11 Homeostasis and the kidney
The kidney and urinary tract
Examination
Palpate the kidneys and feel for tenderness in the loins. Inspect the back looking for a naevus that could overlie a spinal cord anomaly. Check the lower-limb reflexes. The external genitalia should be examined, but tact is needed and you may decide the examination should be deferred unless the history specifically suggests a genital problem. The examination is not complete without testing the urine with a multiple reagent stick (see Table 11.1).
| Test | Significance |
|---|---|
| pH | Normal urine pH ranges from 5 to 7. Inappropriately alkaline urine is found in some infections, and in renal tubular acidosis |
| Protein | Proteinuria is seen in a range of renal diseases, and is the hallmark of nephrotic syndrome. The presence of proteinuria may be indicated by very frothy urine |
| Blood | See Box 11.1 for causes of haematuria |
| Nitrites and leucocytes | Nitrites are products of bacterial decomposition of urea and suggest urinary tract infection. Their presence may be suggested by the smell of ammonia. The findings of leucocytes in urine also suggests infection or inflammation |
| Glucose and ketones | Glycosuria indicates blood glucose levels above the renal threshold for glucose reabsorption. The presence of glycosuria and ketonuria together strongly suggests type 1 diabetes |
| Bilirubin and urobilinogen | Urobilinogen is responsible for the normal yellow colour of urine, but if detectable on a reagent strip implies enhanced red cell destruction, as in haemolytic anaemia, or impaired clearance in bile, most commonly due to hepatitis. The finding of bilirubin in urine suggests hepatocellular dysfunction or obstructive jaundice. Urobilinogen may be absent from urine in obstructive jaundice, although the urine is dark due to the presence of conjugated bilirubin |
Genitourinary defects
Undescended testes
If both testes are undescended, intersex needs to be considered (see Chapter 12, p. 151).
Hypospadias
In hypospadias, the urethral meatus opens onto the ventral shaft of the penis or scrotum, or exceptionally, the perineum. In 60% the meatus is distal (glandular or coronal hypospadias) and in 15% proximal (perineoscrotal or perineal hypospadias), with the remainder affecting the penile shaft (subcoronal or mid-penile hypospadias). Hypospadias affects the urinary stream and may be associated with bowing of the erect penis, ‘chordee’, which will affect sexual function in later life. Proximal hypospadias may be associated with urinary tract abnormalities, and a micturating cystogram should be performed. Hypospadias with an undescended testis may be seen in intersex states (see Chapter 12, p. 151) and genetic sex determination should be performed. Surgical correction in infancy is straightforward. The parents should be advised not to have their son circumcised as the foreskin may be used as part of the surgical repair.
Congenital anomalies of the urinary tract
Congenital anomalies of the urinary tract may affect the kidney, urinary collecting system, bladder or urine outflow tract (as in Case 11.1).
Abnormalities of the ureter include obstruction at the level of the renal pelvis (pelvi-ureteric junction obstruction) or bladder, normally due to a ureterocele – a cystic dilatation of the terminal ureter with a pinhole ureteral orifice. These result in proximal dilatation and predispose to infection and renal injury. Treatment is surgical. Occasionally, the ureter may be partially or completely duplicated. Typically, the ureter draining the upper pole inserts ectopically and is commonly obstructed, whereas the lower pole ureter inserts non-obliquely into the bladder, predisposing to reflux. In girls, an ectopic ureter commonly empties into the vagina leading to constant dribbling incontinence. In boys, the insertion is commonly into the prostatic utricle, seminal vesicle or vas deferens and thus leads to obstruction (see Figure 11.1).
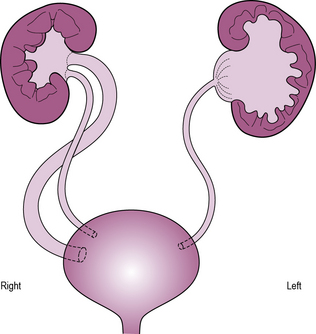
See Table 11.2 for congenital anomalies of the kidney and urinary tract.
