Hirschsprung Disease
Hirschsprung disease (HD), also known as ‘congenital megacolon’ is characterized by the absence of ganglion cells in the myenteric and submucosal plexuses of the intestine. The first known description of this condition was by ancient Hindu surgeons in the Shushruta Samheta,1 and the first descriptions in the modern medical literature were from the 17th century.2 In 1887, Harald Hirschsprung, a pediatrician from Copenhagen, described two cases of the condition that ultimately bore his name.3 At that time most children with congenital megacolon died from malnutrition and enterocolitis. As the underlying pathological basis of the disease was unknown, surgeons removed the massively dilated proximal bowel and created a colostomy. Attempts at re-anastomosis were uniformly unsuccessful.4
Although the absence of ganglion cells in the distal colon of a child with HD was first noted by Tittel in 19015 and subsequent publications repeated this observation, it took many decades for clinicians caring for these children to become aware. The first recognition of aganglionosis by a surgeon as the cause of congenital megacolon was by Ehrenpreis in 1946.6 This was followed in 1949 by Swenson’s first description of a reconstructive operation for HD.7 Although Swenson’s operation was originally performed without a colostomy, technical difficulties in small infants and the debilitated and malnourished state in which many children presented were reasons most surgeons adopted a multi-staged approach with colostomy as the initial step.8 In recent years, improvements in operative technique and earlier diagnosis have resulted in an evolution toward one-stage and minimal access procedures. These advances have resulted in significantly improved morbidity and mortality in infants with HD.
Incidence and Spectrum of Disease
A number of syndromes are associated with HD including trisomy 21, congenital central hypoventilation syndrome, Goldberg–Shprintzen syndrome, Smith–Lemli–Opitz syndrome, neurofibromatosis, and neuroblastoma (Box 34-1).
Etiology and Genetic Basis of Disease
Ganglion cells are derived from the neural crest. By 13 weeks post-conception, the neural crest cells have migrated from proximal to distal through the gastrointestinal tract, after which they differentiate into mature ganglion cells.9 There are two main theories why this process is disturbed in children with HD. The first possibility is that the neural crest cells never reach the distal intestine due to early maturation or differentiation into ganglion cells. Data supporting this theory come from animal models showing spontaneous aganglionosis10,11 and from studies of normal neural crest cell migration performed in chick embryos and human fetuses.12,13 The second possibility is that the neural crest cells reach their destination, but fail to survive or differentiate into ganglion cells due to an inhospitable microenvironment.14,15 It is likely that HD is actually a heterogeneous group of diseases with multiple genetic causes and etiologies.
A genetic basis for HD has long been suspected because of the presence of a family history in many cases and the known association with trisomy 21 and other genetically based conditions. Over the past two decades, an increasing number of researchers have made significant progress in identifying and elucidating the complex array of genetic mutations and mechanisms responsible for this disease.16–18 The first and most common gene to be identified is the RET proto-oncogene, which encodes a tyrosine kinase receptor. Many mutations of this gene and related genes, such as neurturin and glial cell line-derived neurotrophic factor (GDNF), have been described. It remains unclear how these mutations result in aganglionosis, but there is evidence that early neuronal cell death may be a prominent mechanism.19,20 RET abnormalities are most commonly found in patients with familial and long-segment involvement. Mutations in the endothelin family of genes, particularly endothelin-3 and the endothelin-B receptor, are also commonly associated with HD. Many of these children have other neurocristopathies such as dysfunction of melanocytes, congenital deafness, central hypoventilation, and neuroblastoma. From animal models, there is evidence that mutations in the endothelin and SOX-10 genes may produce early maturation or differentiation of neural crest cells, which decreases the number of available progenitor cells and prevents the neural crest cells from migrating any further.21,22 Other genes associated with HD include S1P1 (now known as ZFHX1B), Phox2B, and the Hedgehog/Notch complex.23
Clinical Presentation and Diagnosis
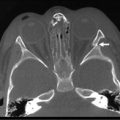
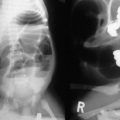
Prenatal diagnosis of HD is rare, and is usually due to total colonic disease resulting in ultrasound (US) findings of fetal intestinal obstruction.24 Most affected patients present during the neonatal period with abdominal distension, bilious vomiting, and feeding intolerance. Delayed passage of meconium beyond the first 24 hours is present in approximately 90%. Occasionally, cecal or appendiceal perforation may be the initial event.25 Plain radiographs characteristically show dilated bowel loops throughout the abdomen. The next step is a water-soluble contrast enema. The pathognomonic finding of HD is a transition zone between the normal and aganglionic bowel (Fig. 34-1), although approximately 10% of neonates with HD may not have a demonstrable radiological transition zone.26 Occasionally, false positive studies occur.27 It is also important to obtain a plain radiograph 24 hours later. Retention of the contrast is very suggestive for HD (Fig. 34-2). It is also important to use a water-soluble material as the enema potentially may be a definitive treatment for other conditions in the differential diagnosis, such as meconium ileus and meconium plug syndrome. Once the diagnosis of HD is suspected, the diagnosis must be confirmed by rectal biopsy, which in the neonate can be done at the bedside without sedation and using a suction technique.
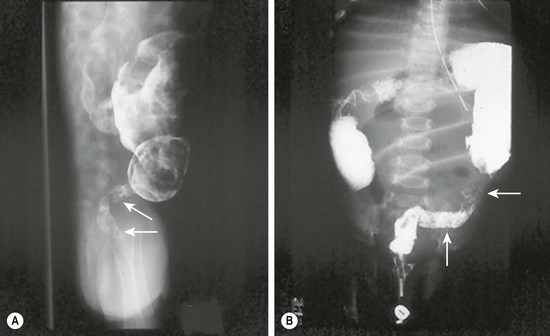
FIGURE 34-1 These two barium enema examinations in different infants demonstrate Hirschsprung disease. The aganglionic rectum (arrows) in both studies is small and contracted. The proximal ganglionic colon is dilated. A transition zone between the aganglionic and ganglionic colon is nicely seen in both studies.
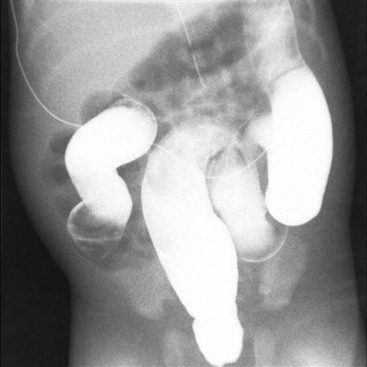
FIGURE 34-2 Retention of contrast is seen on this post-evacuation film which was obtained 24 hours after the contrast enema.
Patients presenting later in childhood have chronic severe constipation. As constipation is common in children, it can be difficult to differentiate HD from more common causes. Clinical features pointing to the diagnosis include delayed passage of meconium at birth, failure to thrive, abdominal distention, and dependence on enemas without significant encopresis.28 Although a contrast enema usually demonstrates a transition zone in older children, a false negative study may be due to massive rectal distension in combination with a very short aganglionic segment. Reversal of the usual rectosigmoid ratio and retention of contrast on a 24-hour postevacuation film also support the diagnosis. Anorectal manometry is another useful screening technique, in which the presence of a recto-anal inhibitory reflex (reflex relaxation of the internal anal sphincter in response to balloon distension of the rectum) essentially rules out HD (Fig. 34-3). In older children, the suction rectal biopsy may be less reliable because of a higher risk of sampling error. Full-thickness biopsies, usually under general anesthesia, may be necessary in these patients.
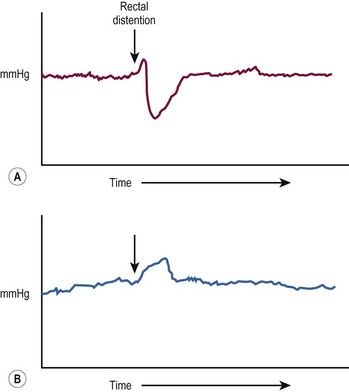
FIGURE 34-3 (A) In the child without Hirschsprung disease undergoing anorectal manometry, the recto-anal inhibitory reflex is normal. Note the drop in the internal sphincter pressure with rectal distention. (B) A child with Hirschsprung disease is seen to have abnormally increased contraction of the anal canal and no relaxation of the internal sphincter with rectal distention. (The arrow points to the initiation of rectal distention in both A and B.)
