[level-membership-for-surgery-category]4
Hereditary, Developmental, and Environmental Influences on the Formation of Dentofacial Deformities
• Development of the Head and Neck
• Syndromes and Anomalies that Include Dentofacial Deformities
• Hereditary Tendencies in the Development of Dentofacial Deformities
• Environmental and Neuromotor Effects on Skeletal Growth
• Effects of Trauma on Skeletal Growth
The term dentofacial deformity is generally used to describe a significant disproportion of the jaws in association with malocclusion. It is estimated that, at a minimum, 5% of the population will have a discrepancy that falls into the developmental dentofacial deformity category. Another 45% of the population will fall into a much larger group of those with malocclusion but without what is considered a “notable” jaw discrepancy component.2,3,5,56,65,72 After jaw growth is complete (i.e., 14 to 16 years of age in girls and 16 to 18 years of age in boys), the definitive reconstruction of a dentofacial deformity can go forward.
Development of the Head and Neck
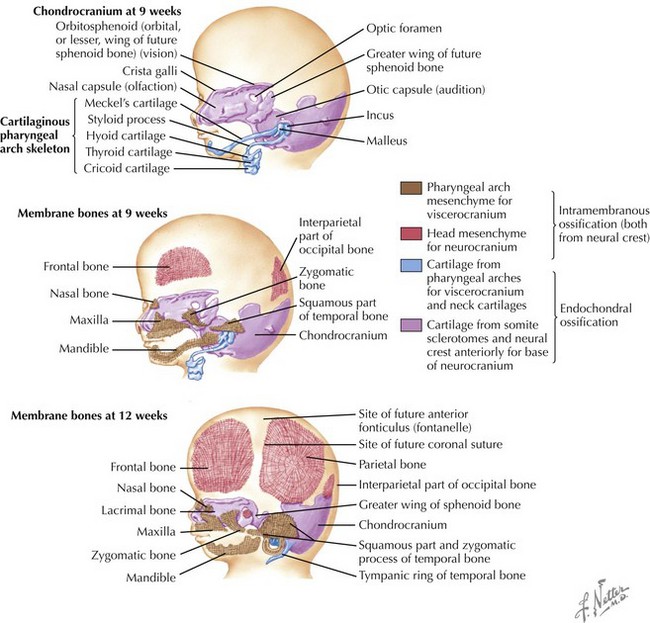
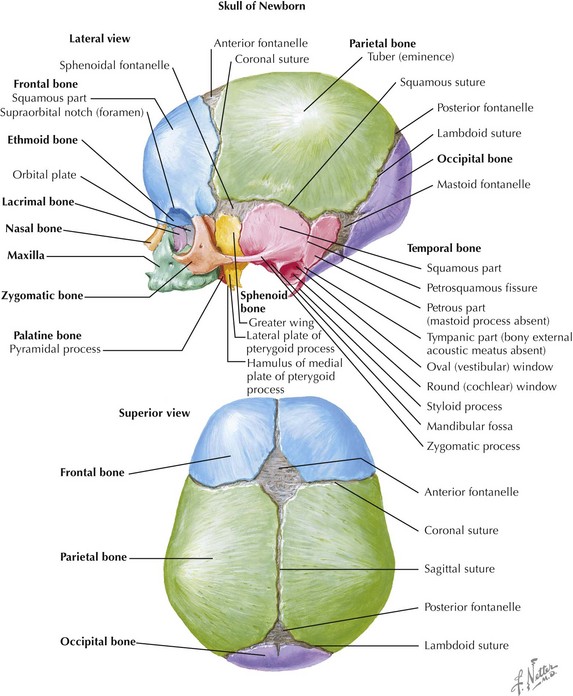
Skull
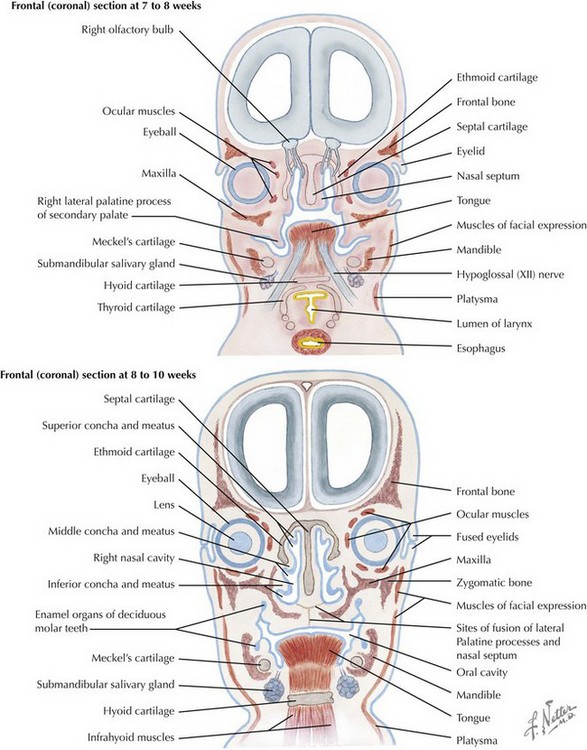
The skull is formed from the lateral plate mesoderm (the neck region), the paraxial mesoderm, and the neural crest (Fig. 4-1). The bony skull is formed by one of two mechanisms: intramembranous ossification or endochondral ossification. Skull development is divided into two parts: the viscerocranium and the neurocranium. The viscerocranium forms the bones of the face, whereas the neurocranium forms the bones of the cranial base and the cranial vault. The neurocranium can be divided into the membranous neurocranium and the cartilaginous neurocranium. The anterior fontanel (bregma) has a wide range of expected closure between 4 and 26 months of age. The posterior fontanel (lambda) generally closes between 1 and 2 months of age.
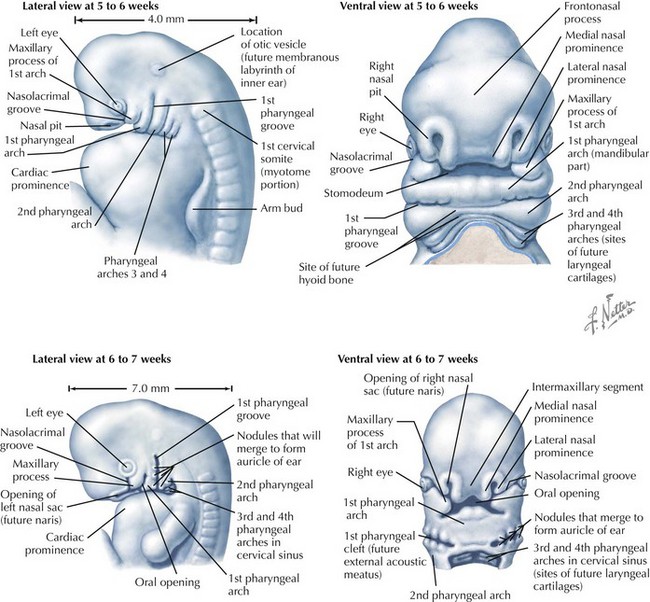
Face
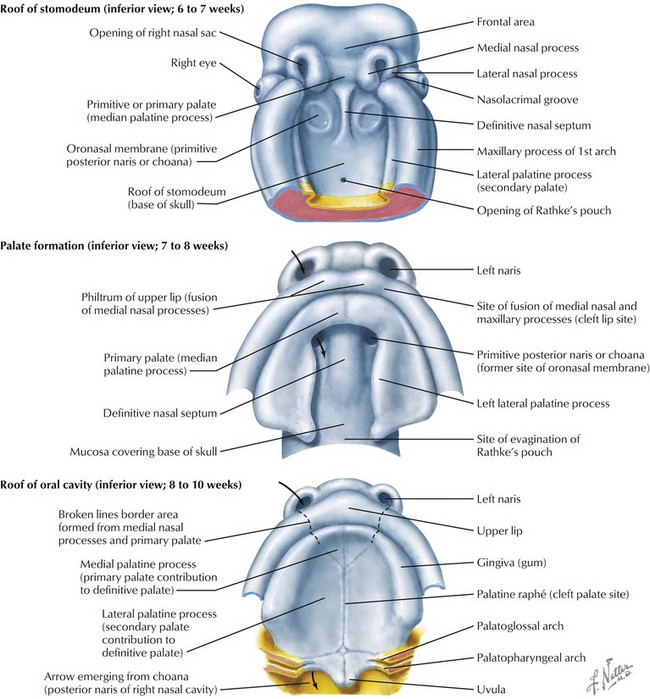
The face is formed mainly from the neural crest, which makes three “swellings” that surround the stomodeum (Fig. 4-2). The three swellings are the frontonasal prominence, the maxillary prominence (from the first pharyngeal arch), and the mandibular prominence (from the first pharyngeal arch).
