richardson, 1991; Rao et al., 2009) | Tinetti Balance & Gait (Tinetti, 1986; Zinzi et al., 2007). |
| Physical Performance Test (PPT) (Reuben & Siu, 1990; Zinzi et al., 2007) | ||
| Activities Specific Balance Confidence Scale (Busse et al., 2009; Grimbergen et al., 2008; Powell & Myers, 1995). Modified Falls Efficacy Scale (Quinn & Rao, 2002) | Functional Reach Test (Rao et al., 2009) | |
| Four square step test (Dite & Temple, 2002) |
Rationale for physiotherapy intervention
Physiotherapy is recognized as a health-care profession, which utilizes ‘physical approaches to promote, maintain and restore physical, psychological and social well-being’ (CSP, 2002). The physiotherapist aims to promote quality of life and independence by encouraging activity and providing support within functional tasks (Royal Dutch Society for Physical Therapy (KGNF), 2004). Physiotherapy is also focused on safety and interventions may be aimed at the prevention of falls (KGNF, 2004).
The beneficial role of physiotherapy within basal ganglia disorders has been previously illustrated within Parkinson’s disease (PD), with two recent systematic reviews reporting that physiotherapy can improve multiple factors including physical functioning, health-related quality of life (HR-QoL), strength, balance and gait (Goodwin et al., 2008; Nieuwboer et al., 2007). The literature in support of physiotherapy for people with HD is, however, less clear. Two systematic reviews have noted that there is a small amount of evidence supporting physiotherapy within HD, but this is somewhat overshadowed by poor methodological rigour, small sample sizes, unclear selection criteria resulting in potential heterogeneity in participant groups, and a lack of follow-up (Bilney et al., 2003b; Busse & Rosser, 2007). More supportive evidence is continually becoming available. For example, a before-after trial with a sample size of 40 found an intensive rehabilitation programme of six sessions per week, held over 3 weeks demonstrated an improvement in motor function over a 2-year period (Zinzi et al., 2007). The positive findings from environmental enrichment studies in mice provides support for active interventions, such as physiotherapy, in people with HD. Mice with HD, placed within an environment providing physical, mental and social stimulation, have a slower disease progression, and maintain motor function for longer (Dobrossy & Dunnett, 2005; Hockly et al., 2002). Enhanced voluntary physical activity (in the pre-symptomatic stages) has also been found to contribute to positive effects of environmental stimulation (van Dellen et al., 2008).
Based on these studies and the biological rationale, there is a place for physiotherapy within the management of people with HD, although in-depth efficacy studies are still required. Small (n=1–10) studies imply that cognitive strategies, external cueing techniques, balance training as well as a combination of muscle strengthening, stretching and cardiovascular exercise are recommended to be appropriate interventions (Bilney et al., 2003b; Churchyard et al., 2001).
Principles and practice of physiotherapy intervention
In terms of a general strategy for physiotherapy interventions, it is important to consider HD as a spectrum; physiotherapy management of people with HD should be modified according to individual problems and to the stage of the disease (Busse et al., 2008b).
Referral and assessment should take place early in the disease; `or even in the pre-manifest stage (Busse et al., 2008b). Current intervention focuses on symptomatic management. However, there is increasing support for early intervention where an impact may be made on biological processes with the potential to influence the natural history of the condition. In addition, early referral to physiotherapy for people with HD may be beneficial in a number of ways. It enables practitioners to ascertain a baseline for the person with HD, supports the establishment of a therapeutic relationship between the person with HD, practitioner and caregivers, and ensures early intervention to try and maintain mobility and function for as long as possible (Busse et al., 2008b).
The main goals of physiotherapy intervention will generally change over time; interventions will initially be preventative, and gradually may become restorative (see Figure 7.4). Management during the different stages are outlined below and illustrated further by the Case Histories.
Early and middle stage management
The focus of gait training should be on identifying those aspects of gait which are functionally limiting (Rao et al., 2008), and then designing an intervention plan that is aimed at ameliorating or compensating for the gait impairments, and providing training so that the patient can reach their ambulation goals. The influence of the practice environment on learning and carryover, and interaction with the environment directly affects the movement that emerges (Bassile & Bock, 1995). Structuring practice of walking within an environment that is realistic to the current life situation of the person with HD is likely to yield the best results.
Balance activities should be task specific (Shumway-Cook & Woollacott, 2001). Progression from a wide to narrower base of support, from static to dynamic activities, from a low to high centre of gravity, and increasing the degrees of freedom that must be controlled can be considered; however, the key principle is that the balance demand of a specific task should be assessed and addressed. Task-specific training is particularly appropriate for people with movement disorders (Bilney et al., 2003a, 2003b) because motor disturbances are typically context-dependent, and are typically seen in complex, well-learned tasks such as walking and reaching. People should be taught to deliberately prepare in advance for forthcoming threats to balance, or to focus their attention on maintaining balance before a task in which equilibrium is challenged is initiated. It is thought this strategy allows people to use frontal cortical systems to regulate stability, rather than rely upon the impaired basal ganglia mechanisms (Bilney et al., 2003a). Training the person with HD to step in response to perturbations, with an emphasis on speed and accuracy of the stepping strategy, is also recommended. Balance training may be enhanced through the use of auditory and visual cueing. To address inability to attend to more than one task at a time, interventions could include instruction to attend to one task at a time (Delval et al., 2008b). People at high risk of falls should be taught to break down complex activities into simple tasks and to focus their attention on performing each task separately; alternatively, having people with HD practise doing two activities at the same time under various practice and context conditions, may be a beneficial component to balance training (Delval et al., 2008b).
