© Springer Japan 2016
Kazunari Kaneko (ed.)Molecular Mechanisms in the Pathogenesis of Idiopathic Nephrotic Syndrome10.1007/978-4-431-55270-3_1
1. History of Research on Pathogenesis of Idiopathic Nephrotic Syndrome
(1)
Department of Pediatrics, Kansai Medical University, 2-5-1 Shin-machi, Hirakata Osaka, 573-1010, Japan
Abstract
Although nephrotic syndrome (NS) is a common kidney disease, its pathogenesis remains unclear. It is classified into idiopathic and secondary, while congenital is a third category for children. The adjective “idiopathic” is used in medicine to describe a disease or condition that has no known cause. The pathogenesis of idiopathic nephrotic syndrome (INS) remains elusive. INS is grouped into the three histological variants: minimal change NS (MCNS), focal segmental glomerulosclerosis (FSGS), and membranous nephropathy (MN). MCNS, FSGS, and MN respectively account for approximately 75–80 %, 20 %, and <3 % of INS in children, whereas each accounts for one third of INS in adults. In the past decade, advances in molecular biology have both improved our understanding of the pathogenesis of INS and created confusion. The candidate active molecules in INS, other than cytokines, include: reactive oxygen species, nuclear factor-kappa B, hemopexin, CD80 (also known as B7.1), and angiopoietin-like 4; mammalian target of rapamycin complex 1 in MCNS; cardiotrophin-like cytokine-1 and soluble urokinase-type plasminogen activator receptor in FSGS; and M-type phospholipase A2 receptor and cationic bovine serum albumin in IMN. In this review, we briefly discuss the historical background of the research on pathogenesis of INS.
Keywords
Idiopathic nephrotic syndromeMinimal change nephrotic syndromeFocal segmental glomerulosclerosisMembranous nephropathyPodocyteCytokine
1.1 Introduction
Nephrotic syndrome (NS) is characterized by heavy proteinuria (daily urinary protein ≥3.0–3.5 g in adults [1] or urine protein–creatinine ratio ≥2000 mg/g or ≥300 mg/dL or 3+ protein on urine dipstick in children [2]). Leakage of massive amounts of serum proteins into the urine leads to a hypoalbuminemia, edema hypercoagulable state, higher rate of infectious disease, and dysregulation of fluid balance. Although glucocorticoid therapy achieves complete remission in 80–90 % of patients with childhood NS, it induces remission in far fewer adults with NS. Adult NS is classified into idiopathic and secondary, while congenital is a third category in childhood NS. Secondary NS is defined as NS associated with a systemic disease, such as diabetic nephropathy, lupus nephritis in adults, or Henoch–Schnlein purpura nephritis in children.
Idiopathic NS (INS) is commonly grouped into the three histological variants: minimal change NS (MCNS), focal segmental glomerulosclerosis (FSGS), and membranous nephropathy (MN) [3, 4]. MCNS, FSGS, and MN respectively account for approximately 75–80 %, 20 %, and <3 % [5] of INS in children, whereas each accounts for one third of INS in adults. Not all cases of MCNS, FSGS, or MN are idiopathic. MCNS can occur in association with lymphoid tumors or immunomodulatory drugs. FSGS is the most common histological variant in patients with HIV nephropathy. Renal lesions resembling idiopathic FSGS may also be present in proteinuric patients with other primary renal disorders, such as chronic glomerulonephritis, reflux nephropathy, and oligomeganephronia [3]. Similarly, idiopathic MN (IMN) is usually a diagnosis of exclusion of secondary causes. Approximately 25 % of adults with MN have underlying systemic diseases, such as autoimmune disease, hepatitis B virus infection, or malignancy [5]. Certain drugs (gold, penicillamine, and nonsteroidal anti-inflammatory drugs) are also known to induce secondary MN.