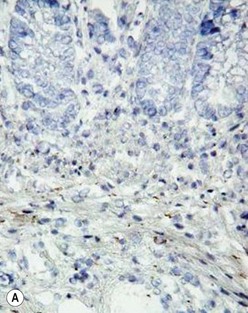
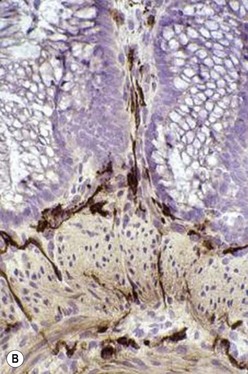
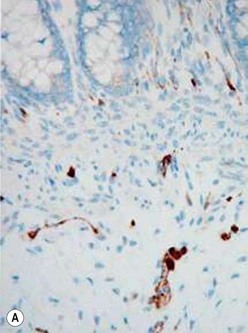
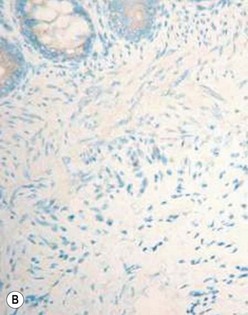
The gold standard for the diagnosis is the absence of ganglion cells in the submucosal and myenteric plexuses on histological examination (Fig. 34-4A). Most patients will also have evidence of hypertrophied nerve trunks (Fig. 34-4B), although this finding is not always present, particularly in children with total colonic disease or a very short aganglionic segment. As there is normally a paucity of ganglion cells in the area 0.5–1.0 cm above the dentate line, the rectal biopsy should be taken at least 1.0–1.5 cm above it. However, a biopsy too proximal may miss a short aganglionic segment. In addition to hematoxylin and eosin, many pathologists also stain for acetylcholinesterase, which has a characteristic pattern in the submucosa and mucosa in children with HD (Fig. 34-5). Recently, it has been shown that immunochemical staining for calretinin is almost always absent in patients with HD (Fig. 34-6).29
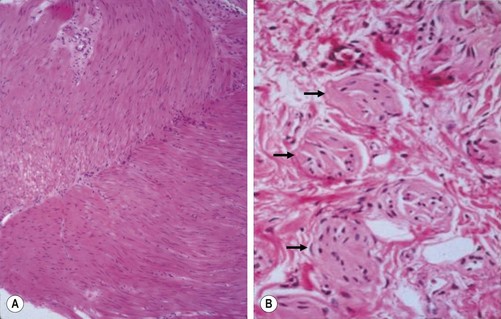
FIGURE 34-4 Histological findings in children with Hirschsprung disease. (A) Absence of ganglion cells in the myenteric plexus. (B) Hypertrophied nerve trunks are marked with arrows.
Preoperative Preparation
Some physicians have advocated nonoperative long-term management of short segment HD using enemas and laxatives. Others have suggested that simple myectomy may be adequate.30 However, these approaches do not provide a good quality of life for most infants and children with HD, and most pediatric surgeons recommend a pull-through procedure even for short segment disease.
Surgical Management
The goals of surgical management for HD are to remove the aganglionic bowel and reconstruct the intestinal tract by bringing the normally innervated bowel down to the anus while preserving normal sphincter function. The most commonly performed operations are the Swenson, Duhamel and Soave procedures, although a number of other operations, such as the Rebhein and State procedures, have been described and are still performed in some centers.31 As there are very few prospective studies comparing operations, the best operation for an individual patient is the one that the surgeon has been trained to do and does frequently.
Although Swenson’s operation was initially developed as a one-stage procedure, the relatively high incidence of stricture, leak, and other adverse outcomes led to the adoption of a routine preliminary colostomy, with definitive pull-through performed three to 12 months later.32 In the 1980s, a number of surgeons reported series of single-stage pull-through operations even in small infants.33,34 Over the following ten to 15 years, many reports suggested that a one-stage approach was safe, avoided the morbidity of stomas in infants, and was more cost effective.35–37 However, a stoma may still be needed for infants and children with severe enterocolitis, perforation, malnutrition, or massively dilated proximal bowel, and in situations when it is not possible to reliably identify the transition zone on frozen section.
Swenson Procedure
The goal of the Swenson pull-through is to remove the entire aganglionic colon, with an end-to-end anastomosis above the anal sphincter. The operation was originally performed via a laparotomy, with the anastomosis being performed from the perineum after everting the aganglionic rectum (Fig. 34-7). It is important to keep the dissection in the correct plane along the rectal wall to avoid injury to the deep pelvic nerves, vessels and other structures such as the vagina, prostate, vas deferens, and seminal vesicles. Despite the theoretical risks inherent in the deep pelvic dissection, long-term functional outcomes after the Swenson procedure are excellent.38
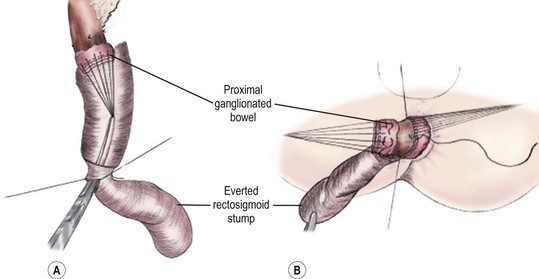
Soave Procedure
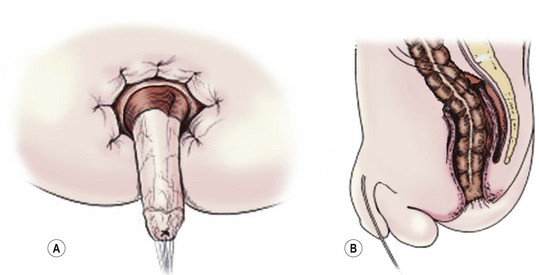
The Soave procedure, subsequently modified by Boley, was designed to avoid the risk of injury to important pelvic structures by performing a submucosal endorectal dissection and positioning the pull-through bowel within an aganglionic muscular ‘cuff’ (Fig. 34-8). Despite concerns by some that the Soave procedure may result in long-term constipation due to incomplete excision of the aganglionic rectum,39 most late follow-up studies have reported similar outcomes between the Soave and Swenson operation.40
Duhamel Procedure
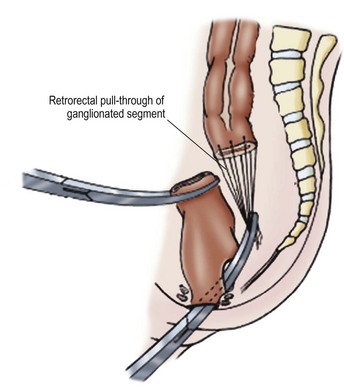
The Duhamel procedure involves bringing the normal colon down through the bloodless plane between the rectum and sacrum, and joining the two walls with a linear stapler to create a new lumen which is aganglionic anteriorly and normally innervated posteriorly (Fig. 34-9). The Duhamel operation is felt by many surgeons to be easier and safer than the Swenson or Soave procedures. Also, it results in a very large anastomosis, which reduces the risk of stricture. Reported longterm results of the Duhamel procedure have been similar to those with the other two operations, although recent studies suggest that outcomes from the Duhamel procedure are inferior to those of the transanal pull-through.41,42
Laparoscopic Pull-through
Georgeson first described the laparoscopic approach for HD in 1995.43 With this technique, a biopsy is initially performed to identify the transition zone, the rectum is mobilized below the peritoneal reflection, and a short mucosal dissection is performed through a perineal approach (Figs 34-10 to 34-14). The rectum is then prolapsed through the anus and the anastomosis performed from below. This procedure is associated with a shorter time in the hospital, and both early and mid-term results appear to be equivalent to those reported for the other procedures.44 Laparoscopic approaches have been also described for the Duhamel and Swenson operations, with excellent short term results.45,46
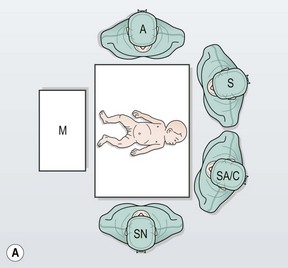
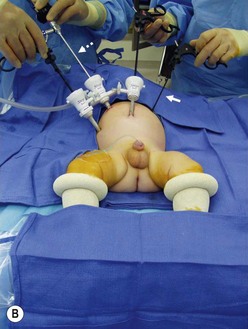
FIGURE 34-10 (A) The surgeon (S) and surgical assistant/camera holder (SA/C) stand above the patient’s head with the monitor (M) positioned beyond the infant’s feet. The scrub nurse (SN) can be positioned according to the surgeon’s preference, although being positioned at the foot of the operating table appears to be ideal. A, anesthesiologist. (B) The photograph shows port placement for this operation. Usually three or four ports are required. The umbilical port is inserted using an open technique, and the other ports are introduced under direct visualization. The telescope (dotted arrow) is placed through the 5 mm port in the right upper abdomen. The surgeon’s two primary working ports are the umbilical port for the left hand and the right lower abdominal port for the right hand. A retracting instrument (solid arrow) is often helpful and can be inserted through a stab incision in the infant’s left upper abdomen. A urinary catheter has been introduced to help decompress the bladder. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
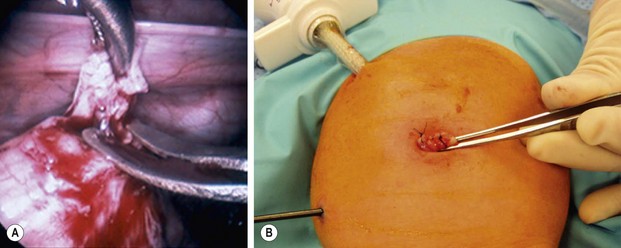