A major aim for therapeutic intervention in HD is for therapists to teach people with HD how to effectively bypass damaged basal ganglia structures and use frontal neural pathways to control movement (Bilney et al., 2003a, 2003b). This involves accessing the motor system by the patient responding to a visual or auditory input (which occurs through frontal pathways), rather than self-initiating movement (which occurs through the basal ganglia-cortical pathway). Sensory cueing involves the use of augmented sensory information, typically in the form of external visual, auditory or manual cueing, to improve performance in a task. There is some controversy as to whether individuals with HD can benefit from rhythmic auditory cues to improve gait characteristics (Delval et al., 2008a; Thaut et al., 1999). Synchronizing gait to rhythmic cues from a metronome (but not music) has been shown to assist the modulation of gait speed in people with HD (Thaut et al., 1999); however, under dual task conditions metronome cues may be less helpful due to attentional deficits in people with HD (Delval et al., 2008a).
Later stage management
As the disease progresses, compensatory strategies such as using sensory cues or attentional strategies can be implemented. Assistive devices (walker with wheels is preferred) and safety equipment, such as a helmet or elbow and knee protectors, may be recommended. Because cognitive impairments also increase in number and severity as the disease progresses, additional compensatory strategies, such as providing cues with goal-directed feedback, teaching skills using one-step commands and providing treatment in a quiet, non-distracting environment should also be considered. Practice should take place through repetition, allowing sufficient time for the person with HD to understand what is required of them and with key points continually being reinforced (Rao et al., 2005).
Assessment of the home, work and community environments in which the person with HD must function should be conducted and modifications made accordingly; in addition, prescription of assistive devices, home-adaptive equipment or modifications may be required. It is critical that the physiotherapist considers the nature of the disease process when considering devices and adaptive equipment. As functional status may be constantly changing, assessment should incorporate both the immediate and the longer-term need. It is important for the therapist to consider equipment that is adaptable to a patient’s changing condition. Standard walkers and four-point canes are difficult to manoeuvre and can interfere with natural gait pattern. As balance impairments progress in the middle stages of the disease, a rollator walker with four swivel wheels, and either hand or push-down breaks is often recommended. Walking sticks can be hazardous for patients with excessive choreic movements. Specialized seating needs should be considered; this may include increased seat-back height and depth, tilt and appropriate foot support. Hard surfaces and edges of assistive devices and wheelchairs should be protected with padding where necessary. Choosing the right kind of adaptive equipment is a collaborative process. Balancing independence and safety requires special consideration for each person’s individual needs (Rao et al., 2005). Use of certain devices and equipment, such as those described above, may provide the necessary support to maximize a person’s functional abilities.
Respiratory problems may develop at any stage but are more likely to occur in the late-middle and late stages of the disease when mobility becomes more limited. Rigidity, bradykinesia and dystonia eventually lead to immobility and dependence for all care in people with HD (Ribchester et al., 2004). Immobility and postural changes may result in decreased respiratory function (Lin et al., 2006). As with many progressive neurological conditions, the major cause of death in HD is respiratory failure (Lanska et al., 1988; Sørensen & Fenger, 1992) due to factors such as weakened respiratory muscles and/or swallowing problems.
Respiratory interventions, such as deep breathing exercises, body positioning to optimize ventilation–perfusion,modified postural drainage, and airway clearance techniques may be required for specific respiratory problems (see Ch. 15). Caregivers may be taught assisted coughing techniques and a suction machine should be made available for patients who have difficulty clearing secretions. Educating caregivers regarding the signs of aspiration is also important.
Palliative care
Palliative care services have an important role in helping people with HD and their families to prepare for the later stages of HD (Moskowitz & Marder 2001; Travers et al., 2007). The ultimate aim for palliative care is to achieve the best possible quality of life for patients and their families. To this end, coordinated management of the physical and cognitive effects of disease progression is essential. The physiotherapist can advise on seating and positioning, respiratory management, as well as relaxation techniques. It should be noted that the use of feeding tubes in patients with dementia is controversial (Cervo et al., 2006); a recent report provides a comprehensive review of the issues involved in the use of feeding tubes for patients with neurological disease which should be patient centred and not based on the convenience of staff or carers (Royal College of Physicians and British Society of Gastroenterolgists, 2010). Obtaining a patient’s views of feeding tubes and end-stage care is better discussed and recorded at a time before they lose capacity (Simpson, 2007).
Overall physiotherapy management of the person with Huntington’s disease
Case histories
CASE HISTORY 1: Early-stage Huntington’s disease
The following case history is modified with permission from Smith et al. (2007).
Subjective assessment
Objective assessment
Impairments in body structure and function
CASE HISTORY 2: Home-based physiotherapy in mid-stage Huntington’s disease
The following case history is modified from Quinn and Rao (2002).
Subjective assessment
Patient information and demographics
Medical/surgical history – no significant medical history prior to diagnosis of HD.