The adjective “idiopathic” is used in medicine to describe diseases or conditions that have no known cause. The pathogenesis of INS, that is, MCNS, primary FSGS, and IMN, remains elusive despite many years of research. In the past decade, however, advances in molecular biology, in conjunction with genomic studies on immune cells and renal visceral glomerular epithelial cells (podocytes), have both improved our understanding of the pathogenesis of INS and created confusion.
In this review, we discuss the historical background of the research on the pathogenesis of INS.
1.2 Old Paradigm for the Pathogenesis of INS
Research on the pathogenesis of INS started with an epoch-making paper entitled “Pathogenesis of lipoid nephrosis: a disorder of T-cell function” by Shalhoub, which appeared in the Lancet in 1974 [6], when no clear distinction was made between MCNS and primary FSGS. In those days, they were considered together as lipoid nephrosis, which is synonymous with INS. However, there is still debate as to whether MCNS and FSGS represent opposite ends of one pathophysiological process or distinct disease entities [7, 8]. Shalhoub proposed that INS was a disorder of T-cell function, resulting in increased plasma levels of lymphocyte-derived permeability factor (Shalhoub’s hypothesis) [6]. This hypothesis was based on the absence of immune complexes in the glomeruli, rapid response to steroid therapy, association of INS with Hodgkin’s disease, and the observation that measles infection often induces remission of INS. Therefore, the massive proteinuria and hypoalbuminemia that characterize INS were thought to result from increased permeability of the glomerular capillary wall due to T-cell activation triggered by stimuli such as viral infection or allergens.
The most compelling evidence also came from experience with renal allografts: NS disappeared when MCNS kidneys were transplanted into patients without NS [9]. The following clinical findings support the concept that vascular permeability factors produced from activated T cells play an important role in MCNS: in patients with MCNS, there is a risk of recurrence of the disease when transplanted [10]; placental transfer of proinflammatory cytokines from a mother to a newborn results in neonatal nephrotic syndrome [11]; and the potential of apheresis monotherapy to induce and maintain complete remission of MCNS suggests that circulating factors have an important role in the pathogenesis of MCNS [12].
Based on Shalhoub’s hypothesis, researchers have tried to identify the circulating factors released from T cells that increase the glomerular permeability to serum proteins, and some have confirmed that the capillary permeability factor is detectable in patients with INS [13, 14]. Among various putative factors increasing the glomerular permeability to serum proteins, cytokines are considered to be the most likely pathogenic factors [4, 15]. Cytokines are small proteins (molecular weight 8–80 kDa) that function as soluble mediators in an autocrine or paracrine manner, which are produced by both immune and nonimmune cells. Relapsed patients with INS (mostly MCNS) were found to have increased levels of various cytokines in the serum or urine including interleukin (IL)-2 [16], soluble IL-2 receptor [16–19], interferon (IFN)-γ [16, 20], IL-4 [20, 21], IL-12 [22], IL-18 [23], tumor necrosis factor (TNF)-α [24], and vascular endothelial growth factor (VEGF) [25]. Isolation of peripheral blood mononuclear cells from patients with INS relapse and measurement of the in vitro mitogen-stimulated production of cytokines in the cultured cell supernatants demonstrate increased production of various cytokines including IL-1 [26], IL-2 [20, 27], IL-4 [20, 21], IL-10 [27], IL-12 [28], IL-18 [23], and TNF-α [29]. Yap et al. also reported increased expression of IL-13 mRNA in patients with MCNS relapse [30]. To date, however, the factor itself has not been identified, among possible capillary permeability factors including cytokines, which cause protein leakage from serum to urine, and the various alterations in cytokine production are not in agreement at all. This disagreement may result from the different immunogenetic characteristics of the patients or the heterogeneity of the stimulated cells in nonphysiological environments. The complex interactions among cytokines also make it difficult to determine which cytokine is increased first. Furthermore, lack of documentation of biopsy findings, inclusion of steroid-treated patients, and differences in methodology make it difficult to determine the factors associated with glomerular permeability. The aberrant populations in T cells in INS have also been vigorously studied with conflicting results, e.g., predominance of T-helper (Th) type 2 cells over Th1 cells [31, 32] based on the high comorbidity of atopy and allergy [33, 34], which are caused by Th2 immunological responses. However, others do not support this hypothesis [15, 35]. Recent clinical reports that there is remission after depletion of B cells using monoclonal antibodies or the anti-CD20 drug rituximab also contradict Shalhoub’s hypothesis, which focuses on the T-cell disorder in INS [36, 37].