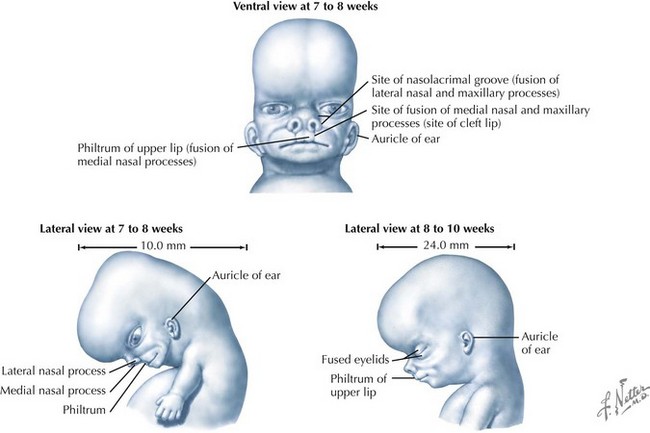
Palate
The palate is formed by the primary palate (intermaxillary segment) and the secondary palate (protrusions from the lateral prominences) (Fig. 4-3). The intermaxillary segment (primary palate) is the initial portion of the palate to develop. It contains the central and lateral incisors. Swellings of the maxillary prominence form shelves that project medially but that are separated by the tongue. When the tongue no longer occupies the space between the palatal shelves, these processes fuse together to form the secondary palate. The primary and secondary palatal tissues all meet at the incisal foramen. The primary and secondary palates and the nasal septum fuse to form the definitive palate.
Syndromes and Anomalies that Include Dentofacial Deformities
Many of the syndromes and anomalies that involve the cranio-orbito-zygomatic (upper face) region also affect the maxillomandibular (lower face) location.1,20,124 These conditions may involve any of the tissue components, including the skin, the muscles, the nerves, the fat, the vessels, the bone, the teeth, the cartilage, and the associated viscera (i.e., brain, ears, eyes, salivary glands, sinuses). Known syndromes and anomalies comprise only a small proportion of the dentofacial deformity group (see Chapters 27 through 31).19 These conditions can be further subdivided according to etiology or tissue of origin.
Deficiencies of Midline Tissue Structures
Holoprosencephalies involve variable deficiencies of midline tissues. These structural deficiencies have been documented to occur early during embryologic development.16,18,23,27 They may range from severe brain anomalies to simple absence or hypoplasia of the corpus callosum. Severe facial anomalies such as cyclopia, ethmocephaly, and cebocephaly are always associated with severe holoprosencephaly, and they are incompatible with life. This can also be true of premaxillary agenesis; however, in some instances, patients may survive for several months or even years. There are reported cases of premaxillary agenesis with normal head circumference and no brain abnormalities (i.e., without holoprosencephaly), although these patients sometimes have mild mental deficiencies. Syntelencephaly (the absence or hypoplasia of the corpus callosum or absent or hypoplastic olfactory tracts and bulbs) is compatible with life and should thus be treated. Associated facial anomalies include retinal colobomas, cleft lip, cleft palate, single maxillary central incisor, and midface deficiencies. Holoprosencephaly has multiple causes and includes many chromosomal types, many monogenetic disorders, and at least three teratogens.
Deficiencies and Anomalies of Neural Crest Origin
Most of the tissues of the face, including the muscular and skeletal components, are derived from the mesoderm. Interestingly, throughout the rest of the body, these components (i.e., muscle and skeleton) are derived from an ectodermal origin.19 The facial mesodermal elements develop from the neural crest and then migrate downward beside the neural tube and laterally under the surface ectoderm. When the neural crest cells have completed migration to their specific location, then facial development is dominated by specific growth centers, the formation of visceral structures (e.g., brain, eyes, sinuses), and the differentiation of the tissue layers. Facial anomalies of neural crest origin are thought to arise when the neuroepithelium has apoptosis (i.e., the cells die).25 This results in less neuroepithelium to migrate through into the facial locations and to ultimately differentiate. Current knowledge of Treacher Collins syndrome indicates that this is not a migration problem but simply a lack of cells to migrate (Figs. 4-4 and 4-5).
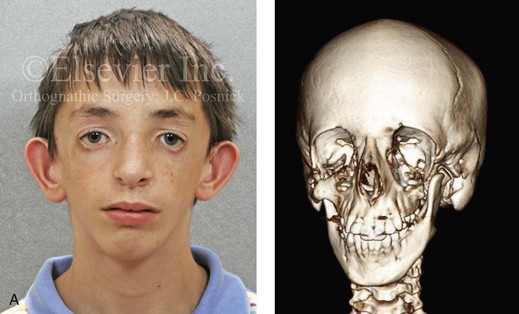
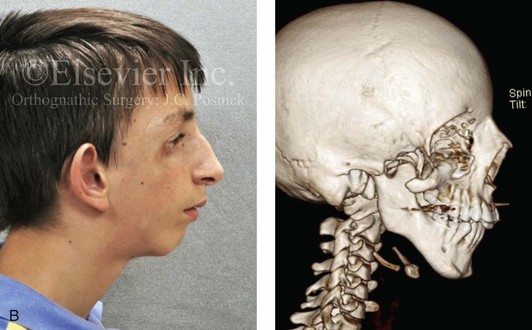
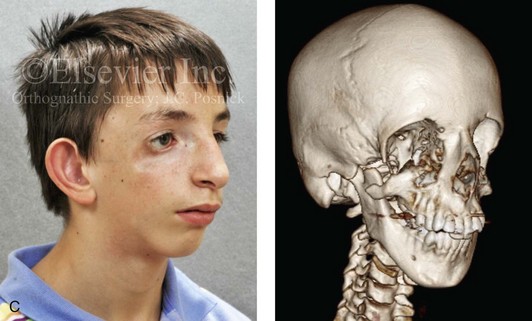
Figure 4-4 An 11-year-old boy who was born with Treacher Collins syndrome has typical malformations of the soft tissues and the skeletal structures within the first and second branchial arches on both sides. There is significant hypoplasia of each zygomatic complex, the orbits, and the maxillomandibular structures (Kaban type II-A mandible). The soft tissues of the adnexal region are deficient. The external ears are malformed, but all parts are present. The central part of the face is fully formed. A, Frontal facial and computed tomography scan views. B, Profile facial and computed tomography scan views. C, Oblique facial and computed tomography scan views.
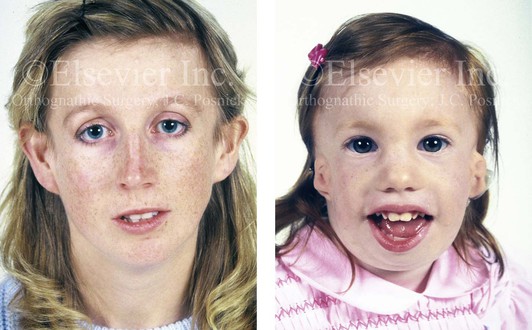
Figure 4-5 This mother and daughter demonstrate the extent of variation of expression of Treacher Collins syndrome within a family. The mother was not aware that she carried the Treacher Collins gene until after the birth of her daughter. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment. J Oral Maxillofac Surg 55:1120–1133, 1997.
Teratogens that are known to result in head and neck (neural crest) congenital anomalies include cytomegalovirus (microcephaly, hydrocephaly, microophthalmia); Dilantin (cleft lip and palate); vitamin D excess (premature suture closure); Valium (cleft lip and palate); Rubella virus (microophthalmia, cataracts, deafness); and thalidomide (variations of hemifacial microsomia and Treacher Collins syndrome). Despite these known relationships, teratogenic agents are not the most frequent causes of these syndromes.1
The branchial arch syndromes are neural crest anomalies that comprise an etiologically heterogeneous group of disorders.14 They account for only a small percentage of the patients who present to the surgeon or orthodontist in need of jaw reconstruction. The best known of these are hemifacial microsomia, its variant Goldenhar syndrome, and Treacher Collins syndrome.14 Hemifacial microsomia affects aural, zygomatic, and mandibular growth at a minimum (Fig. 4-6). The disorder may be mild or severe, and it is generally limited to one side of the face. However, bilateral involvement, with more severe expression on one side, is also known to occur. Goldenhar syndrome, which is a variant of hemifacial microsomia, also includes epibulbar dermoids and vertebral anomalies. Involvement is often but not always limited to the face. There may be cardiac, renal, skeletal, and central nervous system anomalies (see Chapter 28). At a minimum, patients with Treacher Collins syndrome demonstrate the physical findings of bilateral zygomatic hypoplasia, down-slanting palpebral fissures, malformed ears, and micrognathia (see Chapter 27). Inheritance occurs in an autosomal-dominant fashion, and expressivity varies (see Figs. 4-4 and 4-5). The syndrome maps to 5q32-q33.1, and mutations in the TCOF1 gene are of the nonsense, insertion, deletion, or splice-site types.14,30,139
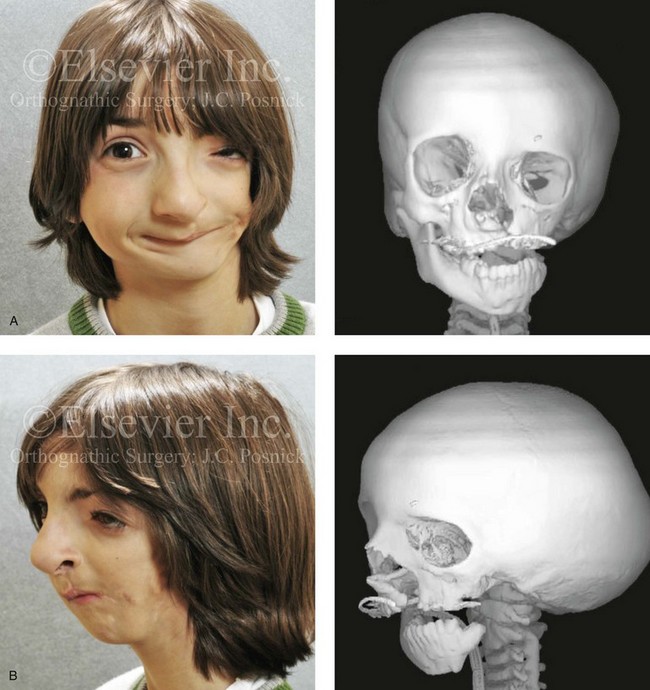
Figure 4-6 A 7-year-old boy born with hemifacial microsomia and unilateral cleft lip and palate (left side) has typical malformations of the soft tissues and skeletal structures within the first and second branchial arches on the left side. There is an absence of the left zygomatic complex and hypoplasia of the orbit, the anterior cranial vault, and the maxilla. The mandibular malformation is a Kaban type III. The soft tissues of the left external ear, the ear canal adnexal structures, and the cheek region are markedly deficient. A, Frontal facial and computed tomography scan views. B, Oblique facial and computed tomography scan views.
