[level-membership-for-internal-medicine-category]Retroviral genomes include both coding and noncoding sequences (Fig. 225e-2). In general, noncoding sequences are important recognition signals for DNA or RNA synthesis or processing events and are located in the 5′ and 3′ terminal regions of the genome. All retroviral genomes are terminally redundant, containing identical sequences called long terminal repeats (LTRs). The ends of the retroviral RNA genome differ slightly in sequence from the integrated retroviral DNA. In the latter, the LTR sequences are repeated in both the 5′ and the 3′ terminus of the virus. The LTRs contain sequences involved in initiating the expression of the viral proteins, the integration of the provirus, and the polyadenylation of viral RNAs. The primer binding site, which is critical for the initiation of reverse transcription, and the viral packaging sequences are located outside the LTR sequences. The coding regions include the gag (group-specific antigen, core protein), pol (RNA-dependent DNA polymerase), and env (envelope) genes. The gag gene encodes a precursor polyprotein that is cleaved to form three to five capsid proteins; a fraction of the Gag precursor proteins also contain a protease responsible for cleaving the Gag and Pol polyproteins. A Gag-Pol polyprotein gives rise to the protease that is responsible for cleaving the Gag-Pol polyprotein. The pol gene encodes three proteins: the reverse transcriptase, the integrase, and the protease. The reverse transcriptase copies the viral RNA into the double-strand DNA provirus, which inserts itself into the host cell DNA via the action of integrase. The protease cleaves the Gag-Pol polyprotein into smaller protein products. The env gene encodes the envelope glycoproteins: one protein that binds to specific surface receptors and determines what cell types can be infected and a smaller transmembrane protein that anchors the complex to the envelope. Fig. 225e-3 shows how the retroviral gene products make up the virus structure.
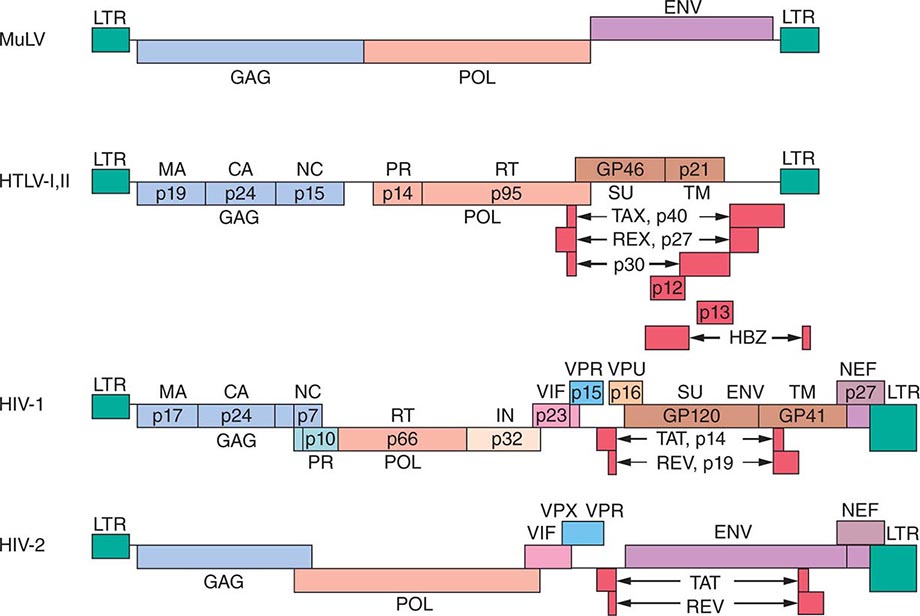
FIGURE 225e-2 Genomic structure of retroviruses. The murine leukemia virus MuLV has the typical three structural genes: gag, pol, and env. The gag region gives rise to three proteins: matrix (MA), capsid (CA), and nucleic acid–binding (NC) proteins. The pol region encodes both a protease (PR) responsible for cleaving the viral polyproteins and a reverse transcriptase (RT). In addition, HIV pol encodes an integrase (IN). The env region encodes a surface protein (SU) and a small transmembrane protein (TM). The human retroviruses have additional gene products translated in each of the three possible reading frames. HTLV-1 and HTLV-2 have tax and rex genes with exons on either side of the env gene. HIV-1 and HIV-2 have six accessory gene products: tat, rev, vif, nef, vpr, and either vpu (in HIV-1) or vpx (in HIV-2). The genes for these proteins are located mainly between the pol and env genes. GP, glycoprotein; HBZ, HTLV-1 basic leucine zipper domain–containing protein; LTR, long terminal repeat.
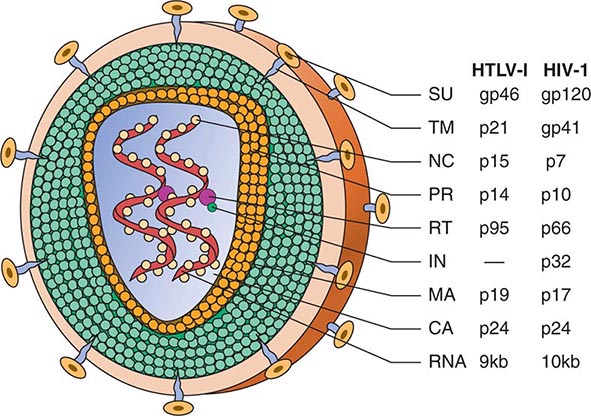
FIGURE 225e-3 Schematic structure of human retroviruses. The surface glycoprotein (SU) is responsible for binding to receptors of host cells. The transmembrane protein (TM) anchors SU to the virus. NC is a nucleic acid–binding protein found in association with the viral RNA. A protease (PR) cleaves the polyproteins encoded by the gag, pol, and env genes into their functional components. RT is reverse transcriptase, and IN is an integrase present in some retroviruses (e.g., HIV-1) that facilitates insertion of the provirus into the host genome. The matrix protein (MA) is a Gag protein closely associated with the lipid of the envelope. The capsid protein (CA) forms the major internal structure of the virus, the core shell.
HTLVs have a region between env and the 3′ LTR that encodes several proteins and transcripts in overlapping reading frames (Fig. 225e-2). Tax is a 40-kDa protein that does not bind to DNA but induces the expression of host cell transcription factors that alter host cell gene expression and is capable of inducing cell transformation under certain circumstances. Rex is a 27-kDa protein that regulates the expression of viral mRNAs. Other transcripts from this region (p12, p13, p30) tend to restrict expression of viral genes and diminish the immunogenicity of infected cells. The protein of HBZ, a product of the complementary proviral DNA strand, interacts with many cellular transcription factors and signaling proteins. It stimulates proliferation of infected cells and is the only viral product universally expressed in HTLV-1-infected tumor cells. These proteins are produced from messages that are similar but that are spliced differently from overlapping but distinct exons.
The lentiviruses in general, and HIV-1 and -2 in particular, contain a larger genome than other pathogenic retroviruses. They contain an untranslated region between pol and env that encodes portions of several proteins, varying with the reading frame into which the mRNA is spliced. Tat is a 14-kDa protein that augments the expression of virus from the LTR. The Rev protein of HIV-1, similar to the Rex protein of HTLV, regulates RNA splicing and/or RNA transport. The Nef protein downregulates CD4, the cellular receptor for HIV; alters host T cell–activation pathways; and enhances viral infectivity. The Vif protein is necessary for the proper assembly of the HIV nucleoprotein core in many types of cells; without Vif, proviral DNA is not efficiently produced in these infected cells. In addition, the Vif protein targets APOBEC (apolipoprotein B mRNA-editing enzyme catalytic polypeptide, a cytidine deaminase that mutates the viral sequence) for proteasomal degradation, thus blocking its virus-suppressing effect. Vpr, Vpu (HIV-1 only), and Vpx (HIV-2 only) are viral proteins encoded by translation of the same message in different reading frames. As noted above, oncogenic retroviruses depend on cell proliferation for their replication; lentiviruses can infect nondividing cells, largely through effects mediated by Vpr. Vpr facilitates transport of the provirus into the nucleus and can induce other cellular changes, such as G2 growth arrest and differentiation of some target cells. Vpx is structurally related to Vpr, but its functions are not fully defined. Vpu promotes the degradation of CD4 in the endoplasmic reticulum and stimulates the release of virions from infected cells.
Retroviruses can be either exogenously acquired (by infection with an infected cell or a free virion capable of replication) or transmitted in the germline as endogenous virus. Endogenous retroviruses are often replication defective. The human genome contains endogenous retroviral sequences, but there are no known replication-competent endogenous retroviruses in humans.
In general, viruses that contain only the gag, pol, and env genes either are not pathogenic or take a long time to induce disease; these observations indicate the importance of the other regulatory genes in viral disease pathogenesis. The pathogenesis of neoplastic transformation by retroviruses relies on the chance integration of the provirus at a spot in the genome resulting in the expression of a cellular gene (protooncogene) that becomes transforming by virtue of its unregulated expression. For example, avian leukosis virus causes B cell leukemia by inducing the expression of myc. Some retroviruses possess captured and altered cellular genes near their integration site, and these viral oncogenes can transform the infected host cell. Viruses that have oncogenes often have lost a portion of their genome that is required for replication. Such viruses need helper viruses to reproduce, a feature that may explain why these acute transforming retroviruses are rare in nature. All human retroviruses identified to date are exogenous and are not acutely transforming (i.e., they lack a transforming oncogene).
These remarkable properties of retroviruses have led to experimental efforts to use them as vectors to insert specific genes into particular cell types, a process known as gene therapy or gene transfer. The process could be used to repair a genetic defect or to introduce a new property that could be used therapeutically; for example, a gene (e.g., thymidine kinase) that would make a tumor cell susceptible to killing by a drug (e.g., ganciclovir) could be inserted. One source of concern about the use of retroviral vectors in humans is that replication-competent viruses might rescue endogenous retroviral replication, with unpredictable results. This concern is not merely hypothetical: the detection of proteins encoded by endogenous retroviral sequences on the surface of cancer cells implies that the genetic events leading to the cancer were able to activate the synthesis of these usually silent genes.
HUMAN T CELL LYMPHOTROPIC VIRUS
HTLV-1 was isolated in 1980 from a T cell lymphoma cell line from a patient originally thought to have cutaneous T cell lymphoma. Later it became clear that the patient had a distinct form of lymphoma (originally reported in Japan) called adult T cell leukemia/lymphoma (ATL). Serologic data have determined that HTLV-1 is the cause of at least two important diseases: ATL and tropical spastic paraparesis, also called HTLV-1-associated myelopathy (HAM). HTLV-1 may also play a role in infective dermatitis, arthritis, uveitis, and Sjögren’s syndrome.
Two years after the isolation of HTLV-1, HTLV-2 was isolated from a patient with an unusual form of hairy cell leukemia that affected T cells. Epidemiologic studies of HTLV-2 failed to reveal a consistent disease association. Similarly, HTLV-3 and HTLV-4 have been identified but have no known disease association.
BIOLOGY AND MOLECULAR BIOLOGY
Because the biology of HTLV-1 and that of HTLV-2 are similar, the following discussion will focus on HTLV-1.
Human glucose transporter protein 1 (GLUT-1) functions as a receptor for HTLV-1, probably acting together with neuropilin-1 (NRP1) and heparan sulfate proteoglycans. Generally, only T cells are productively infected, but infection of B cells and other cell types is occasionally detected. The most common outcome of HTLV-1 infection is latent carriage of randomly integrated provirus in CD4+ T cells. HTLV-1 does not contain an oncogene and does not insert into a unique site in the genome. Indeed, most infected cells express no viral gene products. The only viral gene product that is routinely expressed in tumor cells transformed by HTLV-1 in vivo is hbz. The tax gene is thought to be critical to the transformation process but is not expressed in the tumor cells of many ATL patients, possibly because of the immunogenicity of tax-expressing cells. Cells transformed in vitro, by contrast, actively transcribe HTLV-1 RNA and produce infectious virions. Most HTLV-1-transformed cell lines are the result of the infection of a normal host T cell in vitro. It is difficult to establish cell lines derived from authentic ATL cells.
Although tax does not itself bind to DNA, it does induce the expression of a wide range of host cell gene products, including transcription factors (especially c-rel/NF-κB, ets-1 and -2, and members of the fos/jun family), cytokines (e.g., interleukin [IL] 2, granulocyte-macrophage colony-stimulating factor, and tumor necrosis factor), membrane proteins and receptors (major histocompatibility [MHC] molecules and IL-2 receptor α), and chromatin remodeling complexes. The genes activated by tax are generally controlled by transcription factors of the c-rel/NF-κB and cyclic AMP response element binding (CREB) protein families. It is unclear how this induction of host gene expression leads to neoplastic transformation; tax can interfere with G1 and mitotic cell-cycle checkpoints, block apoptosis, inhibit DNA repair, and promote antigen-independent T cell proliferation. Induction of a cytokine–autocrine loop has been proposed; however, IL-2 is not the crucial cytokine. The involvement of IL-4, IL-7, and IL-15 has been proposed.
In light of the irregular expression of tax in ATL cells, it has been suggested that tax is important in the early phases of transformation but is not essential for the maintenance of the transformed state. The maintenance role is thought to be due to hbz expression. As is clear from the epidemiology of HTLV-1 infection, transformation of an infected cell is a rare event and may depend on heterogeneous second, third, or fourth genetic hits. No consistent chromosomal abnormalities have been described in ATL; however, aneuploidy is common and individual cases with p53 mutations and translocations involving the T cell receptor genes on chromosome 14 have been reported. Tax may repress certain DNA repair enzymes, permitting the accumulation of genetic damage that would normally be repaired. However, the molecular pathogenesis of HTLV-1-induced neoplasia is not fully understood.
FEATURES OF HTLV-1 INFECTION
Epidemiology HTLV-1 infection is transmitted in at least three ways: from mother to child, especially via breast milk; through sexual activity, more commonly from men to women; and through the blood—via contaminated transfusions or contaminated needles. The virus is most commonly transmitted perinatally. Compared with HIV, which can be transmitted in cell-free form, HTLV-1 is less infectious, and its transmission usually requires cell-to-cell contact.
![]() HTLV-1 is endemic in southwestern Japan and Okinawa, where >1 million persons are infected. Antibodies to HTLV-1 are present in the serum of up to 35% of Okinawans, 10% of residents of the Japanese island of Kyushu, and <1% of persons in nonendemic regions of Japan. Despite this high prevalence of infection, only ~500 cases of ATL are diagnosed in this area each year. Clusters of infection have been noted in other areas of the Orient, such as Taiwan; in the Caribbean basin, including northeastern South America; in northwestern South America; in central and southern Africa; in Italy, Israel, Iran, and Papua New Guinea; in the Arctic; and in the southeastern part of the United States (Fig. 225e-4). An estimated 5–10 million persons have HTLV-1 infection worldwide.
HTLV-1 is endemic in southwestern Japan and Okinawa, where >1 million persons are infected. Antibodies to HTLV-1 are present in the serum of up to 35% of Okinawans, 10% of residents of the Japanese island of Kyushu, and <1% of persons in nonendemic regions of Japan. Despite this high prevalence of infection, only ~500 cases of ATL are diagnosed in this area each year. Clusters of infection have been noted in other areas of the Orient, such as Taiwan; in the Caribbean basin, including northeastern South America; in northwestern South America; in central and southern Africa; in Italy, Israel, Iran, and Papua New Guinea; in the Arctic; and in the southeastern part of the United States (Fig. 225e-4). An estimated 5–10 million persons have HTLV-1 infection worldwide.
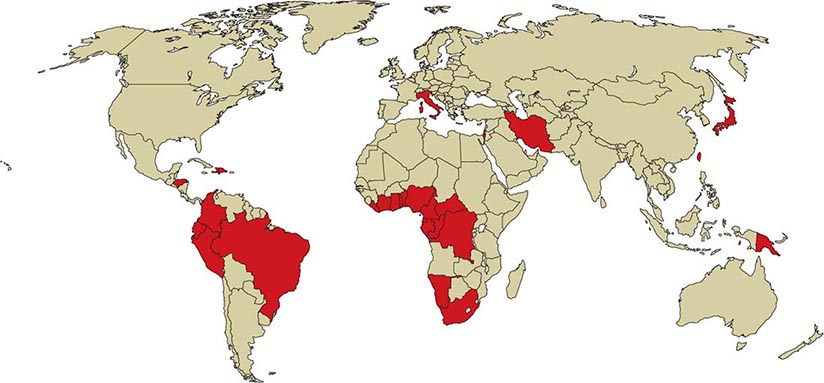
FIGURE 225e-4 Global distribution of HTLV-1 infection. Countries with a prevalence of HTLV-1 infection of 1–5% are shaded darkly. Note that the distribution of infected patients is not uniform in endemic countries. For example, the people of southwestern Japan and northeastern Brazil are more commonly affected than those in other regions of those countries.
Progressive spastic or ataxic myelopathy developing in an individual who is HTLV-1 positive (i.e., who has serum antibodies to HTLV-1) may be due to direct infection of the nervous system with the virus, but destruction of the pyramidal tracts appears to involve HTLV-1-infected CD4+ T cells; a similar disorder may result from infection with HIV or HTLV-2. In rare instances, patients with HAM are seronegative but have detectable antibody to HTLV-1 in cerebrospinal fluid (CSF).
The cumulative lifetime risk of developing ATL is 3% among HTLV-1-infected patients, with a threefold greater risk among men than among women; a similar cumulative risk is projected for HAM (4%), but with women more commonly affected than men. The distribution of these two diseases overlaps the distribution of HTLV-1, with >95% of affected patients showing serologic evidence of HTLV-1 infection. The latency period between infection and the emergence of disease is 20–30 years for ATL. For HAM, the median latency period is ~3.3 years (range, 4 months to 30 years). The development of ATL is rare among persons infected by blood products; however, ~20% of patients with HAM acquire HTLV-1 from contaminated blood. ATL is more common among perinatally infected individuals, whereas HAM is more common among persons infected via sexual transmission.
Associated Diseases • ATL Four clinical types of HTLV-1-induced neoplasia have been described: acute, lymphomatous, chronic, and smoldering. All of these tumors are monoclonal proliferations of CD4+ postthymic T cells with clonal proviral integrations and clonal T cell receptor gene rearrangements.
ACUTE ATL About 60% of patients who develop malignancy have classic acute ATL, which is characterized by a short clinical prodrome (~2 weeks between the first symptoms and the diagnosis) and an aggressive natural history (median survival period, 6 months). The clinical picture is dominated by rapidly progressive skin lesions, pulmonary involvement, hypercalcemia, and lymphocytosis with cells containing lobulated or “flower-shaped” nuclei (see Fig. 134-10). The malignant cells have monoclonal proviral integrations and express CD4, CD3, and CD25 (low-affinity IL-2 receptors) on their surface. Serum levels of CD25 can be used as a tumor marker. Anemia and thrombocytopenia are rare. The skin lesions may be difficult to distinguish from those in mycosis fungoides. Lytic bone lesions, which are common, do not contain tumor cells but rather are composed of osteolytic cells, usually without osteoblastic activity. Despite the leukemic picture, bone marrow involvement is patchy in most cases.
The hypercalcemia of ATL is multifactorial; the tumor cells produce osteoclast-activating factors (tumor necrosis factor α, IL-1, lymphotoxin) and can also produce a parathyroid hormone–like molecule. Affected patients have an underlying immunodeficiency that makes them susceptible to opportunistic infections similar to those seen in patients with AIDS (Chap. 226). The pathogenesis of the immunodeficiency is unclear. Pulmonary infiltrates in ATL patients reflect leukemic infiltration half the time and opportunistic infections with organisms such as Pneumocystis and other fungi the other half. Gastrointestinal symptoms are nearly always related to opportunistic infection. Strongyloides stercoralis is a gastrointestinal parasite that has a pattern of endemic distribution similar to that of HTLV-1. HTLV-1-infected persons also infected with this parasite may develop ATL more often or more rapidly than those without Strongyloides infections. Serum concentrations of lactate dehydrogenase and alkaline phosphatase are often elevated in ATL. About 10% of patients have leptomeningeal involvement leading to weakness, altered mental status, paresthesia, and/or headache. Unlike other forms of central nervous system (CNS) lymphoma, ATL may be accompanied by normal CSF protein levels. The diagnosis depends on finding ATL cells in the CSF (Chap. 134).
LYMPHOMATOUS ATL The lymphomatous type of ATL occurs in ~20% of patients and is similar to the acute form in its natural history and clinical course, except that circulating abnormal cells are rare and lymphadenopathy is evident. The histology of the lymphoma is variable but does not influence the natural history. In general, the diagnosis is suspected on the basis of the patient’s birthplace (see “Epidemiology,” above) and the presence of skin lesions and hypercalcemia. The diagnosis is confirmed by the detection of antibodies to HTLV-1 in serum.
CHRONIC ATL Patients with the chronic form of ATL generally have normal serum levels of calcium and lactate dehydrogenase and no involvement of the CNS, bone, or gastrointestinal tract. The median duration of survival for these patients is 2 years. In some cases, chronic ATL progresses to the acute form of the disease.
SMOLDERING ATL Fewer than 5% of patients have the smoldering form of ATL. In this form, the malignant cells have monoclonal proviral integration; <5% of peripheral blood cells exhibit typical morphologic abnormalities; hypercalcemia, adenopathy, and hepatosplenomegaly do not develop; the CNS, the bones, and the gastrointestinal tract are not involved; and skin lesions and pulmonary lesions may be present. The median survival period for this small subset of patients appears to be ≥5 years.
HAM (TROPICAL SPASTIC PARAPARESIS) In contrast to ATL, in which there is a slight predominance of male patients, HAM affects female patients disproportionately. HAM resembles multiple sclerosis in certain ways (Chap. 458). The onset is insidious. Symptoms include weakness or stiffness in one or both legs, back pain, and urinary incontinence. Sensory changes are usually mild, but peripheral neuropathy may develop. The disease generally takes the form of slowly progressive and unremitting thoracic myelopathy; one-third of patients are bedridden within 10 years of diagnosis, and one-half are unable to walk unassisted by this point. Patients display spastic paraparesis or paraplegia with hyperreflexia, ankle clonus, and extensor plantar responses. Cognitive function is usually spared; cranial nerve abnormalities are unusual.
Magnetic resonance imaging (MRI) reveals lesions in both the white matter and the paraventricular regions of the brain as well as in the spinal cord. Pathologic examination of the spinal cord shows symmetric degeneration of the lateral columns, including the corticospinal tracts; some cases involve the posterior columns as well. The spinal meninges and cord parenchyma contain an inflammatory infiltrate with myelin destruction.
HTLV-1 is not usually found in cells of the CNS but may be detected in a small population of lymphocytes present in the CSF. In general, HTLV-1 replication is greater in HAM than in ATL, and patients with HAM have a stronger immune response to the virus. Antibodies to HTLV-1 are present in the serum and appear to be produced in the CSF of HAM patients, where titers are often higher than in the serum. The pathophysiology of HAM may involve the induction of autoimmune destruction of neural cells by T cells with specificity for viral components such as Tax or Env proteins. One theory is that susceptibility to HAM may be related to the presence of human leukocyte antigen (HLA) alleles capable of presenting viral antigens in a fashion that leads to autoimmunity. Insufficient data are available to confirm an HLA association. However, antibodies in the sera of HAM patients have been shown to bind a neuron-specific antigen (heteronuclear ribonuclear protein A1 [hnRNP A1]) and to interfere with neurotransmission in vitro.
It is unclear what factors influence whether HTLV-1 infection will cause disease and, if it does, whether it will induce a neoplasm (ATL) or an autoimmune disorder (HAM). Differences in viral strains, the susceptibility of particular MHC haplotypes, the route of HTLV-1 infection, the viral load, and the nature of the HTLV-1-related immune response are putative factors, but few definitive data are available.
OTHER PUTATIVE HTLV-1-RELATED DISEASES Even in the absence of the full clinical picture of HAM, bladder dysfunction is common in HTLV-1-infected women. In areas where HTLV-1 is endemic, diverse inflammatory and autoimmune diseases have been attributed to the virus, including uveitis, dermatitis, pneumonitis, rheumatoid arthritis, and polymyositis. However, a causal relationship between HTLV-1 and these illnesses has not been established.
Prevention Women in endemic areas should not breast-feed their children, and blood donors should be screened for serum antibodies to HTLV-1. As in the prevention of HIV infection, the practice of safe sex and the avoidance of needle sharing are important.
FEATURES OF HTLV-2 INFECTION
![]() Epidemiology HTLV-2 is endemic in certain Native American tribes and in Africa. It is generally considered to be a New World virus that was brought from Asia to the Americas 10,000–40,000 years ago during the migration of infected populations across the Bering land bridge. The mode of transmission of HTLV-2 is probably the same as that of HTLV-1 (see above). HTLV-2 may be less readily transmitted sexually than HTLV-1.
Epidemiology HTLV-2 is endemic in certain Native American tribes and in Africa. It is generally considered to be a New World virus that was brought from Asia to the Americas 10,000–40,000 years ago during the migration of infected populations across the Bering land bridge. The mode of transmission of HTLV-2 is probably the same as that of HTLV-1 (see above). HTLV-2 may be less readily transmitted sexually than HTLV-1.
Studies of large cohorts of injection drug users with serologic assays that reliably distinguish HTLV-1 from HTLV-2 indicated that the vast majority of HTLV-positive cohort members were infected with HTLV-2. The seroprevalence of HTLV in a cohort of 7841 injection drug users from drug treatment centers in Baltimore, Chicago, Los Angeles, New Jersey (Asbury Park and Trenton), New York City (Brooklyn and Harlem), Philadelphia, and San Antonio was 20.9%, with >97% of cases due to HTLV-2. The seroprevalence of HTLV-2 was higher in the Southwest and the Midwest than in the Northeast. In contrast, the seroprevalence of HIV-1 was higher in the Northeast than in the Southwest or the Midwest. Approximately 3% of the cohort members were infected with both HTLV-2 and HIV-1. The seroprevalence of HTLV-2 increased linearly with age. Women were significantly more likely to be infected with HTLV-2 than were men; the virus is thought to be more efficiently transmitted from male to female than from female to male.
Associated Diseases Although HTLV-2 was isolated from a patient with a T cell variant of hairy cell leukemia, this virus has not been consistently associated with a particular disease and in fact has been thought of as “a virus searching for a disease.” However, evidence is accumulating that HTLV-2 may play a role in certain neurologic, hematologic, and dermatologic diseases. These data require confirmation, particularly in light of the previous confusion regarding the relative prevalences of HTLV-1 and HTLV-2 among injection drug users.
Prevention Avoidance of needle sharing, adherence to safe-sex practices, screening of blood (by assays for HTLV-1, which also detect HTLV-2), and avoidance of breast-feeding by infected women are important principles in the prevention of spread of HTLV-2.
HUMAN IMMUNODEFICIENCY VIRUS
HIV-1 and HIV-2 are members of the lentivirus subfamily of Retroviridae and are the only lentiviruses known to infect humans. The lentiviruses are slow-acting by comparison with viruses that cause acute infection (e.g., influenza virus) but not by comparison with other retroviruses. The features of acute primary infection with HIV resemble those of more classic acute infections. The characteristic chronicity of HIV disease is consistent with the designation lentivirus. For a detailed discussion of HIV, see Chap. 226.
226 |
Human Immunodeficiency Virus Disease: AIDS and Related Disorders |
AIDS was first recognized in the United States in the summer of 1981, when the U.S. Centers for Disease Control and Prevention (CDC) reported the unexplained occurrence of Pneumocystis jiroveci (formerly P. carinii) pneumonia in five previously healthy homosexual men in Los Angeles and of Kaposi’s sarcoma (KS) with or without P. jiroveci pneumonia and other opportunistic infections in 26 previously healthy homosexual men in New York, San Francisco, and Los Angeles. The disease was soon recognized in male and female injection drug users; in hemophiliacs and blood transfusion recipients; among female sexual partners of men with AIDS; and among infants born to mothers with AIDS. In 1983, human immunodeficiency virus (HIV) was isolated from a patient with lymphadenopathy, and by 1984 it was demonstrated clearly to be the causative agent of AIDS. In 1985, a sensitive enzyme-linked immunosorbent assay (ELISA) was developed; this led to an appreciation of the scope and evolution of the HIV epidemic at first in the United States and other developed nations and ultimately among developing nations throughout the world (see “HIV Infection and AIDS Worldwide,” below). The staggering worldwide evolution of the HIV pandemic has been matched by an explosion of information in the areas of HIV virology, pathogenesis (both immunologic and virologic), treatment of HIV disease, treatment and prophylaxis of the opportunistic diseases associated with HIV infection, and prevention of HIV infection. The information flow related to HIV disease is enormous and continues to expand, and it has become almost impossible for the health care generalist to stay abreast of the literature. The purpose of this chapter is to present the most current information available on the scope of the epidemic; on its pathogenesis, treatment, and prevention; and on prospects for vaccine development. Above all, the aim is to provide a solid scientific basis and practical clinical guidelines for a state-of-the-art approach to the HIV-infected patient.
DEFINITION
The current U.S. CDC classification system for HIV infection and AIDS categorizes people on the basis of clinical conditions associated with HIV infection and CD4+ T lymphocyte measurement. A confirmed HIV case can be classified in one of five HIV infection stages (0, 1, 2, 3, or unknown). If there was a negative HIV test within 6 months of the first HIV infection diagnosis, the stage is 0, and remains 0 until 6 months after diagnosis. Advanced HIV disease (AIDS) is classified as stage 3 if one or more specific opportunistic illness has been diagnosed (Table 226-1). Otherwise, the stage is determined by CD4 test results and immunologic criteria (Table 226-2). If none of these criteria apply (e.g., because of missing information on CD4 test results), the stage is U (unknown).
|
CDC STAGE 3 (AIDS)-DEFINING OPPORTUNISTIC ILLNESSES IN HIV INFECTION |
aOnly among children age <6 years.b Only among adults, adolescents, and children age ≥6 years.
Source: MMWR 63(No. RR-03), April 11, 2014.
|
CDC HIV INFECTION STAGES 1–3 BASED ON AGE-SPECIFIC CD4+ T LYMPHOCYTE COUNT OR CD4+ T LYMPHOCYTE PERCENTAGE OF TOTAL LYMPHOCYTESa |

The definition and staging criteria of AIDS are complex and comprehensive and were established for surveillance purposes rather than for the practical care of patients. Thus, the clinician should not focus on whether the patient fulfills the strict definition of AIDS, but should view HIV disease as a spectrum ranging from primary infection, with or without the acute syndrome, to the asymptomatic stage, to advanced stages associated with opportunistic diseases (see “Pathophysiology and Pathogenesis,” below).
ETIOLOGIC AGENT
HIV is the etiologic agent of AIDS; it belongs to the family of human retroviruses (Retroviridae) and the subfamily of lentiviruses (Chap. 225e). Nononcogenic lentiviruses cause disease in other animal species, including sheep, horses, goats, cattle, cats, and monkeys. The four retroviruses known to cause human disease belong to two distinct groups: the human T lymphotropic viruses (HTLV)-1 and HTLV-2, which are transforming retroviruses; and the human immunodeficiency viruses, HIV-1 and HIV-2, which cause cytopathic effects either directly or indirectly (Chap. 225e). The most common cause of HIV disease throughout the world, and certainly in the United States, is HIV-1, which comprises several subtypes with different geographic distributions (see “Molecular Heterogeneity of HIV-1,” below). HIV-2 was first identified in 1986 in West African patients and was originally confined to West Africa. However, a number of cases that generally can be traced to West Africa or to sexual contacts with West Africans have been identified throughout the world. The currently defined groups of HIV-1 (M, N, O, P) and the HIV-2 groups A through H each are likely derived from a separate transfer to humans from a nonhuman primate reservoir. HIV-1 viruses likely came from chimpanzees and/or gorillas, and HIV-2 from sooty mangabeys. The AIDS pandemic is primarily caused by the HIV-1 M group viruses. Although HIV-1 group O and HIV-2 viruses have been found in numerous countries, including those in the developed world, they have caused much more localized epidemics. The taxonomic relationship between primate lentiviruses is shown in Fig. 226-1.
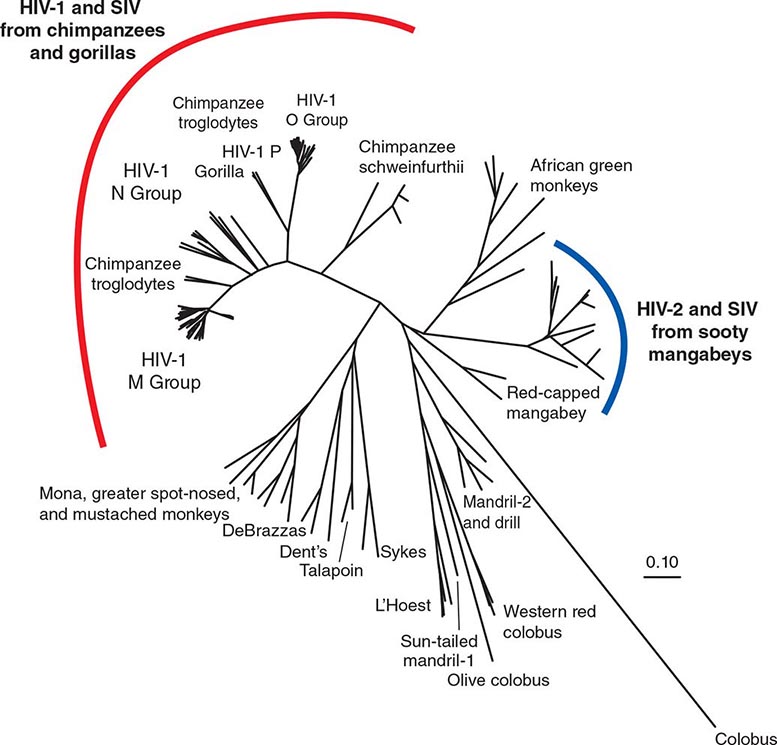
FIGURE 226-1 A phylogenetic tree based on the complete genomes of primate immunodeficiency viruses. The scale (0.10) indicates a 10% difference at the nucleotide level. (Prepared by Brian Foley, PhD, of the HIV Sequence Database, Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; additional information at www.hiv.lanl.gov/content/sequence/HelpDocs/subtypes.html.)
MORPHOLOGY OF HIV
Electron microscopy shows that the HIV virion is an icosahedral structure (Fig. 226-2) containing numerous external spikes formed by the two major envelope proteins, the external gp120 and the transmembrane gp41. The HIV envelope exists as a trimeric heterodimer. The virion buds from the surface of the infected cell and incorporates a variety of host proteins into its lipid bilayer. The structure of HIV-1 is schematically diagrammed in Fig. 226-2B.
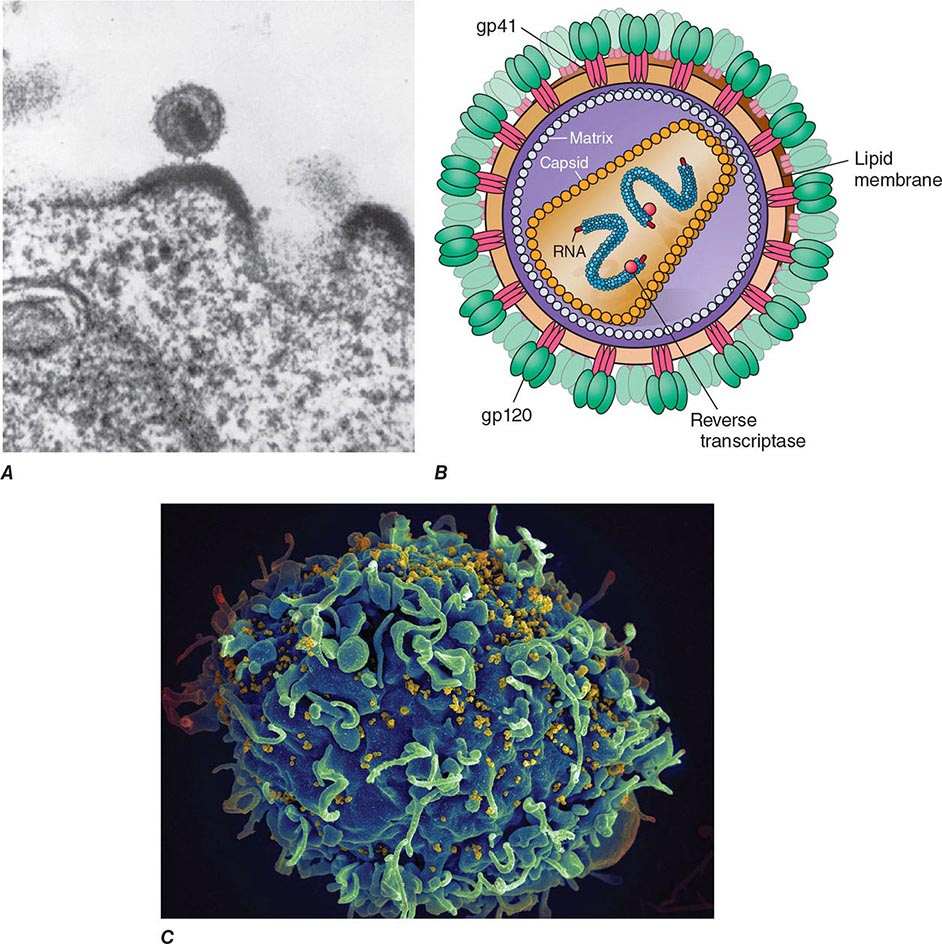
FIGURE 226-2 A. Electron micrograph of HIV. Figure illustrates a typical virion following budding from the surface of a CD4+ T lymphocyte, together with two additional incomplete virions in the process of budding from the cell membrane. B. Structure of HIV-1, including the gp120 envelope, gp41 transmembrane components of the envelope, genomic RNA, enzyme reverse transcriptase, p18(17) inner membrane (matrix), and p24 core protein (capsid). (Copyright by George V. Kelvin.) (Adapted from RC Gallo: Sci Am 256:46, 1987.) C. Scanning electron micrograph of HIV-1 virions infecting a human CD4+ T lymphocyte. The original photograph was imaged at 8000× magnification. (Courtesy of Elizabeth R. Fischer, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases; with permission.)
REPLICATION CYCLE OF HIV
HIV is an RNA virus whose hallmark is the reverse transcription of its genomic RNA to DNA by the enzyme reverse transcriptase. The replication cycle of HIV begins with the high-affinity binding of the gp120 protein via a portion of its V1 region near the N terminus to its receptor on the host cell surface, the CD4 molecule (Fig. 226-3). The CD4 molecule is a 55-kDa protein found predominantly on a subset of T lymphocytes that are responsible for helper function in the immune system (Chap. 372e). It is also expressed on the surface of monocytes/macrophages and dendritic/Langerhans cells. Once it binds to CD4, the gp120 protein undergoes a conformational change that facilitates binding to one of two major co-receptors. The two major co-receptors for HIV-1 are CCR5 and CXCR4. Both receptors belong to the family of seven-transmembrane-domain G protein–coupled cellular receptors, and the use of one or the other or both receptors by the virus for entry into the cell is an important determinant of the cellular tropism of the virus. Certain dendritic cells (DCs) express a diversity of C-type lectin receptors on their surface—one of which is called DC-SIGN—that also bind with high affinity to the HIV gp120 envelope protein, allowing DCs to facilitate virus spread to CD4+ T cells. Following binding of the envelope protein to the CD4 molecule associated with the above-mentioned conformational change in the viral envelope gp120, fusion with the host cell membrane occurs via the newly exposed gp41 molecule penetrating the plasma membrane of the target cell and then coiling upon itself to bring the virion and target cell together (Fig. 226-4). Following fusion, uncoating of the capsid protein shell is initiated—a step that facilitates reverse transcription and leads to formation of the preintegration complex, composed of viral RNA, enzymes, and accessory proteins and surrounded by capsid and matrix proteins (Fig. 226-3). As the preintegration complex traverses the cytoplasm to reach the nucleus, the viral reverse transcriptase enzyme catalyzes the reverse transcription of the genomic RNA into DNA, resulting in the formation of double-stranded proviral HIV-DNA. At the preintegration steps of the replication cycle, the viral genome is vulnerable to cellular factors that can block the progression of infection. In particular, the cytoplasmic tripartite motif-containing protein 5-α (TRIM5-α) is a host restriction factor that interacts with retroviral capsids (Fig. 226-3). Although the exact mechanisms of action of TRIM5-α remain unclear, the HIV-1 capsid is not recognized by the human form of TRIM5-α. Thus this host factor is not effective in restricting HIV-1 replication in human cells. The apolipoprotein B mRNA editing enzyme (catalytic polypeptide-like 3 [APOBEC3]) family of cellular proteins also inhibits progression of virus infection after virus has entered the cell and prior to entering the nucleus (Fig. 226-3). APOBEC3 proteins, which are incorporated into virions and released into the cytoplasm of a newly infected cell, bind to the single minus-strand DNA intermediate and deaminate viral cytidine, causing hypermutation of retroviral genomes. HIV has evolved a powerful strategy to protect itself from APOBEC. The viral protein Vif targets APOBEC3 for proteasomal degradation.