1.3 New Paradigm for the Pathogenesis of INS
A new paradigm for the pathogenesis of INS has emerged since the discovery by Tryggvason et al. in 1998 that mutations in the gene NPHS1, which encodes the podocyte-expressed immunoglobulin superfamily protein nephrin, cause congenital NS in humans [38]. This landmark study led to a substantial increase in our understanding of glomerular biology and physiology and that podocytes (visceral glomerular epithelial cells) have attracted particular attention as a key player in the pathogenesis of INS [39, 40]. Podocytes are terminally differentiated cells that line the outer aspect of the glomerular basement membrane. Podocytes form the final barrier to urinary protein loss by the formation and maintenance of podocyte foot processes (FPs) and the interposed slit diaphragms (SDs) [41]. The SDs are the main selectively permeable barrier in the kidney [42]. Podocyte FPs contain a contractile and dynamic apparatus consisting of actin, myosin II, α-actinin-4, talin, vinculin, and synaptopodin [43, 44]. The FPs are anchored to the glomerular basement membrane via α3β1-integrin [45] and dystroglycans [46]. Our knowledge of SD structure is based on genetic studies of familial NS, which led to the identification of SD proteins such as podocin, nephrin, α-actinin-4, and transient receptor potential channel C6. The genes for these proteins may be mutated in inherited NS [47].
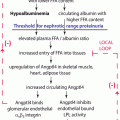
Based on this background, several hypotheses have been proposed in the past decade that focus on the role of the podocyte and the related molecules in the mechanism underlying the proteinuria in INS. The candidate molecules as active factors in INS, other than cytokines, include reactive oxygen species [48], nuclear factor-κB [49], hemopexin [50], CD80 (also known as B7.1) [51, 52], and angiopoietin-like 4 [53, 54]; mammalian target of rapamycin complex 1 in MCNS [55]; cardiotrophin-like cytokine-1 [56] and soluble urokinase-type plasminogen activator receptor [57, 58] in FSGS; and M-type phospholipase A2 receptor [59] and cationic bovine serum albumin in IMN [60]. Furthermore, the recent findings suggest that the molecules expressed by podocytes are therapeutic targets for the immunosuppressive agents used for the treatment of INS such as glucocorticoids, cyclosporin A, and rituximab, although these drugs have been considered to act by correcting lymphocyte dysfunction, especially of T cells [61–64].
1.4 Conclusions
The rapid pace of scientific progress occasionally sinks to a state of chaos. Currently, the study of the pathogenesis of INS appears to correspond to such a status. As mentioned above, INS was historically thought to be caused by T-cell dysfunction. However, recent evidence suggests that not only T cells but also other immune cells including those associated with innate immunity and podocytes are involved in the pathogenesis of this condition and that INS develops by interactions between humoral factors and podocytes in most cases. To organize the distinct pathogenesis based on various molecules that are newly identified as active factors, the following questions should be clarified: (1) When and how was the molecule identified as the pathogenetic factor in INS? (2) Could the molecule be a novel biomarker to distinguish the histological differences among INS, i.e., MCNS, FSGS, or IMN? (3) How does the molecule play a role in the pathogenesis of INS? (4) Is it expected that the molecule can be the target for new drugs in INS in future? (5) Is there any possible association of the molecule with other putative pathogenetic molecules reported by other researchers? If these questions are answered clearly, the complex pathogenesis of INS would be unraveled.