Clefting of the Lip and Palate
The most prevalent congenital defect of dentofacial development is clefting of the lip, the palate, or both (Figures 4-7 through 4-19). This condition occurs in approximately 1 in 1000 whites, 1 in 500 Asians, and 1 in 2000 blacks.27 The etiology of clefts is often complex and multifactorial. Evidence supports the view that genetic factors are associated with orofacial clefting.37 In twins with cleft lips and palates, concordance is far greater among monozygotic twins (40%) as compared with dizygotic twins (4.2%). In twins with isolated cleft palate, concordance is also higher among monozygotic twins (35%) as compared with dizygotic twins (7.8%).138 Nevertheless, orofacial clefting is heterogeneous and variable, and it is likely determined by a number of major genes, minor genes, environmental factors, and a developmental threshold.37 Some syndromes associated with clefting in which specific gene mutations have been identified include van der Woude syndrome (IRF6), popliteal pterygium syndrome (IRF6), autosomal-recessive cleft palate/ectodermal dysplasia (PVRL1), hypodontia/clefting (MSX1), Hay-Wells syndrome (TP63), and cleft palate/ankyloglossia (TBX22). These mutations explain only about 5% of clefting cases, with an additional 10% to 15% of cases explained by variations involving the IRF6 gene.110 Although orofacial clefting is caused by a malformation, the development of secondary deformities of the jaws after birth is well known (see Chapters 32, 33, and 34). For the most part, maxillary hypoplasia in patients with orofacial clefts is felt to be the result of palate surgical interventions carried out during infancy and early childhood and not from the primary congenital anomaly. Although van der Woude and Stickler are just 2 of more than 100 clefting syndromes, they demonstrate the importance of having an awareness of associated anomalies to assist with family counseling and to achieve effective reconstruction.
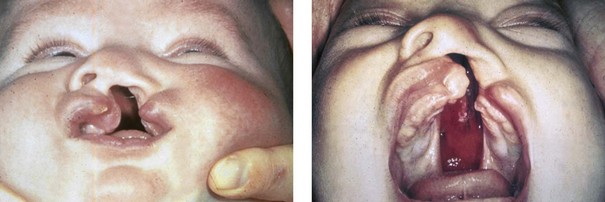
Figure 4-7 Cleft lip and palate represents a spectrum of morphologic findings that are initially dependent on the individual’s specific anomalies but that are then affected by surgical intervention during growth. A newborn with a complete (left side) unilateral cleft lip and palate is shown.
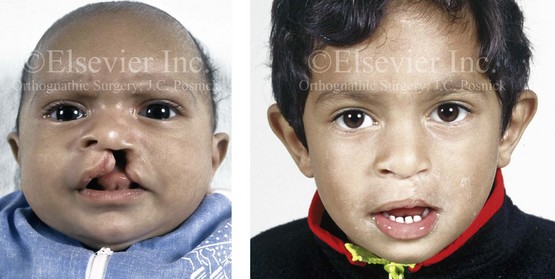
Figure 4-8 A Hispanic child was born with complete clefting of the left primary lip and palate. He was adopted when he was approximately 1 year old, before repair. He then underwent single-stage primary lip, nasal, and palate repair. He is shown before repair and then at the age of 2 years after single-stage repair.
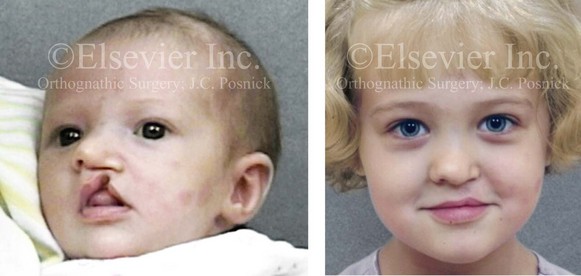
Figure 4-9 A child was born with an incomplete cleft of the left lip that also involved the alveolar ridge back to the incisal foramen. She is shown before primary lip and nasal reconstruction and then at the age of 5 years before alveolar cleft grafting.
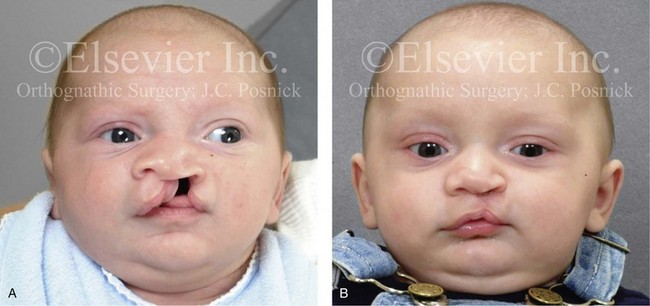
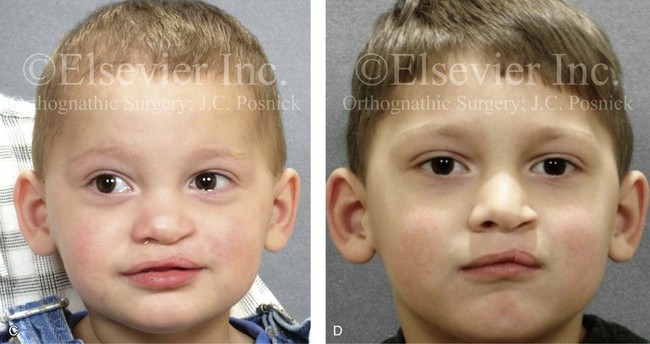
Figure 4-10 A child was born with a complete (left side) unilateral cleft lip and palate. He is shown before and at intervals after primary lip and palate repair. A, He is shown before primary lip and nasal reconstruction and B, at the age of 10 months, just before cleft palate repair. C, The same child at the age of 3 years and D, at the age of 8 years just before mixed dentition bone grafting.
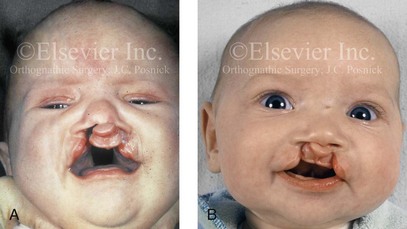
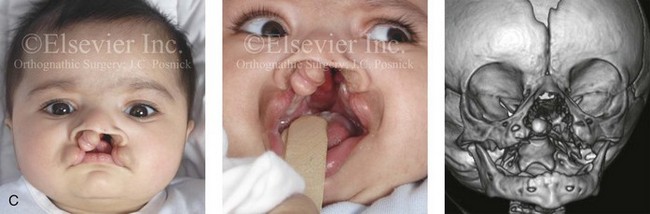
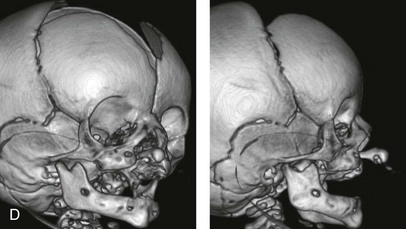
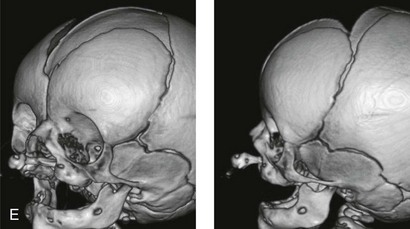
Figure 4-11 Three newborns with bilateral cleft lip and palate (BCLP) are shown. A, A newborn with complete BCLP. B, A newborn with BCLP but with partial attachment of the lip and the nasal floor on the left side. C, A 3-month-old infant with complete BCLP is shown. The premaxilla is attached to the septum of the nose; it is forwardly projecting without attachment to the lateral lip segments. The wide separation of the palatal shelves is also demonstrated by the intraoral view. D and E, A series of computed tomography scan views of the maxillofacial complex of the infant shown in C demonstrate the skeletal anatomy associated with BCLP.
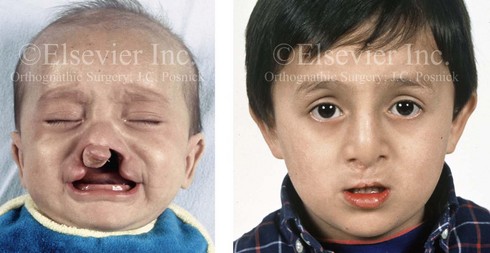
Figure 4-12 A child born with complete bilateral cleft lip and palate. He is shown before and then 7 years after primary lip and palate repair, just before mixed dentition bone grafting.