FIGURE 34-11 (A) An intracorporeal biopsy is being performed on the sigmoid colon. A fine-tipped grasping forceps has been used to grasp the biopsy site, and Metzenbaum scissors are used to obtain the biopsy specimen. (B) This biopsy was performed through the umbilical incision. One port and another instrument have been introduced through the infant’s abdominal wall. A site on the colon for the biopsy was visualized and delivered just under the umbilical cannula. The umbilical cannula was removed, and this portion of the colon was grasped and exteriorized. An extracorporeal biopsy was obtained and the biopsy site was closed. This is an alternative means for obtaining the biopsy. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
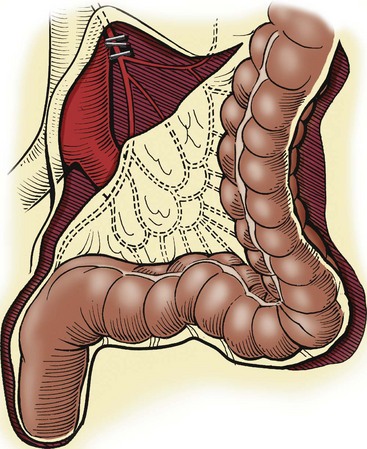
FIGURE 34-12 When the transition zone is proximal to the mid-sigmoid colon, a pedicled colon flap must be developed for the endorectal pull-through. In this situation, the pull-through colon will derive its vascular supply from the marginal artery. Therefore, to mobilize the descending colon and splenic flexure, it is necessary to ligate and divide either the inferior mesenteric artery just distal to its origin from the aorta (as seen in this drawing) or the left colic artery just after it arises from the inferior mesenteric artery. By ligating these vessels at these sites, the arterial supply through the marginal artery is not compromised. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
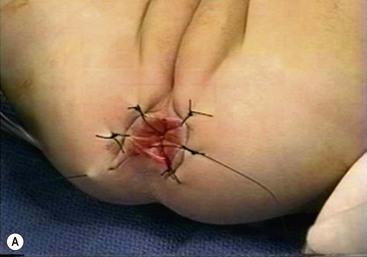
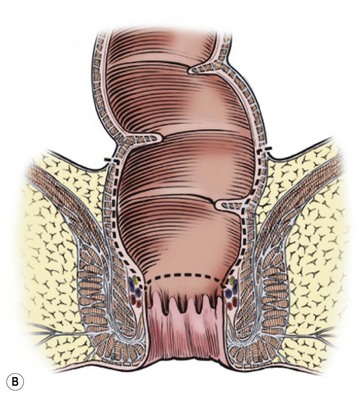
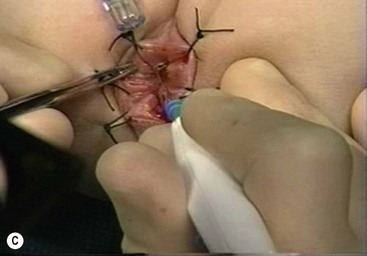
FIGURE 34-13 (A) The perineal dissection begins with the placement of circumferential 2-0 silk traction sutures from the dentate line to the perineum 2 to 3 cm outward from the anus. (B, C) A needle-tipped electrocautery is used to circumferentially incise the rectal mucosa approximately 5 mm proximal to the anal columns. Fine silk traction sutures are then placed in the rectal mucosa to help retract the mucosa during circumferential dissection. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
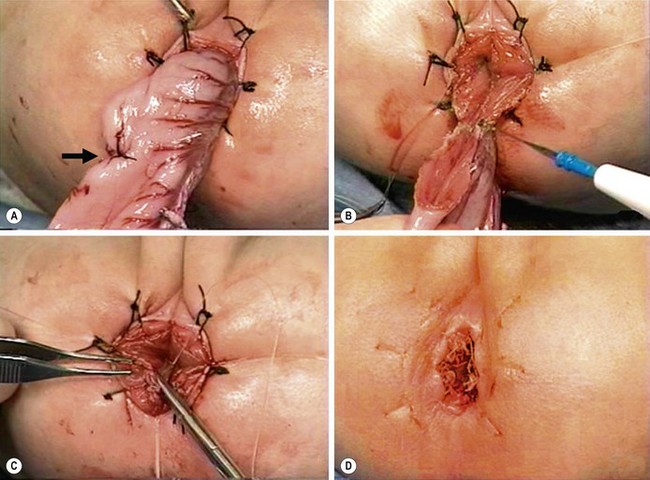
FIGURE 34-14 (A) The muscular cuff of the rectum has been divided and the ganglionic colon has been exteriorized through the anal canal. Note that the anastomosis will be performed proximal to the biopsy site (arrow). (B) The pull-through colon is being completely transected above the biopsy site and made ready for the coloanal anastomosis. (C) The anastomosis is being performed with interrupted 4-0 absorbable sutures. (D) The everting stay sutures have been cut, allowing the anastomosis to retract cephalad. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
Transanal (Perineal) Pull-through
The transanal approach was first described by de la Torre in 199847 and by Langer in 1999,48 and has been adopted and reported by an increasing number of surgeons. The operation can be performed in the prone or lithotomy positions.49 A mucosal incision is made 0.5–1.0 cm above the dentate line, depending on the size of the child, and the mucosa is stripped from the underlying muscle as in the Soave operation. The length of the submucosal dissection varies according to surgeon, although a shorter rectal cuff may be associated with a lower incidence of enterocolitis and the need for dilatation.50 Some surgeons do not perform any submucosal dissection, and effectively perform a transanal Swenson procedure.51
The rectal muscle is incised circumferentially, and the dissection is continued on the rectal wall, dividing the vessels as they enter the rectum. The entire rectum and part of the sigmoid colon can be delivered through the anus. When the transition zone is reached, the anastomosis is performed from below (Fig. 34-15). In patients with a more proximal transition zone (usually above the proximal sigmoid colon), laparoscopy or a small umbilical incision is needed to mobilize the left colon and/or splenic flexure to achieve an adequate length of ganglionated colon for pull-through.52 A transanal approach can also be used if the patient has already had a colostomy by using the stoma as the end of the pull-through bowel and performing the rectal excision using the transanal approach.
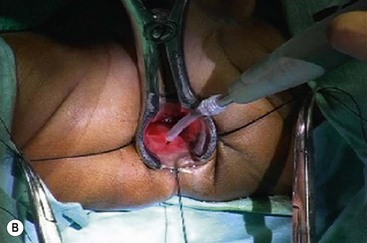
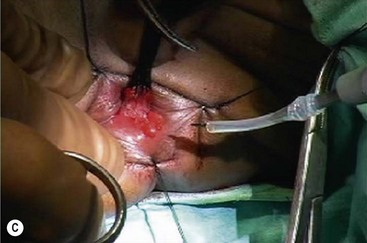
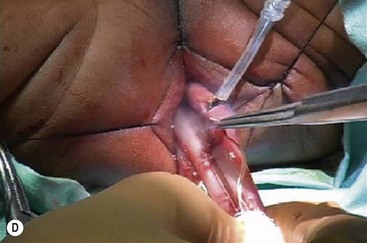
FIGURE 34-15 The salient points for a transanal Soave pull-through are depicted. (A) An umbilical incision is used for a preliminary biopsy. A Hegar dilator is used to push the sigmoid colon into the umbilical incision. (B) Eversion sutures are placed in the anus, and a nasal speculum is used to provide exposure to the anal canal. A circumferential incision is made 5 mm above the dentate line. (C) The submucosal dissection is carried 2–3 cm. (D) The pull-through bowel is divided at least 2 cm above the biopsy site that has ganglion cells, and the anastomosis is performed. Care must be taken to perform the anastomosis to the rectal mucosa, not to the transitional epithelium. Otherwise, normal sensation will be lost and the risk of incontinence will be increased.
There is controversy around whether the histological transition zone should be defined prior to beginning the anal dissection. This controversy centers on the fact that approximately 8–10% of children who have a rectosigmoid transition zone on contrast study actually have a more proximal histological transition zone.53,54 This concern is particularly important for surgeons who perform a different operation for long-segment disease than for rectosigmoid disease. A preliminary biopsy does not have a deleterious effect on postoperative outcomes such as time to feeding, pain, or length of hospitalization.50,55
The transanal approach has a low complication rate, requires minimal postoperative analgesia, and is associated with early feeding and discharge.55,56 Although there have not been any studies comparing the transanal and laparoscopic approaches, the transanal pull-through can be accomplished by most pediatric surgeons, including those without laparoscopic skills, and by pediatric surgeons in parts of the world where access to laparoscopic equipment is limited.
Long-Segment Aganglionosis
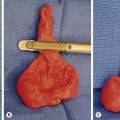
Long-segment HD is usually defined as a transition zone which is proximal to the mid-transverse colon. The most common form is total colonic aganglionosis, which usually also includes some of the distal ileum (Fig. 34-16). In rare cases of near-total intestinal aganglionosis, most of the small bowel is aganglionic as well. Most neonates with long-segment disease present with a distal small bowel obstruction, although occasionally children with long-segment disease may not present until after weaning from breast milk. The contrast enema typically shows a shortened, relatively narrow ‘question mark’ colon (Fig. 34-17).57 There may also be a transition zone in the small bowel. The rectal biopsy shows absence of ganglion cells, but in many cases there are no hypertrophic nerves or abnormalities on acetylcholinesterase staining.
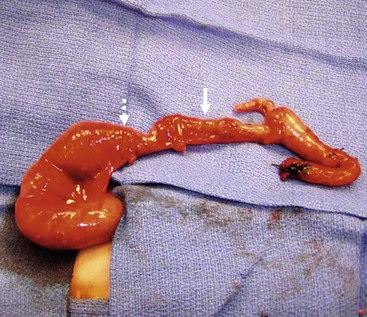
FIGURE 34-16 This neonate has total colon aganglionosis. The solid arrow marks the contracted aganglionic terminal ileum and the transition zone to ganglionated ileum is marked by the dotted arrow.