Declaration of Competing Interests
None to declare.
References
1.
Kodner C. Nephrotic syndrome in adults: diagnosis and management. Am Fam Physician. 2009;80:1129–34.PubMed
2.
KDIGO Clinical Practice Guideline for Glomerulonephritis. Chapter 3: Steroid-sensitive nephrotic syndrome in children. Kidney Int Suppl. 2012;2:163–71. doi:10.1038/kisup.2012.16.
3.
Eddy AA, Symons JM. Nephrotic syndrome in childhood. Lancet. 2003;362:629–39. doi:10.1016/s0140-6736(03)14184-0.CrossRefPubMed
4.
Zhang S, Audard V, Fan Q, Pawlak A, Lang P, Sahali D. Immunopathogenesis of idiopathic nephrotic syndrome. Contrib Nephrol. 2011;169:94–106. doi:10.1159/000313947.CrossRefPubMed
5.
KDIGO Clinical Practice Guideline for Glomerulonephritis. Chapter 7: Idiopathic membranous nephropathy. Kidney Int Suppl. 2012;2:186–97. doi:10.1038/kisup.2012.20.
6.
7.
Ahmad H, Tejani A. Predictive value of repeat renal biopsies in children with nephrotic syndrome. Nephron. 2000;84:342–6. doi:10.1159/000045609.CrossRefPubMed
8.
Neuhaus TJ, Fay J, Dillon MJ, Trompeter RS, Barratt TM. Alternative treatment to corticosteroids in steroid sensitive idiopathic nephrotic syndrome. Arch Dis Child. 1994;71:522–6.PubMedCentralCrossRefPubMed
9.
10.
11.
Assadi F. Neonatal nephrotic syndrome associated with placental transmission of proinflammatory cytokines. Pediatr Nephrol. 2011;26:469–71. doi:10.1007/s00467-010-1700-1.CrossRefPubMed
12.
13.
14.
Savin VJ, Sharma R, Sharma M, McCarthy ET, Swan SK, Ellis E, et al. Circulating factor associated with increased glomerular permeability to albumin in recurrent focal segmental glomerulosclerosis. N Engl J Med. 1996;334:878–83. doi:10.1056/nejm199604043341402.CrossRefPubMed
15.
Araya CE, Wasserfall CH, Brusko TM, Mu W, Segal MS, Johnson RJ, et al. A case of unfulfilled expectations. Cytokines in idiopathic minimal lesion nephrotic syndrome. Pediatr Nephrol. 2006;21:603–10. doi:10.1007/s00467-006-0026-5.CrossRefPubMed
16.
Daniel V, Trautmann Y, Konrad M, Nayir A, Scharer K. T-lymphocyte populations, cytokines and other growth factors in serum and urine of children with idiopathic nephrotic syndrome. Clin Nephrol. 1997;47:289–97.PubMed
17.
18.
19.
Mandreoli M, Beltrandi E, Casadei-Maldini M, Mancini R, Zucchelli A, Zucchelli P. Lymphocyte release of soluble IL-2 receptors in patients with minimal change nephropathy. Clin Nephrol. 1992;37:177–82.PubMed
20.
Neuhaus TJ, Wadhwa M, Callard R, Barratt TM. Increased IL-2, IL-4 and interferon-gamma (IFN-gamma) in steroid-sensitive nephrotic syndrome. Clin Exp Immunol. 1995;100:475–9.PubMedCentralCrossRefPubMed
21.
22.
Lin CY, Chien JW. Increased interleukin-12 release from peripheral blood mononuclear cells in nephrotic phase of minimal change nephrotic syndrome. Acta Paediatr Taiwan. 2004;45:77–80.PubMed
23.
Matsumoto K, Kanmatsuse K. Elevated interleukin-18 levels in the urine of nephrotic patients. Nephron. 2001;88:334–9. doi:10.1159/000046017.CrossRefPubMed
24.
25.