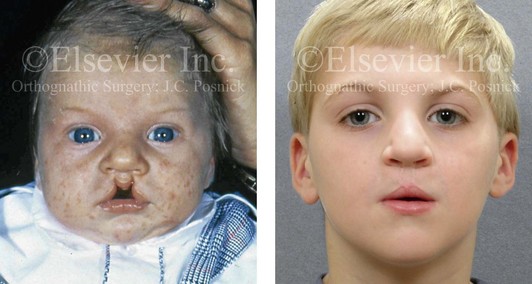
Figure 4-13 A child born with incomplete bilateral cleft lip and palate. He is shown before and then 8 years after primary lip and palate repair, just before mixed dentition bone grafting.
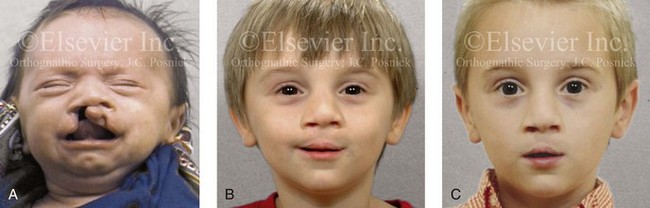
Figure 4-14 A child born with a complete cleft of the right lip and palate and incomplete clefting of the left lip. He is shown A, before and then B, at 2 and C, 8 years after primary lip and palate repair. The last photo was taken just before mixed dentition bone grafting.
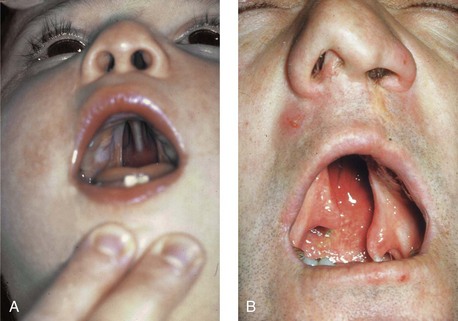
Figure 4-15 A, A child born with complete clefting of the secondary palate (i.e., incisal foramen through the uvula) is shown. B, An adult born with a complete unilateral cleft lip and palate is also shown. He underwent lip repair during childhood, but the cleft palate was neglected. Note the severe septal deviation that obstructs the right nasal valve.
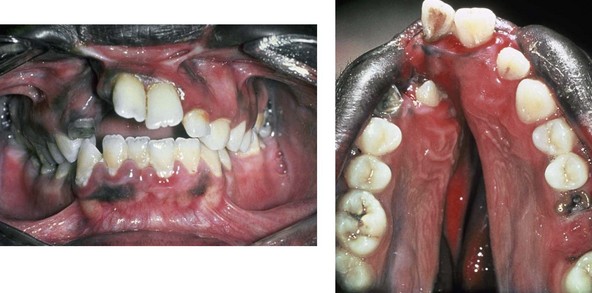
Figure 4-16 A teenage boy from Jamaica who was born with a bilateral cleft lip and a unilateral cleft of the alveolar ridge and palate. He underwent rudimentary lip repair as a child. The unrepaired clefted alveolus and palatal anatomy are shown. Note the normal growth parameters of the upper jaw when the palate is unrepaired. With a lack of continuity of the alveolar ridge, there has been typical distorted growth of the maxilla.
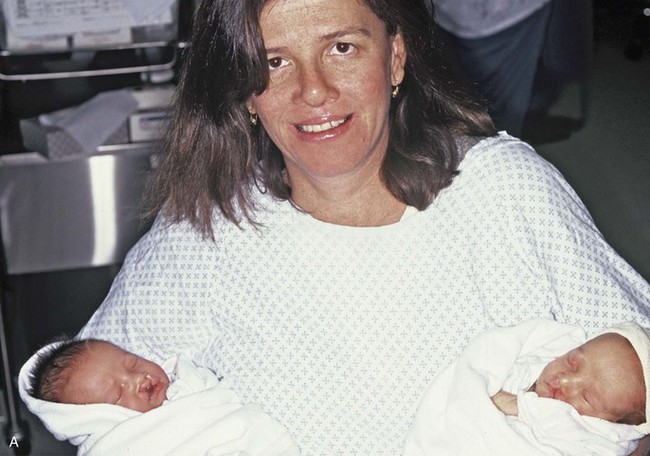
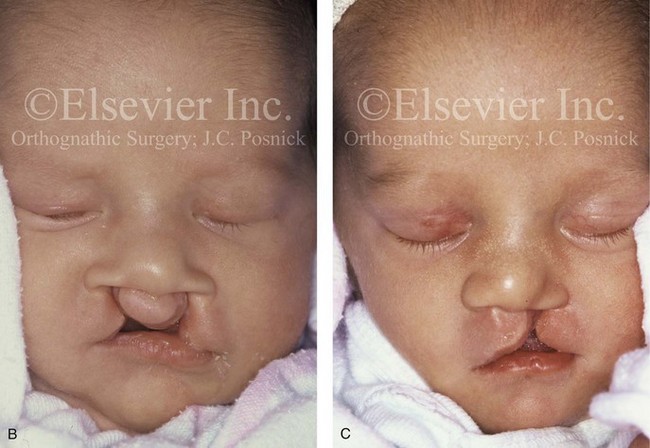
Figure 4-17 A woman who was born with bilateral cleft lip and palate and pitting of the lower (central) lip. At the time of her pregnancy, an ultrasound confirmed twins, one of which was suspected of having bilateral cleft lip and palate and the other of having unilateral cleft lip and palate. This was documented at the time of delivery. A, A family with van der Woude syndrome, including a mother with a repaired bilateral cleft lip and palate and newborn twins. B, Twin A with bilateral cleft lip and palate. C, Twin B with unilateral cleft lip and palate.

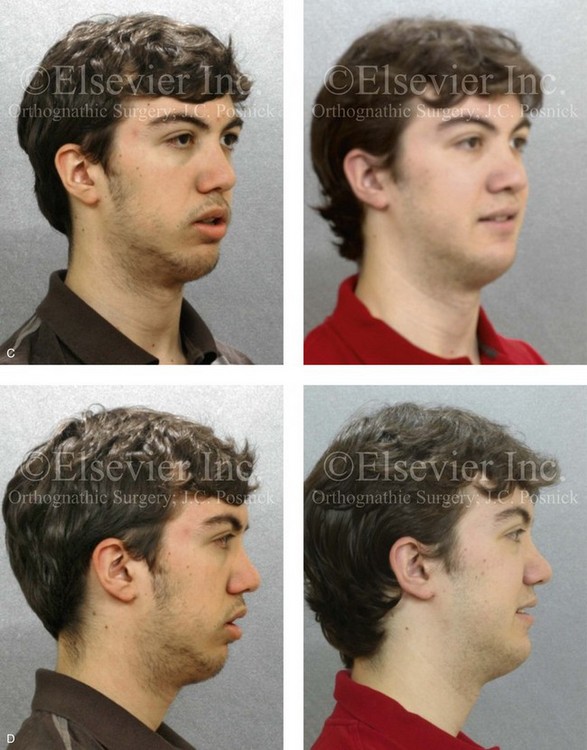
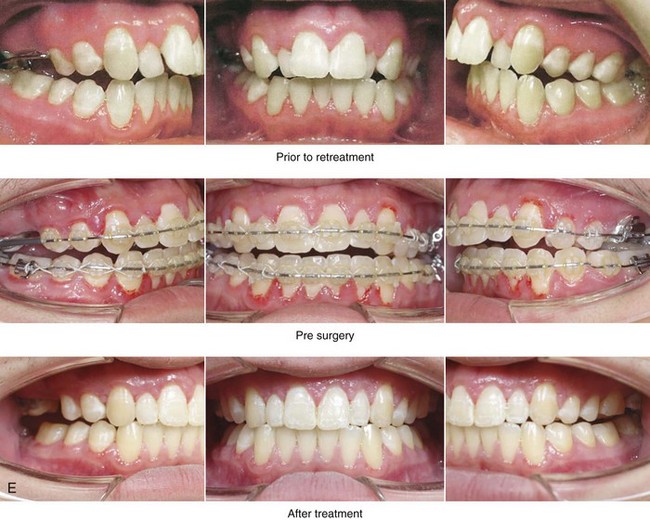
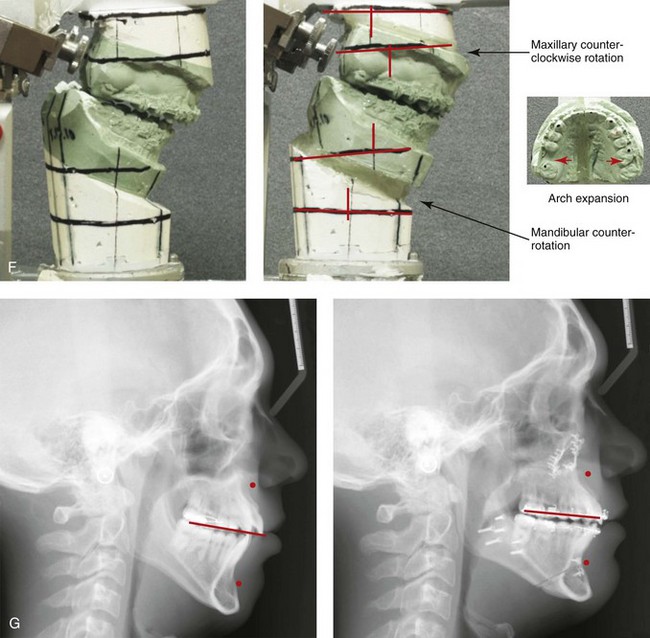
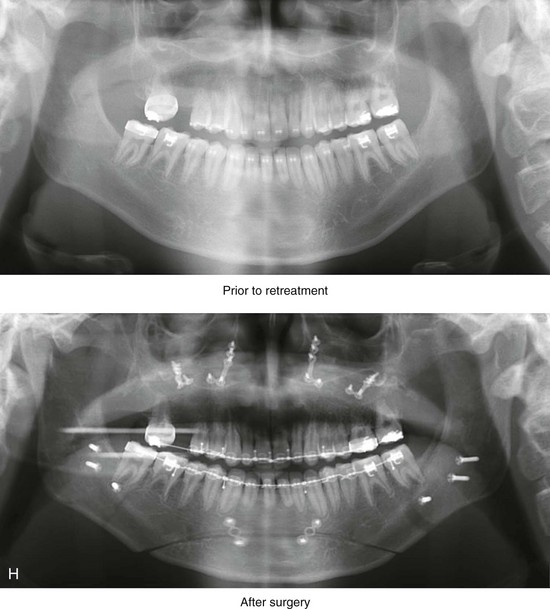
Figure 4-18 A 16-year-old boy who was born with Stickler syndrome (type II collagen mutation). At the time of birth, Pierre Robin sequence was appreciated. The patient underwent repair of the cleft palate before he was 1 year old. He had positive eye findings, and he required the treatment of a retinal detachment during his teenage years. A small cataract is being followed. He arrived for the evaluation of a jaw deformity and malocclusion characterized by maxillomandibular deficiency with anterior open-bite malocclusion. This was combined with chronic obstructive nasal breathing and a long face growth pattern. Attempted growth modification and camouflage orthodontics earlier during life were ineffective. There was generalized root deficiency throughout the maxillary and mandibular dentition, likely as a result of the collagenopathy. A, Frontal views in repose before and after treatment. B, Frontal views with smile before and after treatment. The patient agreed to an orthodontic and surgical approach. Further orthodontic (dental) decompensation was cautiously carried out as a result of the compromised periodontal apparatus. The procedures included maxillary Le Fort I osteotomy (vertical intrusion, horizontal advancement, counterclockwise rotation, and arch expansion); bilateral sagittal split ramus osteotomies (horizontal advancement and counterclockwise rotation); osseous genioplasty (vertical shortening and horizontal advancement); and septoplasty, inferior turbinate reduction, and nasal floor recontouring. C, Oblique facial views before and after treatment. D, Profile views before and after treatment. E, Occlusal views before retreatment, with orthodontics in progress, and after treatment. F, Articulated dental casts that indicate analytic model planning. G, Lateral cephalometric radiographs before and after treatment. H, Panorex radiographs before and after treatment that demonstrate generalized limited root formation. The right maxillary first molar was lost 5 years earlier as a result of limited roots. The right maxillary first molar was lost 1½ years after surgery as a result of limited root support; the right mandibular first molar will likely be lost to similar pathology. Periodontal evaluation and treatment are ongoing. Future plans are for the placement of dental implants in the right posterior maxilla and mandible.
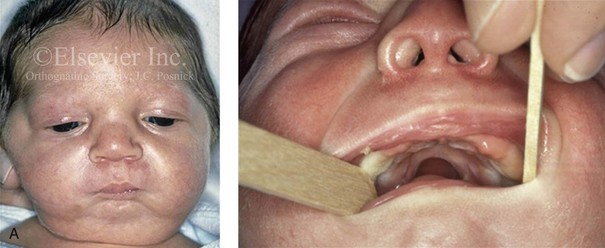
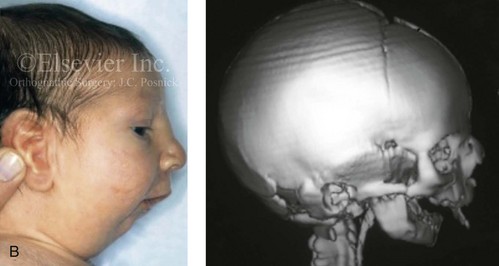
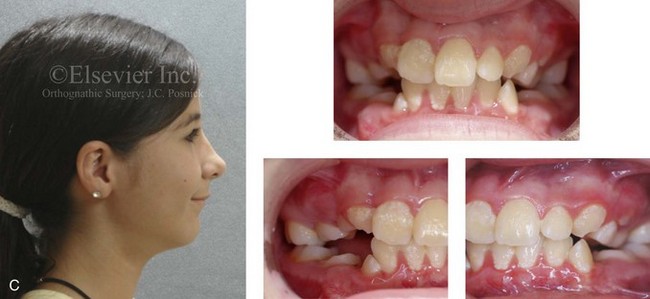