Table 11.2 Congenital anomalies of the kidney and urinary tract
| Anomaly | Clinical consequence | |
|---|---|---|
| Kidney | Agenesis ormulticystic dysplasia Infantile polycystickidney diseaseRenal ectopia andhorseshoe kidney | Bilateral: lethal pulmonary hypoplasia (Potter’s syndrome)Unilateral: associated congenital abnormalitiesChronic kidney disease Association with othercongenital anomalies |
| Ureter | Pelvi-ureteric junctionobstructionUreterocele Ectopic ureter Vesico-ureteric reflux Duplex ureter | Hydronephrosis Hydronephrosis and hydro-ureterHydronephrosis or dribbling incontinenceRisk of infection, renal scarringOften associated with a combination of reflux and obstruction |
| Bladder | Bladder or cloacalexstrophy Neuropathic bladder | Major reconstructive surgery requiredIncontinence, chronic kidney diseaseIncontinence, chronic kidney disease insufficiency |
| Urethra | Posterior urethralvalves (boys) | Chronic kidney disease, vesico-ureteric reflux |
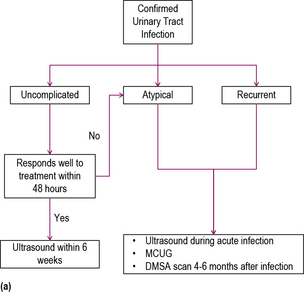
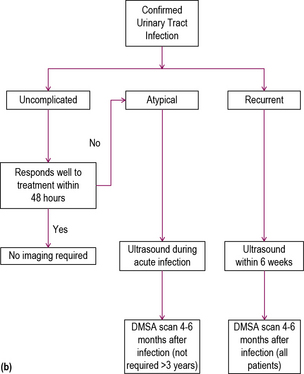
Urinary tract infection
The link between UTI and urinary symptoms is not always clear-cut. Even if the diagnosis seems clinically apparent (as in Case 11.2) the culture may be negative. In some cases the symptoms may be non-specific (irritability or failure-to-thrive) or severe enough, as in Case 11.3, to mimic septicaemia. Hyponatraemia due to renal salt loss may complicate urinary tract infection.
Investigation
Vesico-ureteric reflux describes the backflow of urine from the bladder up the ureter due to a failure of the functional valve where the ureter penetrates the bladder wall. Reflux can be severe enough to distend the renal pelvis and produce backflow into the renal collecting ducts. It is a worrying finding in infants as it may be associated with renal scarring (see Figure 11.2).
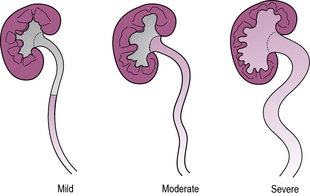
An MCUG is used in young children to determine the presence of reflux, whereas in older children MAG3 scanning is used. MCUG is often very distressing, and should not be used after infancy, with rare exceptions. It has the advantage of demonstrating abnormalities of the bladder or ureters such as the presence of bladder diverticula, ectopic ureters and duplex systems, not readily apparent by other imaging modalities. Antibiotic cover is required following MCUG to prevent ascending infection. MAG3 scanning is useful to differentiate reflux from obstruction and to determine the anatomical site of obstruction (see Figure 11.3).
Treatment
Treatment is directed to reducing the risk of renal injury. This requires prompt recognition and treatment of UTIs, with an appropriate antibiotic. High rates of resistance to trimethoprim and amoxicillin make these unsuitable choices, unless antibiotic susceptibility has been confirmed. Good first-line choices are co-amoxiclav or a cephalosporin such as cefalexin. Prophylaxis of UTI is no longer recommended. Urinary tract obstruction requires surgery. If vesico-ureteric reflux is severe, and renal function declines, then a ‘Sting’ procedure (as in Case 11.3) is usually performed, in which an inert substance, such as Deflux™), is injected just below the ureteral orifice. This usually alleviates reflux. If the Sting procedure fails, ureteric reimplantation may be effective. New scarring after the age of 2 years is uncommon and extremely rare after the age of 5 years.