Objective assessment
Impairments in body structure and function
Functional abilities
CASE HISTORY 3: Late-stage Huntington’s disease
Subjective assessment
Objective assessment
Treatment planning
Outcome measures and goal setting
American Thoracic Society. ATS/ERS Statement on Respiratory Muscle Testing. Am. J. Respir. Crit. Care Med.. 2002;166:518-624.
The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group Statement (ASHG/AMCG). Laboratory guidelines for Huntington disease genetic testing. The American College of Medical Genetics/American Society of Human Genetics Huntington Disease Genetic Testing Working Group 1998. Am. J. Hum. Genet.. 1998;62(5):1243-1247.
Albin R.L., Young A.B., Penny J.B. The functional anatomy of disorders of the basal ganglia. Trends Neurosci.. 1989;12(10):366-374.
Aubeeluck A., Wilson E. Huntington’s disease. Part 1: essential background and management. Br. J. Nurs.. 2008;17(3):146-151.
Bates G., Harper P., Jones L. Huntington’s Disease, third ed. Oxford: Oxford University Press, 2002.
Bassile C., Bock C. Gait training. In: Craik R., Oatis C., editors. Gait Analysis: Theory and Application. St. Louis: Mosby, 1995.
Berg K.O., Wood-Dauphinee S., Williams J.L., et al. Measuring balance in the elderly: validation of an instrument. Can. J. Public Health. 1992;83(Suppl. 2):S7-S11.
Bilney B.M., Morris M., Denisenko S. Physiotherapy for people with movement disorders arising from basal ganglia dysfunction. NZ. J. Physiother.. 2003;31(2):94-100.
Bilney B.M., Morris M., Perry A. Effectiveness of physiotherapy, occupational therapy, and speech pathology for people with Huntington’s disease: a systematic review. Neurorehabil. Neural. Repair.. 2003;17(1):12-24.
Busse M.E., Hughes G., Wiles C.M., et al. Use of hand-held dynamometry in the evaluation of lower limb muscle strength in people with Huntington’s disease. J. Neurol.. 2008;255(10):1534-1540.
Busse M.E., Khalil H., Quinn L., et al. Physical Therapy Intervention for Patients with Huntington’s disease. Phys. Ther.. 2008;88(7):820-831.
Busse M.E., Rosser A.E. Can directed activity improve mobility in Huntington’s disease? Brain Res. Bull.. 2007;72(2–3):172-174.
Busse M.E., Wiles C.M., Rosser A.E. Mobility and Falls in Huntington’s Disease. J. Neurol. Neurosurg. Psychiatry. 2009;80(1):88-90.
Cervo F.A., Bryan L., Farber S. To PEG or not to PEG. A review of evidence for placing feeding tubes in advanced dementia and the decision-making process. Geriatrics. 2006;61(6):30-35.
Chartered Society of Physiotherapy (CSP). Curriculum Framework for Qualifying Programmes in Physiotherapy. London: Chartered Society of Physiotherapy, 2002.
Churchyard A.J., Morris M., Georgiou N., et al. Gait dysfunction in Huntington’s disease: parkinsonism and a disorder of timing. Implications for movement rehabilitation. Adv. Neurol.. 2001;87:375-385.
Conneally P.M. Huntington disease: genetics and epidemiology. Am. J. Hum. Genet.. 1984;36:506-526.
Davies S.W., Turmaine M., Cozens B.A., et al. Formation of neuronal intranuclear inclusions underlies the neuronal dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537-548.
Delval A., Krystkowiak P., Blatt J.L., et al. Role of hypokinesia and bradykinesia in gait disturbances in Huntington’s disease: a biomechanical study. J. Neurol.. 2006;253(1):73-80.
Delval A., Krystkowiak P., Blatt J.L., et al. A biomechanical study of gait initiation in Huntington’s disease. Gait Posture. 2007;25(2):279-288.
Delval A., Krystkowiak P., Delliaux M., et al. Effect of external cueing on gait in Huntington’s disease. Mov. Disord.. 2008;23(10):1446-1452.
Delval A., Krystkowiak P., Delliaux M., et al. Role of attentional resources on gait performance in Huntington’s disease. Mov. Disord.. 2008;23(5):684-689.
DiFiglia M., Sapp E., Chase K.O., et al. Aggregation of huntingtin in neuronal inclusion and dystrophic neurites in brain. Science. 1997;277:1990-1993.
Dite W., Temple V.A. A clinical test of stepping and change of direction to identify multiple falling older adults. Arch. Phys. Med. Rehabil.. 2002;83(11):1566-1571.
Dobrossy M.D., Dunnett S.B. Optimising plasticity: environmental and training associated factors in transplant-mediated brain repair. Rev. Neurosci.. 2005;16(1):1-21.
Foroud T., Gray J Ivashina J., et al. Differences in duration of Huntington’s disease based on age at onset. J. Neurol. Neurosurg. Psychiatry. 1999;66:52-56.
Goodwin V.A., Richards S.H., Taylor R.S., et al. The effectiveness of exercise interventions for people with Parkinson’s disease: a systematic review and meta-analysis. Mov. Disord.. 2008;23:631-640.
Grimbergen Y., Knol M., Bloem B.R., et al. Falls and gait disturbances in Huntington’s disease. Mov. Disord.. 2008;23(7):970-976.
Grimbergen Y., Munneke M., Bloem B.R. Falls in Parkinson’s disease. Curr. Opin. Neurol.. 2004;17(4):405-415.
