156 Hereditary haemorrhagic telangiectasia (Rendu–Osler–Weber disease)
Instruction
Examine the patient’s face and obtain a relevant history.
Salient features
History
• Does it run in the family (autosomal dominant)?
• Is there a history of GI bleeding?
• Is there a history of epistaxis?
• Is there a history of repeated blood transfusions?
• Is there a history of dyspnoea, fatigue, cyanosis or polycythaemia (pulmonary arteriovenous malformations)?
• Is there a history of headaches, subarachnoid haemorrhage (cerebral arteriovenous malformations)?
Examination
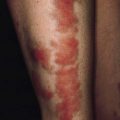
• Punctiform lesions and dilated small vessels present on the face, in particular around the mouth (Fig. 156.1)
• The patient may be pale (as a result of iron-deficiency anaemia).
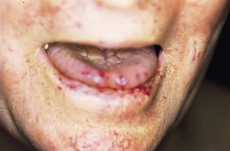
• Look into the patient’s mouth and inspect the tongue and palate for telangiectasia.
• Examine the nail beds, arms, trunk for telangiectasia.
• Examine the chest for bruits (pulmonary arteriovenous malformations with a predilection for lower lobes).
• Look for signs of cardiac failure caused by left-to-right shunting and hepatic bruits (both from hepatic arteriovenous malformations) (N Engl J Med 2000;343:938–52).
Questions
Advanced-level questions
What do you know about the pathology of the condition?
Skin
• The small telangiectasias are focal dilatations of postcapillary venules with prominent stress fibres in the pericytes along the luminal border.
• In fully developed telengiectasia, there is marked dilatation of the venules, which are also convoluted. They extend along the entire dermis, with excessive layers of smooth muscle devoid of elastic fibres. These often are directly connected to dilated arterioles.
• Mononuclear cells, predominantly lymphocytes, accumulate in the perivascular space.
What are the clinical criteria for diagnosing hereditary haemorrhagic telangiectasia?
What are the complications of hereditary telangiectasia?
• Epistaxis (usually begins by the age of 10 years and by age 21 in most; it becomes more severe in later decades in about two-thirds of affected patients) (N Engl J Med 1995;333:918–24)
• GI haemorrhage (usually does not manifest until the fifth or sixth decade). Arteriovenous malformations, angiodysplasias and telangiectasias are present in the stomach, duodenum, small bowel, colon and liver
• Symptomatic liver involvement: the typical clinical presentations include high-output heart failure, portal hypertension and biliary disease (N Engl J Med 2000;343:931)
• Haemoptysis, cyanosis, clubbing, cerebral abscess and embolic stroke from the pulmonary arteriovenous malformations
• Headache and subarachnoid haemorrhage
• High-output cardiac failure is almost always associated with shunts from the hepatic artery to the hepatic veins (N Engl J Med 2000;343:931–6).
How would you manage such patients?
• Anaemia: ferrous sulfate, multiple blood transfusions
• Epistaxis: oestrogen, cauterization, septal dermatoplasty, laser ablation and transcatheter embolotherapy of arteries leading to the nasal mucosa
• Cutaneous telangiectasia: cosmetic therapy with topical agents, laser ablation
• Pulmonary arteriovenous malformations: embolotherapy, surgical resection or ligation of arterial supply
• GI telangiectasia: blood transfusions, photocoagulation, oestrogen–progestogen therapy
• Brain and spinal cord arteriovenous malformations: embolotherapy, neurosurgery, stereotactic surgery
• Active bleeding: ε-aminocaproic acid (N Engl J Med 1994;330:1789, N Engl J Med 1994;330:j1822)
• Anti-vascular endothelial growth factor antibody, bevacizumab (N Engl J Med 2009;360:2143).