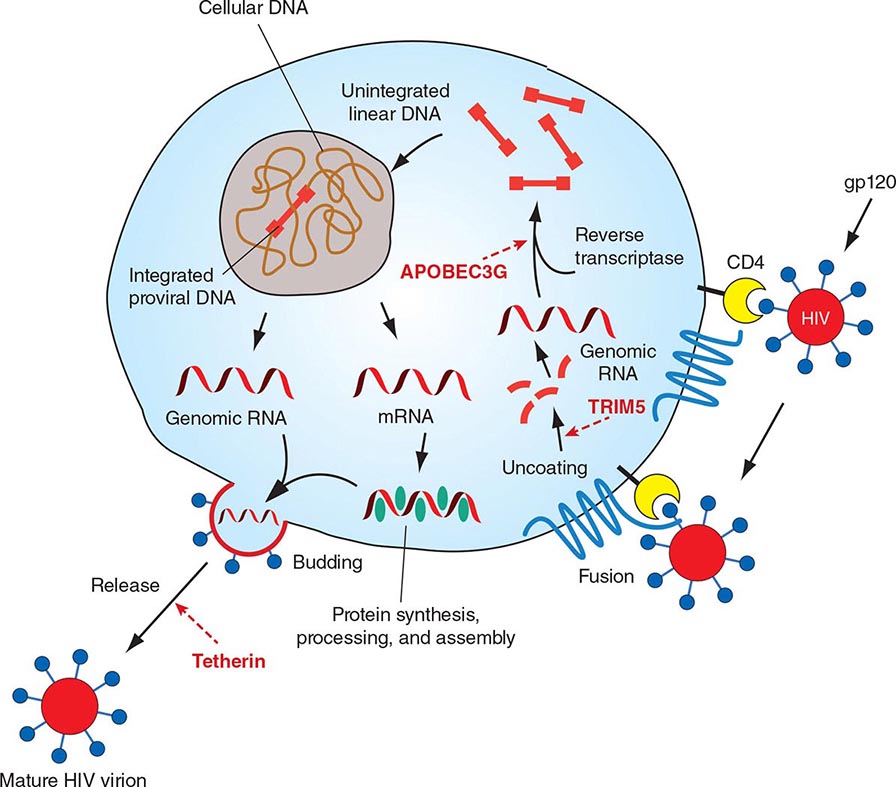
FIGURE 226-3 The replication cycle of HIV. See text for description. (Adapted from AS Fauci: Nature 384:529, 1996.)
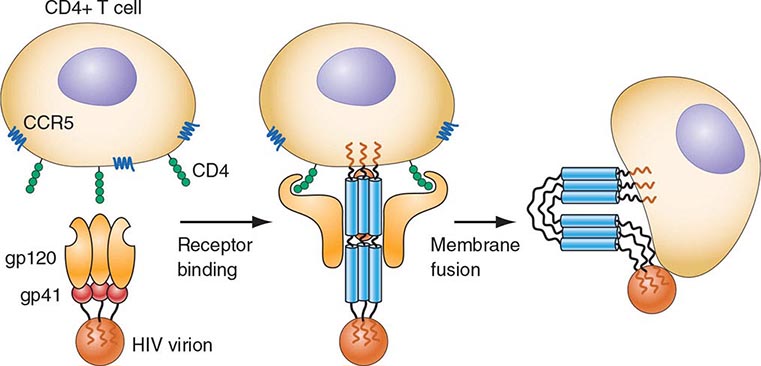
FIGURE 226-4 Binding and fusion of HIV-1 with its target cell. HIV-1 binds to its target cell via the CD4 molecule, leading to a conformational change in the gp120 molecule that allows it to bind to the co-receptor CCR5 (for R5-using viruses). The virus then firmly attaches to the host cell membrane in a coiled-spring fashion via the newly exposed gp41 molecule. Virus-cell fusion occurs as the transitional intermediate of gp41 undergoes further changes to form a hairpin structure that draws the two membranes into close proximity (see text for details). (Adapted from D Montefiori, JP Moore: Science 283:336, 1999; with permission.)
With activation of the cell, the viral DNA accesses the nuclear pore and is exported from the cytoplasm to the nucleus, where it is integrated into the host cell chromosomes through the action of another virally encoded enzyme, integrase (Fig. 226-3). HIV provirus (DNA) integrates into the nuclear DNA preferentially within introns of active genes and regional hotspots. This provirus may remain transcriptionally inactive (latent) or it may manifest varying levels of gene expression, up to active production of virus.
Cellular activation plays an important role in the replication cycle of HIV and is critical to the pathogenesis of HIV disease (see “Pathogenesis and Pathophysiology,” below). Following initial binding, fusion, and internalization of the nucleic acid contents of virions into the target cell, incompletely reverse-transcribed DNA intermediates are labile in quiescent cells and do not integrate efficiently into the host cell genome unless cellular activation occurs shortly after infection. Furthermore, some degree of activation of the host cell is required for the initiation of transcription of the integrated proviral DNA into either genomic RNA or mRNA. This latter process may not necessarily be associated with the detectable expression of the classic cell-surface markers of activation. In this regard, activation of HIV expression from the latent state depends on the interaction of a number of cellular and viral factors. Following transcription, HIV mRNA is translated into proteins that undergo modification through glycosylation, myristoylation, phosphorylation, and cleavage. The viral particle is formed by the assembly of HIV proteins, enzymes, and genomic RNA at the plasma membrane of the cells. Budding of the progeny virion through the lipid bilayer of the host cell membrane is the point at which the core acquires its external envelope and where the host restriction factor tetherin can inhibit the release of budding particles (Fig. 226-3). Tetherin is an interferon (IFN)-induced type II transmembrane protein that interferes with virion detachment, although the HIV accessory protein Vpu counteracts the effect through direct interactions with tetherin. During or soon after budding, the virally encoded protease catalyzes the cleavage of the gag-pol precursor to yield the mature virion. Progression through the virus replication cycle is profoundly influenced by a variety of viral regulatory gene products. Likewise, each point in the replication cycle of HIV is a real or potential target for therapeutic intervention. Thus far, the reverse transcriptase, protease, and integrase enzymes as well as the process of virus–target cell binding and fusion have proved clinically to be susceptible to pharmacologic disruption.
HIV GENOME
Figure 226-5 illustrates schematically the arrangement of the HIV genome. Like other retroviruses, HIV-1 has genes that encode the structural proteins of the virus: gag encodes the proteins that form the core of the virion (including p24 antigen); pol encodes the enzymes responsible for protease processing of viral proteins, reverse transcription, and integration; and env encodes the envelope glycoproteins. However, HIV-1 is more complex than other retroviruses, particularly those of the nonprimate group, in that it also contains at least six other genes (tat, rev, nef, vif, vpr, and vpu), which code for proteins involved in the modification of the host cell to enhance virus growth and the regulation of viral gene expression. Several of these proteins are thought to play a role in the pathogenesis of HIV disease; their various functions are listed in Fig. 226-5. Flanking these genes are the long terminal repeats (LTRs), which contain regulatory elements involved in gene expression (Fig. 226-5). The major difference between the genomes of HIV-1 and HIV-2 is the fact that HIV-2 lacks the vpu gene and has a vpx gene not contained in HIV-1.
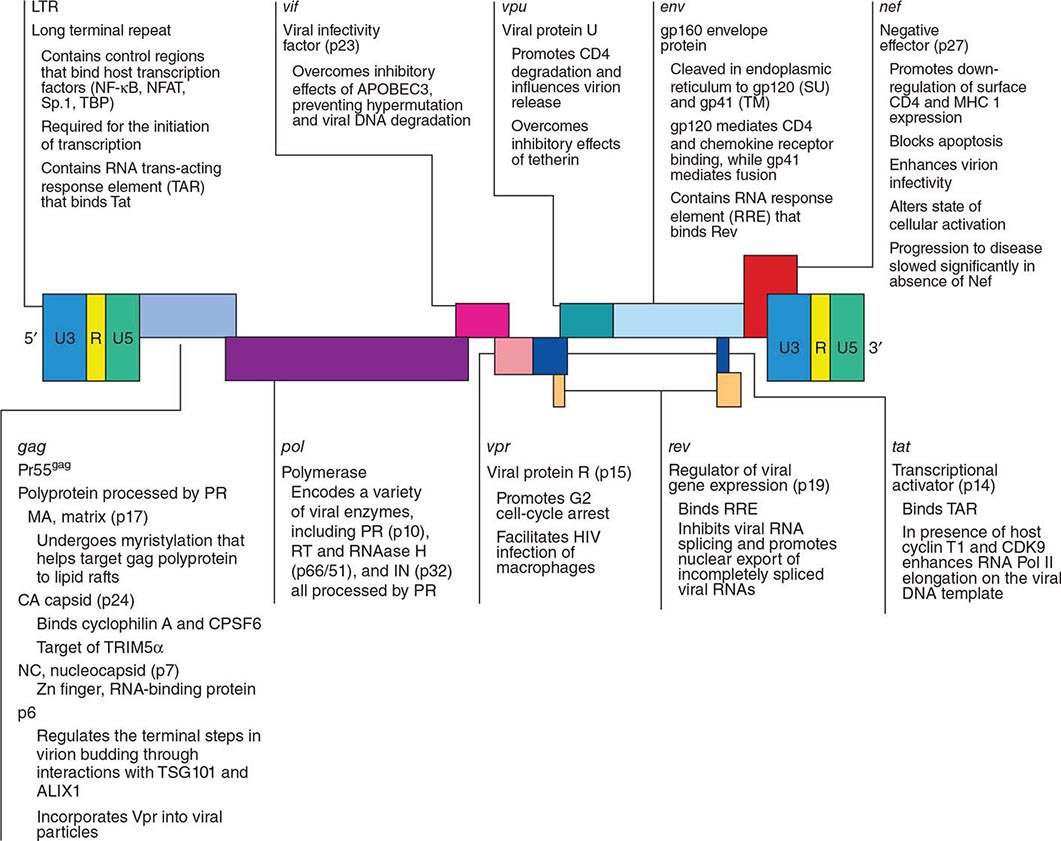
FIGURE 226-5 Organization of the genome of the HIV provirus together with a summary description of its 9 genes encoding 15 proteins. (Adapted from WC Greene, BM Peterlin: Nat Med 8:673, 2002.)
MOLECULAR HETEROGENEITY OF HIV-1
![]() Molecular analyses of HIV isolates reveal varying levels of sequence diversity over all regions of the viral genome. For example, the degree of difference in the coding sequences of the viral envelope protein ranges from a few percent (very close, among isolates from the same infected individual) to more than 50% (extreme diversity, between isolates from the different groups of HIV-1: M, N, O, and P). The changes tend to cluster in hypervariable regions. HIV can evolve by several means, including simple base substitution, insertions and deletions, recombination, and gain and loss of glycosylation sites. HIV sequence diversity arises directly from the limited fidelity of the reverse transcriptase. The balance of immune pressure and functional constraints on proteins influences the regional level of variation within proteins. For example, Envelope, which is exposed on the surface of the virion and is under immune selective pressure from both antibodies and cytolytic T lymphocytes, is extremely variable, with clusters of mutations in hypervariable domains. In contrast, reverse transcriptase, with important enzymatic functions, is relatively conserved, particularly around the active site. The extraordinary variability of HIV-1 contrasts markedly with the relative stability of HTLV-1 and -2.
Molecular analyses of HIV isolates reveal varying levels of sequence diversity over all regions of the viral genome. For example, the degree of difference in the coding sequences of the viral envelope protein ranges from a few percent (very close, among isolates from the same infected individual) to more than 50% (extreme diversity, between isolates from the different groups of HIV-1: M, N, O, and P). The changes tend to cluster in hypervariable regions. HIV can evolve by several means, including simple base substitution, insertions and deletions, recombination, and gain and loss of glycosylation sites. HIV sequence diversity arises directly from the limited fidelity of the reverse transcriptase. The balance of immune pressure and functional constraints on proteins influences the regional level of variation within proteins. For example, Envelope, which is exposed on the surface of the virion and is under immune selective pressure from both antibodies and cytolytic T lymphocytes, is extremely variable, with clusters of mutations in hypervariable domains. In contrast, reverse transcriptase, with important enzymatic functions, is relatively conserved, particularly around the active site. The extraordinary variability of HIV-1 contrasts markedly with the relative stability of HTLV-1 and -2.
The four groups (M, N, O and P) of HIV-1 are the result of four separate chimpanzee-to-human (or possibly gorilla-to-human for groups O and P) transfers. Group M (major), which is responsible for most of the infections in the world, has diversified into subtypes and intersubtype recombinant forms, due to “sub-epidemics” within humans after one of those transfers.
Among primate lentiviruses, HIV-1 is most closely related to viruses isolated from chimpanzees and gorillas (Fig. 226-1). The chimpanzee subspecies Pan troglodytes troglodytes has been established to be the natural reservoir of the HIV-1 M and N groups. The rare viruses of the HIV-1 O and P groups are most closely related to viruses found in Cameroonian gorillas. The M group comprises nine subtypes, or clades, designated A, B, C, D, F, G, H, J, and K, as well as more than 60 known circulating recombinant forms (CRFs) and numerous unique recombinant forms. Intersubtype recombinants are generated by infection of an individual with two subtypes that then recombine and create a virus with a selective advantage. These CRFs range from highly prevalent forms such as CRF01_AE, common in southeast Asia, and CRF02_AG from west and central Africa, to a large number of CRFs that are relatively rare, either because they are of a more recent origin (newly recombined) or because they have not broken out into a major population. The subtypes and CRFs create the major lineages of the M group of HIV-1. HIV-1 M group subtype C dominates the global pandemic, and there is much speculation that it is more transmissible than other subtypes, but solid data on transmissibility variations between subtypes are rare. Human population densities, access to prevention and treatment, prevalence of genital ulcers, iatrogenic transmissions, and other confounding host factors are all possible reasons why one subtype has spread more than another.
Figure 226-6 schematically diagrams the worldwide distribution of HIV-1 subtypes by region. Seven strains account for the vast majority of HIV infections globally: HIV-1 subtypes A, B, C, D, G and two of the CRFs, CRF01_AE and CRF02_AG. Subtype C viruses (of the M group) are by far the most common form worldwide, likely accounting for ~50% of prevalent infections worldwide. In sub-Saharan Africa, home to approximately two-thirds of all individuals living with HIV/AIDS, the majority of infections are caused by subtype C, with smaller proportions of infections caused by subtype A, subtype G, CRF02_AG, and other subtypes and recombinants. In South Africa, the country with the largest number of prevalent infections (6.3 million in 2013), >97% of the HIV-1 isolates sequenced are of subtype C. In Asia, HIV-1 isolates of the CRF01_AE lineage and subtypes C and B predominate. CRF01_AE accounts for most infections in south and southeast Asia, while >95% of infections in India, home to an estimated 2.1 million HIV-infected individuals, are of subtype C (see “HIV Infection and AIDS Worldwide,” below). Subtype B viruses are the overwhelmingly predominant viruses seen in the United States, Canada, certain countries in South America, western Europe, and Australia. It is thought that, purely by chance, subtype B was seeded into the United States and Europe in the late 1970s, thereby establishing an overwhelming founder effect. Many countries have co-circulating viral subtypes that are giving rise to new CRFs. Sequence analyses of HIV-1 isolates from infected individuals indicate that recombination among viruses of different clades likely occurs as a result of infection of an individual with viruses of more than one subtype, particularly in geographic areas where subtypes overlap, and more often in sub-epidemics driven by IV drug use than in those driven by sexual transmission.
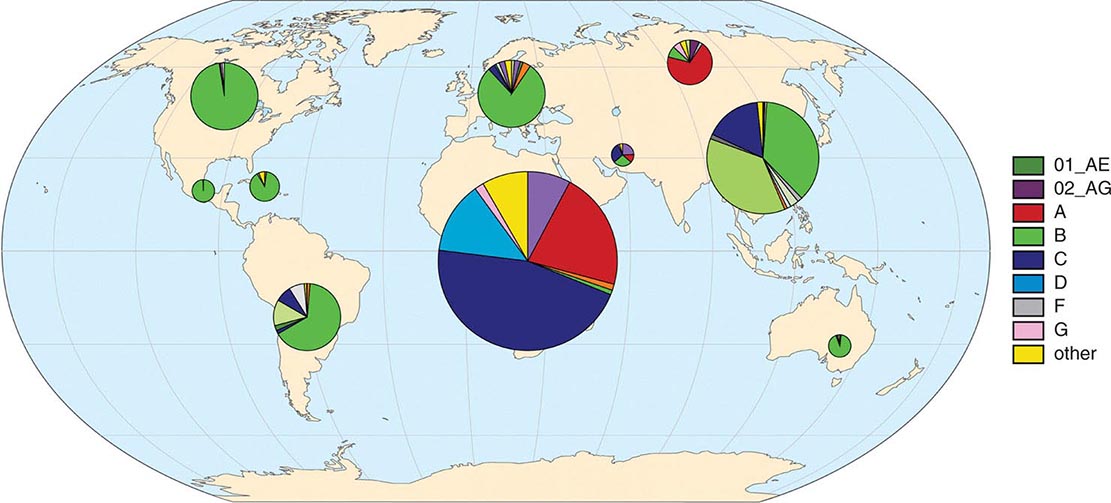
FIGURE 226-6 Global geographic distribution of HIV-1 subtypes and recombinant forms. Distributions derived from relative frequency of subtypes among >500,000 HIV genomic sequences in the Los Alamos National Laboratory HIV Sequence Database. (Additional information available at www.hiv.lanl.gov/components/sequence/HIV/geo/geo.comp)
The extraordinary diversity of HIV, reflected by the presence of multiple subtypes, circulating recombinant forms, and continuous viral evolution, has implications for possible differential rates of transmission, rates of disease progression, responses to therapy, and the development of resistance to antiretroviral drugs. This diversity is also a formidable obstacle to HIV vaccine development, as a broadly useful vaccine would need to induce protective responses against a wide range of viral strains.
TRANSMISSION
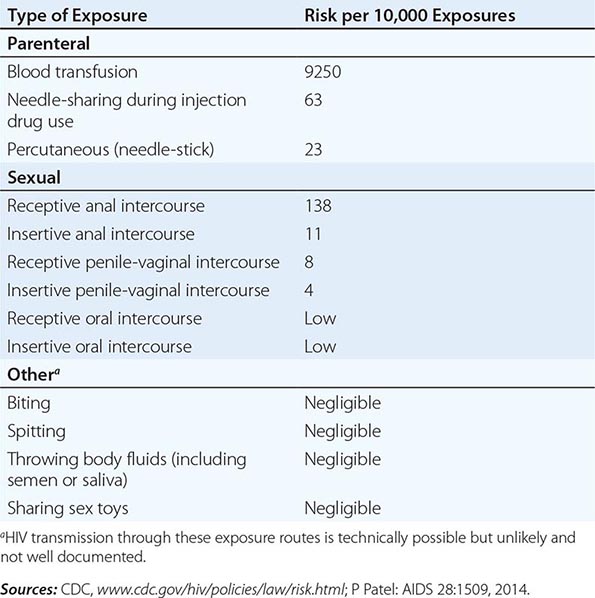
HIV is transmitted primarily by sexual contact (both heterosexual and male to male); by blood and blood products; and by infected mothers to infants intrapartum, perinatally, or via breast milk. After more than 30 years of experience and observations regarding other potential modalities of transmission, there is no evidence that HIV is transmitted by casual contact or that the virus can be spread by insects, such as by a mosquito bite. Table 226-3 lists the estimated risk of HIV transmission for various types of exposures.
|
ESTIMATED PER-ACT PROBABILITY OF ACQUIRING HIV FROM AN INFECTED SOURCE, BY EXPOSURE ACT |
SEXUAL TRANSMISSION
HIV infection is predominantly a sexually transmitted infection (STI) worldwide. By far the most common mode of infection, particularly in developing countries, is heterosexual transmission, although in many western countries a resurgence of male-to-male sexual transmission has occurred. Although a wide variety of factors including viral load and the presence of ulcerative genital diseases influence the efficiency of heterosexual transmission of HIV, such transmission is generally inefficient. A recent systemic review found a low per-act risk of heterosexual transmission in the absence of antiretrovirals: 0.04% for female-to-male transmission and 0.08% for male-to-female transmission during vaginal intercourse in the absence of antiretroviral therapy or condom use (Table 226-3).
HIV has been demonstrated in seminal fluid both within infected mononuclear cells and in cell-free material. The virus appears to concentrate in the seminal fluid, particularly in situations where there are increased numbers of lymphocytes and monocytes in the fluid, as in genital inflammatory states such as urethritis and epididymitis, conditions closely associated with other STIs. The virus has also been demonstrated in cervical smears and vaginal fluid. There is an elevated risk of HIV transmission associated with unprotected receptive anal intercourse (URAI) among both men and women compared to the risk associated with receptive vaginal intercourse. Although data are limited, the per-act risk for HIV transmission via URAI has been estimated to be ~1.4% (Table 226-3). The risk of HIV acquisition associated with URAI is probably higher than that seen in penile-vaginal intercourse because only a thin, fragile rectal mucosal membrane separates the deposited semen from potentially susceptible cells in and beneath the mucosa, and micro-trauma of the mucosal membrane may be associated with anal intercourse. Anal douching and sexual practices that traumatize the rectal mucosa also increase the likelihood of infection. It is likely that anal intercourse provides at least two modalities of infection: (1) direct inoculation into blood in cases of traumatic tears in the mucosa; and (2) infection of susceptible target cells, such as Langerhans cells, in the mucosal layer in the absence of trauma. Insertive anal intercourse also confers an increased risk of HIV acquisition compared to insertive vaginal intercourse. Although the vaginal mucosa is several layers thicker than the rectal mucosa and less likely to be traumatized during intercourse, the virus can be transmitted to either partner through vaginal intercourse. As noted in Table 226-3, male-to-female HIV transmission is usually more efficient than female-to-male transmission. The differences in reported transmission rates between men and women may be due in part to the prolonged exposure to infected seminal fluid of the vaginal and cervical mucosa, as well as the endometrium (when semen enters through the cervical os). By comparison, the penis and urethral orifice are exposed relatively briefly to infected vaginal fluid. Among various cofactors examined in studies of heterosexual HIV transmission, the presence of other STIs has been strongly associated with HIV transmission. In this regard, there is a close association between genital ulcerations and transmission, owing to both susceptibility to infection and infectivity. Infections with microorganisms such as Treponema pallidum (Chap. 206), Haemophilus ducreyi (Chap. 182), and herpes simplex virus (HSV; Chap. 216) are important causes of genital ulcerations linked to transmission of HIV. In addition, pathogens responsible for non-ulcerative inflammatory STIs such as those caused by Chlamydia trachomatis (Chap. 213), Neisseria gonorrhoeae (Chap. 181), and Trichomonas vaginalis (Chap. 254) also are associated with an increased risk of transmission of HIV infection. Bacterial vaginosis, an infection related to sexual behavior, but not strictly an STI, also may be linked to an increased risk of transmission of HIV infection. Several studies suggest that treating other STIs and genital tract syndromes may help prevent transmission of HIV. This effect is most prominent in populations in which the prevalence of HIV infection is relatively low. It is noteworthy that this principle may not apply to the treatment of HSV infections since it has been shown that even following anti-HSV therapy with resulting healing of HSV-related genital ulcers, HIV acquisition is not reduced. Biopsy studies revealed the likely explanation is that HIV receptor–positive inflammatory cells persisted in the genital tissue despite the healing of ulcers, and so HIV-susceptible targets remained at the site.
The quantity of HIV-1 in plasma is a primary determinant of the risk of HIV-1 transmission. In a cohort of heterosexual couples in Uganda discordant for HIV infection and not receiving antiretroviral therapy, the mean serum HIV RNA level was significantly higher among HIV-infected subjects whose partners seroconverted than among those whose partners did not seroconvert. In fact, transmission was rare when the infected partner had a plasma level of <1700 copies of HIV RNA per milliliter, even when genital ulcer disease was present (Fig. 226-7). The rate of HIV transmission per coital act was highest during the early stage of HIV infection when plasma HIV RNA levels were high and in advanced disease as the viral set point increased.
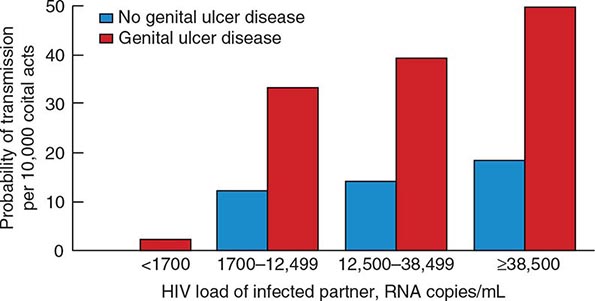
FIGURE 226-7 Probability of HIV transmission per coital act among monogamous, heterosexual, HIV-serodiscordant couples in Uganda. (From RH Gray et al: Lancet 357:1149, 2001.)
Antiretroviral therapy dramatically reduces plasma viremia in most HIV-infected individuals (see “Treatment,” below) and is associated with a reduction in risk of transmission. In a large study of serodiscordant couples, earlier treatment of the HIV-infected partner with antiretroviral therapy rather than treatment delayed until the CD4+ T cells count fell below 250 cells per μL was associated with a 96% reduction in HIV transmission to the uninfected partner. This approach has been widely referred to as treatment as prevention or TasP. Several studies also have suggested a beneficial effect of antiretroviral treatment at the community level.
A number of studies including large, randomized, controlled trials clearly have indicated that male circumcision is associated with a lower risk of acquisition of HIV infection for heterosexual men. Studies are conflicting as to whether circumcision protects against HIV acquisition among men who have sex with men, but data suggest that circumcision is protective in those men who have sex with men who are insertive only. The benefit of circumcision may be due to increased susceptibility of uncircumcised men to ulcerative STIs, as well as to other factors such as microtrauma to the foreskin and glans penis. In addition, the highly vascularized inner foreskin tissue contains a high density of Langerhans cells as well as increased numbers of CD4+ T cells, macrophages, and other cellular targets for HIV. Finally, the moist environment under the foreskin may promote the presence or persistence of microbial flora that, via inflammatory changes, may lead to even higher concentrations of target cells for HIV in the foreskin. In addition, randomized trials have demonstrated that male circumcision also reduces hepatitis C virus (HCV) type 2, human papillomavirus virus (HPV), and genital ulcer disease in men as well as HPV, genital ulcer disease, bacterial vaginosis, and Trichomonas vaginalis infections among female partners of circumcised men. Thus, there may be an added benefit of diminution of risk for HIV acquisition to the female sexual partners of circumcised men.
In some studies the use of oral contraceptives was associated with an increase in incidence of HIV infection over and above that which might be expected by not using a condom for birth control. This phenomenon may be due to drug-induced changes in the cervical mucosa, rendering it more vulnerable to penetration by the virus. Adolescent girls might also be more susceptible to infection upon exposure due to the properties of an immature genital tract with increased cervical ectopy or exposed columnar epithelium.
Oral sex is a much less efficient mode of transmission of HIV than is anal intercourse or vaginal intercourse (Table 226-3). A number of studies have reported that the incidence of transmission of infection by oral sex among couples discordant for HIV was extremely low. However, there have been well-documented reports of HIV transmission that likely resulted from fellatio or cunnilingus. Therefore, the assumption that oral sex is completely safe is not warranted.
The association of alcohol consumption and illicit drug use with unsafe sexual behavior, both homosexual and heterosexual, leads to an increased risk of sexual transmission of HIV. Methamphetamine and other so-called club drugs (e.g., ecstasy, ketamine, and gamma hydroxybutyrate), sometimes taken in conjunction with PDE-5 inhibitors such as sildenafil (Viagra), tadalafil (Cialis), or vardenafil (Levitra), have been associated with risky sexual practices and increased risk of HIV infection, particularly among men who have sex with men.
TRANSMISSION THROUGH INJECTION DRUG USE
HIV can be transmitted to injection drug users (IDUs) who are exposed to HIV while sharing injection paraphernalia such as needles, syringes, the water in which drugs are mixed, or the cotton through which drugs are filtered. Parenteral transmission of HIV during injection drug use does not require IV puncture; SC (“skin popping”) or IM (“muscling”) injections can transmit HIV as well, even though these behaviors are sometimes erroneously perceived as low-risk. Among IDUs, the risk of HIV infection increases with the duration of injection drug use; the frequency of needle sharing; the number of partners with whom paraphernalia are shared, particularly in the setting of “shooting galleries” where drugs are sold and large numbers of IDUs may share a limited number of “works”; comorbid psychiatric conditions such as antisocial personality disorder; the use of cocaine in injectable form or smoked as “crack”; and the use of injection drugs in a geographic location with a high prevalence of HIV infection, such as certain inner-city areas in the United States. As noted in Table 226-3, the per-act risk of transmission from injection drug use with a contaminated needle has been estimated to be approximately 0.6%.
TRANSMISSION BY TRANSFUSED BLOOD AND BLOOD PRODUCTS
HIV can be transmitted to individuals who receive HIV-tainted blood transfusions, blood products, or transplanted tissue. The first cases of AIDS among transfusion recipients and individuals with hemophilia or other clotting disorders were reported in 1982. The vast majority of HIV infections acquired via contaminated blood transfusions, blood components, or transplanted tissue in resource-rich countries occurred prior to the spring of 1985, when mandatory testing of donated blood for HIV-1 was initiated. It is estimated that >90% of individuals exposed to HIV-contaminated blood products become infected (Table 226-3). Although blood screening for HIV is becoming more universal even in the developing world, unfortunately, in some resource-poor countries, HIV continues to be transmitted by blood, blood products, and tissues due to inadequate screening. Transfusions of whole blood, packed red blood cells, platelets, leukocytes, and plasma are all capable of transmitting HIV infection. In contrast, hyperimmune gamma globulin, hepatitis B immune globulin, plasma-derived hepatitis B vaccine, and Rho immune globulin have not been associated with transmission of HIV infection. The procedures involved in processing these products either inactivate or remove the virus.
Currently, in the United States and in most developed countries, the following measures have made the risk of transmission of HIV infection by transfused blood or blood products extremely small: the screening of blood donations for antibodies to HIV-1 and HIV-2 and determination of the presence of HIV nucleic acid usually in minipools of several specimens; the careful selection of potential blood donors with health history questionnaires to exclude individuals with risk behavior; and opportunities for self-deferral and the screening out of HIV-negative individuals with serologic testing for infections that have shared risk factors with HIV, such as hepatitis B and C and syphilis. The chance of infection of a hemophiliac via clotting factor concentrates has essentially been eliminated because of the added layer of safety resulting from heat treatment of the concentrates. It is currently estimated that the risk of infection with HIV in the United States via transfused screened blood is approximately 1 in 2 million units. Therefore, since ~16 million donations are collected in the United States each year, despite the best efforts of science, one cannot completely eliminate the risk of transfusion-related transmission of HIV. In this regard, a case of transfusion-related transmission of HIV was reported in the United States in 2010, which was tracked to a blood donation in 2008; this was the first such reported case since 2002. Transmission of HIV (both HIV-1 and HIV-2) by blood or blood products is still an ongoing threat in certain developing countries, particularly in sub-Saharan Africa, where routine screening of blood is not universally practiced. In other countries, there have been reports of sporadic breakdowns in routinely available screening procedures in which contaminated blood was allowed to be transfused, resulting in small clusters of patients becoming infected. For example, in China in the 1990s, a disturbingly large number of persons became infected by selling blood in situations where the collectors reused needles that were contaminated and, in some instances, mixed blood products from a number of individuals, separated the plasma, and reinfused mixed red blood cells back into the individual donors.
OCCUPATIONAL TRANSMISSION OF HIV: HEALTH CARE WORKERS, LABORATORY WORKERS, AND THE HEALTH CARE SETTING
There is a small but definite occupational risk of HIV transmission to health care workers and laboratory personnel and potentially others who work with HIV-containing materials, particularly when sharp objects are used. An estimated 600,000 to 800,000 health care workers are stuck with needles or other sharp medical instruments in the United States each year. The global number of HIV infections among health care workers attributable to sharps injuries has been estimated to be 1000 cases (range, 200–5000) per year. As of 2010, there had been 57 documented cases of occupational HIV transmission to health care workers in the United States and 143 possible transmissions. There have been no confirmed cases reported since 1999.
Exposures that place a health care worker at potential risk of HIV infection are percutaneous injuries (e.g., a needle stick or cut with a sharp object) or contact of mucous membrane or nonintact skin (e.g., exposed skin that is chapped, abraded, or afflicted with dermatitis) with blood, tissue, or other potentially infectious body fluids. Large, multi-institutional studies have indicated that the risk of HIV transmission following skin puncture from a needle or a sharp object that was contaminated with blood from a person with documented HIV infection is ~0.3% and after a mucous membrane exposure it is 0.09% (see “HIV and the Health Care Worker,” below) if the injured and/or exposed person is not treated within 24 h with antiretroviral drugs. The risk of hepatitis B virus (HBV) infection following a similar type of exposure is ~6–30% in nonimmune individuals; if a susceptible worker is exposed to HBV, postexposure prophylaxis with hepatitis B immune globulin and initiation of HBV vaccine is >90% effective in preventing HBV infection. The risk of HCV infection following percutaneous injury is ~1.8% (Chap. 360).
Rare HIV transmission after nonintact skin exposure has been documented, but the average risk for transmission by this route has not been precisely determined; however, it is estimated to be less than the risk for mucous membrane exposure. Transmission of HIV through intact skin has not been documented. Currently in developed countries, virtually all puncture wounds and mucous membrane exposures in health care workers involving blood from a patient with documented HIV infection are treated prophylactically with combination antiretroviral therapy (cART). This practice, referred to as postexposure prophylaxis or PEP, has dramatically reduced the occurrence of puncture-related transmissions of HIV to health care workers.
In addition to blood and visibly bloody body fluids, semen and vaginal secretions also are considered potentially infectious; however, they have not been implicated in occupational transmission from patients to health care workers. The following fluids also are considered potentially infectious: cerebrospinal fluid, synovial fluid, pleural fluid, peritoneal fluid, pericardial fluid, and amniotic fluid. The risk for transmission after exposure to fluids or tissues other than HIV-infected blood also has not been quantified, but it is probably considerably lower than the risk after blood exposures. Feces, nasal secretions, saliva, sputum, sweat, tears, urine, and vomitus are not considered potentially infectious for HIV unless they are visibly bloody. Rare cases of HIV transmission via human bites have been reported, but not in the setting of occupational exposure.
An increased risk for HIV infection following percutaneous exposures to HIV-infected blood is associated with exposures involving a relatively large quantity of blood, as in the case of a device visibly contaminated with the patient’s blood, a procedure that involves a hollow-bore needle placed directly in a vein or artery, or a deep injury. Factors that might be associated with mucocutaneous transmission of HIV include exposure to an unusually large volume of blood and prolonged contact. In addition, the risk increases for exposures to blood from untreated patients with advanced-stage disease or those patients in the acute stage of HIV infection, owing to the higher levels of HIV in the blood under those circumstances. Since the beginning of the HIV epidemic, there have been rare instances where transmission of infection from a health care worker to patients seemed highly probable. Despite these small number of documented cases, the risk of HIV transmission involving health care workers (infected or not) to patients is extremely low in developed countries—in fact, too low to be measured accurately. In this regard, several epidemiologic studies have been performed tracing thousands of patients of HIV-infected dentists, physicians, surgeons, obstetricians, and gynecologists, and no other cases of HIV transmission that could be linked to the health care providers were identified.
Breaches in infection control and the reuse of contaminated syringes, failure to properly sterilize surgical instruments, and/or hemodialysis equipment have also resulted rarely in the transmission of HIV from patient to patient in hospitals, nursing homes, and outpatient settings. Finally, these very rare occurrences of transmission of HIV as well as HBV and HCV to and from health care workers in the workplace underscore the importance of the use of universal precautions when caring for all patients (see below and Chap. 168).
MOTHER-TO-CHILD TRANSMISSION OF HIV
HIV infection can be transmitted from an infected mother to her fetus during pregnancy, during delivery, or by breast-feeding. This remains an important form of transmission of HIV infection in certain developing countries, where the proportion of infected women to infected men is ~1:1. Virologic analyses of aborted fetuses indicate that HIV can be transmitted to the fetus during the first or second trimesters of pregnancy. However, maternal transmission to the fetus occurs most commonly in the perinatal period. Two studies performed in Rwanda and the Democratic Republic of Congo (then called Zaire) indicated that among all mother-to-child transmissions of HIV, the relative proportions were 23–30% before birth, 50–65% during birth, and 12–20% via breast-feeding.
In the absence of prophylactic antiretroviral therapy to the mother during pregnancy, labor, and delivery, and to the fetus following birth, the probability of transmission of HIV from mother to infant/fetus ranges from 15% to 25% in industrialized countries and from 25% to 35% in developing countries. These differences may relate to the adequacy of prenatal care as well as to the stage of HIV disease and the general health of the mother during pregnancy. Higher rates of transmission have been reported to be associated with many factors—the best documented of which is the presence of high maternal levels of plasma viremia, with the risk increasing linearly with the level of maternal plasma viremia. It is very unlikely that mother-to-child transmission will occur if the mother’s level of plasma viremia is <1000 copies of HIV RNA/mL of blood and extremely unlikely if the level is undetectable (i.e., <50 copies/mL). However, there may not be a lower “threshold” below which transmission never occurs, since certain studies have reported rare transmission by women with viral RNA levels <50 copies/mL. Increased mother-to-child transmission is also correlated with closer human leukocyte antigen (HLA) match between mother and child. A prolonged interval between membrane rupture and delivery is another well-documented risk factor for transmission. Other conditions that are potential risk factors, but that have not been consistently demonstrated, are the presence of chorioamnionitis at delivery; STIs during pregnancy; illicit drug use during pregnancy; cigarette smoking; preterm delivery; and obstetric procedures such as amniocentesis, amnioscopy, fetal scalp electrodes, and episiotomy. In a seminal study conducted in the United States and France in the 1990s, zidovudine treatment of HIV-infected pregnant women from the beginning of the second trimester through delivery and of the infant for 6 weeks following birth dramatically decreased the rate of intrapartum and perinatal transmission of HIV infection from 22.6% in the untreated group to <5%. Today, the rate of mother-to-child transmission has fallen to 1% or less in pregnant women who are receiving combination antiretroviral therapy (cART) for their HIV infection. Such treatment, combined with cesarean section delivery, has rendered mother-to-child transmission of HIV an unusual event in the United States and other developed nations. In this regard, both the United States Public Health Service and the World Health Organization guidelines recommend that all pregnant HIV-infected women should receive cART for the health of the mother and to prevent perinatal transmission regardless of plasma HIV RNA copy number or CD4+ T cell counts.
Breast-feeding is an important modality of transmission of HIV infection in developing countries, particularly where mothers continue to breast-feed for prolonged periods. The risk factors for mother-to-child transmission of HIV via breast-feeding are not fully understood; factors that increase the likelihood of transmission include detectable levels of HIV in breast milk, the presence of mastitis, low maternal CD4+ T cell counts, and maternal vitamin A deficiency. The risk of HIV infection via breast-feeding is highest in the early months of breast-feeding. In addition, exclusive breast-feeding has been reported to carry a lower risk of HIV transmission than mixed feeding. In developed countries, breast feeding of babies by an HIV-infected mother is contraindicated since alternative forms of adequate nutrition, i.e., formulas, are readily available. In developing countries, where breast-feeding may be essential for the overall health of the infant, the continuation of cART in the infected mother during the period of breastfeeding markedly diminishes the risk of transmission of HIV to the infant. In fact, once cART has been initiated in a pregnant woman, many experts recommend that therapy be continued for life.
TRANSMISSION OF HIV BY OTHER BODY FLUIDS
Although HIV can be isolated typically in low titers from saliva of a small proportion of infected individuals, there is no convincing evidence that saliva can transmit HIV infection, either through kissing or through other exposures, such as occupationally to health care workers. Saliva contains endogenous antiviral factors; among these factors, HIV-specific immunoglobulins of IgA, IgG, and IgM isotypes are detected readily in salivary secretions of infected individuals. It has been suggested that large glycoproteins such as mucins and thrombospondin 1 sequester HIV into aggregates for clearance by the host. In addition, a number of soluble salivary factors inhibit HIV to various degrees in vitro, probably by targeting host cell receptors rather than the virus itself. Perhaps the best studied of these, secretory leukocyte protease inhibitor (SLPI), blocks HIV infection in several cell culture systems, and it is found in saliva at levels that approximate those required for inhibition of HIV in vitro. In this regard, higher salivary levels of SLPI in breast-fed infants were associated with a decreased risk of HIV transmission through breast milk. It has also been suggested that submandibular saliva reduces HIV infectivity by stripping gp120 from the surface of virions, and that saliva-mediated disruption and lysis of HIV-infected cells occurs because of the hypotonicity of oral secretions. There have been outlier cases of suspected transmission by saliva, but these have probably been blood-to-blood transmissions. Transmission of HIV by a human bite can occur but is a rare event. Although virus can be identified, if not isolated, from virtually any body fluid, there is no evidence that HIV transmission can occur as a result of exposure to tears, sweat, or urine. However, there have been isolated cases of transmission of HIV infection by body fluids that may or may not have been contaminated with blood. Most of these situations occurred in the setting of a close relative providing intensive nursing care for an HIV-infected person without observing universal precautions, underscoring the importance of adhering to such precautions in the handling of body fluids and wastes from HIV-infected individuals.
EPIDEMIOLOGY
HIV INFECTION AND AIDS WORLDWIDE
![]() HIV infection/AIDS is a global pandemic, with cases reported from virtually every country. At the end of 2013, an estimated 35.0 million individuals were living with HIV infection, according to the Joint United Nations Programme on HIV/AIDS (UNAIDS). An estimated 95% of people living with HIV/AIDS reside in low- and middle-income countries; ~50% are female, and 3.2 million are children <15 years. The distribution of these cases is illustrated in Fig. 226-8. The estimated number of people living with HIV—i.e., the global prevalence—has increased more than fourfold since 1990, reflecting the combined effects of continued high rates of new HIV infections and the life-prolonging impact of antiretroviral therapy (Fig. 226-9). In 2013, the global prevalence rate among persons age 15–49 years was 0.8%, with rates varying widely by country and region as illustrated in Fig. 226-10.
HIV infection/AIDS is a global pandemic, with cases reported from virtually every country. At the end of 2013, an estimated 35.0 million individuals were living with HIV infection, according to the Joint United Nations Programme on HIV/AIDS (UNAIDS). An estimated 95% of people living with HIV/AIDS reside in low- and middle-income countries; ~50% are female, and 3.2 million are children <15 years. The distribution of these cases is illustrated in Fig. 226-8. The estimated number of people living with HIV—i.e., the global prevalence—has increased more than fourfold since 1990, reflecting the combined effects of continued high rates of new HIV infections and the life-prolonging impact of antiretroviral therapy (Fig. 226-9). In 2013, the global prevalence rate among persons age 15–49 years was 0.8%, with rates varying widely by country and region as illustrated in Fig. 226-10.