Matsumoto K, Kanmatsuse K. Elevated vascular endothelial growth factor levels in the urine of patients with minimal-change nephrotic syndrome. Clin Nephrol. 2001;55:269–74.PubMed
26.
27.
28.
Matsumoto K, Kanmatsuse K. Increased IL-12 release by monocytes in nephrotic patients. Clin Exp Immunol. 1999;117:361–7.PubMedCentralCrossRefPubMed
29.
30.
Yap HK, Cheung W, Murugasu B, Sim SK, Seah CC, Jordan SC. Th1 and Th2 cytokine mRNA profiles in childhood nephrotic syndrome: evidence for increased IL-13 mRNA expression in relapse. J Am Soc Nephrol. 1999;10:529–37.PubMed
31.
Kanai T, Shiraishi H, Yamagata T, Ito T, Odaka J, Saito T, et al. Th2 cells predominate in idiopathic steroid-sensitive nephrotic syndrome. Clin Exp Nephrol. 2010;14:578–83. doi:10.1007/s10157-010-0330-z.CrossRefPubMed
32.
van den Berg JG, Weening JJ. Role of the immune system in the pathogenesis of idiopathic nephrotic syndrome. Clin Sci (Lond). 2004;107:125–36. doi:10.1042/cs20040095.CrossRef
33.
34.
Meadow SR, Sarsfield JK, Scott DG, Rajah SM. Steroid-responsive nephrotic syndrome and allergy: immunological studies. Arch Dis Child. 1981;56:517–24.PubMedCentralCrossRefPubMed
35.
36.
Bagga A, Sinha A, Moudgil A. Rituximab in patients with the steroid-resistant nephrotic syndrome. N Engl J Med. 2007;356:2751–2. doi:10.1056/NEJMc063706.CrossRefPubMed
37.
Kimata T, Hasui M, Kino J, Kitao T, Yamanouchi S, Tsuji S, et al. Novel use of rituximab for steroid-dependent nephrotic syndrome in children. Am J Nephrol. 2013;38:483–8. doi:10.1159/000356439.CrossRefPubMed
38.
39.
Chugh SS, Clement LC, Mace C. New insights into human minimal change disease: lessons from animal models. Am J Kidney Dis. 2012;59:284–92. doi:10.1053/j.ajkd.2011.07.024.PubMedCentralCrossRefPubMed
40.
Shimada M, Araya C, Rivard C, Ishimoto T, Johnson RJ, Garin EH. Minimal change disease: a “two-hit” podocyte immune disorder? Pediatr Nephrol. 2011;26:645–9. doi:10.1007/s00467-010-1676-x.CrossRefPubMed
41.
Simic I, Tabatabaeifar M, Schaefer F. Animal models of nephrotic syndrome. Pediatr Nephrol. 2013;28:2079–88. doi:10.1007/s00467-012-2376-5.CrossRefPubMed
42.
43.
Drenckhahn D, Franke RP. Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab Invest. 1988;59:673–82.PubMed
44.
45.
Adler S. Characterization of glomerular epithelial cell matrix receptors. Am J Pathol. 1992;141:571–8.PubMedCentralPubMed
46.
Regele HM, Fillipovic E, Langer B, Poczewki H, Kraxberger I, Bittner RE, et al. Glomerular expression of dystroglycans is reduced in minimal change nephrosis but not in focal segmental glomerulosclerosis. J Am Soc Nephrol. 2000;11:403–12.PubMed
47.
Jalanko H. Congenital nephrotic syndrome. Pediatr Nephrol. 2009;24:2121–8. doi:10.1007/s00467-007-0633-9.PubMedCentralCrossRefPubMed
48.
Bertelli R, Trivelli A, Magnasco A, Cioni M, Bodria M, Carrea A, et al. Failure of regulation results in an amplified oxidation burst by neutrophils in children with primary nephrotic syndrome. Clin Exp Immunol. 2010;161:151–8. doi:10.1111/j.1365-2249.2010.04160.x.PubMedCentralPubMed
49.