International Huntington’s Association/World Federation of Neurology Research Group on Huntington’s chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. J. Med. Genet.. 1994;31:555-559.
Harper P.S. The epidemiology of Huntington’s disease. In: Bates G., Harper P., Jones L., editors. Huntington’s disease 2ed. third ed. Oxford: Oxford University Press; 2002:159-197.
Harper P.S., Quarrell O.W.J., Youngman S. Huntington’s disease: prediction and prevention. Philosophical Transactions of the Royal Society. 1988;319:285-298.
Hausdorff J.M., Cudkowicz M., Firtion R., et al. Gait variability and basal ganglia disorders: stride-to-stride variations of gait cycle timing in Parkinson’s disease and Huntington’s disease. Mov. Disord.. 1998;13(3):428-437.
Henley S.D., Bates G.P., Tabrizi S.J. Biomarkers for neurodegenerative disease. Curr. Opin. Neurol.. 2005;18:698-705.
Hockly E., Cordery P.M., Woodman B., et al. Environmental enrichment slows disease progression in R6/2 Huntington’s disease mice. Ann. Neurol.. 2002;51(2):235-242.
Huntington’s Disease Society of America (HDSA). A Physicians Guide to The Management Of Huntington’s Disease. New York: Huntington’s Disease Society of America, 2000.
Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;26(6):971-983. 72
Huntington Study Group. Unified Huntington’s Disease Rating Scale: reliability and consistency. Mov. Disord.. 1996;11(2):136-142.
Koller W.C., Trimble J. The gait abnormality of Huntington’s disease. Neurology. 1985;35(10):1450-1454.
Lanska D.J., Lavine L., Lanska M.J., et al. Huntington’s disease mortality in the United States. Neurology. 1988;38(5):769-772.
Lin F., Parthasarathy S., Taylor S., et al. Effect of Different SittingPostures on Lung Capacity, Expiratory Flow, and Lumbar Lordosis. Arch. Phys. Med. Rehabil.. 2006;87(4):504-509.
Louis E.D., Lee P., Quinn L., et al. Dystonia in Huntington’s disease: prevalence and clinical characteristics. Mov. Disord.. 1999;14(1):95-101.
Mangarini L., Shasivam K., Seller M., et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493-506.
Marder K., Zhao H., Myers R.H., et al. Rate of functional decline in Huntington’s disease. Huntington Study Group. Neurology. 2000;54(2):452-458.
Miller M., Hankinson J., Brusasco V., et al. Standardisation of Spirometry. Eur. Respir. J.. 2005;26(2):319-338.
Moskowitz C.B., Marder K. Palliative care for people with late-stage Huntington’s Disease. Neurol. Clin.. 2001;19(4):849-865.
Myers A.M., Powell L.E., Maki B.E., et al. Psychological indicators of balance confidence: relationship to actual and perceived abilities. J. Gerontol. A Biol. Sci. Med. Sci.. 1996;51(1):M37-M43.
Nieuwboer A., Kwakkel G., Rochester L., et al. Cueing training in the Home improves gait-related mobility in Parkinson’s disease: the RESCUE trial. J. Neurol. Neurosurg. Psychiatry. 2007;78(2):134-140.
Podsiadlo D., Richardson S. The timed “Up & Go: a test of basic functional mobility for frail elderly persons”. J. Am. Geriatr. Soc.. 1991;39(2):142-148.
Powell L.E., Myers A.M. The Activities-specific Balance Confidence (ABC) Scale. J. Gerontol. A Biol. Sci. Med. Sci.. 1995;50(A1):M28-M34.
Quarrell O. Huntington’s disease – the facts 2ed. Oxford: Oxford University Press, 2009.
Quarrell O., Brewer H.M., Squitieri F., et al. Juvenile Huntington’s Disease and other trinucleotide repeat disorders. Oxford: Oxford University Press, 2009.
Quinn L., Gordon A., Reilmann R., et al. Altered Movement Trajectories and Force control during object transport in Huntington’s disease. Mov. Disord.. 2001;16(3):469-480.
Quinn L., Rao A.K. Physical therapy for people with Huntington disease: current perspectives and case report. Neurology Report. 2002;3:145-153.
Rao A.K., Quinn L., Marder K.S. Reliability of spatiotemporal gait outcome measures in Huntington’s disease. Mov. Disord.. 2005;20(8):1033-1037.
Rao A.K., Muratori L., Louis E.D., et al. Spectrum of Gait impairments in Presymptomatic and Symptomatic Huntington’s disease. Mov. Disord.. 2008;23(8):1100-1107.
Rao A.K., Muratori L., Louis E.D., et al. Clinical measurement of Mobility and Balance impairments in Huntington’s disease: validity and responsiveness. Gait Posture. 2009;29(3):433-436.
Reuben D., Siu A. An Objective measure of Physical function of Elderly outpatients. The Physical Performance Test. J. Am. Geriatr. Soc.. 1990;38(10):1105-1112.
Ribchester R.R., Thomson D., Wood N.I., et al. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur. J. Neurosci.. 2004;20(11):3092-3114.
Roos R.A.C., Hermans J., Vegter-van der Vlis M., et al. Duration of illness of Huntington’s disease is not related to Age at onset. J. Neurol. Neurosurg. Psychiatry. 1993;56:98-100.