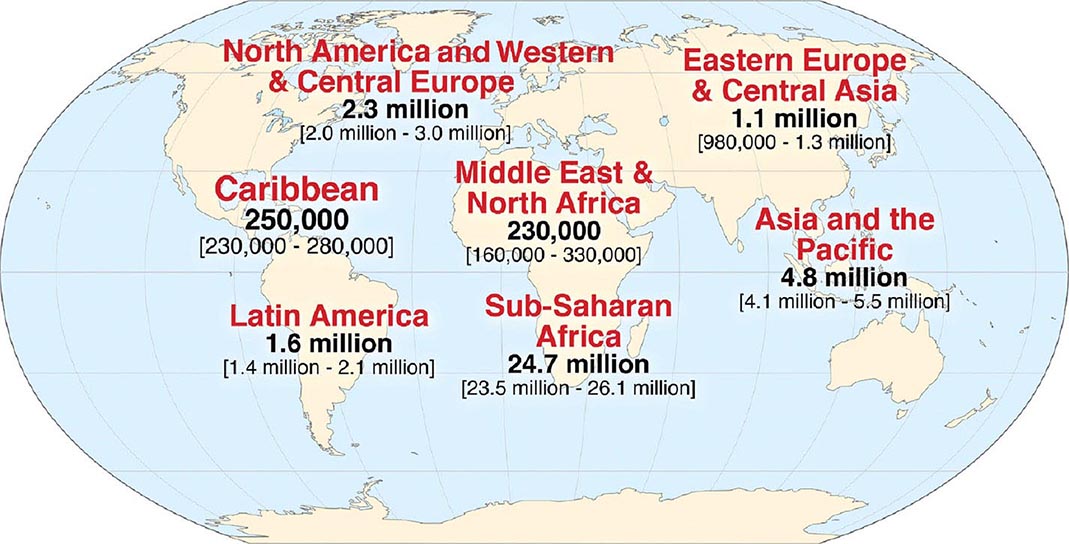
FIGURE 226-8 Estimated number of adults and children living with HIV infection as of December, 2013. Total: 35.0 (33.2 million–37.2 million). (From Joint United Nations Programme on HIV/AIDS [UNAIDS].)
In 2013, an estimated 2.1 million new cases of HIV infection occurred worldwide, including 240,000 among children <15 years; about 40% of new infections were among persons under age 25. Between 2001 and 2013, the estimated number of new HIV infections globally fell by 38% (Fig. 226-9). Recent reductions in global HIV incidence likely reflect progress with HIV prevention efforts and the increased provision to HIV-infected people of antiretroviral therapy, which makes them much less likely to transmit the virus to sexual partners. In 2013, global AIDS deaths totaled 1.5 million (including 190,000 children <15 years), a 35% decrease since 2005 that coincides with a rapid expansion of access to antiretroviral therapy (Fig. 226-9). Since the beginning of the pandemic, an estimated 39 million people have died of an AIDS-related illness.
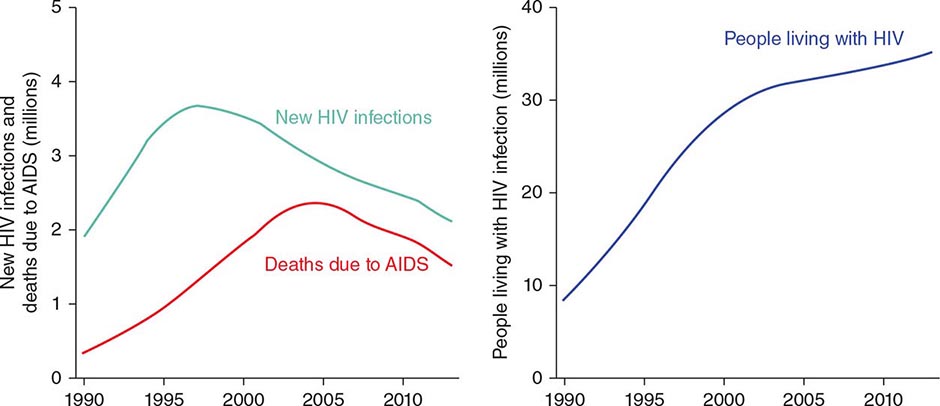
FIGURE 226-9 Global estimates of HIV incidence and AIDS deaths (left) and, HIV prevalence (right), 1990–2013. (From UNAIDS.)
The HIV epidemic has occurred in “waves” in different regions of the world, each wave having somewhat different characteristics depending on the demographics of the country and region in question and the timing of the introduction of HIV into the population. Although the AIDS epidemic was first recognized in the United States and shortly thereafter in Western Europe, it very likely began in sub-Saharan Africa (see above), which has been particularly devastated by the epidemic. More than 70% of all people with HIV infection (~25 million), and nearly 90% of all HIV-infected children live in that region, even though sub-Saharan Africa is home to just 12% of the world’s population (Fig. 226-8). Within the region, southern Africa is worst-affected. In nine southern African countries, seroprevalence data indicate that >10% of the adult population age 15–49 is HIV-infected (Fig. 226-10). In addition, among high-risk individuals (e.g., commercial sex workers, patients attending STI clinics) who live in urban areas of sub-Saharan Africa, seroprevalence is now >50% in some places. Recent data offer promising signs of declining HIV incidence and prevalence in many countries in the region, although frequently at levels that remain high. Heterosexual exposure is the primary mode of HIV transmission in sub-Saharan Africa, with women and girls disproportionately affected, accounting for ~60 percent of all HIV infections in that region. In 2013, an estimated 230,000 people were living with HIV in the Middle East/North Africa region. Cases are largely concentrated among IDUs, men who have sex with men, and sex workers and their clients.
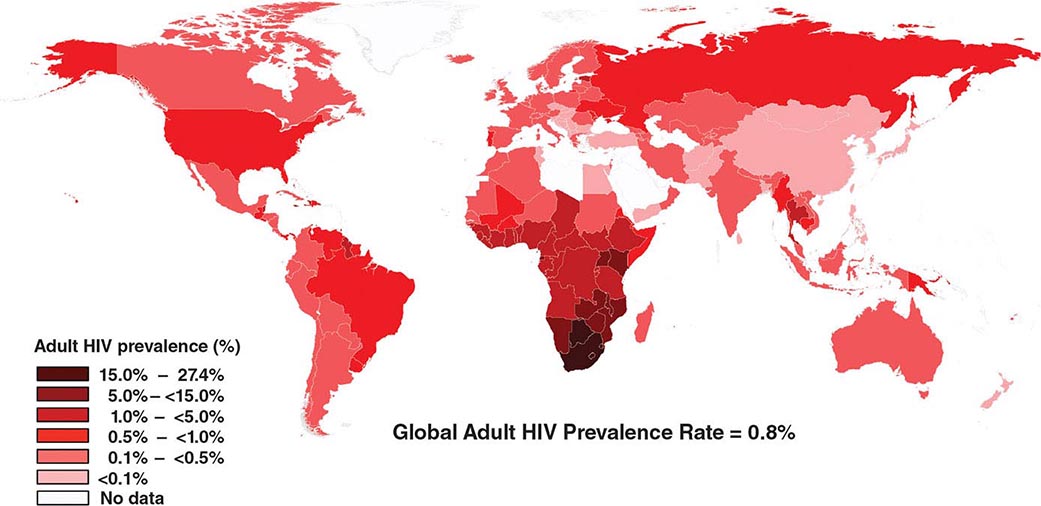
FIGURE 226-10 Global adult HIV prevalence rate, 2013. Data are estimates for adults aged 15-49 years. (From UNAIDS.)
In Asia and the Pacific, an estimated 4.8 million people were living with HIV at the end of 2013. In this region of the world, HIV prevalence is highest in southeast Asian countries, with wide variation in trends between different countries. Among countries in Asia, only Thailand has an adult seroprevalence rate of >1%. However, the populations of many Asian nations are so large (especially India and China) that even low infection and seroprevalence rates result in large numbers of people living with HIV. Although Asia’s epidemic has been concentrated for some time among specific populations—sex workers and their clients, men who have sex with men, and IDUs—it is expanding to the heterosexual partners of those most at risk.
The epidemic is expanding in Eastern Europe and Central Asia, where ~1.1 million people were living with HIV at the end of 2013. The Russian Federation and Ukraine account for the majority of HIV cases in the region. Driven initially by injection drug use and increasingly by heterosexual transmission, the number of new infections in this region has increased dramatically over the past decade.
Approximately 1.9 million people are living with HIV/AIDS in Central and South America and the Caribbean. Brazil is home to the largest number of HIV-infected people in the region. However, the epidemic has been slowed in that country due to successful treatment and prevention efforts. Men who have sex with men account for the largest proportion of HIV infections in Central and South America. The Caribbean region has the highest regional adult seroprevalence rate after Africa. Heterosexual transmission, often tied to sex work, is the main driver of transmission in the region.
Approximately 2.3 million people are living with HIV/AIDS in North America and western and central Europe. The number of new infections among men who have sex with men has increased over the past decade in these mostly high-income areas, while rates of new infections among heterosexuals have stabilized and infections among women and IDUs have fallen.
HIV INFECTION AND AIDS IN THE UNITED STATES
About 1.7 million people have been infected with HIV in the United States since the beginning of the epidemic, of whom >630,000 have died. Approximately 1.1 million individuals in the United States are living with HIV infection, ~16–18% of whom are unaware of their infection, according to recent estimates. As illustrated in Fig. 226-11, only a fraction of HIV-infected people are able to negotiate the steps in the HIV “care continuum,” from diagnosis, to entering into and staying in care, to receiving antiretroviral therapy, and ultimately to achieving a suppressed viral load (see “Treatment,” below).
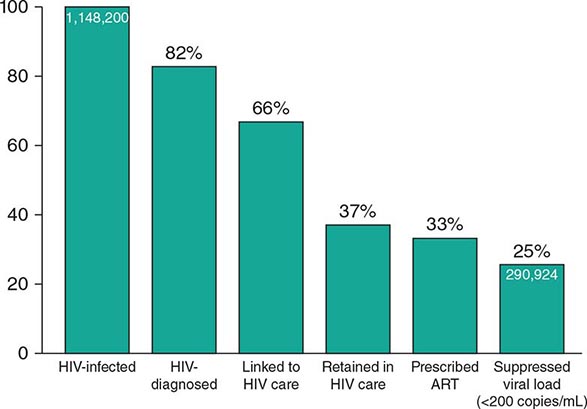
FIGURE 226-11 Estimated percentage of HIV-infected people engaged in selected stages of the continuum of HIV care in the United States. (Adapted from HI Hall et al: JAMA Intern Med 173:1337, 2013.)
More than 60% of those living with HIV/AIDS are Black/African-American or Hispanic/Latino, and more than half are men who have sex with men. The estimated HIV seroprevalence rate among all individuals age 13 years or older in the United States is ~0.5%. Approximately 2% of Black/African-American adults are HIV-infected in the United States, higher than any other group.
The number of new HIV infections in the United States, HIV incidence, peaked at about 130,000 per year in the late 1980s, followed by declines. For more than a decade, HIV incidence has remained stable at approximately 50,000 per year, with the proportion of new infections increasing in recent years among men who have sex with men and falling among women and IDUs. Among adults and adolescents newly diagnosed with HIV infection in 2011 (regardless of stage of infection), ~79% were males and ~21% were women. Of new HIV diagnoses among men, ~79% were attributed to male-to-male sexual contact, ~12% to heterosexual contact, ~6% to injection drug use, and ~4% to a combination of male-to-male sexual contact and injection drug use. Of new HIV diagnoses among women, ~86% were due to heterosexual contact and ~14% to injection drug use (Fig. 226-12). The estimated numbers of new HIV infections in 2011 in the United States for the 10 most affected subpopulations are shown in Fig. 226-13.
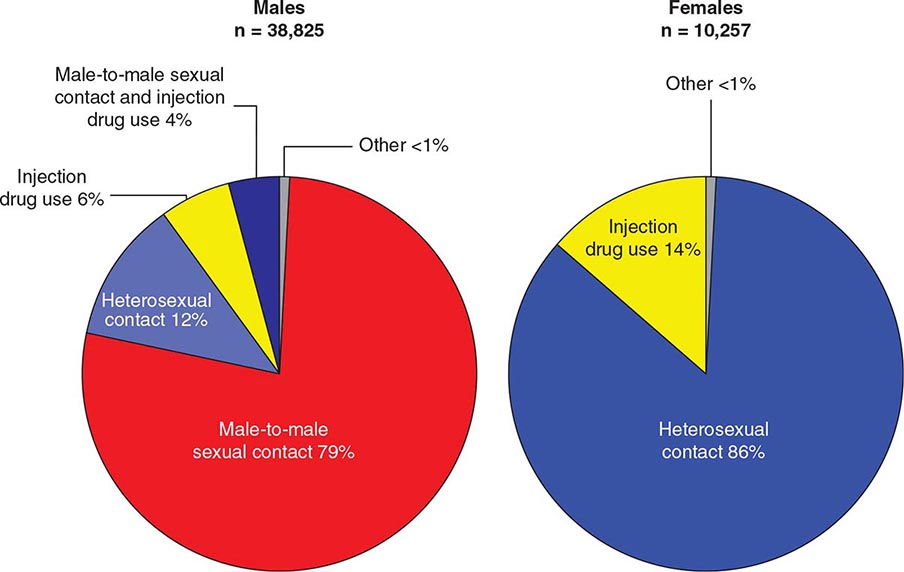
FIGURE 226-12 Transmission categories of adults and adolescents diagnosed with HIV infection (regardless of stage) in 2011, United States. (From CDC.)
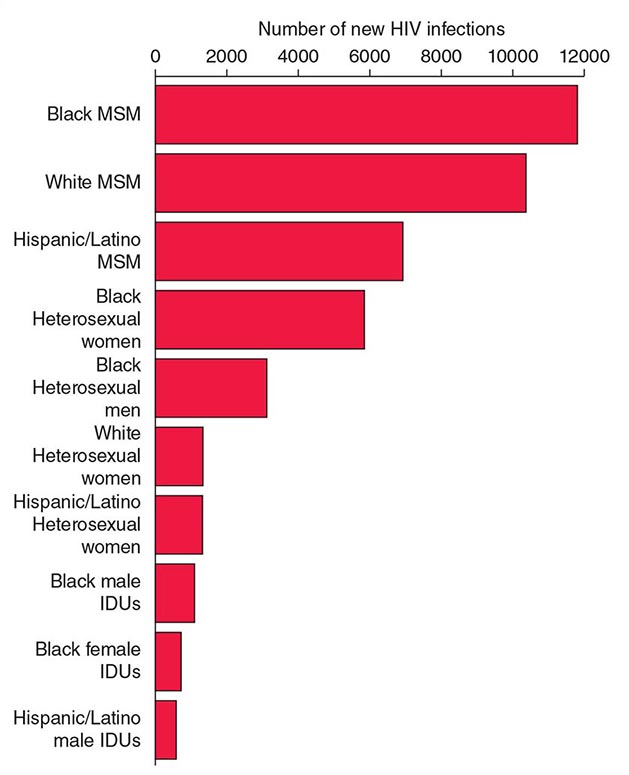
FIGURE 226-13 Estimated number of new HIV infections in the United States, 2011, for the most affected subpopulations. (From CDC.)
Perinatal HIV transmission, from an HIV-infected mother to her baby, has declined significantly in the United States, largely due to the implementation of guidelines for the universal counseling and voluntary HIV testing of pregnant women and the use of antiretroviral therapy for pregnant women and newborn infants to prevent infection (Fig. 226-14). In 2011, fewer than 200 children were diagnosed with HIV infection in the United States.
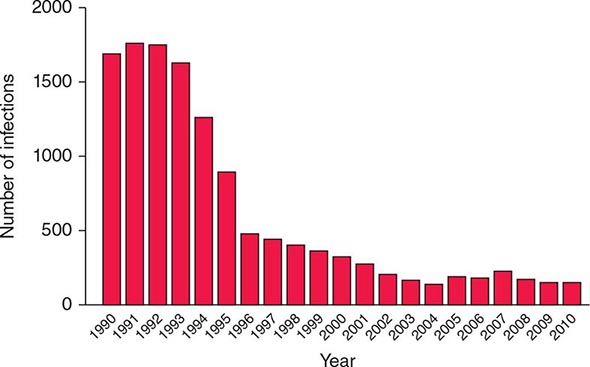
FIGURE 226-14 Estimated number of HIV-infected infants, United States, 1990–2010. (From CDC.)
HIV infection and AIDS have disproportionately affected minority populations in the United States. Among those diagnosed with HIV (regardless of stage of infection) in 2011, 47% percent were Blacks/African Americans, a group that constitutes only 12% of the U.S. population (Fig. 226-15A). The estimated rate of new HIV diagnoses in 2011 by race/ethnicity per 100,000 population in the United States is shown in Fig. 226-15B.
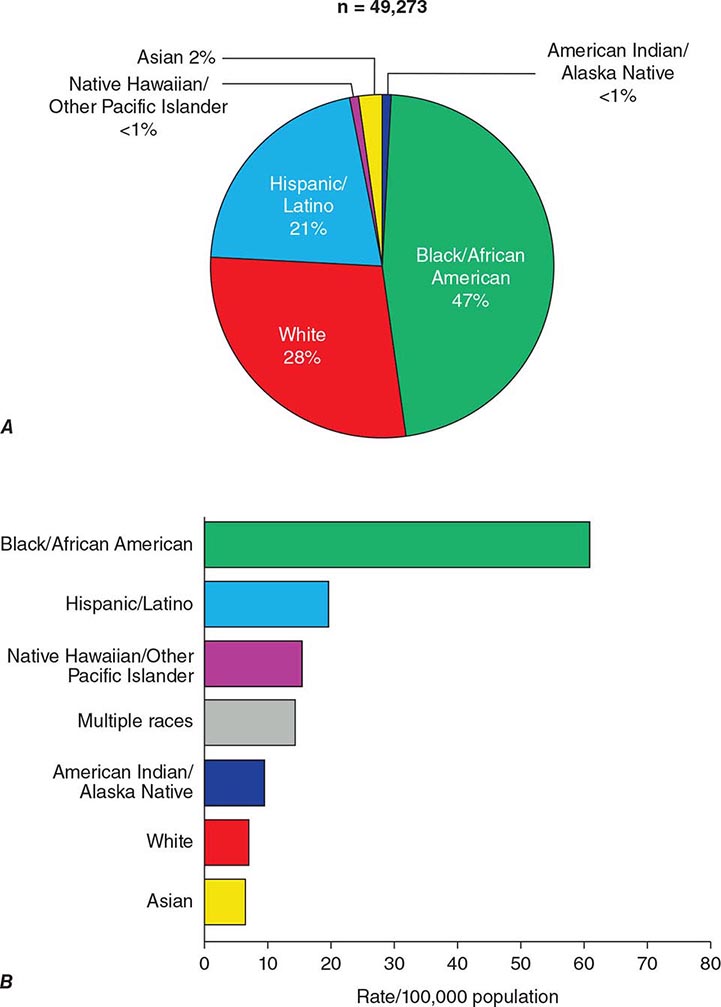
FIGURE 226-15 Race/ethnicity of persons (including children) diagnosed with HIV infection (regardless of stage) during 2011 in the United States. A. Estimated proportion of new infections by race/ethnicity. B. Estimated rate of new infections by race/ethnicity (per 100,000 population). (From CDC.)
The number of individuals diagnosed with AIDS and deaths among persons with AIDS in the United States rose steadily through the 1980s; AIDS cases peaked in 1993 and deaths in 1995 (Fig. 226-16). Since then, the annual numbers of AIDS-related deaths in the United States have fallen ~70%. This trend is due to several factors, including improved prophylaxis and treatment of opportunistic infections, growing experience among the health professions in caring for HIV-infected individuals, improved access to health care, and a decrease in new infections due to saturational effects and prevention efforts. However, the most influential factor clearly has been the increased use of potent antiretroviral drugs, generally administered in a combination of three or four agents.
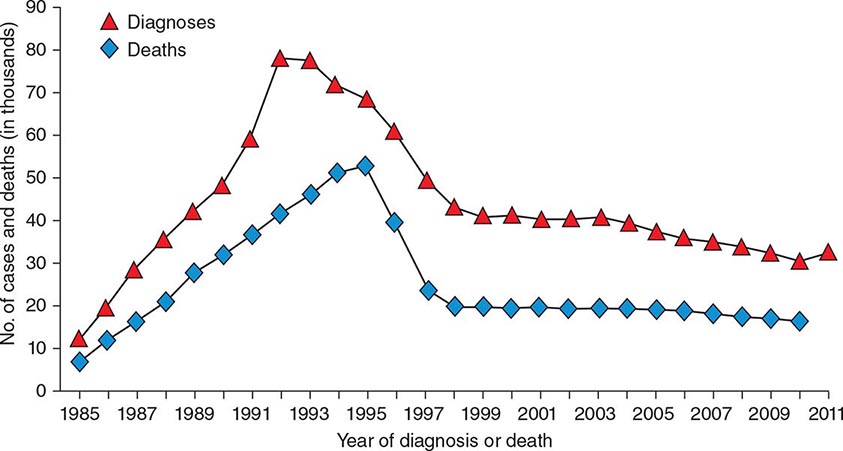
FIGURE 226-16 Estimated number of AIDS cases and AIDS deaths, United States, 1985–2011. (From CDC.)
Although the HIV/AIDS epidemic on the whole is plateauing in the United States, it is spreading rapidly among certain populations, stabilizing in others, and decreasing in others. Similar to other STIs, HIV infection will not spread homogeneously throughout the population of the United States. However, it is clear that anyone who practices high-risk behavior is at risk for HIV infection. In addition, recent increases in infections and AIDS cases among young men who have sex with men as well as the spread in pockets of poverty in both urban and rural regions (particularly among underserved minority populations in the southern United States with inadequate access to health care) testify that the epidemic of HIV infection in the United States remains a public health problem of major proportion.
PATHOPHYSIOLOGY AND PATHOGENESIS
The hallmark of HIV disease is a profound immunodeficiency resulting primarily from a progressive quantitative and qualitative deficiency of the subset of T lymphocytes referred to as helper T cells occurring in a setting of polyclonal immune activation. The helper subset of T cells is defined phenotypically by the presence on its surface of the CD4 molecule (Chap. 372e), which serves as the primary cellular receptor for HIV. A co-receptor must also be present together with CD4 for efficient binding, fusion, and entry of HIV-1 into its target cells (Figs. 226-3 and 226-4). HIV uses two major co-receptors, CCR5 and CXCR4, for fusion and entry; these co-receptors are also the primary receptors for certain chemoattractive cytokines termed chemokines and belong to the seven-transmembrane-domain G protein–coupled family of receptors. A number of mechanisms responsible for cellular depletion and/or immune dysfunction of CD4+ T cells have been demonstrated in vitro; these include direct infection and destruction of these cells by HIV, as well as indirect effects such as immune clearance of infected cells, cell death associated with aberrant immune activation, and immune exhaustion due to aberrant cellular activation with resulting cellular dysfunction. Patients with CD4+ T cell levels below certain thresholds are at high risk of developing a variety of opportunistic diseases, particularly the infections and neoplasms that are AIDS-defining illnesses. Some features of AIDS, such as Kaposi’s sarcoma and certain neurologic abnormalities, cannot be explained completely by the immunodeficiency caused by HIV infection, since these complications may occur prior to the development of severe immunologic impairment.
The combination of viral pathogenic and immunopathogenic events that occurs during the course of HIV disease from the moment of initial (primary) infection through the development of advanced-stage disease is complex and varied. It is important to appreciate that the pathogenic mechanisms of HIV disease are multifactorial and multiphasic and are different at different stages of the disease. Therefore, it is essential to consider the typical clinical course of an untreated HIV-infected individual in order to more fully appreciate these pathogenic events (Fig. 226-17).
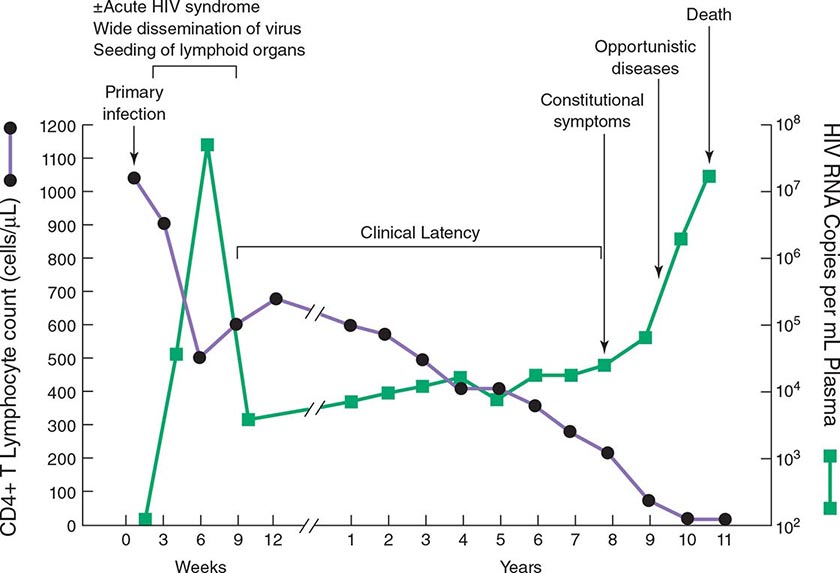
FIGURE 226-17 Typical course of an untreated HIV-infected individual. See text for detailed description. (From G Pantaleo et al: N Engl J Med 328:327, 1993. Copyright 1993 Massachusetts Medical Society. All rights reserved.)
EARLY EVENTS IN HIV INFECTION: PRIMARY INFECTION AND INITIAL DISSEMINATION OF VIRUS
Using mucosal transmission as a model, the earliest events (within hours) that occur following exposure of HIV to the mucosal surface determine whether an infection will be established as well as the subsequent course of events following infection. Although the mucosal barrier is relatively effective in limiting access of HIV to susceptible targets in the lamina propria, the virus can cross the barrier by transport on Langerhans cells, an epidermal type of DC, just beneath the surface or through microscopic rents in the mucosa. Significant disruptions in the mucosal barrier as seen in ulcerative genital disease facilitate viral entry and increase the efficiency of infection. Virus then seeks susceptible targets, which are primarily CD4+ T cells that are spatially dispersed in the mucosa. This spatial dispersion of targets provides a significant obstacle to the establishment of infection. Such obstacles account for the low efficiency of sexual transmission of HIV (see “Sexual Transmission,” above). Both “partially” resting CD4+ T cells and activated CD4+ T cells serve as early amplifiers of infection. Resting CD4+ T cells are more abundant; however, activated CD4+ T cells produce larger amounts of virus. In order for infection to become established, the basic reproductive rate (R0) must become equal to or greater than 1, i.e., each infected cell would infect at least one other cell. Once infection is established, the virus replicates in lymphoid cells in the mucosa, the submucosa, and to some extent the lymphoreticular tissues that drain the gut tissues. For a variable period of time ranging from a few to several days, the virus cannot yet be detected in the plasma. This period is referred to as the “eclipse” phase of infection. As more virus is produced within several days to weeks, it is disseminated, first to the draining lymph nodes and then to other lymphoid compartments where it has easy access to dense concentrations of CD4+ T cell targets, allowing for a burst of high-level viremia that is readily detectable by currently available assays (Fig. 226-18). An important lymphoid organ, the gut-associated lymphoid tissue (GALT), is a major target of HIV infection and the location where large numbers of CD4+ T cells (usually memory cells) are infected and depleted, both by direct viral effects and by activation-associated apoptosis. Once virus replication reaches this threshold and virus is widely disseminated, infection is firmly established and the process is irreversible. It is important to point out that the initial infection of susceptible cells may vary somewhat with the route of infection. Virus that enters directly into the bloodstream via infected blood or blood products (i.e., transfusions, use of contaminated needles for injection drugs, sharp-object injuries, maternal-to-fetal transmission either intrapartum or perinatally, or sexual intercourse where there is enough trauma to cause bleeding) is likely first cleared from the circulation to the spleen and other lymphoid organs, where primary focal infections begin, followed by wider dissemination throughout other lymphoid tissues as described above.
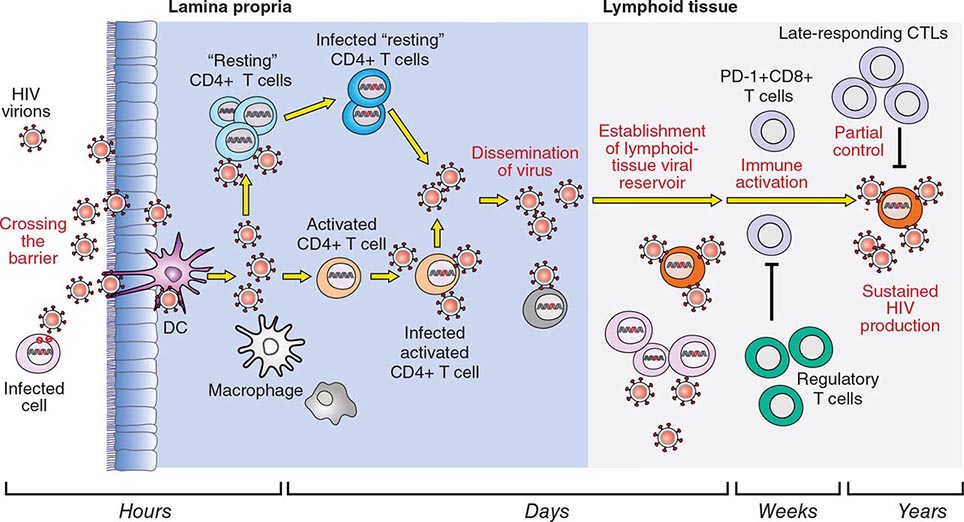
FIGURE 226-18 Summary of early events in HIV infection. See text for detailed description. CTLs, cytolytic T lymphocytes; HIV, human immunodeficiency virus. (Adapted from AT Haase: Nat Rev Immunol 5:783, 2005.)
It has been demonstrated that sexual transmission of HIV is the result of a single infectious event and that a viral genetic bottleneck exists for transmission. In this regard, certain characteristics of the HIV envelope glycoprotein have a major influence on transmission, at least in subtype A and C viruses. Transmitting viruses, often referred to as “founder viruses,” are usually underrepresented in the circulating viremia of the transmitting partner and are less-diverged viruses with signature sequences including shorter V1–V2 loop sequences and fewer predicted N-linked glycosylation sites relative to the major circulating variants. These viruses are almost exclusively R5 strains and are usually sensitive to neutralization by antibody from the transmitting partner. Once replication proceeds in the newly infected partner, the founder virus diverges and accumulates glycosylation sites, becoming progressively more resistant to neutralization (Fig. 226-19).
FIGURE 226-19 As HIV diverges from founder to chronically replicating virus, it accumulates N-linked glycosylation sites. See text for detailed description. (Adapted from CA Derdeyn et al: Science 303:2019, 2004; B Chohan et al: J Virol 79:6528, 2005; and BF Keele et al: Proc Natl Acad Sci USA 105:7552, 2008.)
The acute burst of viremia and wide dissemination of virus in primary HIV infection may be associated with an acute HIV syndrome, which occurs to varying degrees in ~50% of individuals with primary infection (see below). This syndrome is usually associated with high levels of viremia measured in millions of copies of HIV RNA per milliliter of plasma that last for several weeks. Acute mononucleosis-like symptoms are well correlated with the presence of viremia. Virtually all patients develop some degree of viremia during primary infection, which contributes to virus dissemination throughout the lymphoid tissue, even though they may remain asymptomatic or not recall experiencing symptoms. It appears that the initial level of plasma viremia in primary HIV infection does not necessarily determine the rate of disease progression; however, the set point of the level of steady-state plasma viremia after ~1 year does seem to correlate with the slope of disease progression in the untreated patient. The strikingly high levels of viremia observed in many patients with acute HIV infection is felt to be associated with a higher likelihood of transmission of the virus to others by a variety of routes including sexual transmission, shared needles and syringes, and mother-to-child transmission intrapartum, perinatally, or via breast milk.
ESTABLISHMENT OF CHRONIC AND PERSISTENT INFECTION
Persistence of Virus Replication HIV infection is unique among human viral infections. Despite the robust cellular and humoral immune responses that are mounted following primary infection (see “Immune Response to HIV,” below), once infection has been established the virus succeeds in escaping complete immune-mediated clearance, paradoxically seems to thrive on immune activation, and is never eliminated completely from the body. Rather, a chronic infection develops and persists with varying degrees of continual virus replication in the untreated patient for a median of ~10 years before the patient becomes clinically ill (see “Advanced HIV Disease,” below). It is this establishment of a chronic, persistent infection that is the hallmark of HIV disease. Throughout the often protracted course of chronic infection, virus replication can invariably be detected in untreated patients by widely available assays that measure copies of HIV RNA per milliliter of plasma. Levels of virus vary greatly in most untreated patients, ranging from several thousand to a few million copies of HIV RNA per milliliter of plasma. Studies using highly sensitive molecular techniques have demonstrated that even in certain patients in whom plasma viremia is suppressed to below detection (lower limit, 20–50 copies of HIV RNA/mL depending on manufacturer) by cART, there is a continual low level of virus replication. In other human viral infections, with very few exceptions, if the host survives, the virus is completely cleared from the body and a state of immunity against subsequent infection develops. HIV infection very rarely kills the host during primary infection. Certain viruses, such as HSV (Chap. 216), are not completely cleared from the body after infection, but instead enter a latent state; in these cases, clinical latency is accompanied by microbiologic latency. This is not the case with HIV infection as described above. Chronicity associated with persistent virus replication can also be seen in certain cases of HBV and HCV infections (Chap. 362); however, in these infections the immune system is not a target of the virus.
Escape of HIV from Effective Immune System Control Inherent to the establishment of chronicity of HIV infection is the ability of the virus to evade adequate control and elimination by both the cellular and humoral limbs of the immune system. There are a number of mechanisms whereby the virus accomplishes this evasion. Paramount among these is the establishment of a sustained level of replication associated with the generation of viral diversity via mutation and recombination. The selection of mutants that escape control by CD8+ cytolytic T lymphocytes (CTLs) is critical to the propagation and progression of HIV infection. The high rate of virus replication associated with inevitable mutations also contributes to the inability of antibody to neutralize the autologous virus and thus to contain the virus quasispecies present in an individual at any given time. Extensive analyses of sequential HIV isolates and host responses have demonstrated that viral escape from B cell and CD8+ T cell epitopes occurs early after infection and allows the virus to stay one step ahead of effective immune responses. Virus-specific CD8+ CTLs expand greatly during primary HIV infection, and likely represent the high-affinity responses that would be expected to be most efficient in eliminating virus-infected cells; however, the restriction is generally incomplete as viral replication persists at relatively high levels in the majority of individuals. In addition to viral escape from CTLs through high rates of mutation, it is thought that the initially strong response becomes qualitatively dysfunctional owing to the overwhelming immune activation resulting from persistent viral replication, similar to the exhaustion of CD8+ CTLs that has been reported in the murine model of lymphocytic choriomeningitis virus (LCMV) infection. Several studies have indicated that exhaustion of HIV-specific CD8+ T cells during prolonged immune activation is associated with expression of inhibitory receptors, such as programmed death (PD) 1 molecule (of the B7-CD28 family of molecules), as well as loss of polyreactivity and proliferative capacity. Another mechanism contributing to the evasion by HIV of immune system control is the downregulation of HLA class I molecules on the surface of HIV-infected cells by the viral proteins Nef, Tat, and Vpu, resulting in the lack of ability of the CD8+ CTL to recognize and kill the infected target cell. Although this downregulation of HLA class I molecules would favor elimination of HIV-infected cells by natural killer (NK) cells, this latter mechanism does not seem to remove HIV-infected cells effectively (see below).
The principal targets of neutralizing antibodies against HIV are the envelope proteins gp120 and gp41. HIV employs at least three mechanisms to evade neutralizing responses: hypervariability in the primary sequence of the envelope, extensive glycosylation of the envelope, and conformational masking of neutralizing epitopes. Several studies that have followed the humoral immune response to HIV from the earliest points after primary infection indicate that the virus continually mutates to escape the emerging antibody response such that the sequential antibodies that are induced do not neutralize the autologous virus. Broadly neutralizing antibodies capable of neutralizing a wide range of primary HIV isolates in vitro occur in only about 20% of HIV-infected individuals, and, when they do occur, it generally requires 2 to 3 years of infection with continual virus replication to drive the affinity maturation of the antibodies. Unfortunately, by the time these broadly neutralizing antibodies are formed, they are ineffective in containing the virus replication in the patient. Persistent viremia also results in exhaustion of B cells similar to the exhaustion reported for CD4+ T cells, adding to the defects in the humoral response to HIV.
CD4+ T cell help is essential for the integrity of antigen-specific immune responses, both humoral and cell-mediated. HIV preferentially infects activated CD4+ T cells including HIV-specific CD4+ T cells, and so this loss of viral-specific helper T cell responses has profound negative consequences for the immunologic control of HIV replication. Furthermore, this loss occurs early in the course of infection, and animal studies indicate that 40–70% of all memory CD4+ T cells in the GALT are eliminated during acute infection. Another potential means of escape of HIV-infected cells from elimination by CD8+ CTLs is the sequestration of infected cells in immunologically privileged sites such as the central nervous system (CNS).
Finally, the escape of HIV from immune-mediated elimination during primary infection allows the formation of a pool of latently infected cells that may not be recognized or completely eliminated by virus-specific CTLs or by ART (see below). Thus, despite a potent immune response and the marked downregulation of virus replication following primary HIV infection, HIV succeeds in establishing a state of chronic infection with a variable degree of persistent virus replication. During this period most patients make the clinical transition from acute primary infection to variable periods of clinical latency or smoldering disease activity (see below).
The HIV Reservoir: Obstacles to the Eradication of Virus A pool of latently infected, resting CD4+ T cells that serves as at least one component of the persistent reservoir of virus exists in virtually all HIV-infected individuals, including those who are receiving cART. Such cells carry an integrated form of HIV DNA in the genome of the host and can remain in this state until an activation signal drives the expression of HIV transcripts and ultimately replication-competent virus. This form of latency is to be distinguished from preintegration latency, in which HIV enters a resting CD4+ T cell and, in the absence of an activation signal, reverse transcription of the HIV genome occurs to a certain extent but the resulting proviral DNA fails to integrate into the host genome. This period of preintegration latency may last hours to days, and if no activation signal is delivered to the cell, the proviral DNA loses its capacity to initiate a productive infection. If these cells do become activated prior to decay of the preintegration complex, reverse transcription proceeds to completion and the virus continues along its replication cycle (see above and Fig. 226-20). The pool of cells that are in the postintegration state of latency is established early during the course of primary HIV infection. Despite the suppression of plasma viremia to <50 copies of HIV RNA per milliliter by potent regimens of cART administered over several years, this pool of latently infected cells persists and can give rise to replication-competent virus upon cellular activation. Modeling studies built on projections of decay curves have estimated that in such a setting of prolonged suppression, it would require a few to several years for the pool of latently infected cells to be completely eliminated. This has not been documented to occur spontaneously in any patients very likely because the latent viral reservoir is continually replenished by the low levels of persistent virus replication that may remain below the limits of detection of current assays (see below) (Fig. 226-20), even in patients who for the most part are treated successfully. Reservoirs of HIV-infected cells, latent or otherwise, can exist in a number of compartments including the lymphoid tissue, peripheral blood, and the CNS (likely in cells of the monocyte/macrophage lineage) as well as in other unidentified locations. Over the past several years attempts have been made to eliminate HIV in the latent viral reservoir using agents that stimulate resting CD4+ T cells during the course of cART; however, such attempts have been unsuccessful. Thus, this persistent reservoir of infected cells and/or low levels of persistent virus replication are major obstacles to the goal of eradication of virus from infected individuals and hence a “cure,” despite the favorable clinical outcomes that have resulted from cART.
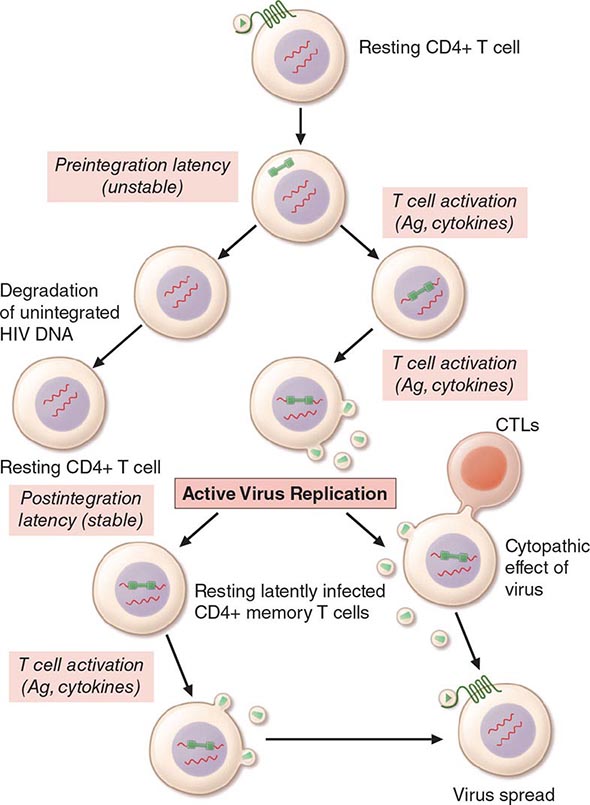
FIGURE 226-20 Generation of latently infected, resting CD4+ T cells in HIV-infected individuals. See text for details. Ag, antigen; CTLs, cytolytic T lymphocytes. (Courtesy of TW Chun; with permission.)
Viral Dynamics The dynamics of viral production and turnover have been quantified using mathematical modeling in the setting of the administration of reverse transcriptase and protease inhibitors to HIV-infected individuals in clinical studies. Treatment with these drugs resulted in a precipitous decline in the level of plasma viremia, which typically fell by well over 90% within 2 weeks. The number of CD4+ T cells in the blood increased concurrently, which suggested that the killing of CD4+ T cells was linked directly to the levels of replicating virus. However, a significant component of the early rise in CD4+ T cell numbers following the initiation of therapy may be due to the redistribution of cells into the peripheral blood from other tissue compartments throughout the body as a consequence of therapy-related diminution in viremia-associated immune system activation. It was determined on the basis of modeling the kinetics of viral decline and the emergence of resistant mutants during therapy that 93–99% of the circulating virus originated from recently infected, rapidly turning over CD4+ T cells and that ~1–7% of circulating virus originated from longer-lived cells, likely monocytes/macrophages. A negligible amount of circulating virus originated from the pool of latently infected cells (Fig. 226-21). It was also determined that the half-life of a circulating virion was ~30–60 min and that of productively infected cells was 1 day. Given the relatively steady level of plasma viremia and of infected cells, it appears that extremely large amounts of virus (~1010–1011 virions) are produced and cleared from the circulation each day. In addition, data suggest that the minimal duration of the HIV-1 replication cycle in vivo is ~2 days. Other studies have demonstrated that the decrease in plasma viremia that results from cART correlates closely with a decrease in virus replication in lymph nodes, further confirming that lymphoid tissue is the main site of HIV replication and the main source of plasma viremia.