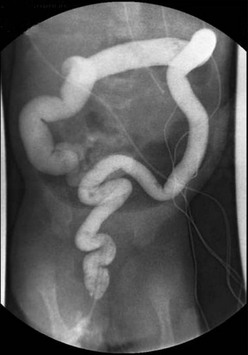
FIGURE 34-17 This contrast enema was performed in a child with total colonic aganglionosis. There is no transition zone in the colon, and the colon is foreshortened with a ‘question mark’ configuration.
The initial operative approach involves sequential colonic biopsies looking for ganglion cells on frozen section. These biopsies can be performed via laparotomy or laparoscopy, or through an umbilical incision. Using the appendix for the initial biopsy can result in a false positive diagnosis of total colonic aganglionosis as there may be a paucity of ganglion cells in the appendix in children with shorter segment disease.58 Thus, a biopsy of the cecum is preferable. Once the level of aganglionosis is identified, most surgeons create a stoma, wait for permanent sections, and perform the definitive reconstructive procedure later. Although primary pull-through without ileostomy for total colonic disease has been performed, this approach requires a high degree of confidence in the pathologist, since it requires performing a total colectomy on the basis of frozen section analysis alone. In addition, there are many reports of ‘skip’ areas in children with total colonic aganglionosis so that permanent sections are advisable before considering total colectomy.59 Finally, some surgeons believe that the outcomes following pull-through are better once the stool has thickened, which usually occurs after the first few months of life.
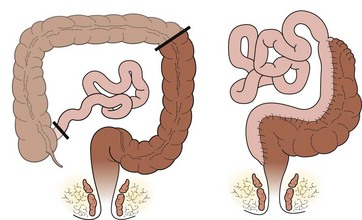
There are three types of operations available for children with long-segment disease: straight pull-through using one of the standard techniques (Swenson, Duhamel, or Soave), colon patch using either the left colon (Martin) (Fig. 34-18) or the right colon (Kimura) (Fig. 34-19), and ileal J-pouch. There are no prospective or well-controlled series reporting long-term results of operations for long-segment HD. Although the colon patch procedures theoretically result in decreased stool output due to better water absorption, the aganglionic colon tends to dilate and many of these patients develop severe enterocolitis, requiring removal of the patch or a permanent stoma. Children undergoing a straight pull-through tend to experience gradually decreasing stool frequency over time, with an acceptable quality of life.60–62
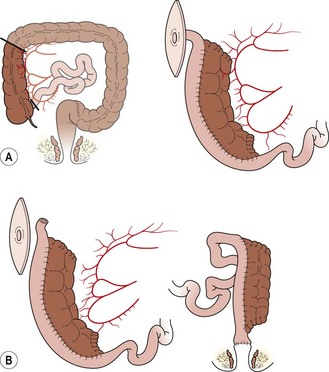
Rarely, almost the entire intestinal tract is aganglionic, usually leaving 10–40 cm of normally innervated jejunum. These children require total parenteral nutrition (TPN) from birth. At the time of the first exploration, the goal is to determine the extent of aganglionosis based on frozen sections, and to create a stoma either at the most distal point that has normally innervated bowel or more distally with aganglionic bowel.63 A central venous catheter should be inserted for TPN, and a gastrostomy should be considered for continuous feeding of breast milk or elemental formula.
These children are best managed by a multidisciplinary group focused on intestinal failure.64 Strict attention to prevention of sepsis, treatment of bacterial overgrowth, use of trophic feedings, and prevention of TPN-related cholestasis using a variety of strategies including omega-3 lipids are all important.65
Subsequent management must be individualized, depending on the length of normally innervated bowel and the clinical status of the child.66 For infants and children who develop significant dilatation of the normally innervated bowel, tapering, imbrication, or bowel lengthening procedures such as the Bianchi or serial transverse enteroplasty (STEP) procedure may be helpful.67,68 The ‘myectomy–myotomy’ procedure popularized by Zeigler involves myectomy of a length of aganglionic small bowel distal to the transition zone.69 For some of these patients, small bowel or combined small bowel-liver transplantation may offer the only chance for survival.70
Postoperative Management
Most children undergoing a laparoscopic or transanal pull-through can be fed immediately and discharged within 24–48 hours. The anastomosis is calibrated with an appropriately sized dilator or finger one to two weeks after the procedure. Although most surgeons instruct the parents to perform daily dilatations, a program of weekly calibration by the surgeon is less traumatic and is associated with similar outcomes.71 The parents should be instructed to protect the buttocks with barrier cream to prevent perineal skin breakdown. In addition, the family and the primary care physician should be educated about the signs and symptoms of postoperative enterocolitis, since this can result in rapid severe illness and even death in a few patients.72
Long-Term Outcomes
Long-term problems in children with HD include ongoing obstructive symptoms, soiling, and enterocolitis.73 It is important for the surgeon to follow these children closely, at least until they are through the toilet training process, in order to identify and provide timely treatment for these problems.40,74,75
Obstructive Symptoms
Obstructive symptoms may take the form of abdominal distension, bloating, vomiting, or ongoing severe constipation. There are five major reasons for these symptoms following a pull-through: mechanical obstruction, recurrent or acquired aganglionosis, disordered motility in the residual colon or small bowel, internal sphincter achalasia, or functional megacolon caused by stool-holding behavior (Box 34-2). The clinician will have much greater success in managing these difficult patients if an organized approach is taken. One proposed algorithm is shown in Figure 34-20.76
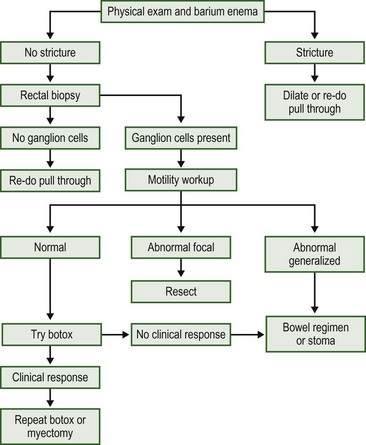
FIGURE 34-20 Algorithm for the investigation and management of the child with obstructive symptoms following a pull-through.
Mechanical Obstruction
The most common cause of mechanical obstruction after a pull-through operation is a stricture. This problem is more common after a Swenson or Soave operation (Fig. 34-21A). Patients undergoing a Duhamel procedure may have a retained ‘spur’ consisting of the anterior aganglionic bowel, which may fill with stool and obstruct the pulled-through bowel (Fig. 34-21B). In other cases, there may be obstruction secondary to a twist in the pulled-through bowel (Fig. 34-21C), or narrowing due to a long muscular cuff in children who have had a Soave operation.
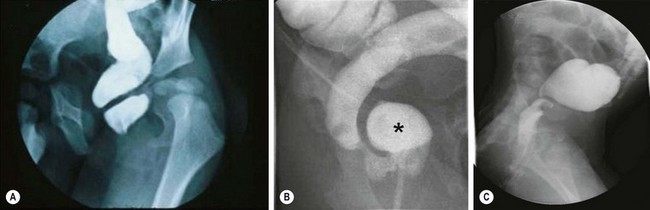
FIGURE 34-21 Causes of mechanical obstruction after a pull-through are shown. (A) Stricture following a Soave procedure. (B) Anterior aganglionic ‘spur’ (asterisk) following a Duhamel procedure. (C) Twisted transanal pull-through.
Obstruction can be discovered with digital rectal examination and a contrast enema. Initial management of an anastomotic stricture consists of repeated dilatation using a finger, dilator, or balloon. For recalcitrant strictures, antegrade dilatation using Tucker dilators,77 intralesional steroid,78 or topical mitomycin C79 can be tried. In some cases, revision of the pull-through is necessary.80–82 Duhamel spurs can be resected from above or managed by extending the staple line from below. Twisted pull-throughs and narrow muscular cuffs usually require a repeat pull-through, although a muscular cuff can occasionally be divided laparoscopically.
Persistent or Acquired Aganglionosis
This problem may be due to an error in histological analysis,83 a transition zone pull-through,84,85 or loss of ganglion cells,86 and can be diagnosed by performing a biopsy above the colo-anal anastomosis.87 The specimen from the original operation should be reviewed and further sections should be taken circumferentially at the resection margin since the transition zone can be asymmetrical in children with HD.88 In most cases, the best treatment for persistent or acquired aganglionosis is a repeat pull-through, which can be accomplished using either a Soave or a Duhamel approach.81
Motility Disorder
Children with HD often have abnormal motility throughout the intestinal tract, including gastroesophageal reflux and delayed gastric emptying.89 These abnormalities may be focal, usually involving the left colon, or may be generalized, and may or may not be associated with other histological abnormalities such as intestinal neuronal dysplasia (IND). Techniques for diagnosing motility disorders include radiological shape study90 radionuclide colon transit study,91 colonic manometry,92 and laparoscopic biopsies looking for evidence of IND.93 If a focal abnormality is found, resection with repeat pull-through using normal bowel is needed. Diffuse dysmotility is best treated with bowel management, which may include antegrade enemas through a cecostomy,94 and the use of prokinetic agents.