Ory V, Fan Q, Hamdaoui N, Zhang SY, Desvaux D, Audard V, et al. c-mip down-regulates NF-kappaB activity and promotes apoptosis in podocytes. Am J Pathol. 2012;180:2284–92. doi:10.1016/j.ajpath.2012.02.008.CrossRefPubMed
50.
Lennon R, Singh A, Welsh GI, Coward RJ, Satchell S, Ni L, et al. Hemopexin induces nephrin-dependent reorganization of the actin cytoskeleton in podocytes. J Am Soc Nephrol. 2008;19:2140–9. doi:10.1681/asn.2007080940.PubMedCentralCrossRefPubMed
51.
Cara-Fuentes G, Wasserfall CH, Wang H, Johnson RJ, Garin EH. Minimal change disease: a dysregulation of the podocyte CD80-CTLA-4 axis? Pediatr Nephrol. 2014;29:2333–40. doi:10.1007/s00467-014-2874-8.CrossRefPubMed
52.
Garin EH, Diaz LN, Mu W, Wasserfall C, Araya C, Segal M, et al. Urinary CD80 excretion increases in idiopathic minimal-change disease. J Am Soc Nephrol. 2009;20:260–6. doi:10.1681/asn.2007080836.PubMedCentralCrossRefPubMed
53.
Clement LC, Avila-Casado C, Mace C, Soria E, Bakker WW, Kersten S, et al. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med. 2011;17:117–22. doi:10.1038/nm.2261.PubMedCentralCrossRefPubMed
54.
Clement LC, Mace C, Avila-Casado C, Joles JA, Kersten S, Chugh SS. Circulating angiopoietin-like 4 links proteinuria with hypertriglyceridemia in nephrotic syndrome. Nat Med. 2014;20:37–46. doi:10.1038/nm.3396.PubMedCentralCrossRefPubMed
55.
Ito N, Nishibori Y, Ito Y, Takagi H, Akimoto Y, Kudo A, et al. mTORC1 activation triggers the unfolded protein response in podocytes and leads to nephrotic syndrome. Lab Invest. 2011;91:1584–95. doi:10.1038/labinvest.2011.135.CrossRefPubMed
56.
McCarthy ET, Sharma M, Savin VJ. Circulating permeability factors in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Clin J Am Soc Nephrol. 2010;5:2115–21. doi:10.2215/cjn.03800609.CrossRefPubMed
57.
Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. 2011;17:952–60. doi:10.1038/nm.2411.PubMedCentralCrossRefPubMed
58.
Wei C, Trachtman H, Li J, Dong C, Friedman AL, Gassman JJ, et al. Circulating suPAR in two cohorts of primary FSGS. J Am Soc Nephrol. 2012;23:2051–9. doi:10.1681/asn.2012030302.PubMedCentralCrossRefPubMed
59.
Beck Jr LH, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11–21. doi:10.1056/NEJMoa0810457.PubMedCentralCrossRefPubMed
60.
Debiec H, Lefeu F, Kemper MJ, Niaudet P, Deschenes G, Remuzzi G, et al. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med. 2011;364:2101–10. doi:10.1056/NEJMoa1013792.CrossRefPubMed
61.
Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J, et al. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. 2008;14:931–8. doi:10.1038/nm.1857.PubMedCentralCrossRefPubMed
62.
Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. 2011;3:85ra46. doi:10.1126/scitranslmed.3002231.PubMedCentralCrossRefPubMed
63.
Mathieson PW. The podocyte as a target for therapies–new and old. Nat Rev Nephrol. 2012;8:52–6. doi:10.1038/nrneph.2011.171.CrossRef
64.
Schonenberger E, Ehrich JH, Haller H, Schiffer M. The podocyte as a direct target of immunosuppressive agents. Nephrol Dial Transplant. 2011;26:18–24. doi:10.1093/ndt/gfq617.CrossRefPubMed