Royal College of Physicians and British Society of Gastroenterology. Oral feeding difficulties and dilemmas: A guide to practical care, particularly towards the end of life. London: Royal College of Physicians, 2010.
Royal Dutch Society for Physical Therapy (KGNF). Guidelines for Physical Therapy in Patients with Parkinson’s Disease. Dutch Journal of Physiotherapy. 2004;114:S3.
Shoulson I., Fahn S. Huntington disease: Clinical care and Evaluation. Neurology. 1979;29(1):1-3.
Shumway-Cook A., Woollacott M. Clinical Management of the Patient With a Postural Control Disorder. In: Biblis M., editor. Motor control: theory and application. Baltimore: Lippincott Williams and Wilkins; 2001:397-445.
Simpson S.A. Late stage care in Huntington’s disease. Brain Res. Bull.. 2007;72(2–3):179-181.
Smith M., Danoff J., Jain M., Long T. Genetic Disorders: Implications for Allied Health Professionals: Two Case Studies. Internet Journal of Allied Health Sciences and Practice. 2007. http://ijahsp.nova.edu. Vol 5, No 4 ISSN 1540-580X’
Snowden J.S., Craufurd D., Griffiths H.L., Neary D. Awareness involuntary movements in Huntington’s disease. Arch. Neurol.. 1998;55(6):801-805.
Sorensen S., Fenger K. Causes of death in patients with Huntington’s disease and in unaffected first degree relatives. J. Med. Genet.. 1992;29(12):911-914.
Tassicker R.J., Teltscher B., Trembath M.K., et al. Problems assessing uptake of Huntington disease predictive testing and a proposed solution. Eur. J. Hum. Genet.. 2009;17:66-70.
Thaut M.H., Miltner R., Lange H.W., et al. Velocity modulation and rhythmic synchronization of gait in Huntington’s disease. Mov. Disord.. 1999;14(5):808-819.
Tinetti M.E. Performance-oriented assessment of mobility problems in elderly patients. J. Am. Geriatr. Soc.. 1986;34(2):119-126.
Travers E., Jones K., Nichol J. Palliative care provision in Huntington’s disease. Int. J. Palliat. Nurs.. 2007;13(3):125-130.
van Dellen A., Cordery P., Spires T.L., et al. Wheel running from a juvenile age delays onset of specific motor deficits but does not alter protein aggregate density in a mouse model of Huntington’s disease. BMC Neurosci.. 2008;9:34.
Walker F.O. Huntington’s disease. Lancet. 2007;369(9557):218-228.
Ware J.J., Sherbourne C. The MOS 36-item Short-form Health survey (SF-36). I. Conceptual Framework and Item Selection. Med. Care.. 1992;30(6):473-483.
Watson M. Refining the Ten-metre walking test for use with neurologically impaired People. Physiotherapy. 2002;88(7):386-397.
World Health Organization. International Classification of Functioning, Disability and Health. Geneva: World Health Organization, 2001.
Zinzi P., Salmaso D., De Grandis R., et al. Effects of an Intensive Rehabilitation Programme on Patients with Huntington’s disease: a pilot study. Clin. Rehabil.. 2007;21(7):603-613.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 7 Huntington’s disease
Introduction
Inevitably, a chapter in a physiotherapy textbook will concentrate on the physical aspects of the condition, but for most families and carers a much greater problem is the behavioural change which can lead to irritability, aggression, depression or apathy. Professionals dealing with HD patients need to have an understanding of the neuropsychological and neuro-psychiatric aspects of HD which will help with their management of the patient. Over time the needs of the patient and family change, so professionals need to reassess their management. This chapter will describe the genetic and pathological features before going on to describe the stages of the condition and the associated physiotherapy management. A number of useful texts are available for a wider discussion of the condition including Bates et al. (2002), Quarrell (2009) and Walker (2007).
Prevalence and aspects of natural history
Most UK studies quote a prevalence of 4–10 per 100 000, with similar results from studies from Europe and the US, and low prevalence figures reported from Japan and Finland (Harper, 2002). Whilst HD may be considered a rare disorder it must be remembered that for every affected person there are approximately twice that number who have the mutation and are currently asymptomatic (Conneally, 1984). The median duration of HD is between 16 and 21 years (Foroud et al., 1999; Roos et al., 1993) so this represents a considerable demand on families and carers.
HD can develop at almost any age from under 20 years (and occasionally under 10 years) to over 75 years, but most people develop the condition between the ages of 35 and 55 years, which is after the usual years of reproduction (Harper et al., 1988). One consequence of this is that the 50% risk for an asymptomatic person does not decline appreciably until s/he is going through middle age.
Individuals with an age of onset less than 20 years are described as having the juvenile form of HD. It is difficult to know precisely how many patients have early onset, but 5% is a reasonable estimate. Young people with juvenile HD may develop problems with speech, bradykinesia and dystonic movements earlier in the course in the illness; this topic is reviewed in Quarrell et al. (2009).