Internal Sphincter Achalasia
This term refers to obstructive symptoms caused by the lack of a normal recto-anal inhibitory reflex which is found in all children with HD (see Fig. 34-3). Most children eventually ‘grow out’ of this problem over time, usually by the age of 5 years. The diagnosis can be confirmed by demonstrating a clinical response to intrasphinteric botulinum toxin.95 The traditional operative approach for internal sphincter achalasia has been internal sphincterotomy or myectomy.96,97 However, since this problem may resolve on its own, and there is concern about sphincter-cutting operations impacting continence, we prefer to use chemical sphincterotomy with intrasphincteric botulinum toxin.98–100 In many cases, repeated injection of botulinum toxin, or applications of nitroglycerine paste or topical nifedipine, are necessary while waiting for resolution of the problem.
Functional Megacolon
Functional megacolon is the result of stool-holding behavior, which is very common in normal children.101 This behavior may be even more common in children with HD because of their predisposition to constipation.102 This problem is best treated with a bowel management regimen consisting of laxatives and behavior modification strategies. In severe cases, the child may require a cecostomy for antegrade enemas, or even a proximal stoma. In many cases, the cecostomy or stoma can be reversed when the child reaches adolescence.
Fecal Soiling
There are three broad causes for soiling after a pull-through: abnormal sphincter function, abnormal sensation, or ‘pseudo-incontinence’ (Box 34-3). Abnormal sphincter function may be due to sphincter injury during the pull-through or to a previous myectomy or sphincterotomy, and can usually be identified using anorectal manometry or endorectal ultrasound. There are two forms of abnormal sensation. The first is lack of sensation of a full rectum, which can also be identified using anorectal manometry, and the other is an inability to detect the difference between gas and stool. This problem is usually due to loss of the transitional epithelium because the anastomosis was performed below the dentate line. This distinction is usually evident on physical examination. Neither sphincter weakness nor abnormal sensation are amenable to a surgical solution. Most of these children are best managed using a bowel routine which may include a constipating diet, stimulant laxatives, and rectal or antegrade enemas. Biofeedback training has been advocated, especially for those children with sphincter weakness. In some cases, the child is best served by a colostomy.
If both the sphincter and sensation are intact, the most likely cause of soiling after a pull-through is pseudo-incontinence.103 This may be caused by severe obstipation with a massively distended rectum and overflow of liquid stool. Other patients leak small amounts of stool through the day, creating ‘skid marks’ in the underwear on a constant basis. Other children can suffer from hyperperistalsis of the pulled-through bowel, which results in the inability of the anal sphincter to achieve control despite normal sphincter function.92
Enterocolitis
The etiology of HAEC is unknown, and is probably multi-factorial. Stasis caused by functional obstruction permits bacterial overgrowth with secondary infection. Infectious agents such as Clostridium dificile or rotavirus have been postulated as being causative, but there are few data to support a specific pathogen.104 There is some evidence implicating alterations in intestinal mucin production and the mucosal production of immunoglobulins, which presumably results in loss of intestinal barrier function and allows bacterial invasion.105,106
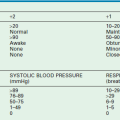
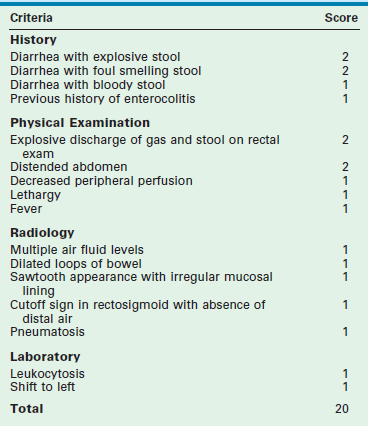
Enterocolitis may be present both before and after operative correction, and can range in severity from mild to life-threatening. HAEC is more common in younger children,107 longer segment disease, and trisomy 21. Clinical presentation includes fever, abdominal distention, diarrhea, elevated leukocyte count, and evidence of intestinal edema on an abdominal film. Because there is overlap between HAEC and other conditions such as gastroenteritis, the true incidence is unknown. A HAEC score has recently been developed, which may be useful in the future in both the clinical and research settings (Table 34-1).108
The treatment of postoperative HAEC involves nasogastric drainage, intravenous fluids, broad-spectrum antibiotics, and decompression of the rectum and colon using rectal stimulation or irrigations. The risk of HAEC may be decreased by using preventive measures such as routine irrigations109 or chronic administration of metronidazole or probiotic agents, particularly in those who are thought to be at higher risk for this problem based on clinical or histological grounds. Since enterocolitis is the most common cause of death in children with HD (Fig. 34-22) and can occur postoperatively even in children who did not have it preoperatively, it is very important that the surgeon educates the family about the risk of this complication and urges early return to the hospital if the child develops concerning symptoms.72
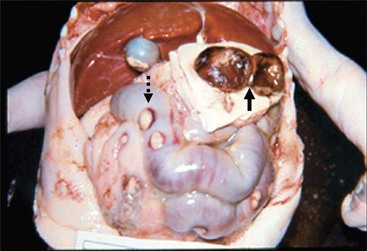
FIGURE 34-22 Enterocolitis can be a major cause of morbidity and mortality, both before any operative procedure for Hirschsprung disease and in the postoperative period. Most of the deaths in the postoperative period are related to enterocolitis. In this autopsy specimen, note the several areas of perforation (dotted arrow) along the colon due to significant enterocolitis. Also note that the patient had undergone a diverting colostomy and had a mucous fistula (solid arrow).
Despite the relatively common occurrence of postoperative problems, most resolve after the first five years of life. Studies of teenagers and adults with HD suggest that sexual function, social satisfaction, and quality of life all appear to be relatively normal in the vast majority of patients once they reach their late teens.74,110 Exceptions include children with long-segment disease, who have a higher risk of enterocolitis, incontinence, and dehydration than children with shorter segment disease, children with trisomy 21 who have a greater risk of enterocolitis and incontinence,111–113 and children with other co-morbidities such as those with congenital central hypoventilation syndrome, congenital heart disease, and syndromes that are associated with mental retardation or other forms of disability.114
Variant Hirschsprung Disease
Variant HD is the term often used to describe children who present with a clinical picture suggestive of HD, but with ganglion cells on rectal biopsy.115 There is a significant amount of controversy surrounding the definitions and features of many of these conditions.116 In some cases, their existence has even been questioned.
Intestinal Neuronal Dysplasia (IND)
This condition was first described by Meier-Ruge in 1971.117 Two types are usually described.118 Type A is less common and is characterized by diminished or absent sympathetic innervation of the myenteric and submucosal plexuses, along with hyperplasia of the myenteric plexus. Type B consists of dysplasia of the submucous plexus with thickened nerve fibers and giant ganglia, increased acetylcholinesterase staining, and identification of ectopic ganglion cells in the lamina propria. Type B can occur independently or concomitant with HD. In addition, IND may be either diffuse or focal.
Despite multiple publications, the topic of IND continues to stimulate a lot of controversy among pediatric surgeons and pediatric pathologists.119,120 Sophisticated histological techniques, including special stains and the use of thick sections, are often necessary for an accurate diagnosis.116 In addition, there is some evidence that the histological finding of IND may in some cases be secondary to chronic obstruction rather than the cause of it. In many cases, there may not be good correlation between the histological finding of IND and the bowel motility.121
Hypoganglionosis
Hypoganglionosis is characterized by sparse and small ganglia, usually in the distal bowel, often associated with abnormalities in acetylcholinesterase distribution. The appropriate treatment is resection of the abnormal colon with a pull-through procedure as one would do for a child with HD.122 This condition must be differentiated from immature ganglia, which are seen in preterm children, and should not be treated surgically.123
Internal Sphincter Achalasia
There are some children who have normal ganglion cells on rectal biopsy, but who lack the recto-anal inhibitory reflex on anorectal manometry and develop symptoms of HD. This condition has been termed internal sphincter achalasia.124 The initial treatment is a bowel management regimen consisting of diet, laxatives, and enemas or irrigations. If this is unsuccessful, some have advocated anal sphincter myectomy.125,126 Because the constipation associated with this condition usually improves over the first five years of life, treatment includes temporary or reversible sphincter-relaxing measures such as botulinum toxin,127 nitroglycerine paste,128 or topical nifedipine, as previously discussed.
‘Ultra-short Segment’ Hirschsprung Disease
Some surgeons use this term to describe children with normal ganglion cells on rectal biopsy, but with absence of the recto-anal inhibitory reflex, which is synonymous with the definition of internal sphincter achalasia. We prefer to reserve it for children who have a documented aganglionic segment of less than 3–4 cm. In children with this condition, the findings of hypertrophic nerves and abnormal cholinesterase staining may be absent.129 The treatment of ultra-short segment HD is controversial. Some authors advocate simple anal sphincter myectomy,130,131 whereas others prefer excision of the aganglionic segment with a pull-through.
Desmosis Coli
This is a rare condition characterized by chronic constipation associated with total or a focal lack of the connective tissue net in the circular and longitudinal muscles and the connective tissue layer of the myenteric plexus, without any abnormalities in the enteric nervous system.132 One family has been described in which HD and desmosis coli coexisted,133 although in most cases they are separate entities.