Figure 4-19 A child born with Robin sequence, which consists of retrognathia with glossoptosis, clefting of the secondary palate, and a degree of respiratory distress. A, Frontal facial and intraoral views demonstrating clefting of the secondary palate. B, Profile facial view indicating retrognathia. Computed tomography scan demonstrating retrognathia but with all of the components of the jaw present. For this child, the small mandible is the result of deforming forces during fetal development rather than the result of a malformation. Catch-up growth of the mandible is anticipated during childhood. C, Profile and occlusal views when the patient was 9 years old, with expected catch-up growth.
Van der Woude Syndrome
In 1845, Demarquay was the first to report the occurrence of congenital sinuses of the lower lip in combination with cleft lip and palate; this condition became known as van der Woude syndrome. This syndrome occurs in roughly 1% to 2% of patients with lip and palatal clefts. Van der Woude syndrome has an autosomal-dominant inheritance pattern; it has approximately 90% penetrance, and it is variably expressed (see Fig. 4-17). Manifestations of the syndrome outside of the oral and facial regions are unusual. Generally, the pits are characterized as depressions that are observed on the vermilion border of the lower lip; they are usually bilateral and symmetrically placed, although an asymmetrical pit or single central pit may occur. Roughly 33% of patients with van der Woude syndrome have pits without clefting, 33% have pits with cleft lip and palate, and 33% have lip pits with isolated cleft palate or submucous cleft palate. Approximately 10% of those individuals with van der Woude syndrome do not exhibit lip pits.
Stickler Syndrome
In 1967, Stickler and Pugh were the first to describe a series of patients who had a combination of eye findings, hearing loss, isolated cleft palate, a Marfanoid habitus, and long-bone changes.1 It is now known that Stickler syndrome is associated with basic defects in collagen. These collagenopathies include mutations on type II collagen and mutations in type XI collagen.25 The inheritance pattern with this syndrome is autosomal dominant with variable expressivity. Cohen and others have reviewed the relationship between high myopia and clefting of the secondary palate and the other features of Stickler syndrome.19 High myopia (i.e., 8 to 18 diopters) is found in 75% of patients by the time they are 5 years old. The eye findings are progressive, with vitreous and chorioretinal degeneration and retinal detachment observed in 70% of patients by the time they are 20 years old. Additional eye findings include astigmatism, cataracts, strabismus, and glaucoma. Individuals with Stickler syndrome may manifest any or all of a range of craniofacial findings, including mild to moderate midface hypoplasia, shallow orbits with eye proptosis, epicanthal folds, a flat nasal bridge, and submucous or full clefting of the secondary palate. Some affected individuals will have progressive degeneration of the dental roots and the associated periodontium. This is primarily due to the inborn error in collagen production, but it may be exacerbated by malocclusion (secondary trauma) and orthodontic manipulations (see Fig. 4-18). Progressive neurosensory high-tone hearing loss has been reported in 80% of patients. Some patients have a Marfanoid body habitus, whereas others have short stature. There may be progressive early joint degeneration by the mid-adult years. Any newborn who is found to have Robin sequence should undergo an initial genetic and ophthalmologic assessment to rule out Stickler syndrome.
Robin Sequence
Robin sequence is commonly defined as cleft palate, micrognathia, and glossoptosis (see Fig. 4-19). This sequence of events can occur as a result of a variety of etiologic and pathogenetic conditions, and it involves a wide spectrum of phenotypes. The distinction must be made between a malformed mandible (micrognathia) and a deformed mandible that is simply retrognathic. The malformed mandible (i.e., Treacher Collins syndrome) may also be missing parts or sections of the jaw (i.e., Kaban types IIB and III) and cannot be expected to “grow out” and self-correct. This is different from a deformed mandible (i.e., retrognathic at birth) caused by fetal malpositioning, which can be expected to “grow out” and become relatively normal. The newborn’s respiratory compromise that occurs with Robin sequence will vary according to the child’s physical findings, and treatment should proceed accordingly. For example, with spondyloepiphyseal dysplasia congenita, the causes of respiratory compromise include a small and mechanically abnormal chest, tracheobronchial malacia, and central apnea as a result of cervical or medullary compression caused by cervical instability.47 This will require a different level of airway management as compared with an isolated retrognathic mandible, which is simply deformed as a result of intrauterine compression. If the deformed (retrognathic) mandible is present at birth, it is expected to “catch up” during growth and basically to self-correct. For these infants, mandibular osteotomies with advancement carried out during childhood should be avoided.