Genetics
As HD is a dominant disorder; each cell contains one normal copy and one abnormal copy of the gene. The gene for HD was cloned in 1993 (Huntington’s Disease Collaborative Research Group, 1993) and the protein for which it codes was named huntingtin. The first part of the HD gene has a sequence CAGCAG…CAG which is repeated a number of times. CAG codes for the amino acid glutamine so that the huntingtin protein contains a sequence called a polyglutamine repeat. The mutation causing HD is an expansion of the number of CAG repeats in the gene and therefore of the polyglutamine repeat in the protein. Up to 35 repeats would be considered normal, although there is a possibility that if a person has a repeat size at the upper end of this range it could expand into the pathological range in future generations; a result between 36 and 39 repeats is abnormal, but it is possible that a person may develop the condition late or even live a full life and not develop HD; 40 or more repeats is considered definitely abnormal (ASHG/AMCG statement, 1998). At a mathematical level, there is a correlation between the average age of onset and the CAG repeat size but there is such a wide variation that it is not possible to predict the age of onset for an individual from knowing the CAG repeat size.
An asymptomatic individual with an affected parent has a prior probability of 50% of inheriting the HD gene. Such a person could have a genetic test at this stage. This is called a predictive test. As there is no effective treatment to alter the natural history of HD such tests are used cautiously in conjunction with counselling and according to widely accepted international guidelines drawn up by an ad hoc committee of the International Huntington’s Association/World Federation of Neurology Research Group on Huntington’s chorea (1994). Less than 20% of adults at risk of HD choose to have a predictive test (Tassiker et al., 2009).
Genetics
Pathology
The gene is widely expressed in the cells of the body but some cells within the brain are very sensitive to abnormal huntingtin and there is selective neurodegeneration. An explanation for the selective neurodegeneration characteristic of HD is unclear. The most striking cell loss occurs in the basal ganglia but other areas of the brain including the cortex are also affected. The anatomy of the basal ganglia is shown in Figure 7.1. To emphasize this point it should be noted that the brain of a patient with HD will be smaller and weigh less than that of an age-matched control. The efferent medium spiny neurons within the caudate and putamen nuclei, which are collectively called the striatum, are especially sensitive to the abnormal huntingtin protein (see Figure 7.2).
Implications of pathology for understanding the movement disorder
The striatum receives excitatory inputs from the cortex. The efferent medium spiny neurons contain inhibitory neurotransmitters which project to the internal globus pallidus via direct and indirect pathways (Albin et al., 1989). Figure 7.3 shows a simplified diagram of the process. In broad terms, damage to the indirect pathway leads to overstimulation of the thalamo-cortical feedback and chorea (random purposeless movements), whereas loss of the direct pathway results in increased inhibition of the thalamus and less activity of the thalamo-cortical feedback producing bradykinesia and rigidity. Although both pathways degenerate, the balance between them is disturbed. The medium spiny neurons of both the direct and indirect pathways contain dopamine receptors, which are consistently reduced in Huntington’s disease. Conventionally, excitatory D1 receptors are on the direct and inhibitory D2 receptors are on the indirect pathway. This means that whilst it is possible to control the chorea of patients with dopa blocker and dopa depleting drugs, this may worsen the bradykinesia and dystonia.
Understanding pathology at the level of the cell
HD should be easy to explain because one gene is involved with one mutation that affects a single protein. Unfortunately, this is not the case because huntingtin is involved in many cellular processes. Apart from man, no natural animal develops HD. Following the identification of the gene a tremendous research effort involved developing animal models. The first of these, and most studied, was the R6 mice which contain multiple copies of the first part of the HD gene (Mangiarini et al., 1996). These mice die before they develop significant cell death. One feature of the cells is that they contain aggregates of the first part of the huntintin protein (Davies, et al., 1997). These aggregates are now recognized as occurring in human HD brains (DiFiglia et al., 1997). It is unclear as to whether these aggregates are directly related to the cellular pathology or represent a defence mechanism within the cell. There are a number of different animal models; details of their similarities and differences are not relevant to this chapter, but animal models have enabled an understanding that neuronal cells are dysfunctional (‘sick’) before they die which gives hope for interventions to alter the natural history of the disease. This natural history is modelled in Figure 7.4 with neuronal dysfunction preceding the onset of symptoms. In the absence of serendipity, an understanding of the pathology at the cellular level using animal and cellular models is the way forward for developing and subsequently testing new treatments for HD.
Figure 7.4 Primary physiotherapy aims and the continuum of Huntington’s disease.
(Modified from the Walker et al., Huntington’s Disease. Lancet 2007;369 (9557): 218-28, with permission from Elsevier.)
Pathology
Life history of the disease
If a person has inherited the gene there will be a period of time where she/he is completely asymptomatic. In time, the neurones become dysfunctional. Onset of HD is insidious with non-specific problems such as mood change or being slightly forgetful, which can be attributed easily to other mundane causes. A person in this prodromal stage may become depressed, but a clinical diagnosis cannot be made confidently until motor signs, such as chorea, appear. These may be infrequent, of low amplitude and may not be noticed by the patient, family or clinician with limited experience of the condition. Indeed even at a time when the chorea is quite obvious it may be denied as a problem by the patient (Snowden et al., 1998).
If a patient is seen when there are obvious signs of HD, the CAG repeat size may be measured from a venous blood sample to confirm the clinical diagnosis. It is important to realize that the result of the genetic test will be the same whether the individual is completely asymptomatic, in the prodromal phase, or in the early, middle or later stages of HD. As with any test the interpretationof the test result depends upon the clinical context. This raises an important issue about the genetic test: it is a ‘trait marker’ but says nothing about the state of the illness. There is a significant research effort to determine other laboratory biomarkers (‘state markers’) which can track the development and progression of the disease (Henley et al., 2005). The boundaries between early, middle and late stages are ill defined, but nonetheless they form a useful way of categorizing the impact of HD.