Future Directions
There are a number of areas in which future research could potentially improve outcomes for children with HD. Perhaps the most important is our rapidly increasing understanding of the genetic basis for aganglionosis, and the tremendous variability in genetic mutations that may result in the full spectrum of clinical disease. The formation of large multidisciplinary research groups like the Hirschsprung Disease Research Collaborative [https://hdrcstudy.org/home] will lead to better correlation between genetic information and pathological and clinical outcomes. This understanding will ultimately lead to more personalized approaches to children with HD, tailoring the operative and postoperative management more precisely to the specific form of the disease.
Another exciting area of future work is the concept of repopulating the aganglionic bowel with neuronal stem cells.134 There is some preliminary work using in vitro systems135 and animal models.136 This research may result in clinical application of this technology, which may be particularly helpful for children with near-total or total intestinal aganglionosis.
References
1. Raveenthiran, V. Knowledge of ancient Hindu surgeons on Hirschsprung disease: Evidence from Sushruta Samhita of circa 1200-600 BC. J Pediatr Surg. 2011; 46:2204–2208.
2. Fiori, MG. Domenico Battini and his description of congenital megacolon: A detailed case report one century before Hirschsprung. J Peripher Nerv Syst. 1998; 3:197–206.
3. Jay, V. Legacy of Harald Hirschsprung. Pediatr Devel Pathol. 2001; 4:203–204.
4. Fraser, J. Surgery of Childhood. New York: William Wood and Company; 1926.
5. Ehrenpreis, T. Hirschsprung’s Disease. Chicago: Year Book Medical Publishers; 1970.
6. Ehrenpreis, T. Some newer aspects on Hirschsprung’s disease and allied disorders. J Pediatr Surg. 1966; 1:329–337.
7. Swenson, O, Rheinlander, HF, Diamond, I. Hirschsprung’s disease: A new concept in etiology-operative results in 34 patients. N Engl J Med. 1949; 241:551–556.
8. Gross, RE. Congenital megacolon (Hirschsprung’s disease). In: Gross RE, ed. The Surgery of Infancy and Childhood. Phildalphia, PA: W.B. Saunders; 1953:330–347.
9. Gariepy, C. Developmental disorders of the enteric nervous system: Genetic and molecular bases. J Pediatr Gastroent Nutr. 2004; 39:5–11.
10. Webster, W. Embryogenesis of the enteric ganglia in normal mice and in mice that develop congenital aganglionic megacolon. J Embryol Exp Morphol. 1973; 30:573–585.
11. Miyahara, K, Kato, Y, Koga, H, et al. Visualization of enteric neural crest cell migration in SOX10 transgenic mouse gutusing time-lapse fluorescence imaging. J Pediatr Surg. 2011; 46:2305–2308.
12. Le Douarin, NM, Teillet, M-A. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J Embryol Exp Morph. 1973; 30:31–48.
13. Paran, TS, Rolle, U, Puri, P. Enteric nervous system and developmental abnormalities in childhood. Pediatr Surg Int. 2006; 22:945–959.
14. Langer, JC, Betti, PA, Blennerhassett, MG. Smooth muscle from aganglionic bowel in Hirschsprung’s disease impairs neuronal development in vitro. Cell Tiss Res. 1994; 276:181–186.
15. Rauch, U, Schafer, K-H. The extracellular matrix and its role in cell migration and development of the enteric nervous system. Eur J Pediatr Surg. 2003; 13:158–162.
16. Parisi, MA, Kapur, RP. Genetics of Hirschsprung disease. Curr Opin Pediatr. 2000; 12:610–617.
17. Amiel, J, Lyonnet, S. Hirschsprung disease, associated syndromes, and genetics: A review. J Med Genet. 2001; 38:729–739.
18. Moore, SW. The contribution of associated congenital anomalies in understanding Hirschsprung’s disease. Pediatr Surg Int. 2006; 22:305–315.
19. Bordeaux, MC, Forcet, C, Granger, L, et al. The RET proto-oncogene induces apoptosis: A novel mechanism for Hirschsprung disease. EMBO J. 2000; 19:4056–4063.
20. Uesaka, T, Nagashimada, M, Yonemura, S, et al. Diminished Ret expression compromises neuronal survival in the colon and causes intestinal aganglionosis in mice. J Clin Invest. 2008; 118:1890–1898.
21. Paratore, C, Eichenberger, C, Suter, U, et al. Sox10 haploinsufficiency affects maintenance of progenitor cells in a mouse model of Hirschsprung disease. Hum Mol Genet. 2002; 11:3075–3085.
22. Gershon, MD. Endothelin and the development of the enteric nervous system. Clin Exp Pharmacol Physiol. 1999; 26:985–988.
23. Ngan, ES, Garcia-Barcelo, MM, Yip, BH, et al. Hedgehog/Notch-induced premature gliogenesis represents a new disease mechanism for Hirschsprung disease in mice and humans. J Clin Invest. 2011; 121:3467–3478.
24. Belin, B, Corteville, JE, Langer, JC. How accurate is prenatal sonography for the diagnosis of imperforate anus and Hirschsprung’s disease? Pediatr Surg Int. 1995; 10:30–32.
25. Newman, B, Nussbaum, A, Kirkpatrick, JA, Jr. Bowel perforation in Hirschsprung’s disease. AJR. 1987; 148:1195–1197.
26. Smith, GHH, Cass, D. Infantile Hirschsprung’s disease—is barium enema useful? Pediatr Surg Int. 1991; 6:318–321.
27. Diamond, IR, Casadiego, G, Traubici, J, et al. The contrast enema for Hirschsprung disease: Predictors of a false-positive result. J Pediatr Surg. 2007; 42:792–795.
28. Lewis, NA, Levitt, MA, Zallen, GS, et al. Diagnosing Hirschsprung’s disease: Increasing the odds of a positive rectal biopsy result. J Pediatr Surg. 2003; 38:412–416.
29. Kapur, RP, Reed, RC, Finn, LS, et al. Calretinin immunohistochemistry versus acetylcholinesterase histochemistry in the evaluation of suction rectal biopsies for Hirschsprung Disease. Pediatr Dev Pathol. 2009; 12:6–15.
30. Kaymakcioglu, N, Yagci, G, Can, MF, et al. Role of anorectal myectomy in the treatment of short segment Hirschsprung’s disease in young adults. Int Surg. 2005; 90:109–112.
31. Visser, R, van de Ven, TJ, van Rooij, IA, et al. Is the Rehbein procedure obsolete in the treatment of Hirschsprung’s disease? Pediatr Surg Int. 2010; 26:1117–1120.
32. Swenson, O. Hirschsprung’s disease: A review. Pediatrics. 2002; 109:914–918.
33. So, HS, Schwartz, DL, Becker, JM, et al. Endorectal ‘pull-through’ without preliminary colostomy in neonates with Hirschsprung’s disease. J Pediatr Surg. 1980; 15:470–471.
34. Cass, DT. Neonatal one-stage repair of Hirschsprung’s disease. Pediatr Surg Int. 1990; 5:341–346.
35. Langer, JC, Fitzgerald, PG, Winthrop, AL, et al. One vs two stage Soave pull-through for Hirschsprung’s disease in the first year of life. J Pediatr Surg. 1996; 31:33–37.
36. Hackam, DJ, Superina, RA, Pearl, RH. Single-stage repair of Hirschsprung’s disease: A comparison of 109 patients over 5 years. J Pediatr Surg. 1997; 32:1028–1031.
37. Bufo, AJ, Chen, MK, Shah, R, et al. Analysis of the costs of surgery for Hirschsprung’s disease: One-stage laparoscopic pull-through versus two-stage Duhamel procedure. Clin Pediatr. 1999; 38:593–596.
38. Sherman, JO, Snyder, ME, Weitzman, JJ, et al. A 40-year multinational retrospective study of 880 Swenson procedures. J Pediatr Surg. 1989; 24:833–838.
39. Swenson, O. Hirschsprung’s disease–a complicated therapeutic problem: Some thoughts and solutions based on data and personal experience over 56 years. J Pediatr Surg. 2004; 39:1449–1453.
40. Moore, SW, Albertyn, R, Cywes, S. Clinical outcome and long-term quality of life after surgical correction of Hirschsprung’s disease. J Pediatr Surg. 1996; 31:1496–1502.
41. Giuliani, S, Betalli, P, Narciso, A, et al. Outcome comparison among laparoscopic Duhamel, laparotomic Duhamel, and transanal endorectal pull-through: A single-center, 18-year experience. J Laparosc Adv Surg Tech. 2011; 21:859–863.
42. Gunnarsdottir, A, Larsson, LT, Arnbjornsson, E. Transanal endorectal vs. Duhamel pull-through for Hirschsprung’s disease. Eur J Pediatr Surg. 2010; 20:242–246.
43. Georgeson, KE, Fuenfer, MM, Hardin, WD. Primary laparoscopic pull-through for Hirschsprung’s disease in infants and children. J Pediatr Surg. 1995; 30:1017–1021.
44. Georgeson, KE, Cohen, RD, Hebra, A, et al. Primary laparoscopic-assisted endorectal colon pull-through for Hirschsprung’s disease: A new gold standard. Ann Surg. 1999; 229:678–683.