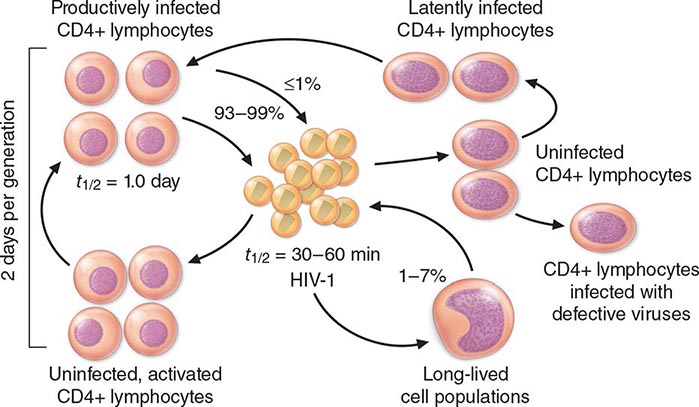
FIGURE 226-21 Dynamics of HIV infection in vivo. See text for detailed description. (From AS Perelson et al: Science 271:1582, 1996.)
The level of steady-state viremia, called the viral set point, at ~1 year following acquisition of HIV infection has important prognostic implications for the progression of HIV disease in the untreated patient. It has been demonstrated that as a group untreated HIV-infected individuals who have a low set point at 6 months to 1 year following infection progress to AIDS much more slowly than individuals whose set point is very high at that time (Fig. 226-22).
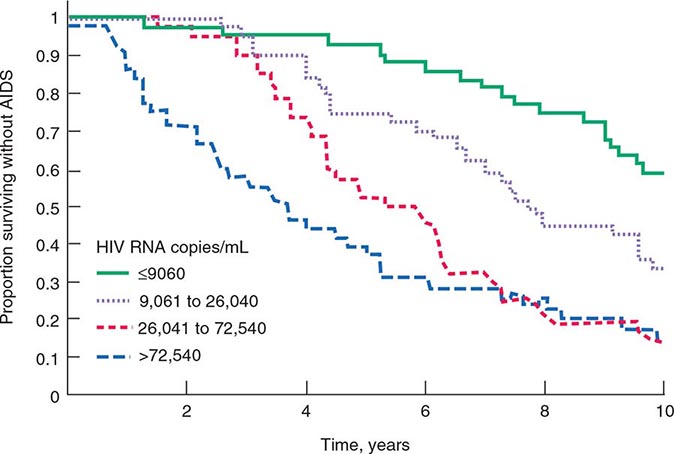
FIGURE 226-22 Relationship between levels of virus and rates of disease progression. Kaplan-Meier curves for AIDS-free survival stratified by baseline HIV-1 RNA categories (copies per milliliter). (From JW Mellors et al: Science 272:1167, 1996.)
Clinical Latency versus Microbiologic Latency With the exception of long-term nonprogressors (see “Long-Term Survivors and Long-Term Nonprogressors,” below), the level of CD4+ T cells in the blood decreases progressively in HIV-infected individuals in the absence of cART. The decline in CD4+ T cells may be gradual or abrupt, the latter usually reflecting a significant spike in the level of plasma viremia. Most patients are relatively asymptomatic while this progressive decline is taking place (see below) and are often described as being in a state of clinical latency. However, this term is misleading; it does not mean disease latency, since progression, although slow in many cases, is generally relentless during this period. Furthermore, clinical latency should not be confused with microbiologic latency, since varying levels of virus replication inevitably occur during this period of clinical latency. Even in those rare patients who have <50 copies of HIV RNA per milliliter in the absence of therapy, there is virtually always some degree of ongoing virus replication.
ADVANCED HIV DISEASE
In untreated patients or in patients in whom therapy has not adequately controlled virus replication, after a variable period, usually measured in years, the CD4+ T cell count falls below a critical level (<200/μL) and the patient becomes highly susceptible to opportunistic disease (Fig. 226-17). For this reason, the CDC case definition of AIDS includes all HIV-infected individuals over 5 years of age with CD4+ T cell counts below this level (Table 226-2). Patients may experience constitutional signs and symptoms or may develop an opportunistic disease abruptly without any prior symptoms, although the latter scenario is unusual. The depletion of CD4+ T cells continues to be progressive and unrelenting in this phase. It is not uncommon for CD4+ T cell counts in the untreated patient to drop to as low as 10/μL or even to zero. In countries where cART and prophylaxis and treatment for opportunistic infections are readily accessible to such patients, survival is increased dramatically even in those patients with advanced HIV disease. In contrast, untreated patients who progress to this severest form of immunodeficiency usually succumb to opportunistic infections or neoplasms (see below).
LONG-TERM SURVIVORS AND LONG-TERM NONPROGRESSORS
It is important to distinguish between the terms long-term survivor and long-term nonprogressor. Long-term nonprogressors are by definition long-term survivors; however, the reverse is not always true. Predictions from one study that antedated the availability of effective cART estimated that ~13% of homosexual/bisexual men who were infected at an early age may remain free of clinical AIDS for >20 years. Many of these individuals may have progressed in their degree of immune deficiency; however, they certainly survived for a considerable period of time. With the advent of effective cART, the survival of HIV-infected individuals has dramatically increased. Early in the AIDS epidemic, prior to the availability of therapy, if a patient presented with a life-threatening opportunistic infection, the median survival was 26 weeks from the time of presentation. Currently, an HIV-infected 20-year-old individual in a high-income country who is appropriately treated with cART can expect to live at least 50 years according to mathematical model projections. In the face of cART, long-term survival is becoming commonplace. Definitions of long-term nonprogressors have varied considerably over the years, and so such individuals constitute a heterogeneous group. Long-term nonprogressors were first described in the 1990s. Originally, individuals were considered to be long-term nonprogressors if they had been infected with HIV for a long period (≥10 years), their CD4+ T cell counts were in the normal range, and they remained stable over years without receiving cART. Approximately 5–15% of HIV-infected individuals fell into this broader nonprogressor category. However, this group was rather heterogenous and over time a significant proportion of these individuals progressed and ultimately required therapy. From this broader group, a much smaller subgroup of “elite” controllers or nonprogressors was identified, and they constituted less than 1% of HIV-infected individuals. These elite controllers, by definition, have extremely low levels of plasma viremia and normal CD4+ T cell counts. It is noteworthy that certain of their HIV-specific immune responses are robust and clearly superior to those of HIV-infected progressors. In this group of elite controllers certain HLA class I haplotypes are overrepresented, particularly HLA-B57-01 and HLA-B27-05. Outside of the subgroup of elite controllers, a number of other genetic factors have been shown to be involved to a greater or lesser degree in the control of virus replication and thus in the rate of HIV disease progression (see “Genetic Factors in HIV-1 and AIDS Pathogenesis,” below).
LYMPHOID ORGANS AND HIV PATHOGENESIS
Regardless of the portal of entry of HIV, lymphoid tissues are the major anatomic sites for the establishment and propagation of HIV infection. Despite the use of measurements of plasma viremia to determine the level of disease activity, virus replication occurs mainly in lymphoid tissue and not in blood; indeed, the level of plasma viremia directly reflects virus production in lymphoid tissue.
Some patients experience progressive generalized lymphadenopathy early in the course of the infection; others experience varying degrees of transient lymphadenopathy. Lymphadenopathy reflects the cellular activation and immune response to the virus in the lymphoid tissue, which is generally characterized by follicular or germinal center hyperplasia. Lymphoid tissue involvement is a common denominator of virtually all patients with HIV infection, even those without easily detectable lymphadenopathy.
Simultaneous examinations of lymph tissue and peripheral blood in patients and monkeys during various stages of HIV and SIV infection, respectively, have led to substantial insight into the pathogenesis of HIV disease. In most of the original human studies, peripheral lymph nodes have been used predominantly as the source of lymphoid tissue. More recent studies in monkeys and humans have also focused on the GALT, where the earliest burst of virus replication occurs associated with marked depletion of CD4+ T cells. In detailed studies of peripheral lymph node tissue, using a combination of polymerase chain reaction (PCR) techniques for HIV DNA and HIV RNA in tissue and HIV RNA in plasma, in situ hybridization for HIV RNA, and light and electron microscopy, the following picture has emerged. During acute HIV infection resulting from mucosal transmission, virus replication progressively amplifies from scattered lymphoid cells in the lamina propria to draining lymphoid tissue, leading to high levels of plasma viremia. The GALT plays a major role in the amplification of virus replication, and virus is disseminated from replication in the GALT to peripheral lymphoid tissue. A profound degree of cellular activation occurs (see below) and is reflected in follicular or germinal center hyperplasia. At this time copious amounts of extracellular virions (both infectious and defective) are trapped on the processes of the follicular dendritic cells (FDCs) in the germinal centers of the lymph nodes. Virions that have bound complement components on their surfaces attach to the surface of FDCs via interactions with complement receptors and likely via Fc receptors that bind to antibodies that are attached to the virions. In situ hybridization reveals expression of virus in individual cells of the paracortical area and, to a lesser extent, the germinal center (Fig. 226-23). The persistence of trapped virus after the transition from acute to chronic infection likely reflects a steady state whereby trapped virus turns over and is replaced by fresh virions that are continually produced. The trapped virus, either as whole virion or shed envelope, serves as a continual activator of CD4+ T cells, thus driving further virus replication.
FIGURE 226-23 HIV in the lymph node of an HIV-infected individual. An individual cell infected with HIV shown expressing HIV RNA by in situ hybridization using a radiolabeled molecular probe. Original ×500. (Adapted from G Pantaleo, AS Fauci et al: Nature 362:355, 1993.)
During early and chronic/asymptomatic stages of HIV disease, the architecture of lymphoid tissues is generally preserved and may even be hyperplastic owing to an increased presence of B cells and specialized CD4+ T cells called follicular helper CD4+ T cells (TFH) in prominent germinal centers. Extracellular virions can be seen by electron microscopy attached to FDC processes. The trapping of antigen is a physiologically normal function for the FDCs, which present antigen to B cells and, along with the action of TFH cells, contribute to the generation of B cell memory. However, in the case of HIV, persistent cellular activation, resulting in the secretion of proinflammatory cytokines such as interleukin (IL) 1β, tumor necrosis factor (TNF) α, IFN-γ, and IL-6, which can induce viral replication (see below) and diminish the effectiveness of the immune response against the virus. In addition, the CD4+ TFH cells that are recruited into the germinal center to provide help to B cells in the generation of an HIV-specific immune response are highly susceptible to infection by virus either trapped on FDC or propagated locally. Thus, in HIV infection, a normal physiologic function of the immune system that contributes to the clearance of virus, as well as to the generation of a specific immune response, can also have deleterious consequences.
As the disease progresses, the architecture of lymphoid tissues begins to show disruption. Confocal microscopy reveals destruction of the fibroblastic reticular cell (FRC) and FDC networks in the T cell zone and B cell follicles, respectively, and an incapacity to replenish naïve cells. The mechanisms of destruction are not completely understood, but they are thought to be associated with collagen deposition causing fibrosis and loss of production of cytokines such as IL-7 and lymphotoxin-α, which are critical to the maintenance of lymphoid tissues and their lymphocyte constituents. As the disease progresses to an advanced stage, there is complete disruption of the architecture of the lymphoid tissues, accompanied by dissolution of the FRC and FDC networks. At this point, the lymph nodes are “burnt out.” This destruction of lymphoid tissue compounds the immunodeficiency of HIV disease and contributes both to the inability to control HIV replication (leading usually to high levels of plasma viremia in the untreated or inadequately treated patient) and to the inability to mount adequate immune responses against opportunistic pathogens. The events from primary infection to the ultimate destruction of the immune system are illustrated in Fig. 226-24. Recently, nonhuman primate studies and some human studies have examined GALT at various stages of HIV disease. Within the GALT, the basal level of activation combined with virus-mediated cellular activation results in the infection and elimination of an estimated 50–90% of CD4+ T cells in the gut. The extent of this early damage to GALT, which constitutes a major component of lymphoid tissue in the body, may play a role in determining the potential for immunologic recovery of the memory cell subset.
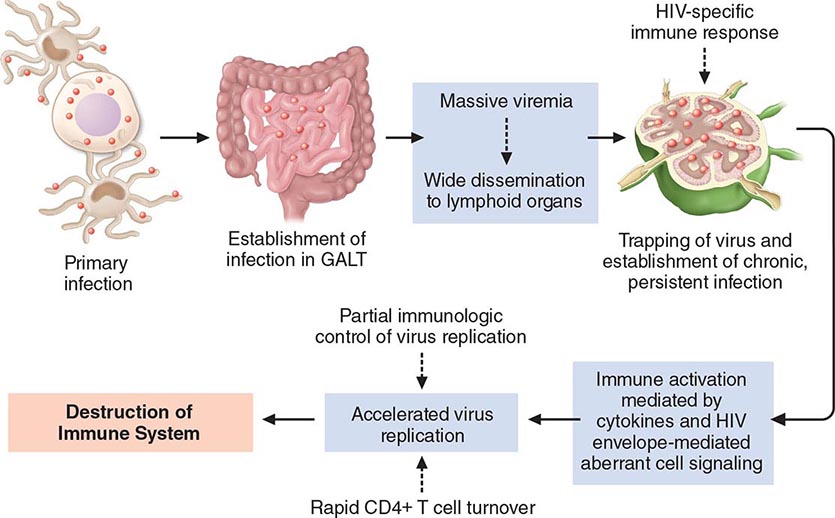
FIGURE 226-24 Events that transpire from primary HIV infection through the establishment of chronic persistent infection to the ultimate destruction of the immune system. See text for details. CTLs, cytolytic T lymphocytes; GALT, gut-associated lymphoid tissue.
IMMUNE ACTIVATION, INFLAMMATION, AND HIV PATHOGENESIS
Activation of the immune system and variable degrees of inflammation are essential components of any appropriate immune response to a foreign antigen. However, immune activation and inflammation, which can be considered aberrant in HIV-infected individuals, play a critical role in the pathogenesis of HIV disease and other chronic conditions associated with HIV disease. Immune activation and inflammation in the HIV-infected individual contribute substantially to (1) the replication of HIV, (2) the induction of immune dysfunction, and (3) the increased incidence of chronic conditions associated with persistent immune activation and inflammation (Table 226-4).
|
CONDITIONS ASSOCIATED WITH PERSISTENT IMMUNE ACTIVATION AND INFLAMMATION IN PATIENTS WITH HIV INFECTION |
INDUCTION OF HIV REPLICATION BY ABERRANT IMMUNE ACTIVATION The immune system is normally in a state of homeostasis, awaiting perturbation by foreign antigenic stimuli. Once the immune response deals with and clears the antigen, the system returns to relative quiescence (Chap. 372e). This is generally not the case in HIV infection where, in the untreated patient, virus replication is invariably persistent with very few exceptions and immune activation is persistent. HIV replicates most efficiently in activated CD4+ T cells; in HIV infection, chronic activation provides the cell substrates necessary for persistent virus replication throughout the course of HIV disease, particularly in the untreated patient and to variable degrees even in certain patients receiving cART whose levels of plasma viremia are suppressed to below the level of detection by standard assays. From a virologic standpoint, although quiescent CD4+ T cells can be infected with HIV, reverse transcription, integration, and virus spread are much more efficient in activated cells. Furthermore, cellular activation induces expression of virus in cells latently infected with HIV. In essence, immune activation and inflammation provide the engine that drives HIV replication. In addition to endogenous factors such as cytokines, a number of exogenous factors such as other microbes that are associated with heightened cellular activation can enhance HIV replication and thus may have important effects on HIV pathogenesis. Co-infection in vivo or in vitro with a range of viruses, such as HSV types 1 and 2, cytomegalovirus (CMV), human herpesvirus (HHV) 6, Epstein-Barr virus (EBV), HBV, adenovirus, and HTLV-1 have been shown to upregulate HIV expression. In addition, infestation with nematodes has been shown to be associated with a heightened state of immune activation that facilitates HIV replication; in certain studies deworming of the infected host has resulted in a decrease in plasma viremia. Two diseases of extraordinary global health significance, malaria and tuberculosis (TB), have been shown to increase HIV viral load in dually infected individuals. Globally, Mycobacterium tuberculosis is the most common opportunistic infection in HIV-infected individuals (Chap. 202). In addition to the fact that HIV-infected individuals are more likely to develop active TB after exposure, it has been demonstrated that active TB can accelerate the course of HIV infection. It has also been shown that levels of plasma viremia are greatly elevated in HIV-infected individuals with active TB who are not on cART, compared with pre-TB levels and levels of viremia after successful treatment of the active TB. The situation is similar in the interaction between HIV and malaria parasites (Chap. 248). Acute infection of HIV-infected individuals with Plasmodium falciparum increases HIV viral load, and the increased viral load is reversed by effective malaria treatment.
MICROBIAL TRANSLOCATION AND PERSISTENT IMMUNE ACTIVATION One proposed mechanism of persistent immune activation involves the disruption of the mucosal barrier in the gut due to HIV replication in and disruption of submucosal lymphoid tissue. As a result of this disruption, there is an increase in the products, particularly lipopolysaccharide (LPS), of bacteria that translocate from the bowel lumen through the damaged mucosa to the circulation, leading to persistent systemic immune activation and inflammation. This effect can persist even after the HIV viral load is brought to <50 copies/mL by cART. Depletion in the GALT of IL-17–producing T cells, which are responsible for defense against extracellular bacteria and fungi, also is thought to contribute to HIV pathogenesis.
PERSISTENT IMMUNE ACTIVATION AND INFLAMMATION INDUCE IMMUNE DYSFUNCTION The activated state in HIV infection is reflected by hyperactivation of B cells leading to hypergammaglobulinemia; increased lymphocyte turnover; activation of monocytes; expression of activation markers on CD4+ and CD8+ T cells; increased activation-associated cellular apoptosis; lymph node hyperplasia, particularly early in the course of disease; increased secretion of proinflammatory cytokines, particularly IL-6; elevated levels of high-sensitivity C-reactive protein, fibrinogen, D-dimer, neopterin, β2-microglobulin, acid-labile interferon, soluble (s) IL-2 receptors (R), sTNFR, sCD27, and sCD40L; and autoimmune phenomena (see “Autoimmune Phenomena,” below). Even in the absence of direct infection of a target cell, HIV envelope proteins can interact with cellular receptors (CD4 molecules and chemokine receptors) to deliver potent activation signals resulting in calcium flux, the phosphorylation of certain proteins involved in signal transduction, co-localization of cytoplasmic proteins including those involved in cell trafficking, immune dysfunction, and, under certain circumstances, apoptosis. From an immunologic standpoint, chronic exposure of the immune system to a particular antigen over an extended period may ultimately lead to an inability to sustain an adequate immune response to the antigen in question. In many chronic viral infections, including HIV infection, persistent viremia is associated with “functional exhaustion” of virus-specific T cells, decreasing their capacity to proliferate and perform effector functions. It has been demonstrated that this phenomenon may be mediated, at least in part, by the upregulation of inhibitory receptors on HIV-specific T cells, such as PD-1 shared by both CD4+ and CD8+ T cells, as well as CTLA-4 on CD4+ and Tim-3, 2B4, and CD106 on CD8+ T cells. Furthermore, the ability of the immune system to respond to a broad spectrum of antigens may be compromised if immunocompetent cells are maintained in a state of chronic activation.
The deleterious effects of chronic immune activation on the progression of HIV disease are well established. As in most conditions of persistent antigen exposure, the host must maintain sufficient activation of antigen (HIV)-specific responses but must also prevent excessive activation and potential immune-mediated damage to tissues. Certain studies suggest that normal immunosuppressive mechanisms that act to keep hyperimmune activation in check, particularly CD4+, FoxP3+, CD25+ regulatory T cells (T-regs), may be dysfunctional or depleted in the context of advanced HIV disease.
MEDICAL CONDITIONS ASSOCIATED WITH PERSISTENT IMMUNE ACTIVATION AND INFLAMMATION IN HIV DISEASE It has become clear, as the survival of HIV-infected individuals has increased, that a number of previously unrecognized medical complications are associated with HIV disease—and that these complications relate to chronic immune activation and inflammation (Table 226-4). These complications can appear even after patients have experienced years of adequate control of viral replication (plasma viremia below detectable levels) for several years. Of particular note are endothelial cell dysfunction and its relationship to cardiovascular disease. Other chronic conditions that have been reported include bone fragility, certain cancers, persistent immune dysfunction, diabetes, kidney and liver disease, and neurocognitive dysfunction, thus presenting an overall picture of accelerated aging.
Apoptosis Apoptosis is a form of programmed cell death that is a normal mechanism for the elimination of effete cells in organogenesis as well as in the cellular proliferation that occurs during a normal immune response (Chap. 372e). Apoptosis is largely dependent on cellular activation, and the aberrant cellular activation associated with HIV disease is correlated with a heightened state of apoptosis. HIV can trigger both Fas-dependent and Fas-independent pathways of apoptosis, the former of which is generally referred to as activation-induced cell death through an extrinsic pathway and involves the upregulation of the death receptor Fas and Fas ligand. Fas-independent pathways can be either extrinsic with different death receptors or intrinsic due to the downregulation of the antiapoptotic proteins such as Bcl-2. More recently, the phenomenon of pyroptosis, an inflammatory form of cell death involving the upregulation of the proinflammatory enzyme caspase-1 and release of the proinflammatory cytokine IL-1 β, has been linked to a bystander effect of HIV replication on CD4+ T cells. Certain viral gene products have been associated with enhanced susceptibility to apoptosis; these include Env, Tat, and Vpr. In contrast, Nef has been shown to possess antiapoptotic properties. A number of studies, including those examining lymphoid tissue, have demonstrated that the rate of apoptosis is elevated in HIV infection and that apoptosis is seen in “bystander” cells such as CD8+ T cells and B cells as well as in uninfected CD4+ T cells. The intensity of apoptosis correlates with the general state of activation of the immune system and not with the stage of disease or with viral burden. It is likely that nonspecific apoptosis of immunocompetent cells related to immune activation contributes to the immune abnormalities in HIV disease.
Autoimmune Phenomena The autoimmune phenomena that are common in HIV-infected individuals reflect, at least in part, chronic immune activation and the dysregulation of B and T cells. Although these phenomena usually occur in the absence of autoimmune disease, a wide spectrum of clinical manifestations that may be associated with autoimmunity have been described (see “Immunologic and Rheumatologic Diseases,” below). Autoimmune phenomena include antibodies against autoantigens expressed on intact lymphocytes and other cells, or against proteins released from dying cells. Antiplatelet antibodies have some clinical relevance in that they may contribute to the thrombocytopenia of HIV disease (see below). Antibodies to nuclear and cytoplasmic components of cells have been reported, as have antibodies to cardiolipin and phospholipids; CD4 molecules; CD43 molecules; C1q-A; variable regions of the T cell receptor α, β, and γ chains; Fas; denatured collagen; and IL-2. In addition, autoantibodies to a range of serum proteins, including albumin, immunoglobulin, and thyroglobulin, have been reported. Molecular mimicry, either from opportunistic pathogens or from HIV itself, is also a trigger or co-factor in autoimmunity. Antibodies against the HIV envelope proteins, especially gp41, often cross-react with host proteins; the best known examples are antibodies directed against the membrane-proximal external region of gp41 that also react with phospholipids and cardiolipin. The phenomenon of polyreactive HIV-specific antibodies may be beneficial to the host (see “Immune Response to HIV,” below).
The increased occurrence and/or exacerbation of certain autoimmune diseases have been reported in HIV infection; these diseases include psoriasis, idiopathic thrombocytopenic purpura, Graves’ disease, antiphospholipid syndrome, and primary biliary cirrhosis. The majority of these manifestations were described prior to the advent of cART and have decreased in frequency since its widespread use. However, with increasing availability of cART, an immune reconstitution inflammatory syndrome (IRIS) has become increasingly common in infected individuals, particularly those with low CD4+ T cell counts. IRIS is an autoimmune-like phenomenon characterized by a paradoxical deterioration of clinical condition, which is usually compartmentalized to a particular organ system, in individuals in whom cART has recently been initiated. It is associated with a decrease in viral load and at least partial recovery of immune competence, which is usually associated with increases in CD4+ T cell counts. The immunopathogenesis is felt to be related to an increase in immune response against the presence of residual antigens that are usually microbial and is commonly seen with underlying Mycobacterium tuberculosis and cryptococcosis. This syndrome is discussed in more detail below.
CYTOKINES AND OTHER SOLUBLE FACTORS IN HIV PATHOGENESIS
The immune system is homeostatically regulated by a complex network of immunoregulatory cytokines, which are pleiotropic and redundant and operate in an autocrine and paracrine manner. They are expressed continuously, even during periods of apparent quiescence of the immune system. On perturbation of the immune system by antigenic challenge, the expression of cytokines increases to varying degrees (Chap. 372e). Cytokines that are important components of this immunoregulatory network are thought to play major roles in HIV disease, during both the early and chronic phases of infection. A potent pro-inflammatory “cytokine storm” is induced during the acute phase of HIV infection, likely a response by inflammatory cells recruited to mucosal tissues where the virus initially replicates at very high levels. Cytokines that are induced during this early phase include IFN-α, IL-15, and the CXC chemokine IP-10 (CXCL10), followed by IL-6, IL-12, and TNF-α, and a delayed peak of the anti-inflammatory cytokine IL-10. Soluble factors of innate immunity are also induced shortly after infection, including neopterin and β-microglobulin. Several of these early-expressed cytokines and factors are not downregulated following the early phase of HIV infection, as seen in self-resolving viral infections, and persist during the chronic phase of infection and contribute to maintaining high levels of immune activation. Among the cytokines and factors associated with early innate immune responses, they are intended to contain viral replication, although most are potent inducers of HIV expression/replication because of their ability to induce immune activation that leads to enhanced viral production and an increase in readily available target cells for HIV (activated CD4+ T cells). The induction of IFN-α, one of the first cytokines induced during primary HIV infection, is thought to play a particularly important role in HIV pathogenesis by inducing a large number of IFN-associated genes that activate the immune system and alter the homeostasis of CD4+ T cells. Other cytokines that are elevated during the chronic phase of HIV infection and linked to immune activation include IFN-γ, the CC-chemokine RANTES (CCL5), macrophage inflammatory protein (MIP)-1β (CCL4), and IL-18.
Several specific cytokines and soluble factors have been associated with HIV pathogenesis at various stages of disease, in various tissues or organs, and in the regulation of HIV replication. Plasma levels of IP-10 are predictive of disease progression, whereas the proinflammatory cytokine IL-6, soluble CD14 (sCD14), and coagulation marker D-dimer are associated with increased risk of all-cause mortality in HIV-infected individuals. In particular, IL-6, sCD14, and D-dimer are associated with increased risk of cardiovascular disease and other causes of death, even in individuals receiving cART. IL-18 has also been shown to play a role in the development of the HIV-associated lipodystrophy syndrome, whereas increased levels of transforming growth factor (TGF)-β are associated with the induction of collagen deposition in lymph nodes (see above). Elevated levels of TNF-α and IL-6 have been demonstrated in plasma and cerebrospinal fluid (CSF), and increased expression of TNF-α, IL-1β, IFN-γ, and IL-6 has been demonstrated in the lymph nodes of HIV-infected individuals. RANTES, MIP-1α (CCL3), and MIP-1β (CCL4) (Chap. 372e) inhibit infection by and spread of R5 HIV-1 strains, while stromal cell–derived factor (SDF) 1 inhibits infection by and spread of X4 strains. The mechanisms whereby the CC-chemokines RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4) inhibit infection of R5 strains of HIV, or SDF-1 blocks X4 strains of HIV, involve blocking of the binding of the virus to its co-receptors, the CC-chemokine receptor CCR5 and the CXC-chemokine receptor CXCR4, respectively. Other soluble factors that have not yet been fully characterized have also been shown to suppress HIV replication, independent of co-receptor usage.
LYMPHOCYTE TURNOVER IN HIV INFECTION
The immune systems of patients with HIV infection are characterized by a profound increase in lymphocyte turnover that is immediately reduced with effective cART. Studies utilizing in vivo or in vitro labeling of lymphocytes in the S-phase of the cell cycle have demonstrated a tight correlation between the degree of lymphocyte turnover and plasma levels of HIV RNA. This increase in turnover is seen in CD4+ and CD8+ T lymphocytes as well as B lymphocytes and can be observed in peripheral blood and lymphoid tissue. Mathematical models derived from these data suggest that one can view the lymphoid pool as consisting of dynamically distinct subpopulations of cells that are differentially affected by HIV infection. A major consequence of HIV infection appears to be a shift in cells from a more quiescent pool to a pool with a higher turnover rate. It is likely that a consequence of a higher rate of turnover is a higher rate of cell death. The role of the thymus in adult human T cell homeostasis and HIV pathogenesis is an area of controversy. While some data point to an important role for the thymus in maintaining T cell numbers and suggest that impairment of thymic function may be responsible for the declines in CD4+ T cells seen in the setting of HIV infection, other studies have concluded that the thymus plays a minor role in HIV pathogenesis. More recently, it has been suggested that the more rapid decline in CD4+ compared to CD8+ T cells may be linked to alterations in inflammatory and homeostatic cytokines that caused increased activation-induced death of CD4+ but not CD8+ T cells (see Table 226-5 for additional mechanisms of depletion).
|
PROPOSED MECHANISMS OF CD4+ T CELL DYSFUNCTION AND DEPLETION |
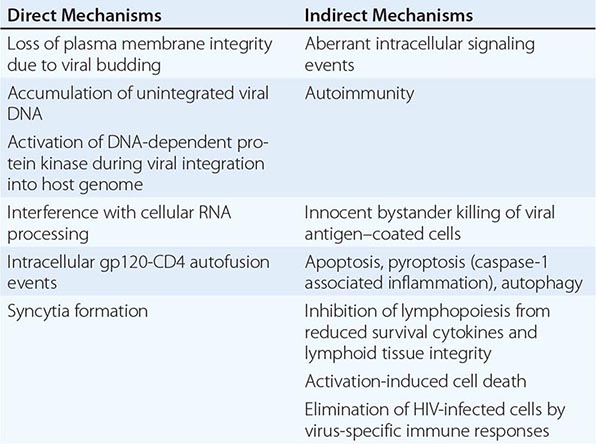
THE ROLE OF VIRAL RECEPTORS AND CO-RECEPTORS IN HIV PATHOGENESIS
As mentioned above, HIV-1 utilizes two major co-receptors along with CD4 to bind to, fuse with, and enter target cells; these co-receptors are CCR5 and CXCR4, which are also receptors for certain endogenous chemokines. Strains of HIV that utilize CCR5 as a co-receptor are referred to as R5 viruses. Strains of HIV that utilize CXCR4 are referred to as X4 viruses. Many virus strains are dual tropic in that they utilize both CCR5 and CXCR4; these are referred to as R5X4 viruses.
The natural chemokine ligands for the major HIV co-receptors can readily block entry of HIV. For example, the CC-chemokines RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4), which are the natural ligands for CCR5, block entry of R5 viruses, whereas SDF-1, the natural ligand for CXCR4, blocks entry of X4 viruses. The mechanism of inhibition of viral entry is a steric inhibition of binding that is not dependent on signal transduction (Fig. 226-25).
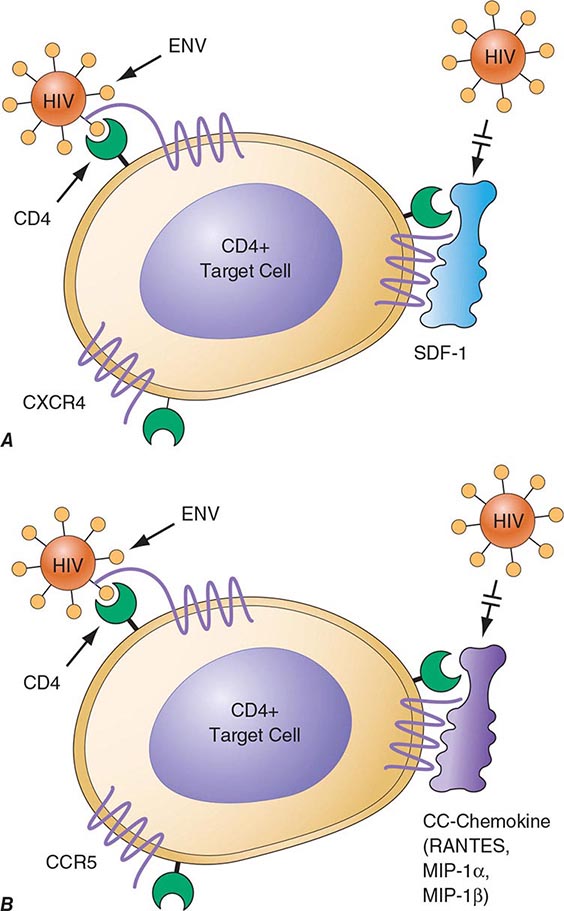
FIGURE 226-25 Model for the role of co-receptors CXCR4 and CCR5 in the efficient binding and entry of X4 (A) and R5 (B) strains of HIV-1, respectively, into CD4+ target cells. Blocking of this initial event in the virus life cycle can be accomplished by inhibition of binding to the co-receptor by the normal ligand for the receptor in question. The ligand for CXCR4 is stromal cell–derived factor (SDF-1); the ligands for CCR5 are RANTES, MIP-1α, and MIP-1β.
The transmitting virus is almost invariably an R5 virus that predominates during the early stages of HIV disease. In ~40% of HIV-infected individuals, there is a transition to a predominantly X4 virus that is associated with a relatively rapid progression of disease. However, at least 60% of infected individuals progress in their disease while maintaining predominance of an R5 virus. It should be pointed out that clade C viruses, unlike other subgroups, almost never switch from CCR5 tropism to CXCR4 tropism; the reason for this difference is unclear.
The basis for the tropism of different envelope glycoproteins for either CCR5 or CXCR4 relates to the ability of the HIV envelope, including the third variable region (V3 loop) of gp120, to interact with these co-receptors. In this regard, binding of gp120 to CD4 induces a conformational change in gp120 that increases its affinity for CCR5 (see above). Finally, R5 viruses are more efficient in infecting monocytes/macrophages and microglial cells of the brain (see “Neuropathogenesis in HIV Disease,” below).
THE INTEGRIN α4β7 AND MUCOSAL TRANSMISSION OF HIV Several “accessory receptors” for HIV have been reported over the years, although only a few have withstood the test of time. These receptors are not necessary for virus binding and fusion to its target CD4+ T cell or for virus replication. However, the integrin α4β7 is an accessory receptor for HIV and it likely plays an important role in the transmission of HIV at mucosal surfaces such as the genital tract and gut. The integrin α4β7, which is the gut homing receptor for peripheral T cells, binds in its activated form to a specific tripeptide in the V2 loop of gp120, resulting in rapid activation of leukocyte function–associated antigen 1 (LFA-1), the central integrin in the establishment of virologic synapses, which facilitate efficient cell-to-cell spread of HIV. It has been demonstrated that α4β7high CD4+ T cells are more susceptible to productive infection than are α4β7low–neg CD4+ T cells because this cellular subset is enriched with metabolically active CD4+ T cells that are CCR5high. These cells are present at the gut and genital tract mucosal surfaces. Importantly, it has been demonstrated that the virus that is transmitted during sexual exposure binds much more efficiently to α4β7 than does the virus that diversifies from the transmitting virus over time by mutation, particularly involving the accumulation of glycosylation sites (see “Early Events in HIV Infection: Primary Infection and Initial Dissemination of Virus,” above).
CELLULAR TARGETS OF HIV
Although the CD4+ T lymphocytes and to a lesser extent CD4+ cells of monocyte lineage are the principal targets of HIV, virtually any cell that expresses the CD4 molecule together with co-receptor molecules (see above and below) can potentially be infected with HIV. Circulating DCs have been reported to express low levels of CD4, and, depending on their stage of maturation, these cells can be infected with HIV. Epidermal Langerhans cells express CD4 and have been infected by HIV in vivo, although, as has been shown in vivo for DCs, FDCs, and B cells, these cells are more likely to bind and transfer virus to activated CD4+ T cells than to be productively infected themselves.
In vitro, HIV has been reported also to infect a wide range of cells and cell lines that express low levels of CD4, no detectable CD4, or only CD4 mRNA. However, since the only cells that have been shown unequivocally to be infected with HIV and to support replication of the virus are CD4+ T lymphocytes and cells of monocyte/macrophage lineage, the physiologic relevance of the in vitro infection of these other cell types is unclear.
Of potentially important clinical relevance is the demonstration that thymic precursor cells, which were assumed to be negative for CD3, CD4, and CD8 molecules, actually do express low levels of CD4 and can be infected with HIV in vitro. In addition, human thymic epithelial cells transplanted into an immunodeficient mouse can be infected with HIV by direct inoculation of virus into the thymus. Since these cells may play a role in the normal regeneration of CD4+ T cells, it is possible that their infection and depletion contribute, at least in part, to the impaired ability of the CD4+ T cell pool to completely reconstitute itself in certain infected individuals in whom cART has suppressed viral replication to <50 copies of HIV RNA per milliliter (see below). In addition, CD34+ monocyte precursor cells have been shown to be infected in vivo in patients with advanced HIV disease. It is likely that these cells express low levels of CD4, and therefore it is not essential to invoke CD4-independent mechanisms to explain the infection.
ABNORMALITIES OF MONONUCLEAR CELLS
CD4+ T Cells The primary immunopathogenic lesion in HIV infection involves CD4+ T cells, and the range of CD4+ T cell abnormalities in advanced HIV infection is broad. The defects are both quantitative and qualitative and ultimately impact virtually every limb of the immune system, indicating the critical dependence of the integrity of the immune system on the inducer/helper function of CD4+ T cells. In advanced HIV disease, most of the observed immune defects can ultimately be explained by the quantitative depletion of CD4+ T cells. However, T cell dysfunction can be demonstrated in patients early in the course of infection, even when the CD4+ T cell count is in the low-normal range. The degree and spectrum of dysfunctions increase as the disease progresses, reflecting the range of CD4+ T cell functional heterogeneity, especially in lymphoid tissues. One of the first sites of intense HIV replication is in the GALT where CD4+ TH17 cells reside; they are important for host defense against extracellular pathogens in the intestinal mucosa and help maintain the integrity of the gut epithelium. In HIV infection, they are depleted by direct and indirect effects of viral replication and cause loss of gut homeostasis and integrity, as well as a shift to a more TH1 phenotype. Studies have shown that even after many years of cART, normalization of the CD4+ T cells in the GALT remains incomplete. In lymph nodes, HIV perturbs another important subset of the CD4+ helper T lineage, namely TFH cells (see “Lymphoid Organs and HIV Pathogenesis,” above). TFH cells, which are either derived directly from naïve CD4+ T cells or other TH precursors, migrate into B follicles during germinal center reactions and provide help to antigen-specific B cells through cell–cell interactions and secretion of cytokines to which B cells respond, the most important of which is IL-21. As with TH17 cells, TFH cells are highly susceptible to HIV infection. However, in contrast to TH17 and most other CD4+ T cell subsets, the number of TFH cells is increased in lymph nodes of HIV-infected individuals, especially those who are viremic. It is unclear whether this increase is helpful to responding B cells, although the likely outcome is that the increase in numbers is detrimental to the quality of the humoral immune response against HIV (see “Immune Response to HIV,” below). In addition, defects of central memory cells are a critical component of HIV immunopathogenesis. The progressive loss of antigen-specific CD4+ T cells has important implications for the control of HIV infection. In this regard, there is a correlation between the maintenance of HIV-specific CD4+ T cell proliferative responses and improved control of infection. Essentially every T cell function has been reported to be abnormal at some stage of HIV infection. Loss of polyfunctional HIV-specific CD4+ T cells, especially those that produce IL-2, occurs early in disease, whereas IFN-producing CD4+ T cells are maintained longer and do not correlate with control of HIV viremia. Other abnormalities include impaired expression of IL-2 receptors, defective IL-2 production, reduced expression of the IL-7 receptor (CD127), and a decreased proportion of CD4+ T cells that express CD28, a major co-stimulatory molecule necessary for the normal activation of T cells, which is also depleted as a result of aging. Cells lacking expression of CD28 do not respond normally to activation signals and may express markers of terminal activation including HLA-DR, CD38, and CD45RO. As mentioned above (“Immune Activation, Inflammation, and HIV Pathogenesis”), a subset of CD4+ T cells referred to as T regulatory cells, or T-regs, may be involved in damping aberrant immune activation that propagates HIV replication. The presence of these T-reg cells correlates with lower viral loads and higher CD4+/CD8+ T cell ratios. A loss of this T-reg capability with advanced disease may be detrimental to the control of virus replication.
It is difficult to explain completely the profound immunodeficiency noted in HIV-infected individuals solely on the basis of direct infection and quantitative depletion of CD4+ T cells. This is particularly apparent during the early stages of HIV disease, when CD4+ T cell numbers may be only marginally decreased. In this regard, it is likely that CD4+ T cell dysfunction results from a combination of depletion of cells due to direct infection of the cell and a number of virus-related but indirect effects on the cell (Table 226-5). Several of these effects have been demonstrated ex vivo and/or by the analysis of cells isolated from the peripheral blood. However, as explained above, many of the defects are related to specialized CD4+ T cells that reside in lymphoid tissues. Furthermore, since the main targets of HIV infection are immunocompetent cells, these responses may contribute to immune cell depletion and immunologic dysfunction by eliminating both infected cells and “innocent bystander” cells. Soluble viral proteins, particularly gp120, can bind with high affinity to the CD4 molecules on uninfected T cells and monocytes; in addition, virus and/or viral proteins can bind to DCs or FDCs. HIV-specific antibody can recognize these bound molecules and potentially collaborate in the elimination of the cells by ADCC. HIV envelope glycoproteins gp120 and gp160 manifest high-affinity binding to the CD4 molecule as well as to various chemokine receptors. Intracellular signals transduced by gp120 through both CD4 and CCR5/CXCR4 have been associated with a number of immunopathogenic processes including anergy, apoptosis, and abnormalities of cell trafficking. The molecular mechanisms responsible for these abnormalities include dysregulation of the T cell receptor–phosphoinositide pathway, p56lck activation, phosphorylation of focal adhesion kinase, activation of the MAP kinase and ras signaling pathways, and downregulation of the co-stimulatory molecules CD40 ligand and CD80.