Clinical assessment
The Unified Huntington’s Disease Rating Scale (UHDRS) (Huntington Study Group, 1996) is a rating system that is used to quantify the severity of Huntington’s disease. It was developed as a clinical rating scale to assess four domains of clinical performance and capacity in individuals with HD: motor function, cognitive function, behavioural abnormalities, and functional capacity. These scores can be calculated by summing the various questions of each section. The UHDRS is useful for tracking changes in the clinical features of HD over time, and appears to be appropriate for repeated administration during clinical studies. Stages in Huntington’s disease are traditionally defined by the Total Functional Capacity scale, developed by Shoulson and Fahn (1979), which classifies patients into one of five stages of disease progression (Marder et al., 2000). The staging evaluates patients based on functioning for ADLs, domestic chores, finances, work and overall care level. Mid-stage is typically defined as levels II-III (see Table 7.1).
Stage I (Early stage)
In the early stages of the disease, the person with HD is able to remain functional and independent, retaining their ability to work and drive (HDSA, 2000). Movement disorders include minor involuntary movements, reduced coordination and linearly progressive motor impersistence or inability to maintain a voluntary muscle contraction at a constant level (Walker, 2007). Even at this stage, it may be possible to detect some slowing of saccadic eye movements (rapidly moving the eye to a new target; this is done by asking the patient to move his/her eye to one corner in response to a cue). Voluntary motor tasks (fine and gross motor skills) may become increasingly difficult as the disease progresses (Aubeeluck & Wilson, 2008). Cognitive tasks also become more difficult with reduced ability to sequence and organize information (HDSA, 2000). Deficits in gait have been identified across the spectrum of HD (Churchyard et al., 2001; Grimbergen et al., 2008; Hausdorff et al., 1998; Koller & Trimble, 1985; Rao et al., 2008). Decreased gait velocity, stride length, and increased time in double support compared to healthy controls may occur (Rao et al., 2008). These impairments appear to worsen with increasing disease severity and can be useful markers of disease progression and treatment efficacy.
Stage II and III (Middle stage)
During the middle stages of HD, involuntary movements such as chorea increase. In the early stages of HD, this is typically seen in the fingers, hands and face muscles, but as the disease progresses, it can be seen throughout thebody, including in all four extremities and the trunk. Chorea does not usually result in a direct impairment of motor function, however voluntary motor impairments may have a functional impact (Walker, 2007).
Dystonic postures (abnormal, sustained posturing) in sitting or standing (Louis et al., 1999) and in movements such as shoulder elevation, foot inversion and supination, and trunk extension might be present. People may experience frequent loss of balance and falls, which interfere with their ability to walk and maintain an upright position (Busse et al., 2009). Common tasks during which patients demonstrate balance problems include those where the base of support is decreased; tandem standing and walking, dual tasks, eyes closed and in response to external perturbations (Bilney et al., 2003b; Quinn & Rao, 2002). Falls are frequent in people with HD; 60% of people with early to mid-stage HD in two separate studies (n=45 & n=24) reported two or more falls in the previous 12 months (Busse et al., 2009; Grimbergen et al., 2008). Falls can have physical and psychological consequences (Myers et al., 1996). The fear of future falls may be a more pervasive problem than falls per se, as fear of falls may lead to people restricting their physical activities. Self-imposed restriction of daily activities leads to immobility and consequently osteoporosis, reduced fitness and social isolation (Grimbergen et al., 2004). Individuals with HD have also been found to have lower-limb muscular weakness compared to age-matched controls (Busse et al., 2008a). Strength can be measured clinically by manual muscle testing or by functional observation of strength during task performance (e.g. stair climbing or evidence of gait deviation during walking).
Stage IV and V (Late stage)
In the advanced stages of the disease motor symptoms continue to progress, severely limiting mobility. Choreic and dystonic movement may further increase (see Figure 7.4), but involuntary movements are often overshadowed by Parkinsonian symptoms (Aubeeluck & Wilson, 2008; HDSA, 2000) such as bradykinesia or slowness of movement. People with HD can also experience pain, particularly in the later stage of the disease. The source of this pain can be unknown; however, dystonia or muscle imbalances can often cause musculoskeletal pain. Excessive chorea can cause pain if people injure themselves by hitting their arm or leg into an object or hard surface.
Weight loss can occur in part due to impaired swallowing function (Aubeeluck & Wilson, 2008; HDSA, 2000). People may no longer be able to work or drive and will need assistance when performing some activities of daily living (ADL) (HDSA, 2000).
Even though people with HD appear to be in constant movement, their underlying volitional movements during task performance have been found to be slower than healthy controls for reaching (Quinn et al., 2001) and ambulation (Delval et al., 2006; Delval et al., 2007). Bradykinesia can typically be evaluated by measuring time to complete a task (movement time). Akinesia or delayed initiation of movement is also seen. In research studies, akinesia is typically measured by reaction time. In the clinical setting, it is often difficult to quantitatively evaluate akinesia, but delays in onset of movement for various tasks can be noted. Impaired speech results in difficulties communicating, and cognitive and psychiatric deterioration may continue, but it is thought that patients retain some comprehension (HDSA, 2000). At this stage most people will require assistance in all aspects of daily living, relying fully on nursing care (HDSA, 2000). Despite the severity of the neurological disorder, the primary causes of death in HD are aspiration pneumonia, cardiovascular disease and complications from falls (Sorensen & Fenger, 1992; Walker, 2007).