45. de Lagausie, P, Berrebi, D, Geib, G, et al. Laparoscopic Duhamel procedure. Management of 30 cases. Surg Endosc. 1999; 13:972–974.
46. Hoffmann, K, Schier, F, Waldschmidt, J. Laparoscopic Swenson’s procedure in children. Eur J Pediatr Surg. 1996; 6:15–17.
47. De la Torre-Mondragon, L, Ortega-Salgado, JA. Transanal endorectal pull-through for Hirschsprung’s disease. J Pediatr Surg. 1998; 33:1283–1286.
48. Langer, JC, Minkes, RK, Mazziotti, MV, et al. Transanal one-stage Soave procedure for infants with Hirschsprung disease. J Pediatr Surg. 1999; 34:148–152.
49. De La Torre, L, Langer, JC. Transanal endorectal pull-through for Hirschsprung disease: Technique, controversies, pearls, pitfalls, and an organized approach to the management of postoperative obstructive symptoms. Semin Pediatr Surg. 2010; 19:96–106.
50. Nasr, A, Langer, JC. Evolution of the technique in the transanal pull-through for Hirschsprung disease: Effect on outcome. J Pediatr Surg. 2007; 42:36–39.
51. Sookpotarom, P, Vejchapipat, P. Primary transanal Swenson pull-through operation for Hirschsprung’s disease. Pediatr Surg Int. 2009; 25:767–773.
52. Sauer, CJE, Langer, JC, Wales, PW. The versatility of the umbilical incision in the management of Hirschsprung’s disease. J Pediatr Surg. 2005; 40:385–389.
53. Proctor, ML, Traubici, J, Langer, JC, et al. Correlation between radiographic transition zone and level of aganglionosis in Hirschsprung’s disease: Implications for surgical approach. J Pediatr Surg. 2003; 38:775–778.
54. Muller, CO, Mignot, C, Belarbi, N, et al. Does the radiographic transition zone correlate with the level of aganglionosis on the specimen in Hirschsprung’s disease? Pediatr Surg Int. 2012; 28:597–601.
55. Langer, JC, Durrant, AC, de la Torre, ML, et al. One-stage transanal Soave pullthrough for Hirschsprung disease: A multicenter experience with 141 children. Ann Surg. 2003; 238:569–576.
56. Kim, AC, Langer, JC, Pastor, AC, et al. Endorectal pull-through for Hirschsprung’s disease-a multicenter, long-term comparison of results: Transanal vs transabdominal approach. J Pediatr Surg. 2010; 45:1213–1220.
57. Stranzinger, E, DiPietro, MA, Teitelbaum, DH, et al. Imaging of total colonic Hirschsprung disease. Pediatr Radiol. 2008; 38:1162–1170.
58. Anderson, KD, Chandra, R. Segmental aganglionosis of the appendix. J Pediatr Surg. 1986; 21:852–854.
59. Burjonrappa, S, Rankin, L. ‘Hop the skip’ with extended segment intestinal biopsy in Hirschsprung’s disease. Intern J Surg Case Reports. 2012; 3:186–189.
60. Shen, C, Song, Z, Zheng, S, et al. A comparison of the effectiveness of the Soave and Martin procedures for the treatment of total colonic aganglionosis. J Pediatr Surg. 2009; 44:2355–2358.
61. Barrena, S, Andres, AM, Burgos, L, et al. Long-term results of the treatment of total colonic aganglionosis with two different techniques. Eur J Pediatr Surg. 2008; 18:375–379.
62. Marquez, TT, Acton, RD, Hess, DJ, et al. Comprehensive review of procedures for total colonic aganglionosis. J Pediatr Surg. 2009; 44:257–265.
63. Travassos, DV, van der Zee, DC. Is complete resection of the aganglionic bowel in extensive total aganglionosis up to the middle ileum always necessary? J Pediatr Surg. 2011; 46:2054–2059.
64. Diamond, IR, de Silva, N, Pencharz, PB, et al. Neonatal short bowel syndrome outcomes after the establishment of the first Canadian multidisciplinary intestinal rehabilitation program: Preliminary experience. J Pediatr Surg. 2007; 42:806–811.
65. Diamond, IR, Sterescu, A, Pencharz, PB, et al. The rationale for the use of parenteral omega-3 lipids in children with short bowel syndrome and liver disease. Pediatr Surg Int. 2008; 24:773–778.
66. Ruttenstock, E, Puri, P. A meta-analysis of clinical outcome in patients with total intestinal aganglionosis. Pediatr Surg Int. 2009; 25:833–839.
67. Wales, PW. Surgical therapy for short bowel syndrome. Pediatr Surg Int. 2004; 20:647–657.
68. Wales, PW, de Silva, N, Langer, JC, et al. Intermediate outcomes after serial transverse enteroplasty in children with short bowel syndrome. J Pediatr Surg. 2007; 42:1804–1810.
69. Ziegler, MM, Royal, RE, Brandt, J, et al. Extended myectomy-myotomy. A therapeutic alternative for total intestinal aganglionosis. Ann Surg. 1993; 218:504–509.
70. Sauvat, F, Grimaldi, C, Lacaille, F, et al. Intestinal transplantation for total intestinal aganglionosis: A series of 12 consecutive children. J Pediatr Surg. 2008; 43:1833–1838.
71. Temple, S, Shawyer, AC, Langer, JC. Is daily dilatation by parents necessary after surgery for Hirschsprung disease and anorectal malformations? J Pediatr Surg. 2012; 47:209–212.
72. Marty, TL, Matlak, ME, Hendrickson, M, et al. Unexpected death from enterocolitis after surgery for Hirschsprung’s disease. Pediatrics. 1995; 96:118–121.
73. Dasgupta, R, Langer, JC. Evaluation and management of persistent problems after surgery for Hirschsprung disease in a child. J Pediatr Gastroenterol Nutr. 2008; 46:13–19.
74. Yanchar, NL, Soucy, P. Long term outcomes of Hirschsprung’s disease: The patients’ perspective. J Pediatr Surg. 1999; 34:1152–1160.
75. Rintala, RJ, Pakarinen, MP. Outcome of anorectal malformations and Hirschsprung’s disease beyond childhood. Semin Pediatr Surg. 2010; 19:160–167.
76. Langer, JC. Persistent obstructive symptoms after surgery for Hirschsprung disease: Development of a diagnostic and therapeutic algorithm. J Pediatr Surg. 2004; 39:1458–1462.
77. Langer, JC, Winthrop, AL. Antegrade dilatation over a string for the management of anastomotic complications after a pull-through procedure. J Am Coll Surg. 1996; 183:411–412.
78. Lucha, PA, Jr., Fticsar, JE, Francis, MJ. The strictured anastomosis: Successful treatment by corticosteroid injections–report of three cases and review of the literature. Dis Colon Rectum. 2005; 48:862–865.
79. Mueller, CM, Beaunoyer, M, St-Vil, D. Topical mitomycin-C for the treatment of anal stricture. J Pediatr Surg. 2010; 45:241–244.
80. van Leeuwen, K, Teitelbaum, DH, Elhalaby, EA, et al. Long-term follow-up of redo pull-through procedures for Hirschsprung’s disease: Efficacy of the endorectal pull-through. J Pediatr Surg. 2000; 35:829–833.
81. Langer, JC. Repeat pullthrough surgery for complicated Hirschsprung disease: Indications, techniques, and results. J Pediatr Surg. 1999; 34:1136–1141.
82. Pena, A, Elicevik, M, Levitt, MA. Reoperations in Hirschsprung disease. J Pediatr Surg. 2007; 42:1008–1013.
83. Shayan, K, Smith, D, Langer, JC. Reliability of intraoperative frozen sections in the management of Hirschsprung disease. J Pediatr Surg. 2004; 39:1345–1348.
84. Ghose, SI, Squire, BR, Stringer, MD, et al. Hirschsprung’s disease: Problems with transition-zone pull-through. J Pediatr Surg. 2000; 35:1805–1809.
85. Coe, A, Collins, MH, Lawal, T, et al. Reoperation for Hirschsprung disease: Pathology of the resected problematic distal pull-through. Pediatr Dev Pathol. 2012; 15:30–38.
86. West, KW, Grosfeld, JL, Rescorla, FJ, et al. Acquired aganglionosis: A rare occurrence following pull-through procedures for Hirschsprung’s disease. J Pediatr Surg. 1990; 25:104–108.
87. Friedmacher, F, Puri, P. Residual aganglionosis after pull-through operation for Hirschsprung’s disease: A systematic review and meta-analysis. Pediatr Surg Int. 2011; 27:1053–1057.
88. White, FV, Langer, JC. Circumferential distribution of ganglion cells in the transition zone of children with Hirschsprung disease. Pediatr Dev Pathol. 2000; 3:216–222.
89. Medhus, AW, Bjornland, K, Emblem, R, et al. Liquid and solid gastric emptying in adults treated for Hirschsprung’s disease during early childhood. Scand J Gastroenterol. 2007; 42:34–40.
90. Zaslavsky, C, da Silveira, TR, Maguilnik, I. Total and segmental colonic transit time with radio-opaque markers in adolescents with functional constipation. J Pediatr Gastroenterol Nutr. 1998; 27:138–142.