The inexorable decline in CD4+ T cell counts that occurs in most HIV-infected individuals may result in part from the inability of the immune system to regenerate over an extended period of time the rapidly turning over CD4+ T cell pool efficiently enough to compensate for both HIV-mediated and naturally occurring attrition of cells. In this regard, the degree and duration of decline of CD4+ T cells at the time of initiation of therapy is an important predictor of the restoration of these cells. A person who maintains a very low CD4+ T cell count for a considerable period of time before the initiation of cART almost invariably has an incomplete reconstitution of such cells. At least two major mechanisms may contribute to the failure of the CD4+ T cell pool to reconstitute itself adequately over the course of HIV infection. The first is the destruction of lymphoid precursor cells, including thymic and bone marrow progenitor cells; the other is the gradual disruption of the lymphoid tissue microenvironment, which is essential for efficient regeneration of immunocompetent cells. Finally, during the advanced stages of CD4+ T lymphopenia, there are increased serum levels of the homeostatic cytokine IL-7. It was initially felt that this elevation was a homeostatic response to the lymphopenia; however, recent findings suggest that the increase in serum IL-7 was a result of reduced utilization of the cytokine related to the loss of cells expressing the IL-7 receptor, CD127, which serves as a normal physiologic regulator of IL-7 production.
CD8+ T Cells A relative CD8+ T lymphocytosis is generally associated with high levels of HIV plasma viremia and likely reflects an immune response to the virus as well as dysregulated homeostasis associated with generalized immune activation. During the late stages of HIV infection, there may be a significant reduction in the numbers of CD8+ T cells despite the presence of high levels of viremia. HIV-specific CD8+ CTLs have been demonstrated in HIV-infected individuals early in the course of disease, and their emergence often coincides with a decrease in plasma viremia—an observation that is a factor in the proposal that virus-specific CTLs can control HIV disease for a finite period of time in a certain percentage of infected individuals. However, emergence of HIV escape mutants that ultimately evade these HIV-specific CD8+ T cells has been described in the majority of HIV-infected individuals who are not receiving cART. In addition, as the disease progresses, the functional capability of these cells gradually decreases, at least in part due to the persistent nature of HIV infection that causes functional exhaustion via the upregulation of inhibitory receptors such as PD-1 on HIV-specific CD8+ T cells (see “Immune Activation, Inflammation, and HIV Pathogenesis,” above). As chronic immune activation persists, there are also systemic effects on CD8+ T cells, such that as a population they assume an abnormal phenotype characterized by expression of activation markers such as HLA-DR and CD38 with an absence of expression of the IL-2 receptor (CD25) and a reduced expression of the IL-7 receptor (CD127). In addition, CD8+ T cells lacking CD28 expression are increased in HIV disease, reflecting a skewed expansion of a less differentiated CD8+ T cell subset. This skewing of subsets is also associated with diminished polyfunctionality, a qualitative difference that distinguishes nonprogressors from progressors. It has been reported that nonprogressors can also be distinguished from progressors by the maintenance in the former of a high proliferative capacity of their HIV-specific CD8+ T cells coupled to increases in perforin expression, characteristics that are markedly diminished in advanced HIV disease. It has been reported that the phenotype of CD8+ T cells in HIV-infected individuals may be of prognostic significance. Those individuals whose CD8+ T cells developed a phenotype of HLA-DR+/CD38– following seroconversion had stabilization of their CD4+ T cell counts, whereas those whose CD8+ T cells developed a phenotype of HLA-DR+/CD38+ had a more aggressive course and a poorer prognosis. In addition to the defects in HIV-specific CD8+ CTLs, functional defects in other MHC-restricted CTLs, such as those directed against influenza and CMV, have been demonstrated. CD8+ T cells secrete a variety of soluble factors that inhibit HIV replication, including the CC-chemokines RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4) as well as potentially a number of unidentified factors. The presence of high levels of HIV viremia in vivo as well as exposure of CD8+ T cells in vitro to HIV envelope, both of which are associated with aberrant immune activation, have been shown to be associated with a variety of cellular functional abnormalities. Furthermore, since the integrity of CD8+ T cell function depends in part on adequate inductive signals from CD4+ T cells, the defect in CD8+ CTLs is likely compounded by the quantitative loss and qualitative dysfunction of CD4+ T cells.
B Cells The predominant defect in B cells from HIV-infected individuals is one of aberrant cellular activation, which is reflected by increased propensity to terminal differentiation and immunoglobulin secretion and increased expression of markers of activation and exhaustion. As a result of activation and differentiation in vivo, B cells from HIV viremic patients manifest a decreased capacity to mount a proliferative response to ligation of the B cell antigen receptor and other B cell stimuli in vitro. B cells from HIV-infected individuals manifest enhanced spontaneous secretion of immunoglobulins in vitro, a process that reflects their highly differentiated state in vivo. There is also an increased incidence of EBV-related B cell lymphomas in HIV-infected individuals that are likely due to combined effects of defective T cell immune surveillance and increased turnover that increases the risk of oncogenesis. Untransformed B cells cannot be infected with HIV, although HIV or its products can activate B cells directly. B cells from patients with high levels of viremia bind virions to their surface via the CD21 complement receptor. It is likely that in vivo activation of B cells by replication-competent or defective virus as well as viral products during the viremic state accounts at least in part for their activated phenotype. B cell subpopulations from HIV-infected individuals undergo a number of changes over the course of HIV disease, including the attrition of resting memory B cells and replacement with several aberrant memory and differentiated B cell subpopulations that collectively express reduced levels of CD21 and either increased expression of activation markers or inhibitory receptors associated with functional exhaustion. The more activated and differentiated B cells are also responsible for increased secretion of immunoglobulins and increased susceptibility to Fas-mediated apoptosis. In more advanced disease, there is also the appearance of immature B cells associated with CD4+ T cell lymphopenia. Cognate B cell–CD4+ T cell interactions are abnormal in viremic HIV-infected individuals in that B cells respond poorly to CD4+ T cell help and CD4+ T cells receive inadequate co-stimulatory signals from activated B cells. In vivo, the aberrant activated state of B cells manifests itself by hypergammaglobulinemia and by the presence of circulating immune complexes and autoantibodies. HIV-infected individuals respond poorly to primary and secondary immunizations with protein and polysaccharide antigens. Using immunization with influenza vaccine, it has been demonstrated that there is a memory B cell defect in HIV-infected individuals, particularly those with high levels of HIV viremia. There is also evidence that responses to HIV and non-HIV antigens in infected individuals, especially those who remain viremic, are enriched in abnormal subsets of B cells that either are highly prone to apoptosis or show signs of functional exhaustion. Taken together, these B cell defects are likely responsible in part for the inadequate response to HIV as well as to decreased response to vaccinations and the increase in certain bacterial infections seen in advanced HIV disease in adults, as well as for the important role of bacterial infections in the morbidity and mortality rates of HIV-infected children, who cannot mount an adequate humoral response to common bacterial pathogens. The absolute number of circulating B cells may be depressed in HIV infection; this phenomenon likely reflects increased activation-induced apoptosis as well as a redistribution of cells out of the circulation and into the lymphoid tissue—phenomena that are associated with ongoing viral replication.
Monocytes/Macrophages Circulating monocytes are generally normal in number in HIV-infected individuals; however, there is evidence of increased activation within this lineage. The increased level of sCD14 and other biomarkers (see above) reported in HIV-infected individuals is an indirect marker of monocyte activation in vivo. A number of other abnormalities of circulating monocytes have been reported in HIV-infected individuals, many of which may be related directly or indirectly to aberrant in vivo immune activation. In this regard, increased levels of lipopolysaccharide (LPS) are found in the sera of HIV-infected individuals due, at least in part, to translocation across the gut mucosal barrier (see above). LPS is a highly inflammatory bacterial product that preferentially binds to macrophages through CD14 and Toll-like receptors, resulting in cellular activation. Functional abnormalities of monocyte/macrophages in HIV disease include decreased secretion of IL-1 and IL-12; increased secretion of IL-10 and IL-18; defects in antigen presentation and induction of T cell responses due to decreased MHC class II expression; and abnormalities of Fc receptor function, C3 receptor–mediated clearance, oxidative burst responses, and certain cytotoxic functions such as ADCC, possibly related to low levels of expression of Fc and complement receptors. Monocytes express the CD4 molecule and several co-receptors for HIV on their surface, including CCR5, CXCR4, and CCR3, and thus are potential targets of HIV infection. The degree of cytopathicity of HIV for cells of the monocyte lineage is low, and HIV can replicate in cells of the monocyte lineage with relatively little cytopathic effect. Hence, monocyte-lineage cells may play a role in the dissemination of HIV in the body and can serve as reservoirs of HIV infection, thus representing an obstacle to the eradication of HIV by antiretroviral drugs. In vivo infection of circulating monocytes is difficult to demonstrate; however, infection of tissue macrophages and macrophage-lineage cells in the brain (infiltrating macrophages or resident microglial cells) and lung (pulmonary alveolar macrophages) can be demonstrated easily. Tissue macrophages are an important source of HIV during the inflammatory response associated with opportunistic infections. Infection of monocyte precursors in the bone marrow may directly or indirectly be responsible for certain of the hematologic abnormalities in HIV-infected individuals. However, as with DCs, monocytes and macrophages express high levels of host restriction factors that likely help explain the low contribution of myeloid cells to the overall viral burden in HIV-infected individuals.
Dendritic and Langerhans Cells DCs and Langerhans cells are thought to play an important role in the initiation of HIV infection by virtue of the ability of HIV to bind to cell-surface C-type lectin receptors, particularly DC-SIGN (see above) and Langerin. This allows efficient presentation of virus to CD4+ T cell targets that become infected; complexes of infected CD4+ T cells and DCs provide an optimal microenvironment for virus replication. There was once considerable disagreement regarding the HIV infectibility and hence the depletion as well as the dysfunction of DCs themselves. However, since the recognition of myeloid (mDC) and plasmacytoid (pDC) subsets, there has been a better appreciation of specific DC dysfunction in HIV disease. pDCs are an important component of the innate immune system and secrete large amounts of IFN-α in response to viral infections. The numbers of circulating pDCs are decreased in HIV infection through mechanisms that remain unclear, and there are conflicting reports regarding the frequency of pDCs in lymphoid tissues, with some studies suggesting that their increased tissue presence and secretion of inflammatory cytokines such as IFN-α contributes to lymphoid hyperplasia. The mDCs or conventional DCs are involved in the initiation of adaptive immunity in draining lymph nodes by presenting antigen to T cells and B cells, as well as by secreting cytokines such as IL-12, IL-15, and IL-18 that activate other immune cells. There are also indications that the relatively low infectibility of DCs may be associated with the expression of host restriction factors, including APOBEC3G (see above).
Natural Killer Cells The role of NK cells is to provide immunosurveillance against virus-infected cells, certain tumor cells, and allogeneic cells (Chap. 372e). There are no convincing data that HIV productively infects NK cells in vivo; however, functional abnormalities in NK cells have been observed throughout the course of HIV disease, and the severity of these abnormalities increases as disease progresses. NK cells are part of the innate immune system and act by direct killing of infected cells and secretion of antiviral cytokines. In early HIV infection there is an increase in the activation of NK cells, and the capacity to secrete IFN-γ is maintained, although they manifest reduced cytotoxic function. During chronic HIV infection, both NK cell cytotoxicity and cytokine secretion become impaired. Given that HIV infection of target cells downregulates HLA-A and -B, but not HLA-C and -D molecules, this may explain in part the relative inability of NK cells to kill HIV-infected target cells. However, the NK cell impairments, especially in patients with high levels of virus replication, are associated with an expansion of an “anergic” CD56–/CD16+ NK cell subset. This abnormal subset of NK cells manifests an increased expression of inhibitory NK cell receptors (iNKRs) and a substantial decrease in expression of natural cytotoxicity receptors (NCRs) and shows a markedly impaired lytic activity. The overrepresentation of this abnormal subset of NK cells may explain in part the observed defects in NK cell function in HIV-infected individuals and likely begins to occur during primary infection. NK cells also serve as important sources of HIV-inhibitory CC-chemokines. NK cells isolated from HIV-infected individuals constitutively produce high levels of MIP-1α (CCL3), MIP-1β (CCL4), and RANTES (CCL5), although the impact of these chemokines on HIV replication in vivo is unclear. Finally, NK cell–DC interactions are important for normal immune function. NK cells and DCs reciprocally modulate each other’s activation and maturation. These interactions are markedly impaired in HIV-infected individuals with high levels of plasma viremia.
GENETIC FACTORS IN HIV-1 AND AIDS PATHOGENESIS
PHENOTYPES OF SUSCEPTIBILITY AND RESPONSE TO HIV INFECTION It is well known that individuals vary in their susceptibility to acquiring HIV infection and that there is wide variation in both the steady-state level of HIV that is established soon after infection (virologic setpoint) as well as the rate at which HIV-infected patients progress to AIDS. Some striking examples include sex workers who remain uninfected despite repeated exposure to HIV; HIV-infected individuals who spontaneously control viral replication in the absence of cART (HIV controllers); patients who resist disease progression for at least 8–10 years, despite viremia; and those progressing to AIDS within 3 years. Investigators have hypothesized that genetic differences may partly explain this interindividual variation in risk of acquiring HIV infection and disease progression rates. In addition to these phenotypes, it has been hypothesized that genetic variation may partly underpin the risk of developing specific AIDS-defining illnesses (e.g., renal and neurologic diseases) and non-AIDS comorbidities (e.g., cardiovascular disease), as well as the variable recovery in CD4+ T cell counts observed while receiving cART.
Candidate gene approaches and genome-wide association studies (GWAS) have demonstrated associations between gene variations and the above-mentioned phenotypes. A list of some of these associations is shown in Table 226-6. While in vitro genome-wide functional scanning using RNA interference has identified hundreds of host factors that may be involved in the HIV life cycle, the association of these genes with HIV susceptibility and/or disease progression remains largely undefined. Below is a discussion of a few key genes with strong associations and their implications for improving clinical care.
|
HOST GENETIC FACTORS THAT INFLUENCE RISK OF HIV-1 ACQUISITION AND RATES OF HIV-1 DISEASE PROGRESSION |
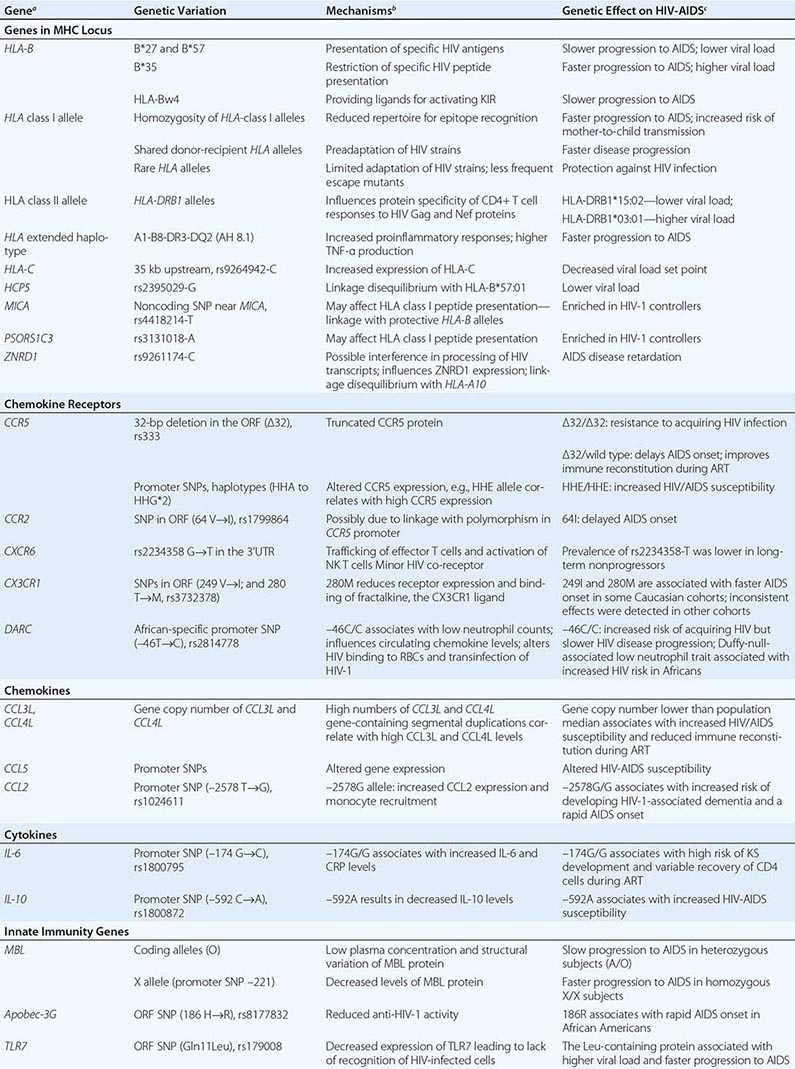
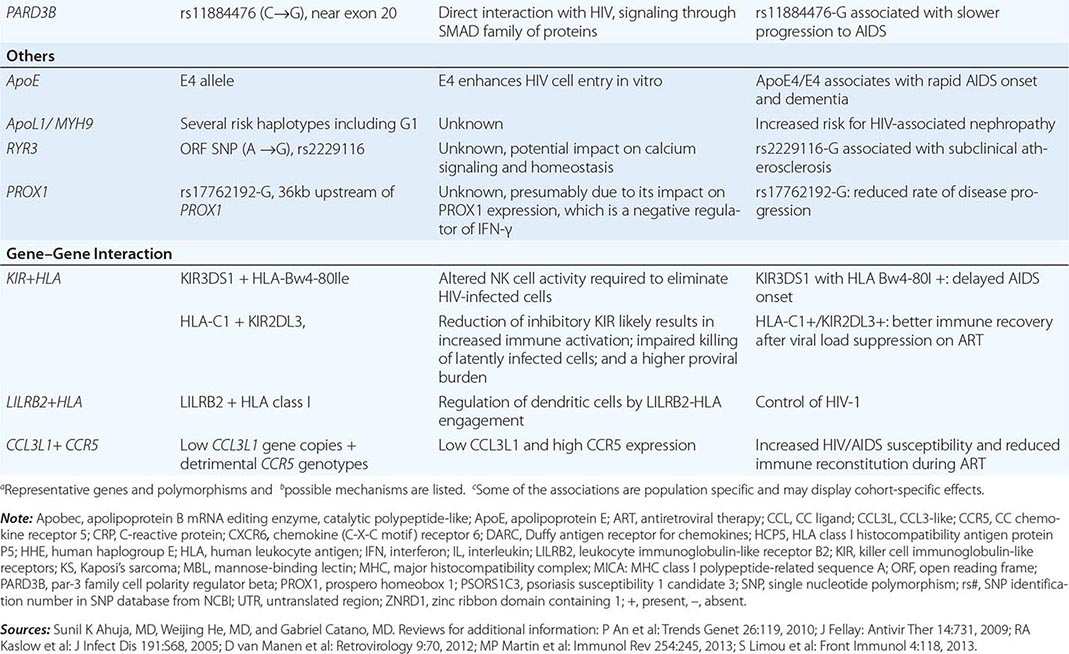
ASSOCIATIONS WITH CCR5 AND TRANSLATION OF GENETIC FINDINGS TO THE CLINIC Possibly, the most dramatic example of the importance of genetic studies for identifying host factors that influence HIV-AIDS pathogenesis is from studies related to the gene that encodes for CC chemokine receptor 5 (CCR5). While in vitro studies established that CCR5 is the major HIV co-receptor for the cell entry of HIV-1 into the host, it was genetic studies that established the seminal in vivo role of this receptor for the initial entry of HIV and AIDS pathogenesis.
Genetic analysis revealed that the in vitro resistance to CCR5-using R5 strains of HIV is in some instances due to carriage of two defective CCR5 alleles. This defect is a 32-bp deletion in the coding sequence (designated as the Δ32 allele). The CCR5 Δ32 allele encodes a truncated protein that is not expressed on the cell surface.
Approximately 1% of individuals of European ancestry are homozygous for the CCR5 Δ32 allele. Depending on the geographic region in Europe, up to 20% of individuals are heterozygous for the CCR5 Δ32 allele. The CCR5 Δ32 allele is either absent or extremely rare in other populations. The evolutionary pressure that resulted in the emergence of the CCR5 Δ32 allele in the European population remains unknown and has been speculated to be secondary to an ancestral pandemic such as the plague.
Individuals homozygous for the CCR5 Δ32 allele (Δ32/Δ32) lack CCR5 surface expression and are highly resistant to acquiring HIV infection. Heterozygosity for the CCR5 Δ32 allele is also associated with a reduced risk of acquiring HIV. Consequently, the frequency of the CCR5 Δ32 allele is enriched in individuals of European descent who remain uninfected despite exposure to the virus. Although the CCR5 Δ32/Δ32 genotype is associated with profound resistance to acquiring HIV, a few individuals with this genotype have become infected with the X4 HIV strain and, in some instances, experienced an accelerated disease course. In general, CCR5 Δ32 heterozygosity is associated with a slower HIV disease course.
Subsequent studies identified single nucleotide polymorphisms (SNPs) in the promoter (regulatory region) of CCR5 that influence its expression levels. Alleles bearing specific cassettes of linked polymorphisms (haplotypes) were identified and designated as human haplogroups A to G*2 (HHA to HHG*2). The CCR5 Δ32 is found on the HHG*2 haplotype. The CCR5 HHE haplotype was associated with higher CCR5 expression, and genetic association studies have shown that homozygosity for the CCR5 HHE haplotype is associated with an increased risk of acquiring HIV, progressing rapidly to AIDS, and reduced immune recovery on cART. The CCR2-64I-bearing HHF*2 haplotype is associated with a slower HIV disease course. The CCR5 HHA haplotype is the ancestral CCR5 haplotype and is associated with a lower CCR5 expression. The HHA haplotype was associated with slower disease progression in African populations and has been speculated to be a basis for why SIV-infected chimpanzees (who all carry the ancestral CCR5 HHA haplotype) may resist disease progression. The CCR5 haplotypes also influence cell-mediated immunity and immune recovery on cART.
The association of variations in the CCR5 gene with HIV-AIDS phenotypes is also an example of how discoveries made in the laboratory (bench) have been translated to improve health outcomes (bedside). The discovery that the CCR5 Δ32/Δ32 genotype is associated with strong resistance to HIV infection, and that uninfected Caucasians bearing this genotype did not appear to have impaired immunity, led to the development of two kinds of therapies. First, it spurred the development of a new class of FDA-approved therapies, entry inhibitors (e.g., maraviroc), that block the interaction of CCR5 with the HIV envelope. Second, it led to the development of novel experimental cellular therapies. An HIV-infected patient with acute myelogenous leukemia was given an allogeneic stem-cell transplantation from an HLA-compatible person whose cells lacked expression of CCR5 due to the Δ32/Δ32 genotype. There has been no evidence of HIV-1 infection in the transplanted patient thus far (6 years). This observation spurred the hope of an HIV cure and led to the development of additional novel cellular therapies involving autologous transplantation of CD4+ T-cells in which the CCR5 gene is inactivated ex vivo using new gene editing procedures.
DISCOVERY OF HLA CLASS I ALLELES THAT ASSOCIATE WITH VIROLOGIC CONTROL OF HIV INFECTION There is a strong association between variations within the HLA-B gene with protective (e.g., HLA-B*57 and -B*27 alleles) or detrimental (e.g., HLA-B*35 allele) outcomes during HIV infection. Carriage of the HLA-B*57 and/or HLA-B*27 alleles is associated with slower disease progression. The beneficial effects of these alleles may relate in part to their consistent associations with a lower virologic setpoint as well as to higher cell-mediated immunity. The protective effect of the HLA-B*57 and -B*27 alleles on HIV disease course is underscored by the finding that the prevalence of these alleles is higher among long-term nonprogressors and HIV elite controllers (see above). On the other hand, the HLA-B*35 allele has been associated with faster progression to AIDS and higher viral load. The prevalence of the HLA-B alleles differs between populations. HLA-B*57:01 in Europeans and HLA-B*57:03 in African Americans are the protective alleles. In some populations (e.g., Japanese) where the HLA-B*57/-B*27 alleles are absent, HLA-B*51 is associated with a protective phenotype.
Possession of the protective HLA-B alleles is associated with broader and stronger CD8+ T cell responses to HIV epitopes. The mechanisms underlying the differential effects of the HLA-B alleles on HIV disease course may relate to differences in the ability of antigen-presenting cells to present immunodominant HIV epitopes to T helper or cytotoxic T lymphocytes in the context of MHC-encoded molecules. This may result in differential immune responses that influence viral replication. In this regard, the HLA-B alleles that impact HIV disease course differ in their amino acid residues in the HLA-B peptide-binding groove—and this may play a critical role in virologic control.
Investigators have also examined the influence of extended HLA haplotypes (linked alleles) on HIV disease course. The extended HLA ancestral haplotype (AH) 8.1 is defined by the presence of HLA-A1, HLA-B8, and HLA-DR3 alleles. AH 8.1 is the most common ancestral haplotype in Caucasians (present in 10%) and is associated with multiple autoimmune diseases in HIV-uninfected persons. These associations of AH 8.1 are thought to be due to a genetically determined hyperresponsiveness characterized by high TNF-α production and lack of complement C4A. Strong epidemiologic data indicate that carriage of AH 8.1 in HIV-infected persons is associated with a rapid decline in CD4+ T cells and faster progression to AIDS development. Gene–gene interactions between HLA alleles and other genes (e.g., killer cell immunoglobulin-like receptors) also may influence HIV disease progression rates.
POLYMORPHISMS IDENTIFIED BY GWAS THAT ASSOCIATE WITH VIROLOGIC CONTROL Large-scale GWASs have been conducted for the phenotype of viral load, including in a large group of HIV controllers. GWAS in HIV-infected persons of European ancestry identified four SNPs in genes in the HLA class I loci that associated with virologic control. These SNPs are within or in the vicinity of HCP5, HLA-C, MICA, and PSORS1C3 genes. The protective effects of the SNPs in HCP5 and MICA may relate to their linkage with known protective HLA-B alleles. The protective HCP5 allele is in linkage disequilibrium with the HLA-B*57:01, and the protective MICA allele tags with the HLA-B*57:01 and B*27:05 alleles. The protective HLA-C SNP is associated with higher HLA-C expression, and this effect is thought to be due to the altered binding of a microRNA to the HLA-C mRNA. Higher HLA-C expression has been associated with beneficial HIV phenotypes. The mechanism associated with the SNP in PSORS1C3 is unknown. GWAS in African Americans identified a SNP that tags the HLA-B*57:03 allele that is known to associate with lower virologic setpoint and slower disease course. Together these GWAS data underscore the importance of variations in HLA class I loci in control of viral replication.
GENETIC ASSOCIATIONS WITH SPECIFIC AIDS AND NON-AIDS CONDITIONS • Carotid artery disease Many of the non-AIDS events in HIV-infected individuals resemble those related to immune senescence and those found in the HIV-uninfected aging population. A functional SNP in the ryanodine receptor 3 (RYR3) gene was found to be associated with an increased risk of common carotid intima–media thickness (cIMT), which is a surrogate for subclinical atherosclerosis. Functional studies on RYR3 and its isoforms demonstrate a major role of these receptors in modulating endothelial function and atherogenesis via calcium signaling pathways, providing a biologically plausible mechanism by which the SNP in RYR3 may associate with increased cIMT risk.
Renal disease HIV-1-associated nephropathy (HIVAN) is a form of focal sclerosing glomerulonephritis caused by direct infection of kidney epithelial cells with HIV. HIVAN is more common in persons of African descent. There is evidence that polymorphisms in the MYH9 gene and in the neighboring APOL1 gene are a strong determinant of susceptibility to HIVAN in African Americans. The effect of carrying two APOL1 risk alleles explains nearly 35% of HIVAN. The mechanisms by which MYH9/APOL1 variants predispose to HIVAN are currently unknown.
HIV-associated neurocognitive disorder HIV-associated neurocognitive disorder (HAND) comprises a spectrum of neurocognitive deficits due to HIV infection. Variations in the apolipoprotein E (ApoE) gene have strong associations with Alzheimer’s disease in the HIV-uninfected population. In HIV-infected persons, possession of the ApoE4 allele has been associated with several cognitive outcomes including dementia, peripheral neuropathy, and impairment in cognition and immediate and delayed verbal memory. Macrophage recruitment and activation plays a central role in the development of many of the HAND syndromes. Variations in chemokines that play an influential role in macrophage activation and recruitment, namely CCL2 (MCP-1) and CCL3 (MIP-1α), have been shown to alter the risk of developing HAND. Variations in mitochondrial genes also have been associated with risk of AIDS and HAND.
ASSOCIATIONS WITH ART-RELATED ADVERSE EVENTS Abacavir, an effective antiretroviral agent, is associated with significant risk of hypersensitivity reactions (2–9% of cases). Interestingly, while the HLA-B*57:01 allele is associated with a slower HIV disease course, possession of this allele is associated with a higher risk of abacavir-associated hypersensitivity. Pharmacogenetic screening for the HLA-B*57:01 allele is recommended before initiation of abacavir treatment.
NEUROPATHOGENESIS IN HIV DISEASE
While there has been a remarkable decrease in the incidence in the severe forms of HIV encephalopathy among those with access to treatment in the era of effective cART, HIV-infected individuals can still experience milder forms of neurocognitive impairment despite adequate cART. A variety of factors may contribute to the neurocognitive decline, which includes lack of complete control of HIV replication in the brain, production of HIV proteins that may be neurotoxic, low CD4+ T cell nadir, chronic immune activation, comorbidities such as drug abuse, and the potential for neurotoxicity of certain of the antiretroviral drugs. HIV has been demonstrated in the brain and CSF of infected individuals with and without neuropsychiatric abnormalities. As opposed to lymphoid tissues, there are no resident lymphocytes in the brain. The main cell types that are infected in the brain in vivo are the perivascular macrophages and the microglial cells; low-level viral replication is also seen in perivascular astrocytes. Monocytes that have already been infected in the blood can migrate into the brain, where they then reside as macrophages, or macrophages can be directly infected within the brain. The precise mechanisms whereby HIV enters the brain are unclear; however, they are thought to relate, at least in part, to the ability of virus-infected and immune-activated macrophages to induce adhesion molecules such as E-selectin and vascular cell adhesion molecule 1 (VCAM-1) on brain endothelium. Other studies have demonstrated that HIV gp120 enhances the expression of intercellular adhesion molecule 1 (ICAM-1) in glial cells; this effect may facilitate entry of HIV-infected cells into the CNS. Virus isolates from the brain are preferentially R5 strains as opposed to X4 strains; in this regard, HIV-infected individuals who are heterozygous for CCR5-Δ32 appear to be relatively protected against the development of HIV encephalopathy. Once HIV enters the brain due to pressures of the local environment, it evolves to develop distinct sequences in the env, tat, and LTR genes. These unique sequences have been associated with neurocognitive dysfunction; however, it is unclear if they are causal (see below).
HIV-infected individuals may manifest white matter lesions as well as neuronal loss. The white matter lesions are due to axonal injury and a disruption of the blood-brain barrier and not due to demyelination. Given the absence of evidence of HIV infection of neurons either in vivo or in vitro, it is highly unlikely that direct infection of these cells accounts for their loss. Rather, the HIV-mediated effects on neurons are thought to involve indirect pathways whereby viral proteins, particularly gp120 and Tat, trigger the release of endogenous neurotoxins from macrophages and to a lesser extent from astrocytes. In addition, it has been demonstrated that both HIV-1 Nef and Tat can induce chemotaxis of leukocytes, including monocytes, into the CNS. Neurotoxins can be released from monocytes as a consequence of infection and/or immune activation. Monocyte-derived neurotoxic factors have been reported to kill neurons via the N-methyl-D-aspartate (NMDA) receptor. In addition, HIV gp120 shed by virus-infected monocytes could cause neurotoxicity by antagonizing the function of vasoactive intestinal peptide (VIP), by elevating intracellular calcium levels, and by decreasing nerve growth factor levels in the cerebral cortex. A variety of monocyte-derived cytokines can contribute directly or indirectly to the neurotoxic effects in HIV infection; these include TNF-α, IL-1, IL-6, TGF-β, IFN-γ, platelet-activating factor, and endothelin. Furthermore, among the CC-chemokines, elevated levels of monocyte chemotactic protein-1 (MCP-1 or CCL-2) in the brain and CSF have been shown to correlate best with the presence and degree of HIV encephalopathy. In addition, infection and/or activation of monocyte-lineage cells can result in increased production of eicosanoids, quinolinic acid, nitric oxide, excitatory amino acids such as L-cysteine and glutamate, arachidonic acid, platelet activating factor, free radicals, TNF-α, and TGF-β, which may contribute to neurotoxicity. Astrocytes may play diverse roles in HIV neuropathogenesis. Reactive gliosis or astrocytosis has been demonstrated in the brains of HIV-infected individuals, and TNF-α and IL-6 have been shown to induce astrocyte proliferation. In addition, astrocyte-derived IL-6 can induce HIV expression in infected cells in vitro. Furthermore, it has been suggested that astrocytes may downregulate macrophage-produced neurotoxins. Treatment with cART leads to improvement in neuropsychiatric manifestations and a decrease in these cytokine levels in CSF, suggesting that they are driven by the virus or by its products. However, even in patients on long-term cART, there may be evidence of persistently activated lymphocytes in the CSF. It is unclear if these lymphocytes may contribute to neuronal injury in the brain or are critical for controlling the CNS viral reservoir. The contribution of host genetic factors to development of neuropsychiatric manifestations of HIV infection has not been well studied. However, evidence supports the role of the E4 allele for apoE in an increased risk of HIV-associated neurocognitive disorders and peripheral neuropathy.
It has also been suggested that the CNS may serve as a relatively sequestered site for a reservoir of latently infected cells that might be a barrier for the eradication of virus by cART (see “Reservoirs of HIV-Infected Cells: Obstacles to the Eradication of Virus,” above).
PATHOGENESIS OF KAPOSI’S SARCOMA
There are at least four distinct epidemiologic forms of KS: (1) the classic form that occurs in older men of predominantly Mediterranean or eastern European Jewish backgrounds with no recognized contributing factors; (2) the equatorial African form that occurs in all ages, also without any recognized precipitating factors; (3) the form associated with organ transplantation and its attendant iatrogenic immunosuppressed state; and (4) the form associated with HIV-1 infection. In the latter two forms, KS is an opportunistic disease; in HIV-infected individuals, unlike typical opportunistic infections, its occurrence is not strictly related to the level of depression of CD4+ T cell counts. The pathogenesis of KS is complex; fundamentally, it is an angioproliferative disease that is not a true neoplastic sarcoma, at least not in its early stages. It is a manifestation of excessive proliferation of spindle cells that are believed to be of vascular origin and have features in common with endothelial and smooth-muscle cells. In HIV disease the development of KS is dependent on the interplay of a variety of factors including HIV-1 itself, human herpes virus 8 (HHV-8), immune activation, and cytokine secretion. A number of epidemiologic and virologic studies have clearly linked HHV-8, which is also referred to as Kaposi’s sarcoma–associated herpesvirus (KSHV), to KS not only in HIV-infected individuals but also in individuals with the other forms of KS. HHV-8 is a γ-herpesvirus related to EBV and herpesvirus saimiri. It encodes a homologue to human IL-6 and, in addition to KS, has been implicated in the pathogenesis of body cavity lymphoma, multiple myeloma, and monoclonal gammopathy of undetermined significance. Sequences of HHV-8 are found universally in the lesions of KS, and patients with KS are virtually all seropositive for HHV-8. HHV-8 DNA sequences can be found in the B cells of 30–50% of patients with KS and 7% of patients with AIDS without clinically apparent KS.
Between 1 and 2% of eligible blood donors are positive for antibodies to HHV-8, while the prevalence of HHV-8 seropositivity in HIV-infected men is 30–35%. The prevalence of HHV-8 seropositivity in HIV-infected women is ~4%. This finding is reflective of the lower incidence of KS in women. It has been debated whether HHV-8 is actually the transforming agent in KS; the bulk of the cells in the tumor lesions of KS are not neoplastic cells. However, it has been demonstrated that endothelial cells can be transformed in vitro by HHV-8. In this regard, HHV-8 possesses a number of genes, including homologues of the IL-8 receptor, Bcl-2, and cyclin D, that can potentially transform the host cell. Despite the complexity of the pathogenic events associated with the development of KS in HIV-infected individuals, HHV-8 is the etiologic agent of this disease. The initiation and/or propagation of KS requires an activated state and is mediated, at least in part, by cytokines. A number of factors, including TNF-α, IL-1β, IL-6, granulocyte-macrophage colony-stimulating factor (GM-CSF), basic fibroblast growth factor, and oncostatin M, function in an autocrine and paracrine manner to sustain the growth and chemotaxis of the KS spindle cells. In this regard, KSHV-derived IL-6 has been demonstrated to induce proliferation of lymphoma cells and to inhibit the cytostatic effects of IFN-α on KSHV-infected lymphoma cells.
IMMUNE RESPONSE TO HIV
As detailed above and below, following the initial burst of viremia during primary infection, HIV-infected individuals mount robust immune responses that in most cases substantially curtail the levels of plasma viremia and likely contribute to delaying the ultimate development of clinically apparent disease for a median of 10 years in untreated individuals. This immune response contains elements of both humoral and cell-mediated immunity involving both innate and adaptive immune responses (Table 226-7; Fig. 226-26). It is directed against multiple antigenic determinants of the HIV virion as well as against viral proteins expressed on the surface of infected cells. Ironically, those CD4+ T cells with T cell receptors specific for HIV are theoretically those CD4+ T cells most likely to be activated—and thus to serve as early targets for productive HIV infection and the cell death or dysfunction associated with infection. Thus, an early consequence of HIV infection is interference with and decrease of the helper T cell population needed to generate an effective immune response.
|
ELEMENTS OF THE IMMUNE RESPONSE TO HIV |
Abbreviation: MHC, major histocompatibility complex.
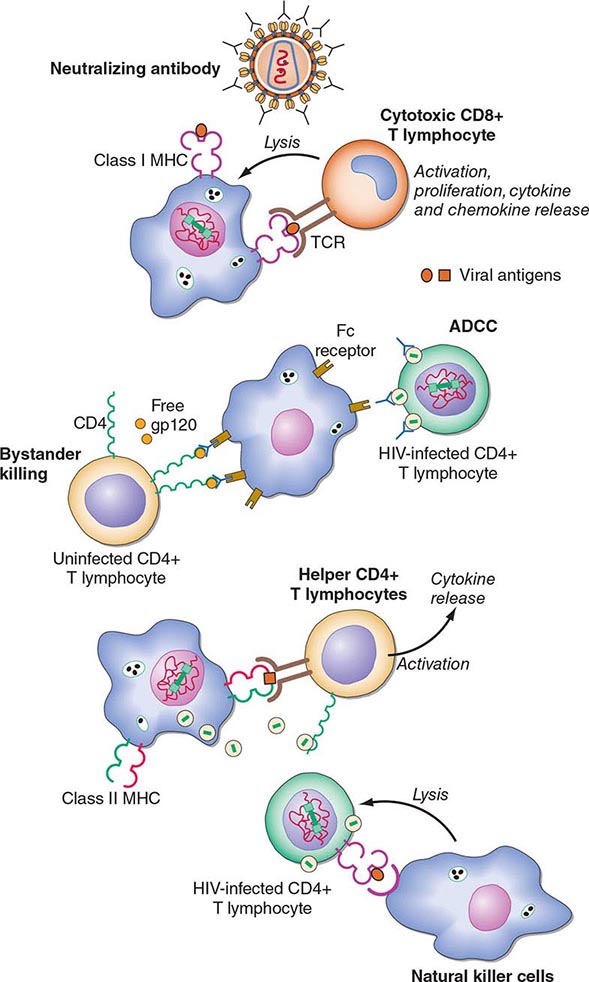
FIGURE 226-26 Schematic representation of the different immunologic effector mechanisms thought to be active in the setting of HIV infection. Detailed descriptions are given in the text. ADCC, antibody-dependent cellular cytotoxicity; MHC, major histocompatibility complex; TCR, T cell receptor.
Although a great deal of investigation has been directed toward delineating and better understanding the components of this immune response, it remains unclear which immunologic effector mechanisms are most important in delaying progression of infection and which, if any, play a role in the pathogenesis of HIV disease. This lack of knowledge has also hampered the ability to develop an effective vaccine for HIV disease.
HUMORAL IMMUNE RESPONSE
Antibodies to HIV usually appear within 3–6 weeks and almost invariably within 12 weeks of primary infection (Fig. 226-27); rare exceptions are in individuals who have defects in the ability to produce HIV-specific antibodies. Detection of these antibodies forms the basis of most diagnostic screening tests for HIV infection. The appearance of HIV-binding antibodies detected by ELISA and Western blot assays occurs prior to the appearance of neutralizing antibodies; the latter generally appear following the initial decreases in plasma viremia and are more closely related to the appearance of HIV-specific CD8+ T lymphocytes. The first antibodies detected are those directed against the immunodominant region of the envelope gp41, followed by the appearance of antibodies to the structural or gag protein p24 and the gag precursor p55. Antibodies to p24 gag are followed by the appearance of antibodies to the outer envelope glycoprotein (gp120), the gag protein p17, and the products of the pol gene (p31 and p66). In addition, one may see antibodies to the low-molecular-weight regulatory proteins encoded by the HIV genes vpr, vpu, vif, rev, tat, and nef. On rare occasion, levels of HIV-specific antibodies may decline during treatment of acute HIV infection.