Haematuria
Often the clinical context will help in discovering the cause of haematuria. Otherwise the haematuria may be an isolated or chance finding (see Table 11.3).
| Diagnosis | Other clinical features |
|---|---|
| Wilms’ tumour | Palpable abdominal mass |
| Acute glomerulonephritis | Oliguria, proteinuria, raised blood urea |
| Henoch–Schönlein purpura | Vasculitic non-blanching rash on legs, arthropathy, abdominal pain |
| Haemolytic-uraemic syndrome | Preceding gastroenteritis, haemolytic anaemia, renal failure |
| Pyelonephritis | Fever, rigors, back pain |
| Renal or ureteric calculi | Renal colic |
| Sickle-cell disease | Anaemia, painful crises |
| Subacute bacterial endocarditis | Congenital heart disease, fever, changing heart murmurs |
| Haemorrhagic cystitis | Heavy haematuria with dysuria |
| Urinary tract infection | Fever, dysuria, irritability |
Incidental haematuria
Polycystic kidney disease (PKD) has two forms – autosomal recessive or infantile, and autosomal dominant (as in Case 11.4). The infantile form may be detected by antenatal ultrasound. The kidneys are enlarged with innumerable cysts, and some degree of renal impairment is usually present at birth. Stage IV chronic kidney disease affects over 50% by the age of 10 years. Autosomal dominant PKD usually presents in adult life but presentation with frank or microscopic haematuria, UTI or hypertension may occur in affected children. Wilms’ tumour may present with haematuria (as in Case 7.14, Chapter 7), usually with a palpable flank mass.
Glomerular disorders
Nephrotic syndrome
Nephrotic syndrome describes the triad of oedema, proteinuria (>40 mg/m2/day) and hypoalbuminaemia (as in Case 11.5). Glomerular protein leak leads to loss of albumin and other plasma proteins. The low plasma oncotic pressure reduces capillary reabsorption of tissue fluid leading to oedema. Reduced plasma volume stimulates antidiuretic hormone (ADH) secretion and the renin–angiotensin system, producing sodium and water retention, compounding the pre-existing oedema.
Nephritis
Post-streptococcal glomerulonephritis is an immune-complex-mediated glomerulonephritis, which follows cutaneous or pharyngeal streptococcal infection (as in Case 11.6). It typically affects children aged 5–12 years. It is very common worldwide, but in developed countries it is less frequent and viral aetiology is as common as streptococcal disease. The severity of renal involvement determines prognosis, with presentations ranging from asymptomatic haematuria to acute renal failure. Greater degrees of renal impairment are associated with hypertension, which, if severe, may lead to hypertensive encephalopathy and heart failure.
Henoch–Schönlein purpura (see Chapter 6, p. 55) is associated with glomerulonephritis in 50% of cases. Children with this condition should have regular urine testing, and renal function and blood pressure checks until resolution. A minority of children develop aggressive glomerulonephritis with a mixed picture of nephritis and nephrotic syndrome, and usually progress to stage IV chronic kidney disease.
Renal tubular disorders
The boy in Case 11.7 has generalized proximal tubular dysfunction – known as renal Fanconi syndrome. Measurement of leucocyte cystine content confirmed cystinosis as the cause. In cystinosis, cystine accumulates in lysosomes and causes progressive renal tubular dysfunction, culminating in renal failure. Cystine accumulation in the cornea causes photophobia. Hypothyroidism, central nervous system (CNS) abnormalities and myopathy occur in later life.
Renal tubular dysfunction may be specific, when just one element of tubular function is impaired, or part of a generalized disorder – renal Fanconi syndrome – of which cystinosis is the commonest cause (see Box 11.2).
Water homeostasis
If homeostasis is overwhelmed this may result in hypernatraemic or hyponatraemic dehydration (see Chapter 13, p. 166).
Primary failure of the control systems is less common but includes inappropriate ADH secretion, diabetes insipidus and aldosterone deficiency (see Chapter 12).
Kidney failure
Introduction
Chronic kidney disease is also associated with:
• Anaemia from chronic disease and erythropoietin deficiency
• Growth failure from several causes
• Renal bone disease from hyperparathyroidism secondary to chronic hypocalcaemia and hyperphosphataemia
• Uraemic toxins leading to impaired cellular immunity
• Bleeding and easy bruising secondary to impaired platelet function.