91. Southwell, BR, Clarke, MC, Sutcliffe, J, et al. Colonic transit studies: Normal values for adults and children with comparison of radiological and scintigraphic methods. Pediatr Surg Int. 2009; 25:559–572.
92. Di Lorenzo, C, Solzi, GF, Flores, AF, et al. Colonic motility after surgery for Hirschsprung’s disease. Am J Gastroenterol. 2000; 95:1759–1764.
93. Mazziottti, MV, Langer, JC. Laparoscopic full-thickness intestinal biopsies in children. J Pediatr Gastroenterol Nutr. 2001; 33:54–57.
94. Yagmurlu, A, Harmon, CM, Georgeson, KE. Laparoscopic cecostomy button placement for the management of fecal incontinence in children with Hirschsprung’s disease and anorectal anomalies. Surg Endosc. 2006; 20:624–627.
95. Minkes, RK, Langer, JC. A prospective study of botulinum toxin for internal anal sphincter hypertonicity in children with Hirschsprung’s disease. J Pediatr Surg. 2000; 35:1733–1736.
96. Abbas Banani, S, Forootan, H. Role of anorectal myectomy after failed endorectal pull-through in Hirschsprung’s disease. J Pediatr Surg. 1994; 29:1307–1309.
97. Wildhaber, BE, Pakarinen, M, Rintala, RJ, et al. Posterior myotomy/myectomy for persistent stooling problems in Hirschsprung’s disease. J Pediatr Surg. 2004; 39:920–926.
98. Koivusalo, AI, Pakarinen, MP, Rintala, RJ. Botox injection treatment for anal outlet obstruction in patients with internal anal sphincter achalasia and Hirschsprung’s disease. Pediatr Surg Int. 2009; 25:873–876.
99. Jiang da, P, Xu, CQ, Wu, B, et al. Effects of botulinum toxin injection on anal achalasia after pull-through operations for Hirschsprung’s disease: A 1-year follow-up study. Inter J Colorectal Dis. 2009; 24:597–598.
100. Patrus, B, Nasr, A, Langer, JC, et al. Intrasphincteric botulinum toxin decreases the rate of hospitalization for postoperative obstructive symptoms in children with Hirschsprung disease. J Pediatr Surg. 2011; 46:184–187.
101. Di Lorenzo, C. Constipation. In: Hyman PE, ed. Pediatric Gastrointestinal Motility Disorders. New York, NY: Academy Professional Information Services; 1994:129–144.
102. Blum, NJ, Taubman, B, Nemeth, N. During toilet training, constipation occurs before stool toileting refusal. Pediatrics. 2004; 113:e520–e522.
103. Levitt, M, Pena, A. Update on pediatric faecal incontinence. Eur J Pediatr Surg. 2009; 19:1–9.
104. Wilson-Storey, D, Scobie, WG, McGenity, KG. Microbiological studies of the enterocolitis of Hirschsprung’s disease. Arch Dis Child. 1990; 65:1338–1339.
105. Mattar, AF, Coran, AG, Teitelbaum, DH. MUC-2 mucin production in Hirschsprung’s disease: Possible association with enterocolitis development. J Pediatr Surg. 2003; 38:417–421.
106. Imamura, A, Puri, P, O’Briain, DS, et al. Mucosal immune defence mechanisms in enterocolitis complicating Hirschsprung’s disease. Gut. 1992; 33:801–806.
107. Haricharan, RN, Seo, JM, Kelly, DR, et al. Older age at diagnosis of Hirschsprung disease decreases risk of postoperative enterocolitis, but resection of additional ganglionated bowel does not. J Pediatr Surg. 2008; 43:1115–1123.
108. Pastor, AC, Osman, F, Teitelbaum, DH, et al. Development of a standardized definition for Hirschsprung’s-associated enterocolitis: A Delphi analysis. J Pediatr Surg. 2009; 44:251–256.
109. Marty, TL, Seo, T, Sullivan, JJ, et al. Rectal irrigations for the prevention of postoperative enterocolitis in Hirschsprung’s disease. J Pediatr Surg. 1995; 30:652–654.
110. Menezes, M, Corbally, M, Puri, P. Long-term results of bowel function after treatment for Hirschsprung’s disease: A 29-year review. Pediatr Surg Int. 2006; 22:987–990.
111. Caniano, DA, Teitelbaum, DH, Qualman, SJ. Management of Hirschsprung’s disease in children with trisomy 21. Am J Surg. 1990; 159:402–404.
112. Morabito, A, Lall, A, Gull, S, et al. The impact of Down’s syndrome on the immediate and long-term outcomes of children with Hirschsprung’s disease. Pediatr Surg Int. 2006; 22:179–181.
113. Travassos, D, van Herwaarden-Lindeboom, M, van der Zee, DC. Hirschsprung’s disease in children with Down syndrome: A comparative study. Eur J Pediatr Surg. 2011; 21:220–223.
114. Moore, SW, Tshifularo, N. Hirschsprung’s disease in the neurologically challenged child. Intern J Adolesc Med Health. 2011; 23:223–227.
115. Puri, P. Variant Hirschsprung’s disease. J Pediatr Surg. 1997; 32:149–157.
116. Feichter, S, Meier-Ruge, WA, Bruder, E. The histopathology of gastrointestinal motility disorders in children. Sem Pediatr Surg. 2009; 18:206–211.
117. Meier-Ruge, W. Casuistic of colon disorder with symptoms of Hirschsprung’s disease. Verh Dtsch Ges Pathol. 1971; 55:506–510.
118. Ryan, DP. Neuronal intestinal dysplasia. Sem Pediatr Surg. 1995; 4:22–25.
119. Csury, L, Pena, A. Intestinal neuronal dysplasia: Myth or reality? Pediatr Surg Int. 1995; 10:441–446.
120. Koletzko, S, Jesch, I, Faus-Kebler, T, et al. Rectal biopsy for diagnosis of intestinal neuronal dysplasia in children: A prospective multicentre study on interobserver variation and clinical outcome. Gut. 1999; 44:853–861.
121. Kapur, RP. Neuronal dysplasia: A controversial pathological correlate of intestinal pseudo-obstruction. Am J Med Gen. 2003; 122A:287–293.
122. Zhang, HY, Feng, JX, Huang, L, et al. Diagnosis and surgical treatment of isolated hypoganglionosis. World J Pediatr. 2008; 4:295–300.
123. Tatekawa, Y, Kanehiro, H, Kanokogi, H, et al. The evaluation of meconium disease by distribution of cathepsin D in intestinal ganglion cells. Pediatr Surg Int. 2000; 16:53–55.
124. Davidson, M, Bauer, CH. Studies of distal colonic motility in children IV: Achalasia of the distal rectal segment despite presence of ganglia in the myenteric plexuses of this area. Pediatrics. 1958; 21:746–761.
125. De Caluwe, D, Yoneda, A, Akl, U, et al. Internal anal sphincter achalasia: Outcome after internal sphincter myectomy. J Pediatr Surg. 2001; 36:736–738.
126. Heikkinen, M, Lindahl, HG, Rintala, RJ. Long-term outcome after internal sphincter myectomy for internal sphincter achalasia. Pediatr Surg Int. 2005; 21:84–87.
127. Messineo, A, Codrich, D, Monai, M, et al. The treatment of internal anal sphincter achalasia with botulinum toxin. Pediatr Surg Int. 2001; 17:521–523.
128. Millar, AJ, Steinberg, RM, Raad, J, et al. Anal achalasia after pull-through operations for Hirschsprung’s disease—preliminary experience with topical nitric oxide. Eur J Pediatr Surg. 2002; 12:207–211.
129. Meier-Ruge, W. Ultrashort segment Hirschsprung disease. An objective picture of the disease substantiated by biopsy. Z Kinderchir. 1985; 40:146–150.
130. Osifo, OD, Okolo, CJ. Outcome of trans-anal posterior anorectal myectomy for the ultrashort segment Hirschsprung’s disease–Benin City experience in five years. Niger Postgrad Med J. 2009; 16:213–217.
131. Meier-Ruge, WA, Bruder, E, Holschneider, AM, et al. Diagnosis and therapy of ultrashort Hirschsprung’s disease. Eur J Pediatr Surg. 2004; 14:392–397.
132. Meier-Ruge, WA. Desmosis of the colon: A working hypothesis of primary chronic constipation. Eur J Pediatr Surg. 1998; 8:209–303.
133. Marshall, DG, Meier-Ruge, WA, Chakravarti, A, Langer, JC. Chronic constipation due to Hirschsprung’s disease and desmosis coli in a family. Pediatr Surg Int. 2002; 18:110–114.
134. Hotta, R, Natarajan, D, Thapar, N. Potential of cell therapy to treat pediatric motility disorders. Sem Pediatr Surg. 2009; 18:263–273.
135. Zhang, D, Brinas, IM, Binder, BJ, et al. Neural crest regionalization for enteric nervous system formation: Implications for Hirschsprung’s disease and stem cell therapy. Dev Biol. 2010; 339:280–294.
136. Tsai, YH, Murakami, N, Gariepy, CE. Postnatal intestinal engraftment of prospectively selected enteric neural crest stem cells in a rat model of Hirschsprung disease. Neurogastroenterol Motil. 2011; 23:362–369.