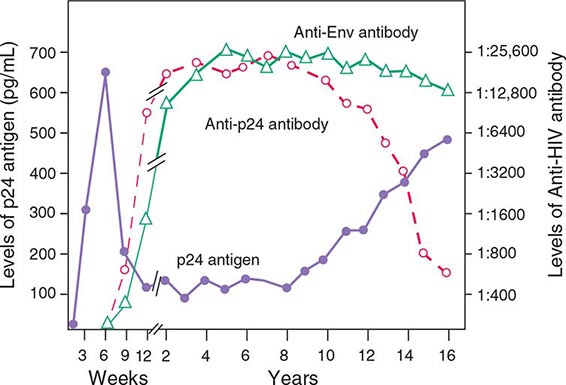
FIGURE 226-27 Relationship between antigenemia and the development of antibodies to HIV. Levels of plasma HIV parallel those of p24 antigen. Antibodies to HIV proteins are generally seen 6–12 weeks following infection and 3–6 weeks after the development of plasma viremia. Late in the course of illness, antibody levels to p24 decline, generally in association with a rising titer of p24 antigen.
While antibodies to multiple antigens of HIV are produced, the precise functional significance of these different antibodies is unclear. The only viral proteins that elicit neutralizing antibodies are the envelope proteins gp120 and gp41. Antibodies directed toward the envelope proteins of HIV have been characterized both as being protective and as possibly contributing to the pathogenesis of HIV disease. Among the protective antibodies are those that function to neutralize HIV directly and prevent the spread of infection to additional cells, as well as those that participate in ADCC. The first neutralizing antibodies are directed against the autologous infecting virus and appear after approximately 12 weeks of infection. Due to its high rate of mutation the virus is usually able to quickly escape these (and subsequent) neutralizing antibodies. One important mechanism of immune escape is the addition of N-linked glycosylation sites, forming a glycan shield that interferes with envelope recognition by these initial antibodies.
A number of broad and potent HIV-neutralizing envelope-specific antibodies have been isolated from HIV-infected individuals in studies designed to better understand the host response to HIV infection. Approximately 20% of patients develop antibodies capable of neutralizing highly diverse strains. These studies have revealed at least five major sites within the HIV envelope that are able to elicit broadly-neutralizing antibodies. These sites include antibodies directed toward the CD4 binding site (CD4bs) of gp120, those binding glycan-dependent epitopes in the V1/V2 region of gp120, those near the base of the V3 region of gp120, those binding to the gp120/gp41 bridge, and those binding to the membrane-proximal region of gp41 (Fig. 226-28). Several of these antibodies contain unique features including high levels of somatic hypermutation, selective germline gene usage (especially for CD4bs antibodies) and long heavy chain complementary determining regions (especially CDRH3). Of note, while these antibodies are broadly neutralizing in vitro, their precise in vivo significance is unclear and the patients from whom they were derived demonstrate evidence of ongoing viral replication unless treated with cART.
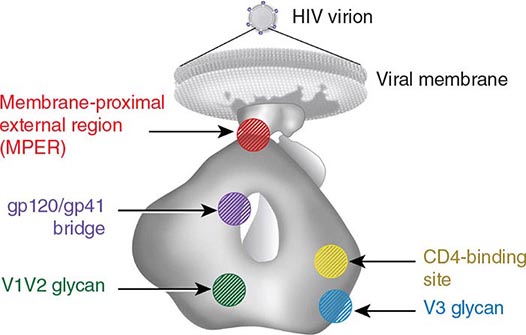
FIGURE 226-28 Known targets of broadly neutralizing antibodies against HIV-1. (Adapted from PD Kwong, JR Mascola: Immunity 37:412, 2012.)
The other major class of protective antibodies are those that participate in ADCC, a form of cell-mediated immunity (Chap. 372e) in which NK cells that bear Fc receptors are armed with specific anti-HIV antibodies that bind to the NK cells via their Fc portion. These armed NK cells then bind to and destroy cells expressing HIV antigens. The levels of anti-envelope antibodies capable of mediating ADCC are highest in the earlier stages of HIV infection. Antibodies to both gp120 and gp41 have been shown to participate in ADCC-mediated killing of HIV-infected cells. In vitro, IL-2 can augment ADCC-mediated killing.
In addition to playing a role in host defense, HIV-specific antibodies have also been implicated in disease pathogenesis. Antibodies directed to gp41, when present in low titer, have been shown in vitro to be capable of facilitating infection of cells through an Fc receptor–mediated mechanism known as antibody enhancement. Thus, the same regions of the envelope protein of HIV that give rise to antibodies capable of mediating ADCC can also elicit the production of antibodies that can facilitate infection of cells in vitro. In addition, it has been postulated that anti-gp120 antibodies that participate in the ADCC killing of HIV-infected cells might also kill uninfected CD4+ T cells if the uninfected cells had bound free gp120, a phenomenon referred to as bystander killing.
One of the most primitive components of the humoral immune system is the complement system (Chap. 372e). This element of innate immunity consists of ~30 proteins that are found circulating in blood or associated with cell membranes. While HIV alone is capable of directly activating the complement cascade, the resulting lysis is weak due to the presence of host cell regulatory proteins captured in the virion envelope during budding. It is possible that complement-opsonized HIV virions have increased infectivity in a manner analogous to antibody-mediated enhancement.
CELLULAR IMMUNE RESPONSE
Given that T cell–mediated immunity is known to play a major role in host defense against most viral infections (Chap. 372e), it is generally thought to be an important component of the host immune response to HIV. T cell immunity can be divided into two major categories: that mediated by helper/inducer CD4+ T cells and that mediated by cytotoxic/immunoregulatory CD8+ T cells.
HIV-specific CD4+ T cells can be detected in the majority of HIV-infected patients through the use of flow cytometry to measure intracellular cytokine production in response to MHC class II tetramers pulsed with HIV peptides or through lymphocyte proliferation assays utilizing HIV antigens such as p24. These cells likely play a critical role in the orchestration of the immune response to HIV by providing help to HIV-specific B cells and CD8+ T cells. They may also be capable of directly killing HIV-infected cells. HIV-specific CD4+ T cells may be preferential targets of HIV infection by HIV-infected antigen-presenting cells during the generation of an immune response to HIV (Fig. 226-26). However, they also are likely to undergo clonal expansions in response to HIV antigens and thus survive as a population of cells. No clear correlations exist between levels of HIV-specific CD4+ T lymphocytes and plasma HIV RNA levels; however, in the setting of high viral loads, CD4+ T cell responses to HIV antigens appear to shift from one of proliferation and IL-2 production to one of IFN-γ production. Thus, while a reverse correlation exists between the level of p24-specific proliferation and levels of plasma HIV viremia, the nature of the causal relationship between these parameters is unclear.
MHC class I–restricted, HIV-specific CD8+ T cells have been identified in the peripheral blood of patients with HIV-1 infection. These cells include CTLs that produce perforins and T cells that can be induced by HIV antigens to express an array of cytokines such as IFN-γ, IL-2, MIP-1β, and TNF-α. CTLs have been identified in the peripheral blood of patients within weeks of HIV infection and prior to the appearance of plasma virus. The selective pressure they exert on the evolution of the population of circulating viruses reflects their potential role in control of HIV infection. These CD8+ T lymphocytes, through their HIV-specific antigen receptors, bind to and cause the lytic destruction of target cells bearing autologous MHC class I molecules presenting HIV antigens. Two types of CTL activity can be demonstrated in the peripheral blood or lymph node mononuclear cells of HIV-infected individuals. The first type directly lyses appropriate target cells in culture without prior in vitro stimulation (spontaneous CTL activity). The other type of CTL activity reflects the precursor frequency of CTLs (CTLp); this type of CTL activity can be demonstrated by stimulation of CD8+ T cells in vitro with a mitogen such as phytohemagglutinin or anti-CD3 antibody.
In addition to CTLs, CD8+ T cells capable of being induced by HIV antigens to express cytokines such as IFN-γ also appear in the setting of HIV-1 infection. It is not clear whether these are the same or different effector pools compared with those cells mediating cytotoxicity; in addition, the relative roles of each in host defense against HIV are not fully understood. It does appear that these CD8+ T cells are driven to in vivo expansion by HIV antigen. There is a direct correlation between levels of CD8+ T cells capable of producing IFN-γ in response to HIV antigens and plasma levels of HIV-1 RNA. Thus, while these cells are clearly induced by HIV-1 infection, their overall ability to control infection remains unclear. Multiple HIV antigens, including Gag, Env, Pol, Tat, Rev, and Nef, can elicit CD8+ T cell responses. Among patients who control viral replication in the absence of antiretroviral drugs are a subset of patients referred to as elite nonprogressors (see “Long-Term Survivors and Long-Term Nonprogressors,” above) whose peripheral blood contains a population of CD8+ T cells that undergo substantial proliferation and perforin expression in response to HIV antigens. It is possible that these cells play an important role in HIV-specific host defense.
At least three other forms of cell-mediated immunity to HIV have been described: non-cytolytic CD8+ T cell–mediated suppression of HIV replication, ADCC, and NK cell activity. Non-cytolytic CD8+ T cell–mediated suppression of HIV replication refers to the ability of CD8+ T cells from an HIV-infected patient to inhibit the replication of HIV in tissue culture without killing infected targets. There is no requirement for HLA compatibility between the CD8+ T cells and the HIV-infected cells. This effector mechanism is thus nonspecific and appears to be mediated by soluble factor(s) including the CC-chemokines RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4). These CC-chemokines are potent suppressors of HIV replication and operate at least in part via blockade of the HIV co-receptor (CCR5) for R5 (macrophage-tropic) strains of HIV-1 (see above). ADCC, as described above in relation to humoral immunity, involves the killing of HIV-expressing cells by NK cells armed with specific antibodies directed against HIV antigens. Finally, NK cells alone have been shown to be capable of killing HIV-infected target cells in tissue culture. This primitive cytotoxic mechanism of host defense is directed toward nonspecific surveillance for neoplastic transformation and viral infection through recognition of altered class I MHC molecules.
DIAGNOSIS AND LABORATORY MONITORING OF HIV INFECTION
The establishment of HIV as the causative agent of AIDS and related syndromes early in 1984 was followed by the rapid development of sensitive screening tests for HIV infection. By March 1985, blood donors in the United States were routinely screened for antibodies to HIV. In 1996, blood banks in the United States added the p24 antigen capture assay to the screening process to help identify the rare infected individuals who were donating blood in the time (up to 3 months) between infection and the development of antibodies. In 2002, the ability to detect early infection with HIV was further enhanced by the licensure of nucleic acid testing (NAT) as a routine part of blood donor screening. These refinements decreased the interval between infection and detection (window period) from 22 days for antibody testing to 16 days with p24 antigen testing and subsequently to 12 days with NAT. The development of sensitive assays for monitoring levels of plasma viremia ushered in a new era of being able to monitor the progression of HIV disease more closely. Utilization of these tests, coupled with the measurement of levels of CD4+ T lymphocytes in peripheral blood, is essential in the management of patients with HIV infection.
DIAGNOSIS OF HIV INFECTION
The CDC has recommended that screening for HIV infection be performed as a matter of routine health care. The diagnosis of HIV infection depends on the demonstration of antibodies to HIV and/or the direct detection of HIV or one of its components. As noted above, antibodies to HIV generally appear in the circulation 3–12 weeks following infection.
The standard blood screening tests for HIV infection are based on the detection of antibodies to HIV. A common platform is the ELISA, also referred to as an enzyme immunoassay (EIA). This solid-phase assay is an extremely good screening test with a sensitivity of >99.5%. Most diagnostic laboratories use commercial kits that contain antigens from both HIV-1 and HIV-2 and thus are able to detect antibodies to either. These kits use both natural and recombinant antigens and are continuously updated to increase their sensitivity to newly discovered species, such as group O viruses (Fig. 226-1). The fourth-generation EIA tests combine detection of antibodies to HIV with detection of the p24 antigen of HIV. EIA tests are generally scored as positive (highly reactive), negative (nonreactive), or indeterminate (partially reactive). While the EIA is an extremely sensitive test, it is not optimal with regard to specificity. This is particularly true in studies of low-risk individuals, such as volunteer blood donors. In this latter population, only 10% of EIA-positive individuals are subsequently confirmed to have HIV infection. Among the factors associated with false-positive EIA tests are antibodies to class II antigens (such as may be seen following pregnancy, blood transfusion, or transplantation), autoantibodies, hepatic disease, recent influenza vaccination, and acute viral infections. For these reasons, anyone suspected of having HIV infection based on a positive or inconclusive EIA result should ideally have the result confirmed with a more specific assay such as the Western blot. One can estimate whether an individual has a recent infection with HIV-1 by comparing the results on a standard EIA test that will score positive for all infected individuals with the results on an assay modified to be less sensitive (“detuned assay”) that will score positive for individuals with established HIV infection and negative for individuals with recent infection. In rare instances, an HIV-infected individual treated early in the course of infection may revert to a negative EIA. This does not indicate clearing of infection; rather, it signifies levels of ongoing exposure to virus or viral proteins insufficient to maintain a measurable antibody response. When these individuals have discontinued therapy, viruses and antibodies have reappeared.
The most commonly used confirmatory test is the Western blot (Fig. 226-29). This assay takes advantage of the fact that multiple HIV antigens of different, well-characterized molecular weights elicit the production of specific antibodies. These antigens can be separated on the basis of molecular weight, and antibodies to each component can be detected as discrete bands on the Western blot. A negative Western blot is one in which no bands are present at molecular weights corresponding to HIV gene products. In a patient with a positive or indeterminate EIA and a negative Western blot, one can conclude with certainty that the EIA reactivity was a false positive. On the other hand, a Western blot demonstrating antibodies to products of all three of the major genes of HIV (gag, pol, and env) is conclusive evidence of infection with HIV. Criteria established by the U.S. Food and Drug Administration (FDA) in 1993 for a positive Western blot state that a result is considered positive if antibodies exist to two of the three HIV proteins: p24, gp41, and gp120/160. Using these criteria, ~10% of all blood donors deemed positive for HIV-1 infection lacked an antibody band to the pol gene product p31. Some 50% of these blood donors were subsequently found to be false positives. Thus, the absence of the p31 band should increase the suspicion that one may be dealing with a false-positive test result. In this setting it is prudent to obtain additional confirmation with an RNA-based test for HIV-1 and/or a follow-up Western blot. By definition, Western blot patterns of reactivity that do not fall into the positive or negative categories are considered “indeterminate.” There are two possible explanations for an indeterminate Western blot result. The most likely explanation in a low-risk individual is that the patient being tested has antibodies that cross-react with one of the proteins of HIV. The most common patterns of cross-reactivity are antibodies that react with p24 and/or p55. The least likely explanation in this setting is that the individual is infected with HIV and is in the process of mounting a classic antibody response. In either instance, the Western blot should be repeated in 1 month to determine whether the indeterminate pattern is a pattern in evolution. In addition, one may attempt to confirm a diagnosis of HIV infection with the p24 antigen capture assay or one of the tests for HIV RNA (discussed below). While the Western blot is an excellent confirmatory test for HIV infection in patients with a positive or indeterminate EIA, it is a poor screening test. Among individuals with a negative EIA and PCR for HIV, 20–30% may show one or more bands on Western blot. While these bands are usually faint and represent cross-reactivity, their presence creates a situation in which other diagnostic modalities (such as DNA PCR, RT-PCR, or p24 antigen capture) must be employed to ensure that the bands do not indicate early HIV infection.
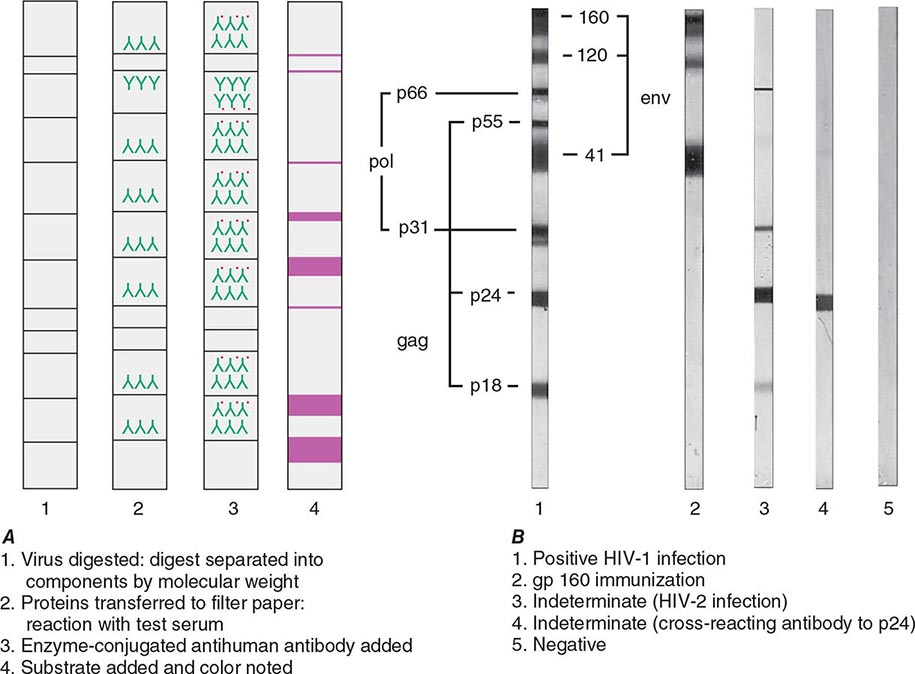
FIGURE 226-29 Western blot assay for detection of antibodies to HIV. A. Schematic representation of how a Western blot is performed. B. Examples of patterns of Western blot reactivity. In each instance the Western blot strip contains antigens to HIV-1. The serum from the patient immunized to the HIV-1 envelope gp160 contains only antibodies to the HIV-1 envelope proteins. The serum from the patient with HIV-2 infection cross-reacts with both reverse transcriptase and gag gene products of HIV-1.
A guideline for the use of these serologic tests in attempting to make a diagnosis of HIV infection is depicted in Fig. 226-30. In patients in whom HIV infection is suspected, the appropriate initial test is the EIA. If the result is negative, unless there is strong reason to suspect early HIV infection (as in a patient exposed within the previous 3 months), the diagnosis is ruled out and retesting should be performed only as clinically indicated. If the EIA is indeterminate or positive, the test should be repeated. If the repeat is negative on two occasions, one can assume that the initial positive reading was due to a technical error in the performance of the assay and that the patient is negative. If the repeat is indeterminate or positive, one should proceed to the HIV-1 Western blot. If the Western blot is positive, the diagnosis is HIV-1 infection. If the Western blot is negative, the EIA can be assumed to have been a false positive for HIV-1 and the diagnosis of HIV-1 infection is ruled out. It would also be prudent at this point to perform specific serologic testing for HIV-2 following the same type of algorithm. If the Western blot for HIV-1 is indeterminate, it should be repeated in 4–6 weeks; in addition, one may proceed to a p24 antigen capture assay, HIV-1 RNA assay, or HIV-1 DNA PCR and specific serologic testing for HIV-2. If the p24 and HIV RNA assays are negative and there is no progression in the Western blot, a diagnosis of HIV-1 is ruled out. If either the p24 or HIV-1 RNA assay is positive and/or the HIV-1 Western blot shows progression, a tentative diagnosis of HIV-1 infection can be made and later confirmed with a follow-up Western blot demonstrating a positive pattern. In addition to these standard laboratory-based assays for detecting antibodies to HIV, an series of point-of-care tests can provide results in 1–60 min. Among the most popular of these is the OraQuick Rapid HIV-1 antibody test that can be run on blood, plasma, or saliva. The sensitivity and specificity of this test is ~99% when run on whole blood. Specificity remains the same but sensitivity drops to 98% when the test is run on saliva. While negative results from this test are adequate to rule out a diagnosis of HIV infection, a positive finding should be considered preliminary and confirmed with standard serologic testing, as described above. Two rapid test kits are licensed for home use. They are the OraQuick In-Home HIV test and the Home Access HIV-1 test system.
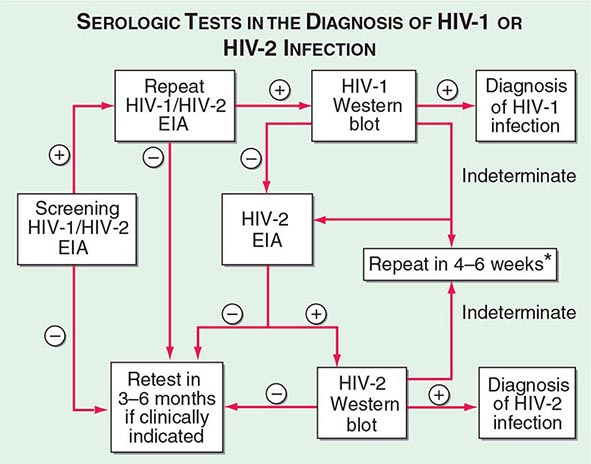
FIGURE 226-30 Algorithm for the use of serologic tests in the diagnosis of HIV-1 or HIV-2 infection. *Stable indeterminate Western blot 4–6 weeks later makes HIV infection unlikely. However, it should be repeated twice at 3-month intervals to rule out HIV infection. Alternatively, one may test for HIV-1 p24 antigen or HIV RNA. EIA, enzyme immunoassay.
A variety of laboratory tests are available for the direct detection of HIV or its components (Table 226-8). These tests may be of considerable help in making a diagnosis of HIV infection when the Western blot results are indeterminate. In addition, the tests detecting levels of HIV RNA can be used to determine prognosis and to assess the response to antiretroviral therapies. The simplest of the direct detection tests is the p24 antigen capture assay. This is an EIA-type assay in which the solid phase consists of antibodies to the p24 antigen of HIV. It detects the viral protein p24 in the blood of HIV-infected individuals where it exists either as free antigen or complexed to anti-p24 antibodies. Overall, ~30% of individuals with untreated HIV infection have detectable levels of free p24 antigen. This increases to ~50% when samples are treated with a weak acid to dissociate antigen-antibody complexes. Throughout the course of HIV infection, an equilibrium exists between p24 antigen and anti-p24 antibodies. During the first few weeks of infection, before an immune response develops, there is a brisk rise in p24 antigen levels (Fig. 226-27). After the development of anti-p24 antibodies, these levels decline. Late in the course of infection, when circulating levels of virus are high, p24 antigen levels also increase, particularly when detected by techniques involving dissociation of antigen-antibody complexes. The p24 antigen capture assay has its greatest use as a screening test for HIV infection in patients suspected of having the acute HIV syndrome, as high levels of p24 antigen are present prior to the development of antibodies. Its use as a stand-alone test for routine blood donor screening for HIV infection has been replaced by use of NAT or “fourth-generation” assays that combine antigen and antibody testing. The ability to measure and monitor levels of HIV RNA in the plasma of patients with HIV infection has been of extraordinary value in furthering our understanding of the pathogenesis of HIV infection, in monitoring the response to cART, and in providing a diagnostic tool in settings where measurements of anti-HIV antibodies may be misleading, such as in acute infection and neonatal infection. Four assays are predominantly used for this purpose. They are reverse transcriptase PCR (RT-PCR; Amplicor); branched DNA (bDNA; VERSANT); transcription-mediated amplification (TMA; APTIMA); and nucleic acid sequence–based amplification (NASBA; NucliSENS). These tests are of value in making a diagnosis of HIV infection, in establishing initial prognosis, and in monitoring the effects of therapy. In addition to these four commercially available tests, the DNA PCR also is employed by research laboratories for making a diagnosis of HIV infection by amplifying HIV proviral DNA from peripheral blood mononuclear cells. The commercially available RNA detection tests have a sensitivity of 40–8100 copies of HIV RNA per milliliter of plasma. Research laboratory–based RNA assays can detect as few as one HIV RNA copy per milliliter, while the DNA PCR tests can detect proviral DNA at a frequency of one copy per 10,000–100,000 cells. Thus, these tests are extremely sensitive. One frequent consequence of a high degree of sensitivity is some loss of specificity, and false-positive results have been reported with each of these techniques. For this reason, a positive EIA with a confirmatory Western blot remains the “gold standard” for a diagnosis of HIV infection, and the interpretation of other test results must be done with this in mind.
|
CHARACTERISTICS OF TESTS FOR DIRECT DETECTION OF HIV |

In the RT-PCR technique, following DNAse treatment, a cDNA copy is made of all RNA species present in plasma. Because HIV is an RNA virus, this will result in the production of DNA copies of the HIV genome in amounts proportional to the amount of HIV RNA present in plasma. This cDNA is then amplified and characterized using standard PCR techniques, employing primer pairs that can distinguish genomic cDNA from messenger cDNA. The bDNA assay involves the use of a solid-phase nucleic acid capture system and signal amplification through successive nucleic acid hybridizations to detect small quantities of HIV RNA. Both tests can achieve a tenfold increase in sensitivity to 40–50 copies of HIV RNA per milliliter with a preconcentration step in which plasma undergoes ultracentrifugation to pellet the viral particles. In the TMA assay, a cDNA copy of viral RNA is made using primers that contain a promoter sequence for T7 RNA polymerase. T7 polymerase is then added to produce multiple copies of RNA amplicon from the DNA template. It is qualified at 100 copies/mL. The NASBA technique involves the isothermal amplification of a sequence within the gag region of HIV in the presence of internal standards and employs the production of multiple RNA copies through the action of T7-RNA polymerase. The resulting RNA species are quantitated through hybridization with a molecular beacon DNA probe that is quenched in the absence of hybridization. The lower limit of detection for the NucliSENS assay is 80 copies/mL.
In addition to being a diagnostic and prognostic tool, RT-PCR and DNA-PCR are also useful for amplifying defined areas of the HIV genome for sequence analysis and have become an important technique for studies of sequence diversity and microbial resistance to antiretroviral agents. In patients with a positive or indeterminate EIA test and an indeterminate Western blot, and in patients in whom serologic testing may be unreliable (such as patients with hypogammaglobulinemia or advanced HIV disease), these tests for quantitating HIV RNA in plasma or detecting proviral DNA in peripheral blood mononuclear cells are valuable tools for making a diagnosis of HIV infection; however, they should be used for diagnosis only when standard serologic testing has failed to provide a definitive result.
LABORATORY MONITORING OF PATIENTS WITH HIV INFECTION
The epidemic of HIV infection and AIDS has provided the clinician with new challenges for integrating clinical and laboratory data to effect optimal patient management. The close relationship between clinical manifestations of HIV infection and CD4+ T cell count has made measurement of CD4+ T cell numbers a routine part of the evaluation of HIV-infected individuals. The discovery of HIV as the cause of AIDS led to the development of sensitive tests that allow one to monitor the levels of HIV in the blood. Determinations of peripheral blood CD4+ T cell counts and measurements of the plasma levels of HIV RNA provide a powerful set of tools for determining prognosis and monitoring response to therapy.
CD4+ T Cell Counts The CD4+ T cell count is the laboratory test generally accepted as the best indicator of the immediate state of immunologic competence of the patient with HIV infection. This measurement, which can be made directly or calculated as the product of the percentage of CD4+ T cells (determined by flow cytometry) and the total lymphocyte count (determined by the white blood cell count [WBC] multiplied by the lymphocyte differential percentage), has been shown to correlate very well with the level of immunologic competence. Patients with CD4+ T cell counts <200/μL are at high risk of disease from P. jiroveci, while patients with CD4+ T cell counts <50/μL are at high risk of disease from CMV, mycobacteria of the M. avium complex (MAC), and/or T. gondii (Fig. 226-31). Once the CD4+ T cell count is <200/μL, patients should be placed on a regimen for P. jiroveci prophylaxis, and once the count is <50/μL, primary prophylaxis for MAC infection is indicated. As with any laboratory measurement, one may wish to obtain two determinations prior to any significant changes in patient management based on CD4+ T cell count alone. Patients with HIV infection should have CD4+ T cell measurements performed at the time of diagnosis and every 3–6 months thereafter. More frequent measurements should be made if a declining trend is noted. For patients who have been on cART for at least 2 years with HIV RNA levels persistently <50 copies/mL, the monitoring of the CD4 count is felt by many to be optional. There are a handful of clinical situations in which the CD4+ T cell count may be misleading. Patients with HTLV-1/HIV co-infection may have elevated CD4+ T cell counts that do not accurately reflect their degree of immune competence. In patients with hypersplenism or those who have undergone splenectomy, and in patients receiving medications that suppress the bone marrow such as IFN-α, the CD4+ T cell percentage may be a more reliable indication of immune function than the CD4+ T cell count. A CD4+ T cell percentage of 15 is comparable to a CD4+ T cell count of 200/μL.
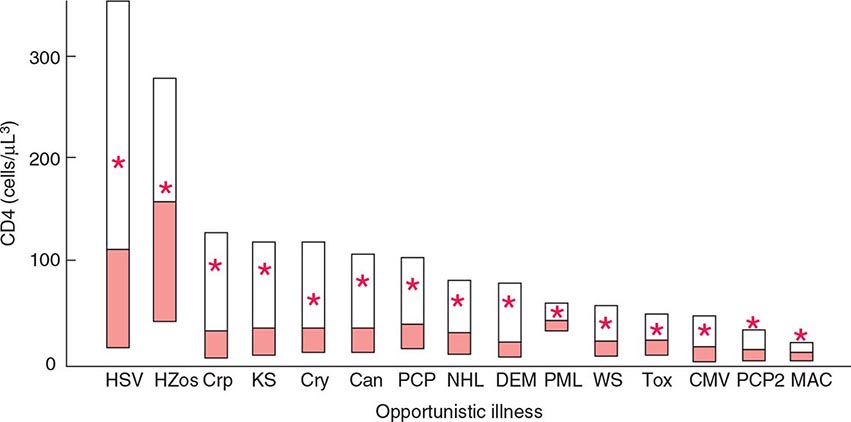
FIGURE 226-31 Relationship between CD4+ T cell counts and the development of opportunistic diseases. Boxplot of the median (line inside the box), first quartile (bottom of the box), third quartile (top of the box), and mean (asterisk) CD4+ lymphocyte count at the time of the development of opportunistic disease. Can, candidal esophagitis; CMV, cytomegalovirus infection; Crp, cryptosporidiosis; Cry, cryptococcal meningitis; DEM, AIDS dementia complex; HSV, herpes simplex virus infection; HZos, herpes zoster; KS, Kaposi’s sarcoma; MAC, Mycobacterium avium complex bacteremia; NHL, non-Hodgkin’s lymphoma; PCP, primary Pneumocystis jiroveci pneumonia; PCP2, secondary P. jiroveci pneumonia; PML, progressive multifocal leukoencephalopathy; Tox, Toxoplasma gondii encephalitis; WS, wasting syndrome. (From RD Moore, RE Chaisson: Ann Intern Med 124:633, 1996.)
HIV RNA Determinations Facilitated by highly sensitive techniques for the precise quantitation of small amounts of nucleic acids, the measurement of serum or plasma levels of HIV RNA has become an essential component in the monitoring of patients with HIV infection. As discussed in “Diagnosis of HIV Infection,” above, the most commonly used technique is the RT-PCR assay. This assay generates data in the form of number of copies of HIV RNA per milliliter of serum or plasma and can reliably detect as few as 40 copies of HIV RNA per milliliter of plasma. Research-based assays can detect down to one copy per milliliter. While it is common practice to describe levels of HIV RNA below these cut-offs as “undetectable,” this is a term that should be avoided as it is imprecise and leaves the false impression that the level of virus is 0. By utilizing more sensitive, nested PCR techniques and by studying tissue levels of virus as well as plasma levels, HIV RNA can be detected in virtually every patient with HIV infection. The one notable exception to this is a patient who underwent cytoreductive therapy followed by a bone marrow transplant from a CCR5Δ32 homozygous donor.
Measurements of changes in HIV RNA levels over time have been of great value in delineating the relationship between levels of virus and rates of disease progression (Fig. 226-22), the rates of viral turnover, the relationship between immune system activation and viral replication, and the time to development of drug resistance. HIV RNA measurements are greatly influenced by the state of activation of the immune system and may fluctuate greatly in the setting of secondary infections or immunization. For these reasons, decisions based on HIV RNA levels should never be made on a single determination. Measurements of plasma HIV RNA levels should be made at the time of HIV diagnosis and every 3–6 months thereafter in the untreated patient. Following the initiation of therapy or any change in therapy, plasma HIV RNA levels should be monitored approximately every 4 weeks until the effectiveness of the therapeutic regimen is determined by the development of a new steady-state level of HIV RNA. In most instances of effective antiretroviral therapy the plasma level of HIV RNA will drop to <50 copies/mL within 6 months of the initiation of treatment. During therapy, levels of HIV RNA should be monitored every 3–6 months to evaluate the continuing effectiveness of therapy.
HIV Resistance Testing The availability of multiple antiretroviral drugs as treatment options has generated a great deal of interest in the potential for measuring the sensitivity of an individual’s HIV virus(es) to different antiretroviral agents. HIV resistance testing can be done through either genotypic or phenotypic measurements. In the genotypic assays, sequence analyses of the HIV genomes obtained from patients are compared with sequences of viruses with known antiretroviral resistance profiles. In the phenotypic assays, the in vivo growth of viral isolates obtained from the patient is compared to the growth of reference strains of the virus in the presence or absence of different antiretroviral drugs. A modification of this phenotypic approach utilizes a comparison of the enzymatic activities of the reverse transcriptase, protease, or integrase genes obtained by molecular cloning of patients’ isolates to the enzymatic activities of genes obtained from reference strains of HIV in the presence or absence of different drugs targeted to these genes. These tests are quite good in identifying those antiretroviral agents that have been utilized in the past and suggesting agents that may be of future value in a given patient. Resistance testing is recommended at the time of initial diagnosis and, if therapy is not initiated at that time, at the time of initiation of cART. Drug resistance testing is also indicated in the setting of virologic failure and should be performed while the patient is still on the failing regimen because of the propensity for the pool of HIV quasispecies to rapidly revert to wild-type in the absence of the selective pressures of cART. In the hands of experts, resistance testing enhances the short-term ability to decrease viral load by ~0.5 log compared with changing drugs merely on the basis of drug history. In addition to the use of resistance testing to help in the selection of new drugs in patients with virologic failure, it may also be of value in selecting an initial regimen for treatment of therapy-naïve individuals. This is particularly true in geographic areas with a high level of background resistance. The patient needs to have an HIV-1 RNA level above 500–1000 copies/mL for an accurate resistance determination. Resistance assays lose their consistency at lower levels of plasma viremia.
Co-Receptor Tropism Assays Following the licensure of maraviroc as the first CCR5 antagonist for the treatment of HIV infection (see below), it became necessary to be able to determine whether a patient’s virus was likely to respond to this treatment. Patients tend to have CCR5-tropic virus early in the course of infection, with a trend toward CXCR4 viruses later in disease. The antiretroviral agent maraviroc is effective only against CCR5-tropic viruses. Because the genotypic determinants of cellular tropism are poorly defined, a phenotypic assay is necessary to determine this property of HIV. Two commercial assays, the Trofile assay (Monogram Biosciences) and the Phenoscript assay (VIRalliance), are available to make this determination. These assays clone the envelope regions of the patient’s virus into an indicator virus that is then used to infect target cells expressing either CCR5 or CXCR4 as their co-receptor. These assays take weeks to perform and are expensive. Another, less costly option is to obtain a genotypic assay of the V3 region of HIV-1 and then employ a computer algorithm to predict viral tropism from the sequence. While this approach is less expensive than the classic phenotypic assay, there are fewer data to validate its predictive value.
Other Tests A variety of other laboratory tests have been studied as potential markers of HIV disease activity. Among these are quantitative culture of replication-competent HIV from plasma, peripheral blood mononuclear cells, or resting CD4+ T cells; circulating levels of β2-microglobulin, soluble IL-2 receptor, IgA, acid-labile endogenous IFN, or TNF-α; and the presence or absence of activation markers such as CD38, HLA-DR, and PD-1 on CD4+ or CD8+ T cells. Nonspecific serologic markers of inflammation and/or coagulation such as IL-6, D-dimer, and sCD14 have been shown to have a high correlation with all-cause mortality (Table 226-9). While these measurements have value as markers of disease activity and help to increase our understanding of the pathogenesis of HIV disease, they do not currently play a major role in the monitoring of patients with HIV infection.
|
ASSOCIATION BETWEEN HIGH-SENSITIVITY CRP, IL-6, AND D-DIMER WITH ALL-CAUSE MORTALITY IN PATIENTS WITH HIV INFECTION |
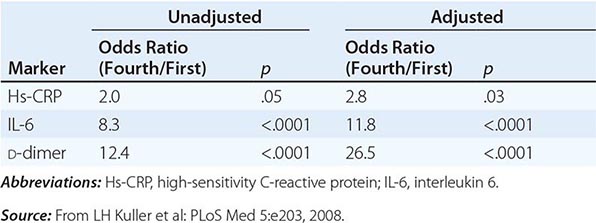
CLINICAL MANIFESTATIONS
The clinical consequences of HIV infection encompass a spectrum ranging from an acute syndrome associated with primary infection to a prolonged asymptomatic state to advanced disease. It is best to regard HIV disease as beginning at the time of primary infection and progressing through various stages. As mentioned above, active virus replication and progressive immunologic impairment occur throughout the course of HIV infection in most patients. With the exception of the rare, true, “elite” virus controllers or long-term nonprogressors (see “Long-Term Survivors and Long-Term Nonprogressors,” above), HIV disease in untreated patients inexorably progresses even during the clinically latent stage. Since the mid-1990s, cART has had a major impact on preventing and reversing the progression of disease over extended periods of time in a substantial proportion of adequately treated patients.
ACUTE HIV INFECTION
It is estimated that 50–70% of individuals with HIV infection experience an acute clinical syndrome ~3–6 weeks after primary infection (Fig. 226-32). Varying degrees of clinical severity have been reported, and although it has been suggested that symptomatic seroconversion leading to the seeking of medical attention indicates an increased risk for an accelerated course of disease, there does not appear to be a correlation between the level of the initial burst of viremia in acute HIV infection and the subsequent course of disease. The typical clinical findings in the acute HIV syndrome are listed in Table 226-10; they occur along with a burst of plasma viremia. It has been reported that several symptoms of the acute HIV syndrome (fever, skin rash, pharyngitis, and myalgia) occur less frequently in those infected by injection drug use compared with those infected by sexual contact. The syndrome is typical of an acute viral syndrome and has been likened to acute infectious mononucleosis. Symptoms usually persist for one to several weeks and gradually subside as an immune response to HIV develops and the levels of plasma viremia decrease. Opportunistic infections have been reported during this stage of infection, reflecting the immunodeficiency that results from reduced numbers of CD4+ T cells and likely also from the dysfunction of CD4+ T cells owing to viral protein and endogenous cytokine-induced perturbations of cells (Table 226-5) associated with the extremely high levels of plasma viremia. A number of immunologic abnormalities accompany the acute HIV syndrome, including multiphasic perturbations of the numbers of circulating lymphocyte subsets. The number of total lymphocytes and T cell subsets (CD4+ and CD8+) are initially reduced. An inversion of the CD4+/CD8+ T cell ratio occurs later because of a rise in the number of CD8+ T cells. In fact, there may be a selective and transient expansion of CD8+ T cell subsets, as determined by T cell receptor analysis (see above). The total circulating CD8+ T cell count may remain elevated or return to normal; however, CD4+ T cell levels usually remain somewhat depressed, although there may be a slight rebound toward normal. Lymphadenopathy occurs in ~70% of individuals with primary HIV infection. Most patients recover spontaneously from this syndrome and many are left with only a mildly depressed CD4+ T cell count that remains stable for a variable period before beginning its progressive decline; in some individuals, the CD4+ T cell count returns to the normal range. Approximately 10% of patients manifest a fulminant course of immunologic and clinical deterioration after primary infection, even after the disappearance of initial symptoms. In most patients, primary infection with or without the acute syndrome is followed by a prolonged period of clinical latency or smoldering low disease activity.
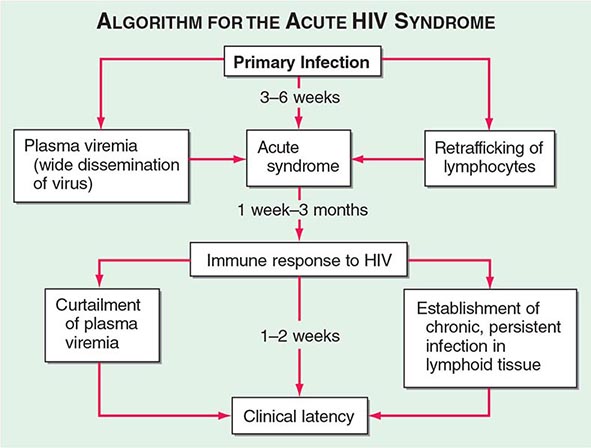
FIGURE 226-32 The acute HIV syndrome. See text for detailed description. (Adapted from G Pantaleo et al: N Engl J Med 328:327, 1993. Copyright 1993 Massachusetts Medical Society. All rights reserved.)
|
CLINICAL FINDINGS IN THE ACUTE HIV SYNDROME |
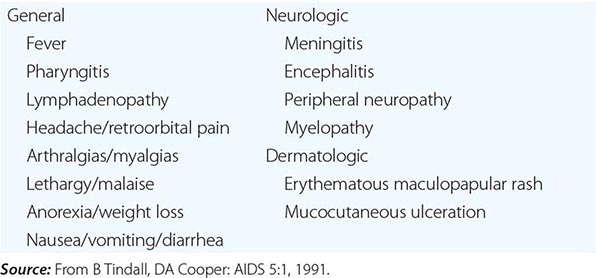
THE ASYMPTOMATIC STAGE—CLINICAL LATENCY
Although the length of time from initial infection to the development of clinical disease varies greatly, the median time for untreated patients is ~10 years. As emphasized above, HIV disease with active virus replication is ongoing and progressive during this asymptomatic period. The rate of disease progression is directly correlated with HIV RNA levels. Patients with high levels of HIV RNA in plasma progress to symptomatic disease faster than do patients with low levels of HIV RNA (Fig. 226-22). Some patients referred to as long-term nonprogressors show little if any decline in CD4+ T cell counts over extended periods of time. These patients generally have extremely low levels of HIV RNA; a subset, referred to as elite nonprogressors, exhibits HIV RNA levels <50 copies/mL. Certain other patients remain entirely asymptomatic despite the fact that their CD4+ T cell counts show a steady progressive decline to extremely low levels. In these patients, the appearance of an opportunistic disease may be the first manifestation of HIV infection. During the asymptomatic period of HIV infection, the average rate of CD4+ T cell decline is ~50/μL per year. When the CD4+ T cell count falls to <200/μL, the resulting state of immunodeficiency is severe enough to place the patient at high risk for opportunistic infection and neoplasms and, hence, for clinically apparent disease.