Achondroplasia
Achondroplasia is the most common condition associated with severe disproportionate short stature.22 The cause of this condition is the failure during development of the primary growth cartilages of the limbs and the cranial base. This results in short arms and legs as well as a characteristic midface deficiency or deformity that is most visually notable at the nasofrontal process.
Most affected individuals do well but attain a final height of only approximately 4 feet. The condition is known to be caused by mutations on the FGFR3 (fibroblast growth factor receptor 3) gene.22 Eighty percent of reported cases are sporadic, whereas 20% are familial. Although craniosynostosis is not a typical feature, three cases with craniosynostosis have been reported.
Premature Suture Closure of the Cranial Vault and Skull Base
The flat bones of the cranial vault develop from mesenchymal condensations over the developing brain (see Fig. 4-1). These bones grow primarily via the apposition of bone at their edges. The regions in which the cranial bony edges collide are called sutures. In addition to appositional growth at the edges (i.e., suture growth), cranial vault development also occurs through the process of remodeling (i.e., resorption and deposition).17 Continued separation of the sutures during the process of the apposition of bone at the edges is also important to the normal growth of the bones of the cranial base and the midface.
Synostosis is a condition of premature fusion (i.e., the arrest of appositional growth at the bone edges) that occurs before the associated visceral structures (e.g., brain, eyes) complete growth.17 When this happens, there is distortion or deformity of the shape of the affected bones and possible compression of the underlying visceral structures (i.e., the brain). Premature closure of the cranial vault sutures (craniosynostosis) that do not extend into the cranial base will have minimal effects on the maxillofacial form (e.g., metopic, unilateral coronal, and sagittal suture synostosis; Figures 4-20, 4-21, and 4-22). However, prematurely fused cranial vault sutures that extend into the cranial base will also affect midfacial growth and development (e.g., Crouzon, Apert, and Pfeiffer syndrome; Figures 4-23 through 4-26; see Chapter 30).
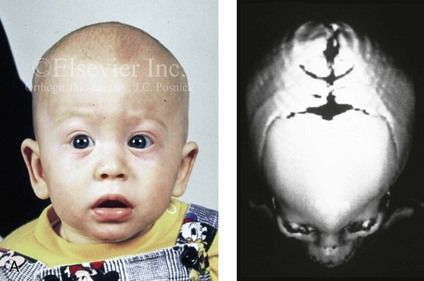
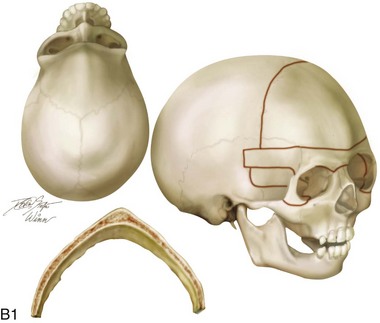
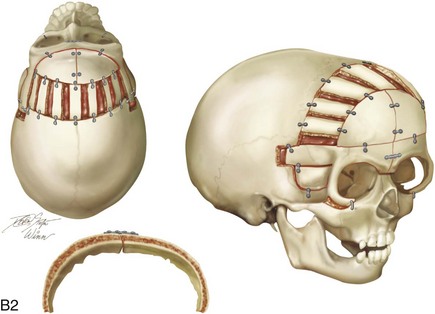
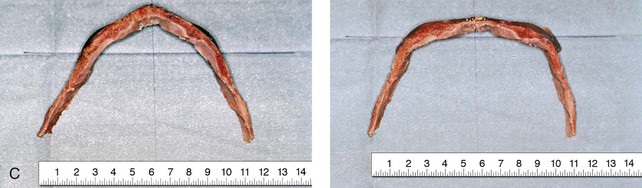
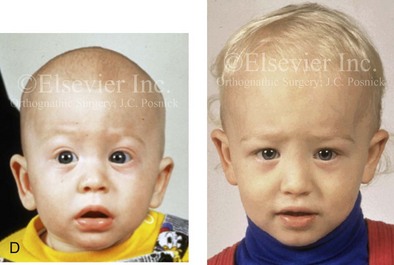

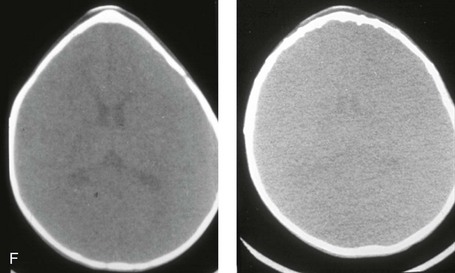
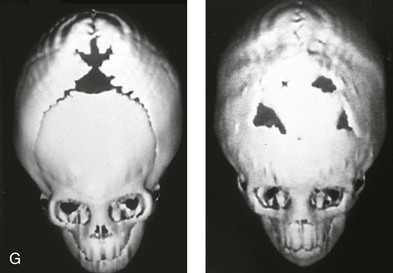
Figure 4-20 A child born with metopic synostosis that resulted in trigonocephaly. He underwent anterior cranial vault and three-quarter orbital osteotomies with reshaping when he was 10 months old. A, Facial and computed tomography scan views before surgery. B, Illustrations of the craniofacial skeleton in a child with metopic synostosis that resulted in trigonocephaly before and after anterior cranial vault and three-quarter orbital osteotomies. C, Bird’s-eye view of removed orbital osteotomy unit before and after reshaping. Part B modified from an original illustration by Bill Winn. D, Frontal views before and 1 year after reconstruction. E, Facial views of patient at 5 years old. F and G, Computed tomography scan views before and after reconstruction.
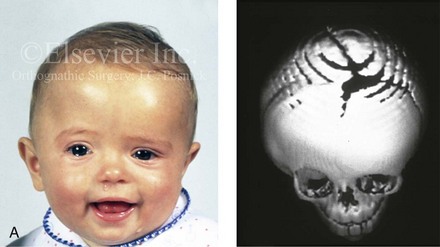
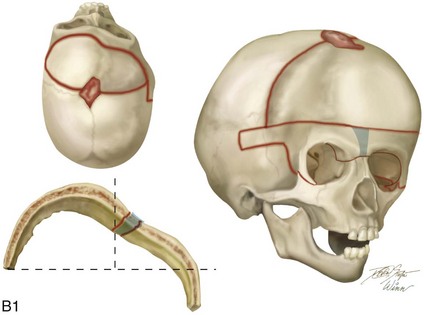
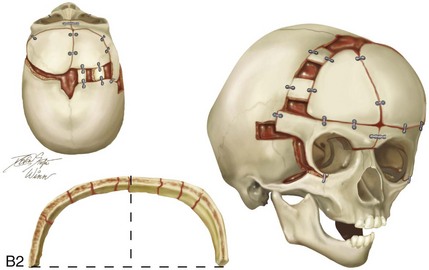
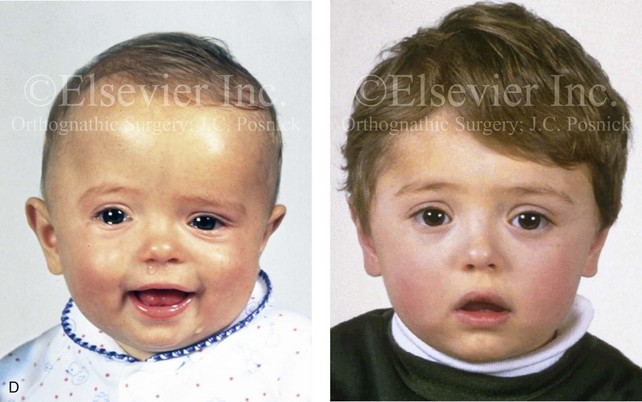
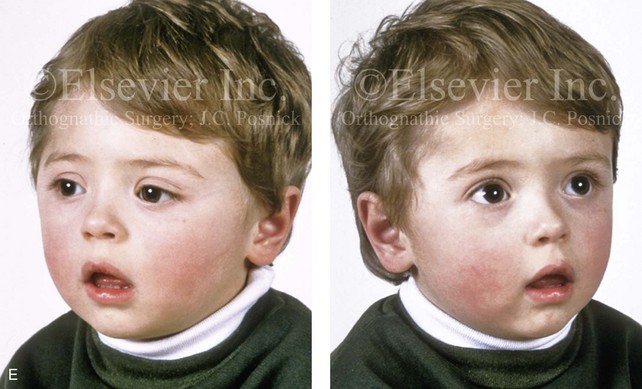
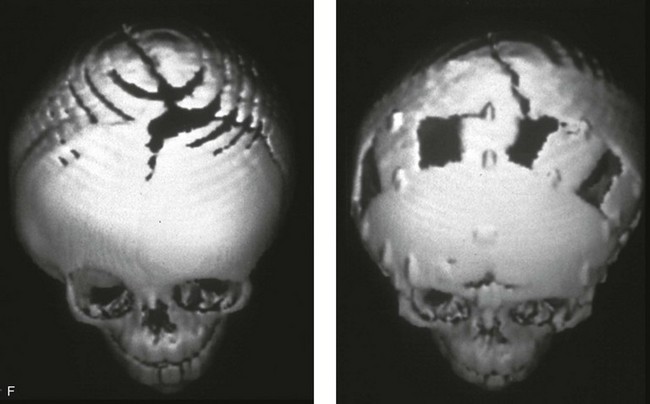
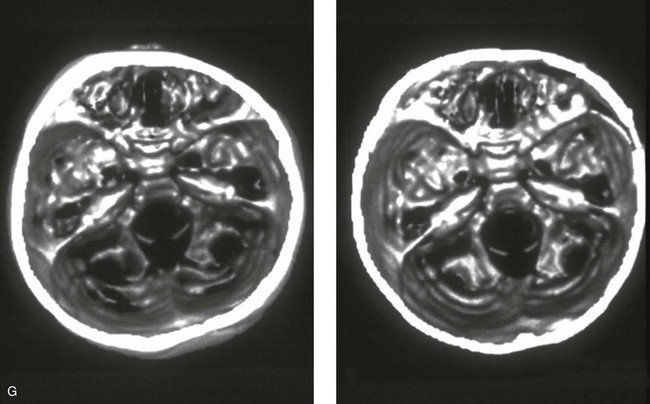
Figure 4-21 A child born with right unilateral coronal synostosis that resulted in anterior plagiocephaly. He underwent anterior cranial vault and three-quarter orbital osteotomies with reshaping when he was 9 months old. A, Facial and computed tomography scan views before reconstruction. B, Illustrations of the craniofacial skeleton in a child with unilateral coronal synostosis that resulted in anterior plagiocephaly before and after anterior cranial vault and three-quarter orbital osteotomies. Part B modified from an original illustration by Bill Winn.C, Bird’s-eye view of removed orbital osteotomy units before and after reshaping. D, Frontal views before and 5 years after reconstruction. E, Oblique views 5 years after reconstruction. F and G, Computed tomography scan views of the cranial vault before and just after reconstruction.