Physiotherapy assessment
The physiotherapy aims should be both anticipatory and responsive to the disease stage (see Figure 7.4) with interventions to assist HD patients to maintain independence, functional capacity and participation in society (Bilney et al., 2003a).
The World Health Organization International Classification of Functioning, Disability and Health (ICF) (WHO, 2001) is useful as an aid in structuring assessment of a person’s functioning and participation (see Figure 7.5), and in allowing consideration of the triad of motor, cognitive and psychiatric symptoms that are often seen in HD. The ICF can be used for functional status assessment, goal setting and treatment planning, and focuses on aspects of a person’s health and health-related wellbeing in terms of activities and participation, i.e. the description of the tasks (activities) and/or life situations (participation) in which the person wishes to engage, and the impact that impaired body function or structure is having on these aspects. The ICF further takes into consideration the contextual factors (both environmental and personal), which may impact on a person’s life, and the relationships with body function/structure, activities and participation. The specific impairments and associated activities and participations that may need consideration when assessing a person with HD are listed in Table 7.2. Documentation of the impact of the noted impairments on functional abilities, activities and participation is important to help highlight specific factors that contribute to a patient’s functional problems and inform the clinical reasoning process in terms of maximizing functional abilities. Qualifiers of the degree of impairment, and the performance and capacity of activities and participation enable essential documentation of disability and health status.
Table 7.2 Specific impairments and associated activities and participations that may need consideration when assessing a person with Huntington’s disease
| Impairments in Body Structure and Function | |
|---|---|
| B1 Global and specific mental functions B3 Voice and speech functions b 440-449 Functions of the respiratory system B7 Neuromusculoskeletal and movement functions b 730-749 Muscle functions b 750-789 Movement functions S1 Structures of the nervous system S7 Structures related to movement |
Cognitive-perceptual: Memory Executive functions Spatial abilities Neuromuscular: Chorea Dystonia Strength Flexibility |
| Functional Abilities | |
|---|---|
| Activities and participation | |
| D1 Learning and applying knowledge D2 General tasks and demands D3 Communication D4 Mobility d 410-429 Changing and maintaining body position d 460-469 Walking and moving D5 Self care D6 Domestic life D7 Interpersonal interactions D8/9 Major life areas and community life For each functional activity, therapists should evaluate and document the level of assistance or caregiver burden needed to complete the task, and also determine the patient’s skill level (e.g. time to complete task, consistency of task performance, etc). This section can include any of the following skills, as appropriate: self-care and home management including activities of daily living (bed mobility, dressing, self care, toileting, bathing, eating, cooking, preparing meals and instrumental activities of daily living) Listing of specific skills assessed (e.g. ambulation, sitting ability, standing ability, sit to stand, wheelchair skills, stair climbing, feeding, bed mobility). Therapists should evaluate and document the functional performance of skills that are pertinent to the person with HD’s independence. |
Speech Feeding, drinking (swallowing, sucking) Bathing, toileting, dressing Household chores Cooking, cleaning Walking (distance, speed, shoes) |
| Environmental Factors | |
|---|---|
| E1 Products and technology e 120 assistive products E3 Support and relationships E4 Attitudes E5 Services and systems For each functional activity, therapists should evaluate and document the level of assistance or caregiver burden needed to complete the task, and also determine the patient’s skill level (e.g. time to complete task, consistency of task performance, etc.). This section can include any of the following skills, as appropriate: Assistive and adaptive devices Environmental, home and work (job/school) barriers |
Equipment: Assistive devices, feeding drinking, dressing equipment Wheelchair, brace, walker |
Specific coding for the domains are shown.
The subjective assessment allows the physiotherapist to evaluate the patient’s presenting problem(s), in the contextof their past medical history and how this may affect their ability to live independently and within society. This also provides the opportunity to build a therapeutic, collaborative relationship, and facilitate realistic goal setting. It must be noted that the patient’s caregiver(s) should be involved, particularly if the patient has difficulties communicating as a result of cognitive or physical impairment. The objective assessment should include assessment of neuromuscular, musculoskeletal (posture, range of motion, pain and muscle strength), cardio-respiratory and cardio-vascular impairments. An overview of the HD specific considerations for the physiotherapy assessment is shown in Table 7.3. These are useful to aid clinical reasoning and goal setting but are not necessarily outcomes that are sensitive to change over time. There are numerous outcome measures that have potential utility in the assessment of efficacy of physiotherapy interventions for people with HD. Measurement tools that have been used in previous HD-related studies are presented in Table 7.4 and are classified according to the WHO ICF. The Berg Balance Scale, Timed Up and Go Test and the Functional Reach Test have all been found to be valid and responsive clinical measures in HD and are considered useful to detect those at risk of falls (Busse et al., 2009; Grimbergen et al., 2008; Rao et al., 2009).
Table 7.3 Specific assessment points for consideration in Huntington’s disease
| Subjective Assessment |
|---|