SYMPTOMATIC DISEASE
Symptoms of HIV disease can appear at any time during the course of HIV infection. Generally speaking, the spectrum of illnesses that one observes changes as the CD4+ T cell count declines. The more severe and life-threatening complications of HIV infection occur in patients with CD4+ T cell counts <200/μL. A diagnosis of AIDS is made in individuals age 6 years and older with HIV infection and a CD4+ T cell count <200/μL (Stage 3, Table 226-2) and in anyone with HIV infection who develops one of the HIV-associated diseases considered to be indicative of a severe defect in cell-mediated immunity (Table 226-1). While the causative agents of the secondary infections are characteristically opportunistic organisms such as P. jiroveci, atypical mycobacteria, CMV, and other organisms that do not ordinarily cause disease in the absence of a compromised immune system, they also include common bacterial and mycobacterial pathogens. Following the widespread use of cART and implementation of guidelines for the prevention of opportunistic infections (Table 226-11), the incidence of these secondary infections has decreased dramatically (Fig. 226-33). Overall, the clinical spectrum of HIV disease is constantly changing as patients live longer and new and better approaches to treatment and prophylaxis are developed. In addition to the classic AIDS-defining illnesses, patients with HIV infection also have an increase in serious non-AIDS illnesses, including non-AIDS related cancers and cardiovascular, renal, and hepatic disease. Non-AIDS events dominate the disease burden for patients with HIV infection receiving cART (Table 226-4). While AIDS-related illnesses are the leading cause of death in patients with HIV infection, they account for fewer than 50% of deaths. Non-AIDS-defining malignancies, liver disease, and cardiovascular disease each account for 10–15% of deaths in patients with HIV infection. The physician providing care to a patient with HIV infection must be well versed in general internal medicine as well as HIV-related opportunistic diseases. In general, it should be stressed that a key element of treatment of symptomatic complications of HIV disease, whether they are primary or secondary, is achieving good control of HIV replication through the use of cART and instituting primary and secondary prophylaxis for opportunistic infections as indicated.
|
NIH/CDC/IDSA 2013 GUIDELINES FOR THE PREVENTION OF OPPORTUNISTIC INFECTIONS IN PERSONS INFECTED WITH HIV |

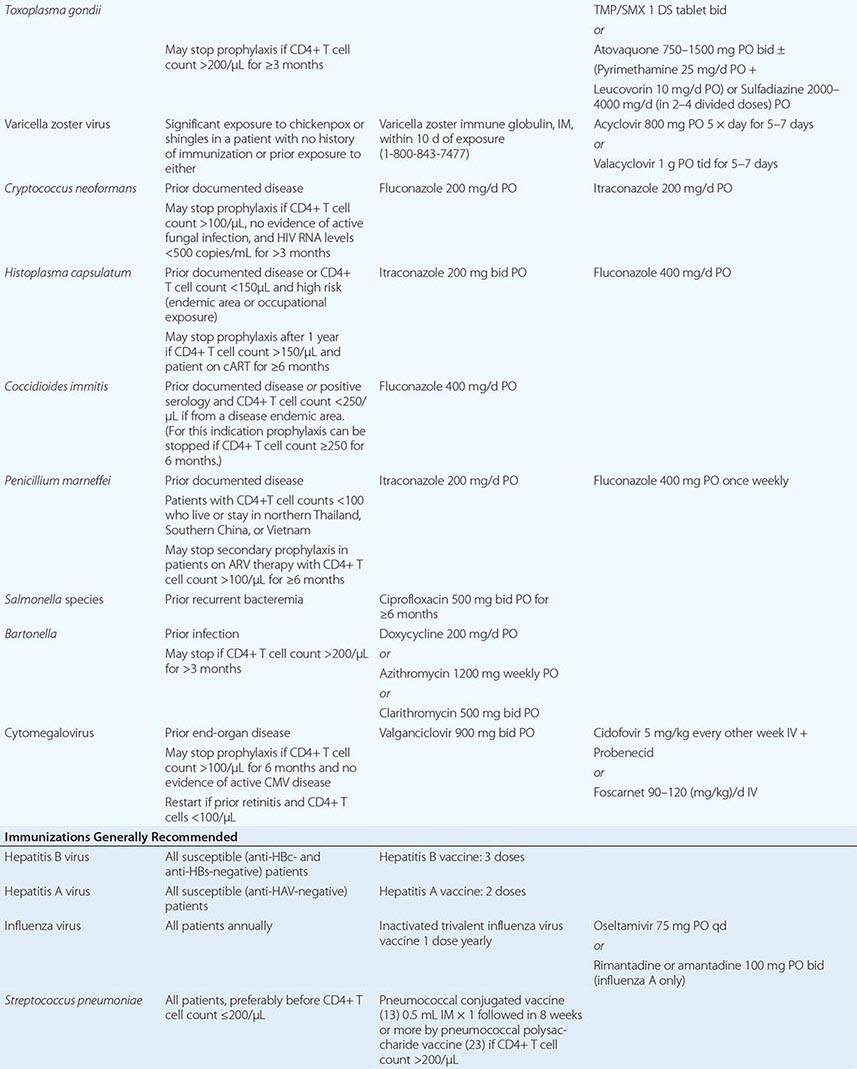
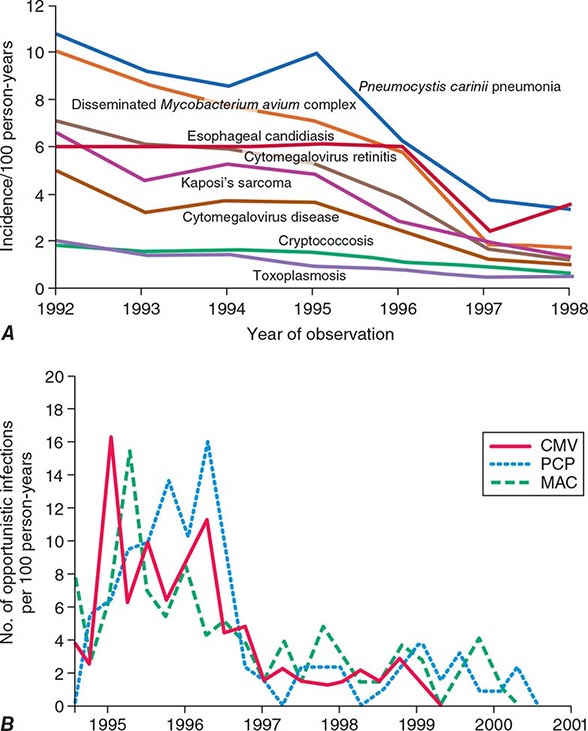
FIGURE 226-33 A. Decrease in the incidence of opportunistic infections and Kaposi’s sarcoma in HIV-infected individuals with CD4+ T cell counts <100/μL from 1992 through 1998. (Adapted and updated from FJ Palella et al: N Engl J Med 338:853, 1998, and JE Kaplan et al: Clin Infect Dis 30[S1]:S5, 2000, with permission.) B. Quarterly incidence rates of cytomegalovirus (CMV), Pneumocystis jiroveci pneumonia (PCP), and Mycobacterium avium complex (MAC) from 1995 to 2001. (From FJ Palella et al: AIDS 16:1617, 2002.)
Diseases of the Respiratory System Acute bronchitis and sinusitis are prevalent during all stages of HIV infection. The most severe cases tend to occur in patients with lower CD4+ T cell counts. Sinusitis presents as fever, nasal congestion, and headache. The diagnosis is made by CT or MRI. The maxillary sinuses are most commonly involved; however, disease is also frequently seen in the ethmoid, sphenoid, and frontal sinuses. While some patients may improve without antibiotic therapy, radiographic improvement is quicker and more pronounced in patients who have received antimicrobial therapy. It is postulated that this high incidence of sinusitis results from an increased frequency of infection with encapsulated organisms such as H. influenzae and Streptococcus pneumoniae. In patients with low CD4+ T cell counts one may see mucormycosis infections of the sinuses. In contrast to the course of this infection in other patient populations, mucormycosis of the sinuses in patients with HIV infection may progress more slowly. In this setting aggressive, frequent local debridement in addition to local and systemic amphotericin B may result in effective treatment.
Pulmonary disease is one of the most frequent complications of HIV infection. The most common manifestation of pulmonary disease is pneumonia. Three of the 10 most common AIDS-defining illnesses are recurrent bacterial pneumonia, tuberculosis, and pneumonia due to the unicellular fungus P. jiroveci. Other major causes of pulmonary infiltrates include other mycobacterial infections, other fungal infections, nonspecific interstitial pneumonitis, KS, and lymphoma.
Bacterial pneumonia is seen with an increased frequency in patients with HIV infection, with 0.8–2.0 cases per 100 person-years. Patients with HIV infection are particularly prone to infections with encapsulated organisms. S. pneumoniae (Chap. 171) and H. influenzae (Chap. 182) are responsible for most cases of bacterial pneumonia in patients with AIDS. This may be a consequence of altered B cell function and/or defects in neutrophil function that may be secondary to HIV disease (see above). Pneumonias due to S. aureus (Chap. 172) and P. aeruginosa (Chap. 189) also are reported to occur with an increased frequency in patients with HIV infection. S. pneumoniae (pneumococcal) infection may be the earliest serious infection to occur in patients with HIV disease. This can present as pneumonia, sinusitis, and/or bacteremia. Patients with untreated HIV infection have a sixfold increase in the incidence of pneumococcal pneumonia and a 100-fold increase in the incidence of pneumococcal bacteremia. Pneumococcal disease may be seen in patients with relatively intact immune systems. In one study, the baseline CD4+ T cell count at the time of a first episode of pneumococcal pneumonia was ~300/μL. Of interest is the fact that the inflammatory response to pneumococcal infection appears proportional to the CD4+ T cell count. Due to this high risk of pneumococcal disease, immunization with the conjugated pneumococcal vaccine followed by booster immunization with the 23-valent pneumococcal polysaccharide vaccine is one of the generally recommended prophylactic measures for patients with HIV infection. This is likely most effective if given while the CD4+ T cell count is >200/μL and, if given to patients with lower CD4+ T cell counts, should be repeated once the count has been above 200 for 6 months. Although clear guidelines do not exist, it also makes sense to repeat immunization every 5 years. The incidence of bacterial pneumonia is cut in half when patients quit smoking.
Pneumocystis pneumonia (PCP), once the hallmark of AIDS, has dramatically declined in incidence following the development of effective prophylactic regimens and the widespread use of cART. It is, however, still the single most common cause of pneumonia in patients with HIV infection in the United States and can be identified as a likely etiologic agent in 25% of cases of pneumonia in patients with HIV infection, with an incidence in the range of 2–3 cases per 100 person-years. Approximately 50% of cases of HIV-associated PCP occur in patients who are unaware of their HIV status. The risk of PCP is greatest among those who have experienced a previous bout of PCP and those who have CD4+ T cell counts of <200/μL. Overall, 79% of patients with PCP have CD4+ T cell counts <100/μL and 95% of patients have CD4+ T cell counts <200/μL. Recurrent fever, night sweats, thrush, and unexplained weight loss also are associated with an increased incidence of PCP. For these reasons, it is strongly recommended that all patients with CD4+ T cell counts <200/μL (or a CD4 percentage <15) receive some form of PCP prophylaxis. The incidence of PCP is approaching zero in patients with known HIV infection receiving appropriate cART and prophylaxis. In the United States, primary PCP is now occurring at a median CD4+ T cell count of 36/μL, while secondary PCP is occurring at a median CD4+ T cell count of 10/μL. Patients with PCP generally present with fever and a cough that is usually nonproductive or productive of only scant amounts of white sputum. They may complain of a characteristic retrosternal chest pain that is worse on inspiration and is described as sharp or burning. HIV-associated PCP may have an indolent course characterized by weeks of vague symptoms and should be included in the differential diagnosis of fever, pulmonary complaints, or unexplained weight loss in any patient with HIV infection and <200 CD4+ T cells/μL. The most common finding on chest x-ray is either a normal film, if the disease is suspected early, or a faint bilateral interstitial infiltrate. The classic finding of a dense perihilar infiltrate is unusual in patients with AIDS. In patients with PCP who have been receiving aerosolized pentamidine for prophylaxis, one may see an x-ray picture of upper lobe cavitary disease, reminiscent of TB. Other less common findings on chest x-ray include lobar infiltrates and pleural effusions. Thin-section CT may demonstrate a patchy ground-glass appearance. Routine laboratory evaluation is usually of little help in the differential diagnosis of PCP. A mild leukocytosis is common, although this may not be obvious in patients with prior neutropenia. Elevation of lactate dehydrogenase is common. Arterial blood-gases may indicate hypoxemia with a decline in Pao2 and an increase in the arterial-alveolar (a–A) gradient. Arterial blood-gas measurements not only aid in making the diagnosis of PCP but also provide important information for staging the severity of the disease and directing treatment (see below). A definitive diagnosis of PCP requires demonstration of the organism in samples obtained from induced sputum, bronchoalveolar lavage, transbronchial biopsy, or open-lung biopsy. PCR has been used to detect specific DNA sequences for P. jiroveci in clinical specimens where histologic examinations have failed to make a diagnosis.
In addition to pneumonia, a number of other clinical problems have been reported in HIV-infected patients as a result of infection with P. jiroveci. Otic involvement may be seen as a primary infection, presenting as a polypoid mass involving the external auditory canal. In patients receiving aerosolized pentamidine for prophylaxis against PCP, one may see a variety of extrapulmonary manifestations of P. jiroveci. These include ophthalmic lesions of the choroid, a necrotizing vasculitis that resembles Burger’s disease, bone marrow hypoplasia, and intestinal obstruction. Other organs that have been involved include lymph nodes, spleen, liver, kidney, pancreas, pericardium, heart, thyroid, and adrenals. Organ infection may be associated with cystic lesions that may appear calcified on CT or ultrasound.
The standard treatment for PCP or disseminated pneumocystosis is trimethoprim/sulfamethoxazole (TMP/SMX). A high (20–85%) incidence of side effects, particularly skin rash and bone marrow suppression, is seen with TMP/SMX in patients with HIV infection. Alternative treatments for mild to moderate PCP include dapsone/trimethoprim, clindamycin/primaquine, and atovaquone. IV pentamidine is the treatment of choice for severe disease in the patient unable to tolerate TMP/SMX. For patients with a Pao2 <70 mmHg or with an a–A gradient >35 mmHg, adjunct glucocorticoid therapy should be used in addition to specific antimicrobials. Overall, treatment should be continued for 21 days and followed by secondary prophylaxis. Prophylaxis for PCP is indicated for any HIV-infected individual who has experienced a prior bout of PCP, any patient with a CD4+ T cell count of <200/μL or a CD4 percentage <15, any patient with unexplained fever for >2 weeks, and any patient with a recent history of oropharyngeal candidiasis. The preferred regimen for prophylaxis is TMP/SMX, one double-strength tablet daily. This regimen also provides protection against toxoplasmosis and some bacterial respiratory pathogens. For patients who cannot tolerate TMP/SMX, alternatives for prophylaxis include dapsone plus pyrimethamine plus leucovorin, aerosolized pentamidine administered by the Respirgard II nebulizer, and atovaquone. Primary or secondary prophylaxis for PCP can be discontinued in those patients treated with cART who maintain good suppression of HIV (<50 copies/mL) and CD4+ T cell counts >200/μL for at least 3 months.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]Retroviral genomes include both coding and noncoding sequences (Fig. 225e-2). In general, noncoding sequences are important recognition signals for DNA or RNA synthesis or processing events and are located in the 5′ and 3′ terminal regions of the genome. All retroviral genomes are terminally redundant, containing identical sequences called long terminal repeats (LTRs). The ends of the retroviral RNA genome differ slightly in sequence from the integrated retroviral DNA. In the latter, the LTR sequences are repeated in both the 5′ and the 3′ terminus of the virus. The LTRs contain sequences involved in initiating the expression of the viral proteins, the integration of the provirus, and the polyadenylation of viral RNAs. The primer binding site, which is critical for the initiation of reverse transcription, and the viral packaging sequences are located outside the LTR sequences. The coding regions include the gag (group-specific antigen, core protein), pol (RNA-dependent DNA polymerase), and env (envelope) genes. The gag gene encodes a precursor polyprotein that is cleaved to form three to five capsid proteins; a fraction of the Gag precursor proteins also contain a protease responsible for cleaving the Gag and Pol polyproteins. A Gag-Pol polyprotein gives rise to the protease that is responsible for cleaving the Gag-Pol polyprotein. The pol gene encodes three proteins: the reverse transcriptase, the integrase, and the protease. The reverse transcriptase copies the viral RNA into the double-strand DNA provirus, which inserts itself into the host cell DNA via the action of integrase. The protease cleaves the Gag-Pol polyprotein into smaller protein products. The env gene encodes the envelope glycoproteins: one protein that binds to specific surface receptors and determines what cell types can be infected and a smaller transmembrane protein that anchors the complex to the envelope. Fig. 225e-3 shows how the retroviral gene products make up the virus structure.
FIGURE 225e-2 Genomic structure of retroviruses. The murine leukemia virus MuLV has the typical three structural genes: gag, pol, and env. The gag region gives rise to three proteins: matrix (MA), capsid (CA), and nucleic acid–binding (NC) proteins. The pol region encodes both a protease (PR) responsible for cleaving the viral polyproteins and a reverse transcriptase (RT). In addition, HIV pol encodes an integrase (IN). The env region encodes a surface protein (SU) and a small transmembrane protein (TM). The human retroviruses have additional gene products translated in each of the three possible reading frames. HTLV-1 and HTLV-2 have tax and rex genes with exons on either side of the env gene. HIV-1 and HIV-2 have six accessory gene products: tat, rev, vif, nef, vpr, and either vpu (in HIV-1) or vpx (in HIV-2). The genes for these proteins are located mainly between the pol and env genes. GP, glycoprotein; HBZ, HTLV-1 basic leucine zipper domain–containing protein; LTR, long terminal repeat.
FIGURE 225e-3 Schematic structure of human retroviruses. The surface glycoprotein (SU) is responsible for binding to receptors of host cells. The transmembrane protein (TM) anchors SU to the virus. NC is a nucleic acid–binding protein found in association with the viral RNA. A protease (PR) cleaves the polyproteins encoded by the gag, pol, and env genes into their functional components. RT is reverse transcriptase, and IN is an integrase present in some retroviruses (e.g., HIV-1) that facilitates insertion of the provirus into the host genome. The matrix protein (MA) is a Gag protein closely associated with the lipid of the envelope. The capsid protein (CA) forms the major internal structure of the virus, the core shell.
HTLVs have a region between env and the 3′ LTR that encodes several proteins and transcripts in overlapping reading frames (Fig. 225e-2). Tax is a 40-kDa protein that does not bind to DNA but induces the expression of host cell transcription factors that alter host cell gene expression and is capable of inducing cell transformation under certain circumstances. Rex is a 27-kDa protein that regulates the expression of viral mRNAs. Other transcripts from this region (p12, p13, p30) tend to restrict expression of viral genes and diminish the immunogenicity of infected cells. The protein of HBZ, a product of the complementary proviral DNA strand, interacts with many cellular transcription factors and signaling proteins. It stimulates proliferation of infected cells and is the only viral product universally expressed in HTLV-1-infected tumor cells. These proteins are produced from messages that are similar but that are spliced differently from overlapping but distinct exons.
The lentiviruses in general, and HIV-1 and -2 in particular, contain a larger genome than other pathogenic retroviruses. They contain an untranslated region between pol and env that encodes portions of several proteins, varying with the reading frame into which the mRNA is spliced. Tat is a 14-kDa protein that augments the expression of virus from the LTR. The Rev protein of HIV-1, similar to the Rex protein of HTLV, regulates RNA splicing and/or RNA transport. The Nef protein downregulates CD4, the cellular receptor for HIV; alters host T cell–activation pathways; and enhances viral infectivity. The Vif protein is necessary for the proper assembly of the HIV nucleoprotein core in many types of cells; without Vif, proviral DNA is not efficiently produced in these infected cells. In addition, the Vif protein targets APOBEC (apolipoprotein B mRNA-editing enzyme catalytic polypeptide, a cytidine deaminase that mutates the viral sequence) for proteasomal degradation, thus blocking its virus-suppressing effect. Vpr, Vpu (HIV-1 only), and Vpx (HIV-2 only) are viral proteins encoded by translation of the same message in different reading frames. As noted above, oncogenic retroviruses depend on cell proliferation for their replication; lentiviruses can infect nondividing cells, largely through effects mediated by Vpr. Vpr facilitates transport of the provirus into the nucleus and can induce other cellular changes, such as G2 growth arrest and differentiation of some target cells. Vpx is structurally related to Vpr, but its functions are not fully defined. Vpu promotes the degradation of CD4 in the endoplasmic reticulum and stimulates the release of virions from infected cells.
Retroviruses can be either exogenously acquired (by infection with an infected cell or a free virion capable of replication) or transmitted in the germline as endogenous virus. Endogenous retroviruses are often replication defective. The human genome contains endogenous retroviral sequences, but there are no known replication-competent endogenous retroviruses in humans.
In general, viruses that contain only the gag, pol, and env genes either are not pathogenic or take a long time to induce disease; these observations indicate the importance of the other regulatory genes in viral disease pathogenesis. The pathogenesis of neoplastic transformation by retroviruses relies on the chance integration of the provirus at a spot in the genome resulting in the expression of a cellular gene (protooncogene) that becomes transforming by virtue of its unregulated expression. For example, avian leukosis virus causes B cell leukemia by inducing the expression of myc. Some retroviruses possess captured and altered cellular genes near their integration site, and these viral oncogenes can transform the infected host cell. Viruses that have oncogenes often have lost a portion of their genome that is required for replication. Such viruses need helper viruses to reproduce, a feature that may explain why these acute transforming retroviruses are rare in nature. All human retroviruses identified to date are exogenous and are not acutely transforming (i.e., they lack a transforming oncogene).
These remarkable properties of retroviruses have led to experimental efforts to use them as vectors to insert specific genes into particular cell types, a process known as gene therapy or gene transfer. The process could be used to repair a genetic defect or to introduce a new property that could be used therapeutically; for example, a gene (e.g., thymidine kinase) that would make a tumor cell susceptible to killing by a drug (e.g., ganciclovir) could be inserted. One source of concern about the use of retroviral vectors in humans is that replication-competent viruses might rescue endogenous retroviral replication, with unpredictable results. This concern is not merely hypothetical: the detection of proteins encoded by endogenous retroviral sequences on the surface of cancer cells implies that the genetic events leading to the cancer were able to activate the synthesis of these usually silent genes.
HUMAN T CELL LYMPHOTROPIC VIRUS
HTLV-1 was isolated in 1980 from a T cell lymphoma cell line from a patient originally thought to have cutaneous T cell lymphoma. Later it became clear that the patient had a distinct form of lymphoma (originally reported in Japan) called adult T cell leukemia/lymphoma (ATL). Serologic data have determined that HTLV-1 is the cause of at least two important diseases: ATL and tropical spastic paraparesis, also called HTLV-1-associated myelopathy (HAM). HTLV-1 may also play a role in infective dermatitis, arthritis, uveitis, and Sjögren’s syndrome.
Two years after the isolation of HTLV-1, HTLV-2 was isolated from a patient with an unusual form of hairy cell leukemia that affected T cells. Epidemiologic studies of HTLV-2 failed to reveal a consistent disease association. Similarly, HTLV-3 and HTLV-4 have been identified but have no known disease association.
BIOLOGY AND MOLECULAR BIOLOGY
Because the biology of HTLV-1 and that of HTLV-2 are similar, the following discussion will focus on HTLV-1.
Human glucose transporter protein 1 (GLUT-1) functions as a receptor for HTLV-1, probably acting together with neuropilin-1 (NRP1) and heparan sulfate proteoglycans. Generally, only T cells are productively infected, but infection of B cells and other cell types is occasionally detected. The most common outcome of HTLV-1 infection is latent carriage of randomly integrated provirus in CD4+ T cells. HTLV-1 does not contain an oncogene and does not insert into a unique site in the genome. Indeed, most infected cells express no viral gene products. The only viral gene product that is routinely expressed in tumor cells transformed by HTLV-1 in vivo is hbz. The tax gene is thought to be critical to the transformation process but is not expressed in the tumor cells of many ATL patients, possibly because of the immunogenicity of tax-expressing cells. Cells transformed in vitro, by contrast, actively transcribe HTLV-1 RNA and produce infectious virions. Most HTLV-1-transformed cell lines are the result of the infection of a normal host T cell in vitro. It is difficult to establish cell lines derived from authentic ATL cells.
Although tax does not itself bind to DNA, it does induce the expression of a wide range of host cell gene products, including transcription factors (especially c-rel/NF-κB, ets-1 and -2, and members of the fos/jun family), cytokines (e.g., interleukin [IL] 2, granulocyte-macrophage colony-stimulating factor, and tumor necrosis factor), membrane proteins and receptors (major histocompatibility [MHC] molecules and IL-2 receptor α), and chromatin remodeling complexes. The genes activated by tax are generally controlled by transcription factors of the c-rel/NF-κB and cyclic AMP response element binding (CREB) protein families. It is unclear how this induction of host gene expression leads to neoplastic transformation; tax can interfere with G1 and mitotic cell-cycle checkpoints, block apoptosis, inhibit DNA repair, and promote antigen-independent T cell proliferation. Induction of a cytokine–autocrine loop has been proposed; however, IL-2 is not the crucial cytokine. The involvement of IL-4, IL-7, and IL-15 has been proposed.
In light of the irregular expression of tax in ATL cells, it has been suggested that tax is important in the early phases of transformation but is not essential for the maintenance of the transformed state. The maintenance role is thought to be due to hbz expression. As is clear from the epidemiology of HTLV-1 infection, transformation of an infected cell is a rare event and may depend on heterogeneous second, third, or fourth genetic hits. No consistent chromosomal abnormalities have been described in ATL; however, aneuploidy is common and individual cases with p53 mutations and translocations involving the T cell receptor genes on chromosome 14 have been reported. Tax may repress certain DNA repair enzymes, permitting the accumulation of genetic damage that would normally be repaired. However, the molecular pathogenesis of HTLV-1-induced neoplasia is not fully understood.
FEATURES OF HTLV-1 INFECTION
Epidemiology HTLV-1 infection is transmitted in at least three ways: from mother to child, especially via breast milk; through sexual activity, more commonly from men to women; and through the blood—via contaminated transfusions or contaminated needles. The virus is most commonly transmitted perinatally. Compared with HIV, which can be transmitted in cell-free form, HTLV-1 is less infectious, and its transmission usually requires cell-to-cell contact.
![]() HTLV-1 is endemic in southwestern Japan and Okinawa, where >1 million persons are infected. Antibodies to HTLV-1 are present in the serum of up to 35% of Okinawans, 10% of residents of the Japanese island of Kyushu, and <1% of persons in nonendemic regions of Japan. Despite this high prevalence of infection, only ~500 cases of ATL are diagnosed in this area each year. Clusters of infection have been noted in other areas of the Orient, such as Taiwan; in the Caribbean basin, including northeastern South America; in northwestern South America; in central and southern Africa; in Italy, Israel, Iran, and Papua New Guinea; in the Arctic; and in the southeastern part of the United States (Fig. 225e-4). An estimated 5–10 million persons have HTLV-1 infection worldwide.
HTLV-1 is endemic in southwestern Japan and Okinawa, where >1 million persons are infected. Antibodies to HTLV-1 are present in the serum of up to 35% of Okinawans, 10% of residents of the Japanese island of Kyushu, and <1% of persons in nonendemic regions of Japan. Despite this high prevalence of infection, only ~500 cases of ATL are diagnosed in this area each year. Clusters of infection have been noted in other areas of the Orient, such as Taiwan; in the Caribbean basin, including northeastern South America; in northwestern South America; in central and southern Africa; in Italy, Israel, Iran, and Papua New Guinea; in the Arctic; and in the southeastern part of the United States (Fig. 225e-4). An estimated 5–10 million persons have HTLV-1 infection worldwide.
FIGURE 225e-4 Global distribution of HTLV-1 infection. Countries with a prevalence of HTLV-1 infection of 1–5% are shaded darkly. Note that the distribution of infected patients is not uniform in endemic countries. For example, the people of southwestern Japan and northeastern Brazil are more commonly affected than those in other regions of those countries.
Progressive spastic or ataxic myelopathy developing in an individual who is HTLV-1 positive (i.e., who has serum antibodies to HTLV-1) may be due to direct infection of the nervous system with the virus, but destruction of the pyramidal tracts appears to involve HTLV-1-infected CD4+ T cells; a similar disorder may result from infection with HIV or HTLV-2. In rare instances, patients with HAM are seronegative but have detectable antibody to HTLV-1 in cerebrospinal fluid (CSF).
The cumulative lifetime risk of developing ATL is 3% among HTLV-1-infected patients, with a threefold greater risk among men than among women; a similar cumulative risk is projected for HAM (4%), but with women more commonly affected than men. The distribution of these two diseases overlaps the distribution of HTLV-1, with >95% of affected patients showing serologic evidence of HTLV-1 infection. The latency period between infection and the emergence of disease is 20–30 years for ATL. For HAM, the median latency period is ~3.3 years (range, 4 months to 30 years). The development of ATL is rare among persons infected by blood products; however, ~20% of patients with HAM acquire HTLV-1 from contaminated blood. ATL is more common among perinatally infected individuals, whereas HAM is more common among persons infected via sexual transmission.
Associated Diseases • ATL Four clinical types of HTLV-1-induced neoplasia have been described: acute, lymphomatous, chronic, and smoldering. All of these tumors are monoclonal proliferations of CD4+ postthymic T cells with clonal proviral integrations and clonal T cell receptor gene rearrangements.
ACUTE ATL About 60% of patients who develop malignancy have classic acute ATL, which is characterized by a short clinical prodrome (~2 weeks between the first symptoms and the diagnosis) and an aggressive natural history (median survival period, 6 months). The clinical picture is dominated by rapidly progressive skin lesions, pulmonary involvement, hypercalcemia, and lymphocytosis with cells containing lobulated or “flower-shaped” nuclei (see Fig. 134-10). The malignant cells have monoclonal proviral integrations and express CD4, CD3, and CD25 (low-affinity IL-2 receptors) on their surface. Serum levels of CD25 can be used as a tumor marker. Anemia and thrombocytopenia are rare. The skin lesions may be difficult to distinguish from those in mycosis fungoides. Lytic bone lesions, which are common, do not contain tumor cells but rather are composed of osteolytic cells, usually without osteoblastic activity. Despite the leukemic picture, bone marrow involvement is patchy in most cases.
The hypercalcemia of ATL is multifactorial; the tumor cells produce osteoclast-activating factors (tumor necrosis factor α, IL-1, lymphotoxin) and can also produce a parathyroid hormone–like molecule. Affected patients have an underlying immunodeficiency that makes them susceptible to opportunistic infections similar to those seen in patients with AIDS (Chap. 226). The pathogenesis of the immunodeficiency is unclear. Pulmonary infiltrates in ATL patients reflect leukemic infiltration half the time and opportunistic infections with organisms such as Pneumocystis and other fungi the other half. Gastrointestinal symptoms are nearly always related to opportunistic infection. Strongyloides stercoralis is a gastrointestinal parasite that has a pattern of endemic distribution similar to that of HTLV-1. HTLV-1-infected persons also infected with this parasite may develop ATL more often or more rapidly than those without Strongyloides infections. Serum concentrations of lactate dehydrogenase and alkaline phosphatase are often elevated in ATL. About 10% of patients have leptomeningeal involvement leading to weakness, altered mental status, paresthesia, and/or headache. Unlike other forms of central nervous system (CNS) lymphoma, ATL may be accompanied by normal CSF protein levels. The diagnosis depends on finding ATL cells in the CSF (Chap. 134).
LYMPHOMATOUS ATL The lymphomatous type of ATL occurs in ~20% of patients and is similar to the acute form in its natural history and clinical course, except that circulating abnormal cells are rare and lymphadenopathy is evident. The histology of the lymphoma is variable but does not influence the natural history. In general, the diagnosis is suspected on the basis of the patient’s birthplace (see “Epidemiology,” above) and the presence of skin lesions and hypercalcemia. The diagnosis is confirmed by the detection of antibodies to HTLV-1 in serum.
CHRONIC ATL Patients with the chronic form of ATL generally have normal serum levels of calcium and lactate dehydrogenase and no involvement of the CNS, bone, or gastrointestinal tract. The median duration of survival for these patients is 2 years. In some cases, chronic ATL progresses to the acute form of the disease.
SMOLDERING ATL Fewer than 5% of patients have the smoldering form of ATL. In this form, the malignant cells have monoclonal proviral integration; <5% of peripheral blood cells exhibit typical morphologic abnormalities; hypercalcemia, adenopathy, and hepatosplenomegaly do not develop; the CNS, the bones, and the gastrointestinal tract are not involved; and skin lesions and pulmonary lesions may be present. The median survival period for this small subset of patients appears to be ≥5 years.
HAM (TROPICAL SPASTIC PARAPARESIS) In contrast to ATL, in which there is a slight predominance of male patients, HAM affects female patients disproportionately. HAM resembles multiple sclerosis in certain ways (Chap. 458). The onset is insidious. Symptoms include weakness or stiffness in one or both legs, back pain, and urinary incontinence. Sensory changes are usually mild, but peripheral neuropathy may develop. The disease generally takes the form of slowly progressive and unremitting thoracic myelopathy; one-third of patients are bedridden within 10 years of diagnosis, and one-half are unable to walk unassisted by this point. Patients display spastic paraparesis or paraplegia with hyperreflexia, ankle clonus, and extensor plantar responses. Cognitive function is usually spared; cranial nerve abnormalities are unusual.
Magnetic resonance imaging (MRI) reveals lesions in both the white matter and the paraventricular regions of the brain as well as in the spinal cord. Pathologic examination of the spinal cord shows symmetric degeneration of the lateral columns, including the corticospinal tracts; some cases involve the posterior columns as well. The spinal meninges and cord parenchyma contain an inflammatory infiltrate with myelin destruction.
HTLV-1 is not usually found in cells of the CNS but may be detected in a small population of lymphocytes present in the CSF. In general, HTLV-1 replication is greater in HAM than in ATL, and patients with HAM have a stronger immune response to the virus. Antibodies to HTLV-1 are present in the serum and appear to be produced in the CSF of HAM patients, where titers are often higher than in the serum. The pathophysiology of HAM may involve the induction of autoimmune destruction of neural cells by T cells with specificity for viral components such as Tax or Env proteins. One theory is that susceptibility to HAM may be related to the presence of human leukocyte antigen (HLA) alleles capable of presenting viral antigens in a fashion that leads to autoimmunity. Insufficient data are available to confirm an HLA association. However, antibodies in the sera of HAM patients have been shown to bind a neuron-specific antigen (heteronuclear ribonuclear protein A1 [hnRNP A1]) and to interfere with neurotransmission in vitro.
It is unclear what factors influence whether HTLV-1 infection will cause disease and, if it does, whether it will induce a neoplasm (ATL) or an autoimmune disorder (HAM). Differences in viral strains, the susceptibility of particular MHC haplotypes, the route of HTLV-1 infection, the viral load, and the nature of the HTLV-1-related immune response are putative factors, but few definitive data are available.
OTHER PUTATIVE HTLV-1-RELATED DISEASES Even in the absence of the full clinical picture of HAM, bladder dysfunction is common in HTLV-1-infected women. In areas where HTLV-1 is endemic, diverse inflammatory and autoimmune diseases have been attributed to the virus, including uveitis, dermatitis, pneumonitis, rheumatoid arthritis, and polymyositis. However, a causal relationship between HTLV-1 and these illnesses has not been established.
Prevention Women in endemic areas should not breast-feed their children, and blood donors should be screened for serum antibodies to HTLV-1. As in the prevention of HIV infection, the practice of safe sex and the avoidance of needle sharing are important.
FEATURES OF HTLV-2 INFECTION
![]() Epidemiology HTLV-2 is endemic in certain Native American tribes and in Africa. It is generally considered to be a New World virus that was brought from Asia to the Americas 10,000–40,000 years ago during the migration of infected populations across the Bering land bridge. The mode of transmission of HTLV-2 is probably the same as that of HTLV-1 (see above). HTLV-2 may be less readily transmitted sexually than HTLV-1.
Epidemiology HTLV-2 is endemic in certain Native American tribes and in Africa. It is generally considered to be a New World virus that was brought from Asia to the Americas 10,000–40,000 years ago during the migration of infected populations across the Bering land bridge. The mode of transmission of HTLV-2 is probably the same as that of HTLV-1 (see above). HTLV-2 may be less readily transmitted sexually than HTLV-1.
Studies of large cohorts of injection drug users with serologic assays that reliably distinguish HTLV-1 from HTLV-2 indicated that the vast majority of HTLV-positive cohort members were infected with HTLV-2. The seroprevalence of HTLV in a cohort of 7841 injection drug users from drug treatment centers in Baltimore, Chicago, Los Angeles, New Jersey (Asbury Park and Trenton), New York City (Brooklyn and Harlem), Philadelphia, and San Antonio was 20.9%, with >97% of cases due to HTLV-2. The seroprevalence of HTLV-2 was higher in the Southwest and the Midwest than in the Northeast. In contrast, the seroprevalence of HIV-1 was higher in the Northeast than in the Southwest or the Midwest. Approximately 3% of the cohort members were infected with both HTLV-2 and HIV-1. The seroprevalence of HTLV-2 increased linearly with age. Women were significantly more likely to be infected with HTLV-2 than were men; the virus is thought to be more efficiently transmitted from male to female than from female to male.
Associated Diseases Although HTLV-2 was isolated from a patient with a T cell variant of hairy cell leukemia, this virus has not been consistently associated with a particular disease and in fact has been thought of as “a virus searching for a disease.” However, evidence is accumulating that HTLV-2 may play a role in certain neurologic, hematologic, and dermatologic diseases. These data require confirmation, particularly in light of the previous confusion regarding the relative prevalences of HTLV-1 and HTLV-2 among injection drug users.
Prevention Avoidance of needle sharing, adherence to safe-sex practices, screening of blood (by assays for HTLV-1, which also detect HTLV-2), and avoidance of breast-feeding by infected women are important principles in the prevention of spread of HTLV-2.
HUMAN IMMUNODEFICIENCY VIRUS
HIV-1 and HIV-2 are members of the lentivirus subfamily of Retroviridae and are the only lentiviruses known to infect humans. The lentiviruses are slow-acting by comparison with viruses that cause acute infection (e.g., influenza virus) but not by comparison with other retroviruses. The features of acute primary infection with HIV resemble those of more classic acute infections. The characteristic chronicity of HIV disease is consistent with the designation lentivirus. For a detailed discussion of HIV, see Chap. 226.
226 |
Human Immunodeficiency Virus Disease: AIDS and Related Disorders |
AIDS was first recognized in the United States in the summer of 1981, when the U.S. Centers for Disease Control and Prevention (CDC) reported the unexplained occurrence of Pneumocystis jiroveci (formerly P. carinii) pneumonia in five previously healthy homosexual men in Los Angeles and of Kaposi’s sarcoma (KS) with or without P. jiroveci pneumonia and other opportunistic infections in 26 previously healthy homosexual men in New York, San Francisco, and Los Angeles. The disease was soon recognized in male and female injection drug users; in hemophiliacs and blood transfusion recipients; among female sexual partners of men with AIDS; and among infants born to mothers with AIDS. In 1983, human immunodeficiency virus (HIV) was isolated from a patient with lymphadenopathy, and by 1984 it was demonstrated clearly to be the causative agent of AIDS. In 1985, a sensitive enzyme-linked immunosorbent assay (ELISA) was developed; this led to an appreciation of the scope and evolution of the HIV epidemic at first in the United States and other developed nations and ultimately among developing nations throughout the world (see “HIV Infection and AIDS Worldwide,” below). The staggering worldwide evolution of the HIV pandemic has been matched by an explosion of information in the areas of HIV virology, pathogenesis (both immunologic and virologic), treatment of HIV disease, treatment and prophylaxis of the opportunistic diseases associated with HIV infection, and prevention of HIV infection. The information flow related to HIV disease is enormous and continues to expand, and it has become almost impossible for the health care generalist to stay abreast of the literature. The purpose of this chapter is to present the most current information available on the scope of the epidemic; on its pathogenesis, treatment, and prevention; and on prospects for vaccine development. Above all, the aim is to provide a solid scientific basis and practical clinical guidelines for a state-of-the-art approach to the HIV-infected patient.
DEFINITION
The current U.S. CDC classification system for HIV infection and AIDS categorizes people on the basis of clinical conditions associated with HIV infection and CD4+ T lymphocyte measurement. A confirmed HIV case can be classified in one of five HIV infection stages (0, 1, 2, 3, or unknown). If there was a negative HIV test within 6 months of the first HIV infection diagnosis, the stage is 0, and remains 0 until 6 months after diagnosis. Advanced HIV disease (AIDS) is classified as stage 3 if one or more specific opportunistic illness has been diagnosed (Table 226-1). Otherwise, the stage is determined by CD4 test results and immunologic criteria (Table 226-2). If none of these criteria apply (e.g., because of missing information on CD4 test results), the stage is U (unknown).
|
CDC STAGE 3 (AIDS)-DEFINING OPPORTUNISTIC ILLNESSES IN HIV INFECTION |
aOnly among children age <6 years.b Only among adults, adolescents, and children age ≥6 years.