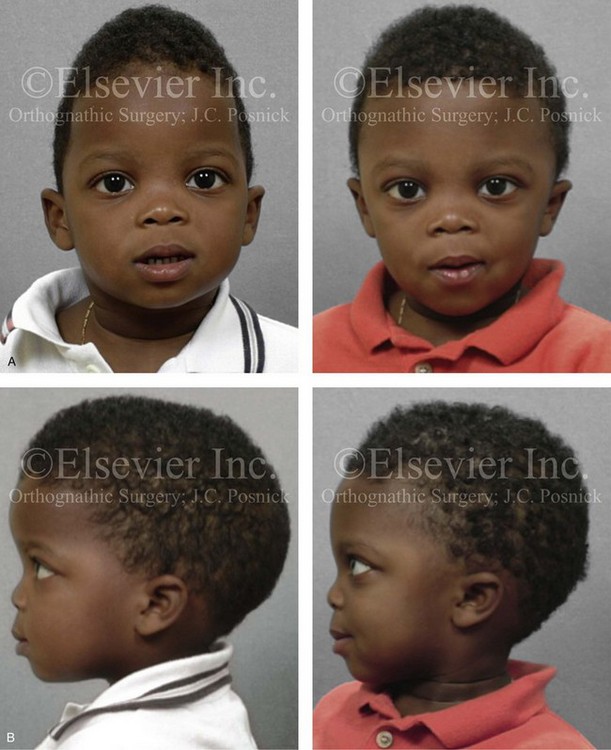
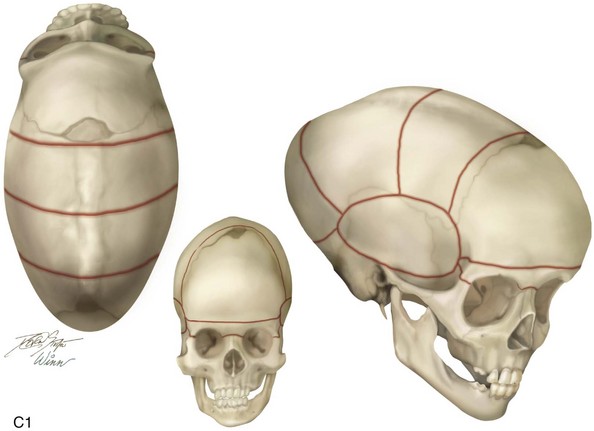
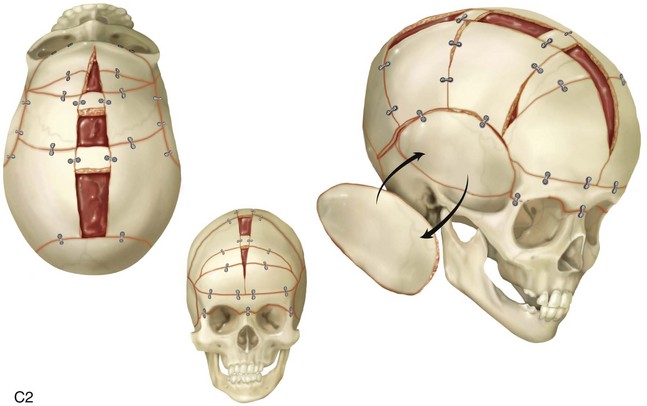
Figure 4-22 A child born with sagittal synostosis that resulted in a scaphocephalic shape of the cranial vault that was characterized by increased anteroposterior length and decreased bitemporal and biparietal width. It was not until the child was 2 years old that he was diagnosed with sagittal synostosis. After a comprehensive evaluation, he underwent total cranial vault reshaping through a coronal (scalp) incision. He is shown before and after a single-stage reconstruction. A, Frontal facial views before and after reconstruction. B, Profile facial views before and after reconstruction. C, Illustrations of the craniofacial skeleton in a child with sagittal synostosis that resulted in scaphocephaly before and after total cranial vault and upper orbital osteotomies with reconstruction. Part C modified from an original illustration by Bill Winn.
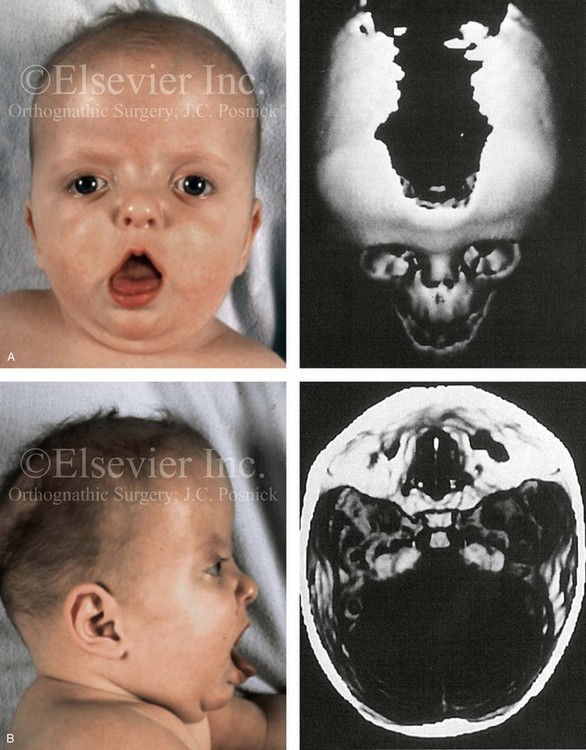
Figure 4-23 A 6-month-old girl who was born with Apert syndrome and bilateral coronal synostosis. A, Facial and computed tomography scan views without intervention. B, Profile and computed tomography scan views without intervention.
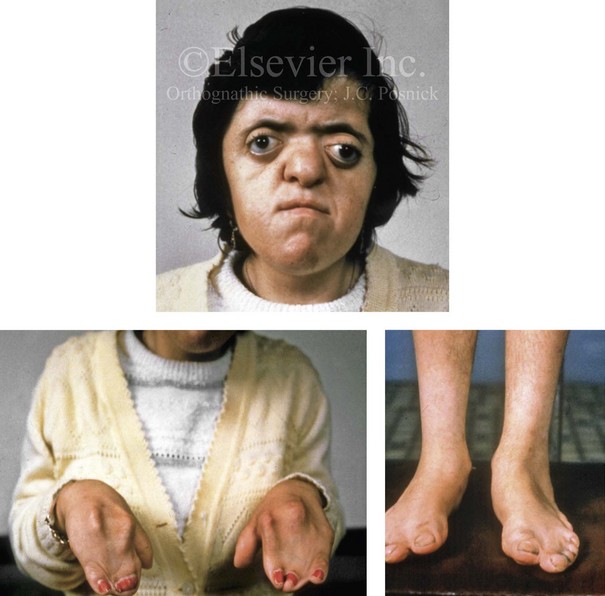
Figure 4-24 A 28-year-old woman with uncorrected Apert syndrome. She was unable to undergo craniofacial or extremity reconstruction in her home country. Facial and extremity views are shown.
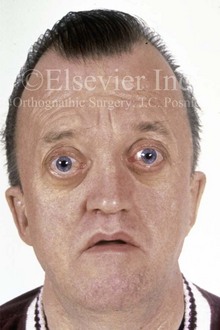
Figure 4-25 A 50-year-old man who was born with Crouzon syndrome. His dysmorphic features include oxycephaly, orbital dystopia with eye proptosis, and midface deficiency with an Angle Class III malocclusion. Despite these difficulties, he is married and employed as a taxicab driver; he also has a son with Crouzon syndrome.