See Table 11.4 for the causes of acute and chronic kidney disease.
| Acute | Chronic | |
|---|---|---|
| Prerenal | Shock, e.g. haemorrhage dehydration, burns, sepsis | |
| Renal | Glomerulonephritis Haemolytic-uraemic syndrome Renal vein thrombosis Liver failure (hepatorenal syndrome) |
Congenital cystic or dysplastic diseases, e.g. medullary cystic kidney Chronic reflux nephropathy Glomerulonephritis Metabolic disease, e.g. cystinosis |
| Post-renal | Acute urinary tract obstruction | Posterior urethral valves Bilateral pelvi-ureteric or vesico-ureteric obstruction Renal calculi Neurogenic bladder, e.g. spina bifida |
Management of renal failure
See Table 11.5 for the treatment of renal failure.
| Problem | Treatment |
|---|---|
| Hyperkalaemia | Calcium polystyrene sulphonate enemas |
| Intravenous or inhaled salbutamol | |
| Intravenous calcium gluconate | |
| Insulin and glucose infusion | |
| Dialysis | |
| Hypertension and/or oedema | Loop diuretics, e.g. furosemide |
| ACE inhibitor Calcium-channel blocker Beta-blocker |
|
| Acidosis | Sodium citrate or bicarbonate |
| Hypocalcaemia | Phosphate binders, e.g. calcium carbonate |
| Parenteral calcium for severe hypocalcaemia | |
| 1α vitamin D, calcium supplements | |
| Hyponatraemia | Sodium supplements |
| Anaemia | Blood transfusion |
| B12 and folate supplementation | |
| Erythropoietin | |
| Growth failure/failure-to-thrive | Optimal nutrition |
| Treatment of anaemia and acidosis | |
| Growth hormone | |
| Renal transplantation | |
| Uraemic encephalopathy | Treatment of hypertension |
| Correction of metabolic abnormalities | |
| Renal transplantation |
Gastroenteritis and renal failure
Haemolytic-uraemic syndrome
Haemolytic-uraemic syndrome (HUS) is the commonest cause of acute renal failure in children. Eighty per cent of cases result from infection with Shiga-toxin-producing Escherichia coli O157:H7 (as in Case 11.8). The infection results from contact with infected domestic animals or ingestion of under-cooked or contaminated meat. The toxin induces widespread endothelial injury with resultant haemolytic anaemia with platelet consumption and renal failure. One in ten children with HUS die, and a further 10% develop stage IV chronic kidney disease.
Regulating calcium and bone metabolism
Hypocalcaemia
Acute hypocalcaemia leads to neuromuscular excitability, which may manifest with seizures (as in Case 11.9), tetany and laryngeal spasm. Chronic hypocalcaemia leads to psychomotor retardation, sometimes with raised intracranial pressure, cataracts and photophobia. Low calcium may occur due to deficiency (hypoparathyroidism) or end-organ resistance to PTH (pseudohypoparathyroidism), or due to vitamin D deficiency. Low magnesium commonly accompanies hypocalcaemia.
In the sick neonate, transient hypoparathyroidism may lead to severe hypocalcaemia, as in Case 11.9. This may also be observed in infants born to mothers with hyperparathyroidism, due to suppression of fetal PTH secretion by maternal hypercalcaemia. Hypoplasia of the parathyroid glands occurs to a variable extent in Di George syndrome (see Chapter 18, p. 272), although in most infants hypocalcaemia is transient. In pseudohypoparathyroidism the PTH receptor is defective. It is associated with a characteristic appearance and mental retardation in some cases. Hypoparathyroidism also occurs as an autoimmune disease, classically in association with chronic mucocutaneous candidiasis and adrenal insufficiency, and other autoimmune disorders, and is inherited in an autosomal dominant manner (polyglandular endocrinopathy).
Hypercalcaemia
Chronic hypercalcaemia, if severe, leads to irritability, muscular weakness, anorexia, constipation, polyuria and nephrocalcinosis. Hypercalcaemia in childhood results from excess parathyroid hormone (from parathyroid adenoma or hyperplasia) or vitamin D intoxication, and is also seen in Williams’ syndrome (see Chapter 18, p. 278), although the cause is unknown. Mild hypercalcaemia may also occur in adrenal insufficiency and thyrotoxicosis.