Source: MMWR 63(No. RR-03), April 11, 2014.
|
CDC HIV INFECTION STAGES 1–3 BASED ON AGE-SPECIFIC CD4+ T LYMPHOCYTE COUNT OR CD4+ T LYMPHOCYTE PERCENTAGE OF TOTAL LYMPHOCYTESa |
The definition and staging criteria of AIDS are complex and comprehensive and were established for surveillance purposes rather than for the practical care of patients. Thus, the clinician should not focus on whether the patient fulfills the strict definition of AIDS, but should view HIV disease as a spectrum ranging from primary infection, with or without the acute syndrome, to the asymptomatic stage, to advanced stages associated with opportunistic diseases (see “Pathophysiology and Pathogenesis,” below).
ETIOLOGIC AGENT
HIV is the etiologic agent of AIDS; it belongs to the family of human retroviruses (Retroviridae) and the subfamily of lentiviruses (Chap. 225e). Nononcogenic lentiviruses cause disease in other animal species, including sheep, horses, goats, cattle, cats, and monkeys. The four retroviruses known to cause human disease belong to two distinct groups: the human T lymphotropic viruses (HTLV)-1 and HTLV-2, which are transforming retroviruses; and the human immunodeficiency viruses, HIV-1 and HIV-2, which cause cytopathic effects either directly or indirectly (Chap. 225e). The most common cause of HIV disease throughout the world, and certainly in the United States, is HIV-1, which comprises several subtypes with different geographic distributions (see “Molecular Heterogeneity of HIV-1,” below). HIV-2 was first identified in 1986 in West African patients and was originally confined to West Africa. However, a number of cases that generally can be traced to West Africa or to sexual contacts with West Africans have been identified throughout the world. The currently defined groups of HIV-1 (M, N, O, P) and the HIV-2 groups A through H each are likely derived from a separate transfer to humans from a nonhuman primate reservoir. HIV-1 viruses likely came from chimpanzees and/or gorillas, and HIV-2 from sooty mangabeys. The AIDS pandemic is primarily caused by the HIV-1 M group viruses. Although HIV-1 group O and HIV-2 viruses have been found in numerous countries, including those in the developed world, they have caused much more localized epidemics. The taxonomic relationship between primate lentiviruses is shown in Fig. 226-1.
FIGURE 226-1 A phylogenetic tree based on the complete genomes of primate immunodeficiency viruses. The scale (0.10) indicates a 10% difference at the nucleotide level. (Prepared by Brian Foley, PhD, of the HIV Sequence Database, Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; additional information at www.hiv.lanl.gov/content/sequence/HelpDocs/subtypes.html.)
MORPHOLOGY OF HIV
Electron microscopy shows that the HIV virion is an icosahedral structure (Fig. 226-2) containing numerous external spikes formed by the two major envelope proteins, the external gp120 and the transmembrane gp41. The HIV envelope exists as a trimeric heterodimer. The virion buds from the surface of the infected cell and incorporates a variety of host proteins into its lipid bilayer. The structure of HIV-1 is schematically diagrammed in Fig. 226-2B.
FIGURE 226-2 A. Electron micrograph of HIV. Figure illustrates a typical virion following budding from the surface of a CD4+ T lymphocyte, together with two additional incomplete virions in the process of budding from the cell membrane. B. Structure of HIV-1, including the gp120 envelope, gp41 transmembrane components of the envelope, genomic RNA, enzyme reverse transcriptase, p18(17) inner membrane (matrix), and p24 core protein (capsid). (Copyright by George V. Kelvin.) (Adapted from RC Gallo: Sci Am 256:46, 1987.) C. Scanning electron micrograph of HIV-1 virions infecting a human CD4+ T lymphocyte. The original photograph was imaged at 8000× magnification. (Courtesy of Elizabeth R. Fischer, Rocky Mountain Laboratories, National Institute of Allergy and Infectious Diseases; with permission.)
REPLICATION CYCLE OF HIV
HIV is an RNA virus whose hallmark is the reverse transcription of its genomic RNA to DNA by the enzyme reverse transcriptase. The replication cycle of HIV begins with the high-affinity binding of the gp120 protein via a portion of its V1 region near the N terminus to its receptor on the host cell surface, the CD4 molecule (Fig. 226-3). The CD4 molecule is a 55-kDa protein found predominantly on a subset of T lymphocytes that are responsible for helper function in the immune system (Chap. 372e). It is also expressed on the surface of monocytes/macrophages and dendritic/Langerhans cells. Once it binds to CD4, the gp120 protein undergoes a conformational change that facilitates binding to one of two major co-receptors. The two major co-receptors for HIV-1 are CCR5 and CXCR4. Both receptors belong to the family of seven-transmembrane-domain G protein–coupled cellular receptors, and the use of one or the other or both receptors by the virus for entry into the cell is an important determinant of the cellular tropism of the virus. Certain dendritic cells (DCs) express a diversity of C-type lectin receptors on their surface—one of which is called DC-SIGN—that also bind with high affinity to the HIV gp120 envelope protein, allowing DCs to facilitate virus spread to CD4+ T cells. Following binding of the envelope protein to the CD4 molecule associated with the above-mentioned conformational change in the viral envelope gp120, fusion with the host cell membrane occurs via the newly exposed gp41 molecule penetrating the plasma membrane of the target cell and then coiling upon itself to bring the virion and target cell together (Fig. 226-4). Following fusion, uncoating of the capsid protein shell is initiated—a step that facilitates reverse transcription and leads to formation of the preintegration complex, composed of viral RNA, enzymes, and accessory proteins and surrounded by capsid and matrix proteins (Fig. 226-3). As the preintegration complex traverses the cytoplasm to reach the nucleus, the viral reverse transcriptase enzyme catalyzes the reverse transcription of the genomic RNA into DNA, resulting in the formation of double-stranded proviral HIV-DNA. At the preintegration steps of the replication cycle, the viral genome is vulnerable to cellular factors that can block the progression of infection. In particular, the cytoplasmic tripartite motif-containing protein 5-α (TRIM5-α) is a host restriction factor that interacts with retroviral capsids (Fig. 226-3). Although the exact mechanisms of action of TRIM5-α remain unclear, the HIV-1 capsid is not recognized by the human form of TRIM5-α. Thus this host factor is not effective in restricting HIV-1 replication in human cells. The apolipoprotein B mRNA editing enzyme (catalytic polypeptide-like 3 [APOBEC3]) family of cellular proteins also inhibits progression of virus infection after virus has entered the cell and prior to entering the nucleus (Fig. 226-3). APOBEC3 proteins, which are incorporated into virions and released into the cytoplasm of a newly infected cell, bind to the single minus-strand DNA intermediate and deaminate viral cytidine, causing hypermutation of retroviral genomes. HIV has evolved a powerful strategy to protect itself from APOBEC. The viral protein Vif targets APOBEC3 for proteasomal degradation.
FIGURE 226-3 The replication cycle of HIV. See text for description. (Adapted from AS Fauci: Nature 384:529, 1996.)
FIGURE 226-4 Binding and fusion of HIV-1 with its target cell. HIV-1 binds to its target cell via the CD4 molecule, leading to a conformational change in the gp120 molecule that allows it to bind to the co-receptor CCR5 (for R5-using viruses). The virus then firmly attaches to the host cell membrane in a coiled-spring fashion via the newly exposed gp41 molecule. Virus-cell fusion occurs as the transitional intermediate of gp41 undergoes further changes to form a hairpin structure that draws the two membranes into close proximity (see text for details). (Adapted from D Montefiori, JP Moore: Science 283:336, 1999; with permission.)
With activation of the cell, the viral DNA accesses the nuclear pore and is exported from the cytoplasm to the nucleus, where it is integrated into the host cell chromosomes through the action of another virally encoded enzyme, integrase (Fig. 226-3). HIV provirus (DNA) integrates into the nuclear DNA preferentially within introns of active genes and regional hotspots. This provirus may remain transcriptionally inactive (latent) or it may manifest varying levels of gene expression, up to active production of virus.
Cellular activation plays an important role in the replication cycle of HIV and is critical to the pathogenesis of HIV disease (see “Pathogenesis and Pathophysiology,” below). Following initial binding, fusion, and internalization of the nucleic acid contents of virions into the target cell, incompletely reverse-transcribed DNA intermediates are labile in quiescent cells and do not integrate efficiently into the host cell genome unless cellular activation occurs shortly after infection. Furthermore, some degree of activation of the host cell is required for the initiation of transcription of the integrated proviral DNA into either genomic RNA or mRNA. This latter process may not necessarily be associated with the detectable expression of the classic cell-surface markers of activation. In this regard, activation of HIV expression from the latent state depends on the interaction of a number of cellular and viral factors. Following transcription, HIV mRNA is translated into proteins that undergo modification through glycosylation, myristoylation, phosphorylation, and cleavage. The viral particle is formed by the assembly of HIV proteins, enzymes, and genomic RNA at the plasma membrane of the cells. Budding of the progeny virion through the lipid bilayer of the host cell membrane is the point at which the core acquires its external envelope and where the host restriction factor tetherin can inhibit the release of budding particles (Fig. 226-3). Tetherin is an interferon (IFN)-induced type II transmembrane protein that interferes with virion detachment, although the HIV accessory protein Vpu counteracts the effect through direct interactions with tetherin. During or soon after budding, the virally encoded protease catalyzes the cleavage of the gag-pol precursor to yield the mature virion. Progression through the virus replication cycle is profoundly influenced by a variety of viral regulatory gene products. Likewise, each point in the replication cycle of HIV is a real or potential target for therapeutic intervention. Thus far, the reverse transcriptase, protease, and integrase enzymes as well as the process of virus–target cell binding and fusion have proved clinically to be susceptible to pharmacologic disruption.
HIV GENOME
Figure 226-5 illustrates schematically the arrangement of the HIV genome. Like other retroviruses, HIV-1 has genes that encode the structural proteins of the virus: gag encodes the proteins that form the core of the virion (including p24 antigen); pol encodes the enzymes responsible for protease processing of viral proteins, reverse transcription, and integration; and env encodes the envelope glycoproteins. However, HIV-1 is more complex than other retroviruses, particularly those of the nonprimate group, in that it also contains at least six other genes (tat, rev, nef, vif, vpr, and vpu), which code for proteins involved in the modification of the host cell to enhance virus growth and the regulation of viral gene expression. Several of these proteins are thought to play a role in the pathogenesis of HIV disease; their various functions are listed in Fig. 226-5. Flanking these genes are the long terminal repeats (LTRs), which contain regulatory elements involved in gene expression (Fig. 226-5). The major difference between the genomes of HIV-1 and HIV-2 is the fact that HIV-2 lacks the vpu gene and has a vpx gene not contained in HIV-1.
FIGURE 226-5 Organization of the genome of the HIV provirus together with a summary description of its 9 genes encoding 15 proteins. (Adapted from WC Greene, BM Peterlin: Nat Med 8:673, 2002.)
MOLECULAR HETEROGENEITY OF HIV-1
![]() Molecular analyses of HIV isolates reveal varying levels of sequence diversity over all regions of the viral genome. For example, the degree of difference in the coding sequences of the viral envelope protein ranges from a few percent (very close, among isolates from the same infected individual) to more than 50% (extreme diversity, between isolates from the different groups of HIV-1: M, N, O, and P). The changes tend to cluster in hypervariable regions. HIV can evolve by several means, including simple base substitution, insertions and deletions, recombination, and gain and loss of glycosylation sites. HIV sequence diversity arises directly from the limited fidelity of the reverse transcriptase. The balance of immune pressure and functional constraints on proteins influences the regional level of variation within proteins. For example, Envelope, which is exposed on the surface of the virion and is under immune selective pressure from both antibodies and cytolytic T lymphocytes, is extremely variable, with clusters of mutations in hypervariable domains. In contrast, reverse transcriptase, with important enzymatic functions, is relatively conserved, particularly around the active site. The extraordinary variability of HIV-1 contrasts markedly with the relative stability of HTLV-1 and -2.
Molecular analyses of HIV isolates reveal varying levels of sequence diversity over all regions of the viral genome. For example, the degree of difference in the coding sequences of the viral envelope protein ranges from a few percent (very close, among isolates from the same infected individual) to more than 50% (extreme diversity, between isolates from the different groups of HIV-1: M, N, O, and P). The changes tend to cluster in hypervariable regions. HIV can evolve by several means, including simple base substitution, insertions and deletions, recombination, and gain and loss of glycosylation sites. HIV sequence diversity arises directly from the limited fidelity of the reverse transcriptase. The balance of immune pressure and functional constraints on proteins influences the regional level of variation within proteins. For example, Envelope, which is exposed on the surface of the virion and is under immune selective pressure from both antibodies and cytolytic T lymphocytes, is extremely variable, with clusters of mutations in hypervariable domains. In contrast, reverse transcriptase, with important enzymatic functions, is relatively conserved, particularly around the active site. The extraordinary variability of HIV-1 contrasts markedly with the relative stability of HTLV-1 and -2.
The four groups (M, N, O and P) of HIV-1 are the result of four separate chimpanzee-to-human (or possibly gorilla-to-human for groups O and P) transfers. Group M (major), which is responsible for most of the infections in the world, has diversified into subtypes and intersubtype recombinant forms, due to “sub-epidemics” within humans after one of those transfers.
Among primate lentiviruses, HIV-1 is most closely related to viruses isolated from chimpanzees and gorillas (Fig. 226-1). The chimpanzee subspecies Pan troglodytes troglodytes has been established to be the natural reservoir of the HIV-1 M and N groups. The rare viruses of the HIV-1 O and P groups are most closely related to viruses found in Cameroonian gorillas. The M group comprises nine subtypes, or clades, designated A, B, C, D, F, G, H, J, and K, as well as more than 60 known circulating recombinant forms (CRFs) and numerous unique recombinant forms. Intersubtype recombinants are generated by infection of an individual with two subtypes that then recombine and create a virus with a selective advantage. These CRFs range from highly prevalent forms such as CRF01_AE, common in southeast Asia, and CRF02_AG from west and central Africa, to a large number of CRFs that are relatively rare, either because they are of a more recent origin (newly recombined) or because they have not broken out into a major population. The subtypes and CRFs create the major lineages of the M group of HIV-1. HIV-1 M group subtype C dominates the global pandemic, and there is much speculation that it is more transmissible than other subtypes, but solid data on transmissibility variations between subtypes are rare. Human population densities, access to prevention and treatment, prevalence of genital ulcers, iatrogenic transmissions, and other confounding host factors are all possible reasons why one subtype has spread more than another.
Figure 226-6 schematically diagrams the worldwide distribution of HIV-1 subtypes by region. Seven strains account for the vast majority of HIV infections globally: HIV-1 subtypes A, B, C, D, G and two of the CRFs, CRF01_AE and CRF02_AG. Subtype C viruses (of the M group) are by far the most common form worldwide, likely accounting for ~50% of prevalent infections worldwide. In sub-Saharan Africa, home to approximately two-thirds of all individuals living with HIV/AIDS, the majority of infections are caused by subtype C, with smaller proportions of infections caused by subtype A, subtype G, CRF02_AG, and other subtypes and recombinants. In South Africa, the country with the largest number of prevalent infections (6.3 million in 2013), >97% of the HIV-1 isolates sequenced are of subtype C. In Asia, HIV-1 isolates of the CRF01_AE lineage and subtypes C and B predominate. CRF01_AE accounts for most infections in south and southeast Asia, while >95% of infections in India, home to an estimated 2.1 million HIV-infected individuals, are of subtype C (see “HIV Infection and AIDS Worldwide,” below). Subtype B viruses are the overwhelmingly predominant viruses seen in the United States, Canada, certain countries in South America, western Europe, and Australia. It is thought that, purely by chance, subtype B was seeded into the United States and Europe in the late 1970s, thereby establishing an overwhelming founder effect. Many countries have co-circulating viral subtypes that are giving rise to new CRFs. Sequence analyses of HIV-1 isolates from infected individuals indicate that recombination among viruses of different clades likely occurs as a result of infection of an individual with viruses of more than one subtype, particularly in geographic areas where subtypes overlap, and more often in sub-epidemics driven by IV drug use than in those driven by sexual transmission.
FIGURE 226-6 Global geographic distribution of HIV-1 subtypes and recombinant forms. Distributions derived from relative frequency of subtypes among >500,000 HIV genomic sequences in the Los Alamos National Laboratory HIV Sequence Database. (Additional information available at www.hiv.lanl.gov/components/sequence/HIV/geo/geo.comp)
The extraordinary diversity of HIV, reflected by the presence of multiple subtypes, circulating recombinant forms, and continuous viral evolution, has implications for possible differential rates of transmission, rates of disease progression, responses to therapy, and the development of resistance to antiretroviral drugs. This diversity is also a formidable obstacle to HIV vaccine development, as a broadly useful vaccine would need to induce protective responses against a wide range of viral strains.
TRANSMISSION
HIV is transmitted primarily by sexual contact (both heterosexual and male to male); by blood and blood products; and by infected mothers to infants intrapartum, perinatally, or via breast milk. After more than 30 years of experience and observations regarding other potential modalities of transmission, there is no evidence that HIV is transmitted by casual contact or that the virus can be spread by insects, such as by a mosquito bite. Table 226-3 lists the estimated risk of HIV transmission for various types of exposures.
|
ESTIMATED PER-ACT PROBABILITY OF ACQUIRING HIV FROM AN INFECTED SOURCE, BY EXPOSURE ACT |
SEXUAL TRANSMISSION
HIV infection is predominantly a sexually transmitted infection (STI) worldwide. By far the most common mode of infection, particularly in developing countries, is heterosexual transmission, although in many western countries a resurgence of male-to-male sexual transmission has occurred. Although a wide variety of factors including viral load and the presence of ulcerative genital diseases influence the efficiency of heterosexual transmission of HIV, such transmission is generally inefficient. A recent systemic review found a low per-act risk of heterosexual transmission in the absence of antiretrovirals: 0.04% for female-to-male transmission and 0.08% for male-to-female transmission during vaginal intercourse in the absence of antiretroviral therapy or condom use (Table 226-3).
HIV has been demonstrated in seminal fluid both within infected mononuclear cells and in cell-free material. The virus appears to concentrate in the seminal fluid, particularly in situations where there are increased numbers of lymphocytes and monocytes in the fluid, as in genital inflammatory states such as urethritis and epididymitis, conditions closely associated with other STIs. The virus has also been demonstrated in cervical smears and vaginal fluid. There is an elevated risk of HIV transmission associated with unprotected receptive anal intercourse (URAI) among both men and women compared to the risk associated with receptive vaginal intercourse. Although data are limited, the per-act risk for HIV transmission via URAI has been estimated to be ~1.4% (Table 226-3). The risk of HIV acquisition associated with URAI is probably higher than that seen in penile-vaginal intercourse because only a thin, fragile rectal mucosal membrane separates the deposited semen from potentially susceptible cells in and beneath the mucosa, and micro-trauma of the mucosal membrane may be associated with anal intercourse. Anal douching and sexual practices that traumatize the rectal mucosa also increase the likelihood of infection. It is likely that anal intercourse provides at least two modalities of infection: (1) direct inoculation into blood in cases of traumatic tears in the mucosa; and (2) infection of susceptible target cells, such as Langerhans cells, in the mucosal layer in the absence of trauma. Insertive anal intercourse also confers an increased risk of HIV acquisition compared to insertive vaginal intercourse. Although the vaginal mucosa is several layers thicker than the rectal mucosa and less likely to be traumatized during intercourse, the virus can be transmitted to either partner through vaginal intercourse. As noted in Table 226-3, male-to-female HIV transmission is usually more efficient than female-to-male transmission. The differences in reported transmission rates between men and women may be due in part to the prolonged exposure to infected seminal fluid of the vaginal and cervical mucosa, as well as the endometrium (when semen enters through the cervical os). By comparison, the penis and urethral orifice are exposed relatively briefly to infected vaginal fluid. Among various cofactors examined in studies of heterosexual HIV transmission, the presence of other STIs has been strongly associated with HIV transmission. In this regard, there is a close association between genital ulcerations and transmission, owing to both susceptibility to infection and infectivity. Infections with microorganisms such as Treponema pallidum (Chap. 206), Haemophilus ducreyi (Chap. 182), and herpes simplex virus (HSV; Chap. 216) are important causes of genital ulcerations linked to transmission of HIV. In addition, pathogens responsible for non-ulcerative inflammatory STIs such as those caused by Chlamydia trachomatis (Chap. 213), Neisseria gonorrhoeae (Chap. 181), and Trichomonas vaginalis (Chap. 254) also are associated with an increased risk of transmission of HIV infection. Bacterial vaginosis, an infection related to sexual behavior, but not strictly an STI, also may be linked to an increased risk of transmission of HIV infection. Several studies suggest that treating other STIs and genital tract syndromes may help prevent transmission of HIV. This effect is most prominent in populations in which the prevalence of HIV infection is relatively low. It is noteworthy that this principle may not apply to the treatment of HSV infections since it has been shown that even following anti-HSV therapy with resulting healing of HSV-related genital ulcers, HIV acquisition is not reduced. Biopsy studies revealed the likely explanation is that HIV receptor–positive inflammatory cells persisted in the genital tissue despite the healing of ulcers, and so HIV-susceptible targets remained at the site.
The quantity of HIV-1 in plasma is a primary determinant of the risk of HIV-1 transmission. In a cohort of heterosexual couples in Uganda discordant for HIV infection and not receiving antiretroviral therapy, the mean serum HIV RNA level was significantly higher among HIV-infected subjects whose partners seroconverted than among those whose partners did not seroconvert. In fact, transmission was rare when the infected partner had a plasma level of <1700 copies of HIV RNA per milliliter, even when genital ulcer disease was present (Fig. 226-7). The rate of HIV transmission per coital act was highest during the early stage of HIV infection when plasma HIV RNA levels were high and in advanced disease as the viral set point increased.
FIGURE 226-7 Probability of HIV transmission per coital act among monogamous, heterosexual, HIV-serodiscordant couples in Uganda. (From RH Gray et al: Lancet 357:1149, 2001.)
Antiretroviral therapy dramatically reduces plasma viremia in most HIV-infected individuals (see “Treatment,” below) and is associated with a reduction in risk of transmission. In a large study of serodiscordant couples, earlier treatment of the HIV-infected partner with antiretroviral therapy rather than treatment delayed until the CD4+ T cells count fell below 250 cells per μL was associated with a 96% reduction in HIV transmission to the uninfected partner. This approach has been widely referred to as treatment as prevention or TasP. Several studies also have suggested a beneficial effect of antiretroviral treatment at the community level.
A number of studies including large, randomized, controlled trials clearly have indicated that male circumcision is associated with a lower risk of acquisition of HIV infection for heterosexual men. Studies are conflicting as to whether circumcision protects against HIV acquisition among men who have sex with men, but data suggest that circumcision is protective in those men who have sex with men who are insertive only. The benefit of circumcision may be due to increased susceptibility of uncircumcised men to ulcerative STIs, as well as to other factors such as microtrauma to the foreskin and glans penis. In addition, the highly vascularized inner foreskin tissue contains a high density of Langerhans cells as well as increased numbers of CD4+ T cells, macrophages, and other cellular targets for HIV. Finally, the moist environment under the foreskin may promote the presence or persistence of microbial flora that, via inflammatory changes, may lead to even higher concentrations of target cells for HIV in the foreskin. In addition, randomized trials have demonstrated that male circumcision also reduces hepatitis C virus (HCV) type 2, human papillomavirus virus (HPV), and genital ulcer disease in men as well as HPV, genital ulcer disease, bacterial vaginosis, and Trichomonas vaginalis infections among female partners of circumcised men. Thus, there may be an added benefit of diminution of risk for HIV acquisition to the female sexual partners of circumcised men.
In some studies the use of oral contraceptives was associated with an increase in incidence of HIV infection over and above that which might be expected by not using a condom for birth control. This phenomenon may be due to drug-induced changes in the cervical mucosa, rendering it more vulnerable to penetration by the virus. Adolescent girls might also be more susceptible to infection upon exposure due to the properties of an immature genital tract with increased cervical ectopy or exposed columnar epithelium.
Oral sex is a much less efficient mode of transmission of HIV than is anal intercourse or vaginal intercourse (Table 226-3). A number of studies have reported that the incidence of transmission of infection by oral sex among couples discordant for HIV was extremely low. However, there have been well-documented reports of HIV transmission that likely resulted from fellatio or cunnilingus. Therefore, the assumption that oral sex is completely safe is not warranted.
The association of alcohol consumption and illicit drug use with unsafe sexual behavior, both homosexual and heterosexual, leads to an increased risk of sexual transmission of HIV. Methamphetamine and other so-called club drugs (e.g., ecstasy, ketamine, and gamma hydroxybutyrate), sometimes taken in conjunction with PDE-5 inhibitors such as sildenafil (Viagra), tadalafil (Cialis), or vardenafil (Levitra), have been associated with risky sexual practices and increased risk of HIV infection, particularly among men who have sex with men.
TRANSMISSION THROUGH INJECTION DRUG USE
HIV can be transmitted to injection drug users (IDUs) who are exposed to HIV while sharing injection paraphernalia such as needles, syringes, the water in which drugs are mixed, or the cotton through which drugs are filtered. Parenteral transmission of HIV during injection drug use does not require IV puncture; SC (“skin popping”) or IM (“muscling”) injections can transmit HIV as well, even though these behaviors are sometimes erroneously perceived as low-risk. Among IDUs, the risk of HIV infection increases with the duration of injection drug use; the frequency of needle sharing; the number of partners with whom paraphernalia are shared, particularly in the setting of “shooting galleries” where drugs are sold and large numbers of IDUs may share a limited number of “works”; comorbid psychiatric conditions such as antisocial personality disorder; the use of cocaine in injectable form or smoked as “crack”; and the use of injection drugs in a geographic location with a high prevalence of HIV infection, such as certain inner-city areas in the United States. As noted in Table 226-3, the per-act risk of transmission from injection drug use with a contaminated needle has been estimated to be approximately 0.6%.
TRANSMISSION BY TRANSFUSED BLOOD AND BLOOD PRODUCTS
HIV can be transmitted to individuals who receive HIV-tainted blood transfusions, blood products, or transplanted tissue. The first cases of AIDS among transfusion recipients and individuals with hemophilia or other clotting disorders were reported in 1982. The vast majority of HIV infections acquired via contaminated blood transfusions, blood components, or transplanted tissue in resource-rich countries occurred prior to the spring of 1985, when mandatory testing of donated blood for HIV-1 was initiated. It is estimated that >90% of individuals exposed to HIV-contaminated blood products become infected (Table 226-3). Although blood screening for HIV is becoming more universal even in the developing world, unfortunately, in some resource-poor countries, HIV continues to be transmitted by blood, blood products, and tissues due to inadequate screening. Transfusions of whole blood, packed red blood cells, platelets, leukocytes, and plasma are all capable of transmitting HIV infection. In contrast, hyperimmune gamma globulin, hepatitis B immune globulin, plasma-derived hepatitis B vaccine, and Rho immune globulin have not been associated with transmission of HIV infection. The procedures involved in processing these products either inactivate or remove the virus.
Currently, in the United States and in most developed countries, the following measures have made the risk of transmission of HIV infection by transfused blood or blood products extremely small: the screening of blood donations for antibodies to HIV-1 and HIV-2 and determination of the presence of HIV nucleic acid usually in minipools of several specimens; the careful selection of potential blood donors with health history questionnaires to exclude individuals with risk behavior; and opportunities for self-deferral and the screening out of HIV-negative individuals with serologic testing for infections that have shared risk factors with HIV, such as hepatitis B and C and syphilis. The chance of infection of a hemophiliac via clotting factor concentrates has essentially been eliminated because of the added layer of safety resulting from heat treatment of the concentrates. It is currently estimated that the risk of infection with HIV in the United States via transfused screened blood is approximately 1 in 2 million units. Therefore, since ~16 million donations are collected in the United States each year, despite the best efforts of science, one cannot completely eliminate the risk of transfusion-related transmission of HIV. In this regard, a case of transfusion-related transmission of HIV was reported in the United States in 2010, which was tracked to a blood donation in 2008; this was the first such reported case since 2002. Transmission of HIV (both HIV-1 and HIV-2) by blood or blood products is still an ongoing threat in certain developing countries, particularly in sub-Saharan Africa, where routine screening of blood is not universally practiced. In other countries, there have been reports of sporadic breakdowns in routinely available screening procedures in which contaminated blood was allowed to be transfused, resulting in small clusters of patients becoming infected. For example, in China in the 1990s, a disturbingly large number of persons became infected by selling blood in situations where the collectors reused needles that were contaminated and, in some instances, mixed blood products from a number of individuals, separated the plasma, and reinfused mixed red blood cells back into the individual donors.
OCCUPATIONAL TRANSMISSION OF HIV: HEALTH CARE WORKERS, LABORATORY WORKERS, AND THE HEALTH CARE SETTING
There is a small but definite occupational risk of HIV transmission to health care workers and laboratory personnel and potentially others who work with HIV-containing materials, particularly when sharp objects are used. An estimated 600,000 to 800,000 health care workers are stuck with needles or other sharp medical instruments in the United States each year. The global number of HIV infections among health care workers attributable to sharps injuries has been estimated to be 1000 cases (range, 200–5000) per year. As of 2010, there had been 57 documented cases of occupational HIV transmission to health care workers in the United States and 143 possible transmissions. There have been no confirmed cases reported since 1999.
Exposures that place a health care worker at potential risk of HIV infection are percutaneous injuries (e.g., a needle stick or cut with a sharp object) or contact of mucous membrane or nonintact skin (e.g., exposed skin that is chapped, abraded, or afflicted with dermatitis) with blood, tissue, or other potentially infectious body fluids. Large, multi-institutional studies have indicated that the risk of HIV transmission following skin puncture from a needle or a sharp object that was contaminated with blood from a person with documented HIV infection is ~0.3% and after a mucous membrane exposure it is 0.09% (see “HIV and the Health Care Worker,” below) if the injured and/or exposed person is not treated within 24 h with antiretroviral drugs. The risk of hepatitis B virus (HBV) infection following a similar type of exposure is ~6–30% in nonimmune individuals; if a susceptible worker is exposed to HBV, postexposure prophylaxis with hepatitis B immune globulin and initiation of HBV vaccine is >90% effective in preventing HBV infection. The risk of HCV infection following percutaneous injury is ~1.8% (Chap. 360).
Rare HIV transmission after nonintact skin exposure has been documented, but the average risk for transmission by this route has not been precisely determined; however, it is estimated to be less than the risk for mucous membrane exposure. Transmission of HIV through intact skin has not been documented. Currently in developed countries, virtually all puncture wounds and mucous membrane exposures in health care workers involving blood from a patient with documented HIV infection are treated prophylactically with combination antiretroviral therapy (cART). This practice, referred to as postexposure prophylaxis or PEP, has dramatically reduced the occurrence of puncture-related transmissions of HIV to health care workers.
In addition to blood and visibly bloody body fluids, semen and vaginal secretions also are considered potentially infectious; however, they have not been implicated in occupational transmission from patients to health care workers. The following fluids also are considered potentially infectious: cerebrospinal fluid, synovial fluid, pleural fluid, peritoneal fluid, pericardial fluid, and amniotic fluid. The risk for transmission after exposure to fluids or tissues other than HIV-infected blood also has not been quantified, but it is probably considerably lower than the risk after blood exposures. Feces, nasal secretions, saliva, sputum, sweat, tears, urine, and vomitus are not considered potentially infectious for HIV unless they are visibly bloody. Rare cases of HIV transmission via human bites have been reported, but not in the setting of occupational exposure.
An increased risk for HIV infection following percutaneous exposures to HIV-infected blood is associated with exposures involving a relatively large quantity of blood, as in the case of a device visibly contaminated with the patient’s blood, a procedure that involves a hollow-bore needle placed directly in a vein or artery, or a deep injury. Factors that might be associated with mucocutaneous transmission of HIV include exposure to an unusually large volume of blood and prolonged contact. In addition, the risk increases for exposures to blood from untreated patients with advanced-stage disease or those patients in the acute stage of HIV infection, owing to the higher levels of HIV in the blood under those circumstances. Since the beginning of the HIV epidemic, there have been rare instances where transmission of infection from a health care worker to patients seemed highly probable. Despite these small number of documented cases, the risk of HIV transmission involving health care workers (infected or not) to patients is extremely low in developed countries—in fact, too low to be measured accurately. In this regard, several epidemiologic studies have been performed tracing thousands of patients of HIV-infected dentists, physicians, surgeons, obstetricians, and gynecologists, and no other cases of HIV transmission that could be linked to the health care providers were identified.
Breaches in infection control and the reuse of contaminated syringes, failure to properly sterilize surgical instruments, and/or hemodialysis equipment have also resulted rarely in the transmission of HIV from patient to patient in hospitals, nursing homes, and outpatient settings. Finally, these very rare occurrences of transmission of HIV as well as HBV and HCV to and from health care workers in the workplace underscore the importance of the use of universal precautions when caring for all patients (see below and Chap. 168).
MOTHER-TO-CHILD TRANSMISSION OF HIV
HIV infection can be transmitted from an infected mother to her fetus during pregnancy, during delivery, or by breast-feeding. This remains an important form of transmission of HIV infection in certain developing countries, where the proportion of infected women to infected men is ~1:1. Virologic analyses of aborted fetuses indicate that HIV can be transmitted to the fetus during the first or second trimesters of pregnancy. However, maternal transmission to the fetus occurs most commonly in the perinatal period. Two studies performed in Rwanda and the Democratic Republic of Congo (then called Zaire) indicated that among all mother-to-child transmissions of HIV, the relative proportions were 23–30% before birth, 50–65% during birth, and 12–20% via breast-feeding.
In the absence of prophylactic antiretroviral therapy to the mother during pregnancy, labor, and delivery, and to the fetus following birth, the probability of transmission of HIV from mother to infant/fetus ranges from 15% to 25% in industrialized countries and from 25% to 35% in developing countries. These differences may relate to the adequacy of prenatal care as well as to the stage of HIV disease and the general health of the mother during pregnancy. Higher rates of transmission have been reported to be associated with many factors—the best documented of which is the presence of high maternal levels of plasma viremia, with the risk increasing linearly with the level of maternal plasma viremia. It is very unlikely that mother-to-child transmission will occur if the mother’s level of plasma viremia is <1000 copies of HIV RNA/mL of blood and extremely unlikely if the level is undetectable (i.e., <50 copies/mL). However, there may not be a lower “threshold” below which transmission never occurs, since certain studies have reported rare transmission by women with viral RNA levels <50 copies/mL. Increased mother-to-child transmission is also correlated with closer human leukocyte antigen (HLA) match between mother and child. A prolonged interval between membrane rupture and delivery is another well-documented risk factor for transmission. Other conditions that are potential risk factors, but that have not been consistently demonstrated, are the presence of chorioamnionitis at delivery; STIs during pregnancy; illicit drug use during pregnancy; cigarette smoking; preterm delivery; and obstetric procedures such as amniocentesis, amnioscopy, fetal scalp electrodes, and episiotomy. In a seminal study conducted in the United States and France in the 1990s, zidovudine treatment of HIV-infected pregnant women from the beginning of the second trimester through delivery and of the infant for 6 weeks following birth dramatically decreased the rate of intrapartum and perinatal transmission of HIV infection from 22.6% in the untreated group to <5%. Today, the rate of mother-to-child transmission has fallen to 1% or less in pregnant women who are receiving combination antiretroviral therapy (cART) for their HIV infection. Such treatment, combined with cesarean section delivery, has rendered mother-to-child transmission of HIV an unusual event in the United States and other developed nations. In this regard, both the United States Public Health Service and the World Health Organization guidelines recommend that all pregnant HIV-infected women should receive cART for the health of the mother and to prevent perinatal transmission regardless of plasma HIV RNA copy number or CD4+ T cell counts.
Breast-feeding is an important modality of transmission of HIV infection in developing countries, particularly where mothers continue to breast-feed for prolonged periods. The risk factors for mother-to-child transmission of HIV via breast-feeding are not fully understood; factors that increase the likelihood of transmission include detectable levels of HIV in breast milk, the presence of mastitis, low maternal CD4+ T cell counts, and maternal vitamin A deficiency. The risk of HIV infection via breast-feeding is highest in the early months of breast-feeding. In addition, exclusive breast-feeding has been reported to carry a lower risk of HIV transmission than mixed feeding. In developed countries, breast feeding of babies by an HIV-infected mother is contraindicated since alternative forms of adequate nutrition, i.e., formulas, are readily available. In developing countries, where breast-feeding may be essential for the overall health of the infant, the continuation of cART in the infected mother during the period of breastfeeding markedly diminishes the risk of transmission of HIV to the infant. In fact, once cART has been initiated in a pregnant woman, many experts recommend that therapy be continued for life.
TRANSMISSION OF HIV BY OTHER BODY FLUIDS
Although HIV can be isolated typically in low titers from saliva of a small proportion of infected individuals, there is no convincing evidence that saliva can transmit HIV infection, either through kissing or through other exposures, such as occupationally to health care workers. Saliva contains endogenous antiviral factors; among these factors, HIV-specific immunoglobulins of IgA, IgG, and IgM isotypes are detected readily in salivary secretions of infected individuals. It has been suggested that large glycoproteins such as mucins and thrombospondin 1 sequester HIV into aggregates for clearance by the host. In addition, a number of soluble salivary factors inhibit HIV to various degrees in vitro, probably by targeting host cell receptors rather than the virus itself. Perhaps the best studied of these, secretory leukocyte protease inhibitor (SLPI), blocks HIV infection in several cell culture systems, and it is found in saliva at levels that approximate those required for inhibition of HIV in vitro. In this regard, higher salivary levels of SLPI in breast-fed infants were associated with a decreased risk of HIV transmission through breast milk. It has also been suggested that submandibular saliva reduces HIV infectivity by stripping gp120 from the surface of virions, and that saliva-mediated disruption and lysis of HIV-infected cells occurs because of the hypotonicity of oral secretions. There have been outlier cases of suspected transmission by saliva, but these have probably been blood-to-blood transmissions. Transmission of HIV by a human bite can occur but is a rare event. Although virus can be identified, if not isolated, from virtually any body fluid, there is no evidence that HIV transmission can occur as a result of exposure to tears, sweat, or urine. However, there have been isolated cases of transmission of HIV infection by body fluids that may or may not have been contaminated with blood. Most of these situations occurred in the setting of a close relative providing intensive nursing care for an HIV-infected person without observing universal precautions, underscoring the importance of adhering to such precautions in the handling of body fluids and wastes from HIV-infected individuals.
EPIDEMIOLOGY
HIV INFECTION AND AIDS WORLDWIDE
![]() HIV infection/AIDS is a global pandemic, with cases reported from virtually every country. At the end of 2013, an estimated 35.0 million individuals were living with HIV infection, according to the Joint United Nations Programme on HIV/AIDS (UNAIDS). An estimated 95% of people living with HIV/AIDS reside in low- and middle-income countries; ~50% are female, and 3.2 million are children <15 years. The distribution of these cases is illustrated in Fig. 226-8. The estimated number of people living with HIV—i.e., the global prevalence—has increased more than fourfold since 1990, reflecting the combined effects of continued high rates of new HIV infections and the life-prolonging impact of antiretroviral therapy (Fig. 226-9). In 2013, the global prevalence rate among persons age 15–49 years was 0.8%, with rates varying widely by country and region as illustrated in Fig. 226-10.
HIV infection/AIDS is a global pandemic, with cases reported from virtually every country. At the end of 2013, an estimated 35.0 million individuals were living with HIV infection, according to the Joint United Nations Programme on HIV/AIDS (UNAIDS). An estimated 95% of people living with HIV/AIDS reside in low- and middle-income countries; ~50% are female, and 3.2 million are children <15 years. The distribution of these cases is illustrated in Fig. 226-8. The estimated number of people living with HIV—i.e., the global prevalence—has increased more than fourfold since 1990, reflecting the combined effects of continued high rates of new HIV infections and the life-prolonging impact of antiretroviral therapy (Fig. 226-9). In 2013, the global prevalence rate among persons age 15–49 years was 0.8%, with rates varying widely by country and region as illustrated in Fig. 226-10.
FIGURE 226-8 Estimated number of adults and children living with HIV infection as of December, 2013. Total: 35.0 (33.2 million–37.2 million). (From Joint United Nations Programme on HIV/AIDS [UNAIDS].)
In 2013, an estimated 2.1 million new cases of HIV infection occurred worldwide, including 240,000 among children <15 years; about 40% of new infections were among persons under age 25. Between 2001 and 2013, the estimated number of new HIV infections globally fell by 38% (Fig. 226-9). Recent reductions in global HIV incidence likely reflect progress with HIV prevention efforts and the increased provision to HIV-infected people of antiretroviral therapy, which makes them much less likely to transmit the virus to sexual partners. In 2013, global AIDS deaths totaled 1.5 million (including 190,000 children <15 years), a 35% decrease since 2005 that coincides with a rapid expansion of access to antiretroviral therapy (Fig. 226-9). Since the beginning of the pandemic, an estimated 39 million people have died of an AIDS-related illness.
FIGURE 226-9 Global estimates of HIV incidence and AIDS deaths (left) and, HIV prevalence (right), 1990–2013. (From UNAIDS.)
The HIV epidemic has occurred in “waves” in different regions of the world, each wave having somewhat different characteristics depending on the demographics of the country and region in question and the timing of the introduction of HIV into the population. Although the AIDS epidemic was first recognized in the United States and shortly thereafter in Western Europe, it very likely began in sub-Saharan Africa (see above), which has been particularly devastated by the epidemic. More than 70% of all people with HIV infection (~25 million), and nearly 90% of all HIV-infected children live in that region, even though sub-Saharan Africa is home to just 12% of the world’s population (Fig. 226-8). Within the region, southern Africa is worst-affected. In nine southern African countries, seroprevalence data indicate that >10% of the adult population age 15–49 is HIV-infected (Fig. 226-10). In addition, among high-risk individuals (e.g., commercial sex workers, patients attending STI clinics) who live in urban areas of sub-Saharan Africa, seroprevalence is now >50% in some places. Recent data offer promising signs of declining HIV incidence and prevalence in many countries in the region, although frequently at levels that remain high. Heterosexual exposure is
[/not-level-membership-for-internal-medicine-category]