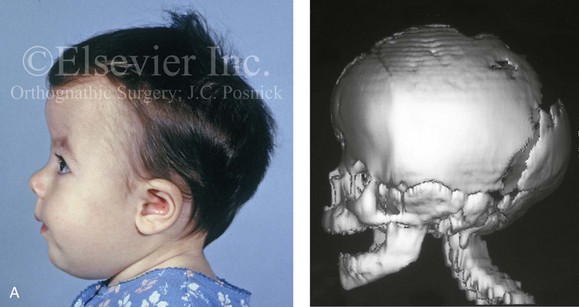
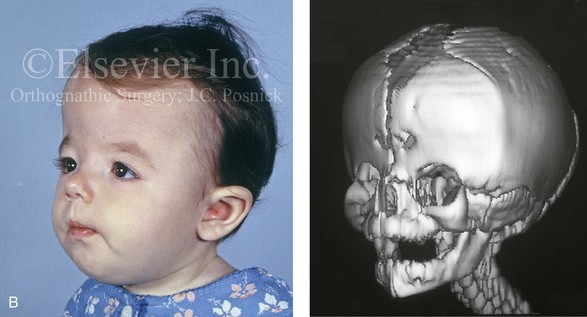
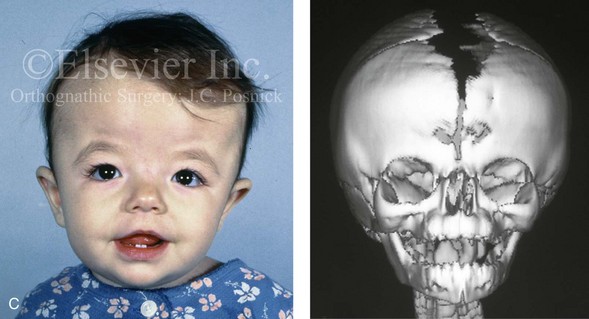
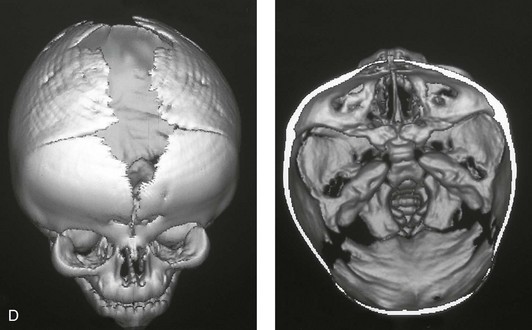
Figure 4-26 A 2-month-old child who was born with Pfeiffer syndrome (type I). She has bilateral coronal synostosis that resulted in brachycephaly but without the suggestion of midface deficiency. A, Profile facial and computed tomography (CT) scan views before intervention. B, Oblique facial and CT scan views before intervention. C, Frontal facial and CT scan views before intervention. D, Additional CT scan views before intervention.
Overgrowth Syndromes and Anomalies
Acromegaly is caused by an anterior pituitary tumor that secretes excessive growth hormone after the normal completion of growth.21 The response of specific growth centers to abnormal serum levels of growth hormone is the overdevelopment of the hands, the feet, the supraorbital ridges, and the mandible. When there is excessive mandibular growth, the position of the teeth within the bone and the occlusion are also effected. An enlarged sella turcica (i.e., the resorption of bone in response to pituitary tumor expansion) can be seen on a lateral cephalometric radiograph or a computed tomography scan (Fig. 4-27). Excess growth hormone causes the proliferation of the condylar cartilage of the mandible, thereby resulting in the bilateral symmetric forward projection of the jaw. This overgrowth continues until the serum growth hormone levels return to normal. Unfortunately, the resultant skeletal deformities persist. Other examples of maxillomandibular overgrowth include hemifacial hypertrophy (see Chapter 31), hemimandibular hyperplasia, and hemimandibular elongation (see Chapter 22).
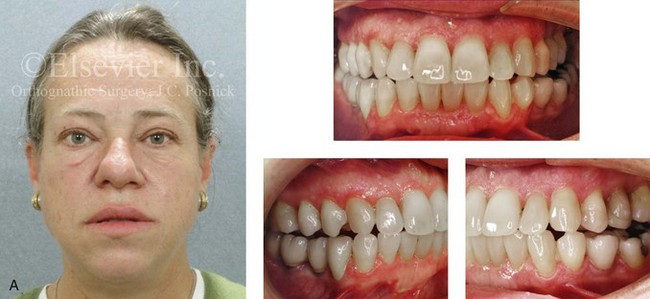
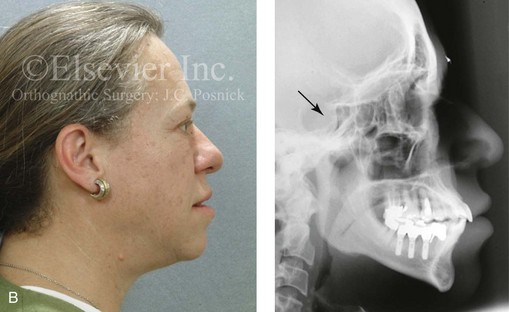
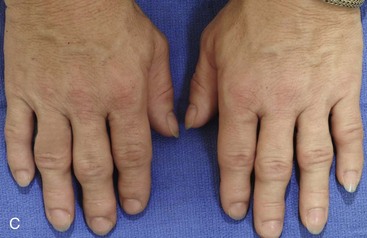
Figure 4-27 A middle-aged woman who presented for the orthodontic evaluation of a progressive malocclusion. She had a normal occlusion, average facial features, and no distortions of the extremities earlier during life. Gradually, over the prior 5 years, malocclusion and changes in facial and extremity morphology had occurred. At presentation to the orthodontist, standard records were taken (lateral cephalometric and Panorex radiographs and dental models). The patient was then seen for surgical evaluation. A review of the lateral cephalometric radiograph showed an enlarged sella turcica. An endocrinology consult confirmed the diagnosis of acromegaly. Appropriate treatment was undertaken without further worsening of the facial or extremity deformities. A, Facial and occlusal views at presentation indicate growth hormone changes. B, Profile and lateral cephalometric views at presentation. Note the enlarged sella turcica. C, A view of the dorsal aspects of hands indicates growth hormone changes.
Hereditary Tendencies in the Development of Dentofacial Deformities
Jaw deformities are thought to be more common among individuals with genetic backgrounds that are crosses between different racial and ethnic groups as compared with those from isolated human populations.10,32,40,57,59,60,62,83,85,86,91,93,108 It has been documented that crossing different breeds of dogs and other mammals can result in striking dentofacial deformities, particularly mandibular prognathism that resembles the human variant.82,117,119 Harris and Johnson examined the longitudinal records from the Bolton Brush Growth Study and concluded that the heritability of maxillomandibular characteristics is relatively high48 (Figures 4-28, 4-29, and 4-30). It is assumed that approximately one third of children who present with a severe Class III malocclusion will have a parent with the same problem and that one sixth of these children will have an affected sibling.73 Interestingly, a short face growth pattern with a deep-bite malocclusion is found more frequently in whites (see Chapter 23). Bimaxillary dento-alveolar protrusion with anterior open-bite malocclusion is more prevalent among blacks (see Chapter 24).112
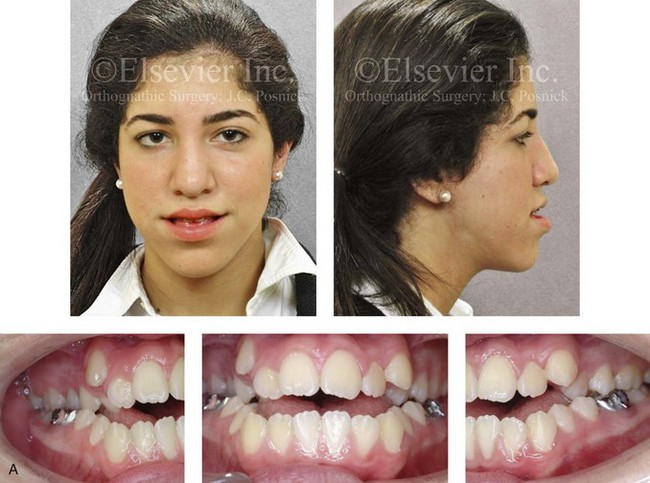
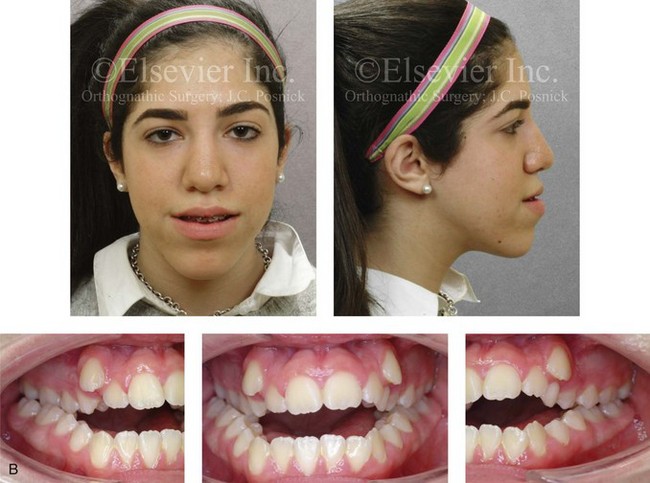
Figure 4-28 Biologic identical twin sisters are shown during their teenage years to confirm a hereditary tendency toward the occurrence of developmental jaw deformities with malocclusion. A, Facial and occlusal views of Twin A demonstrating bimaxillary dental protrusion with jaw disharmony and anterior open-bite malocclusion. B, Facial and occlusal views of Twin B demonstrating bimaxillary dental protrusion with jaw disharmony and anterior open-bite malocclusion.
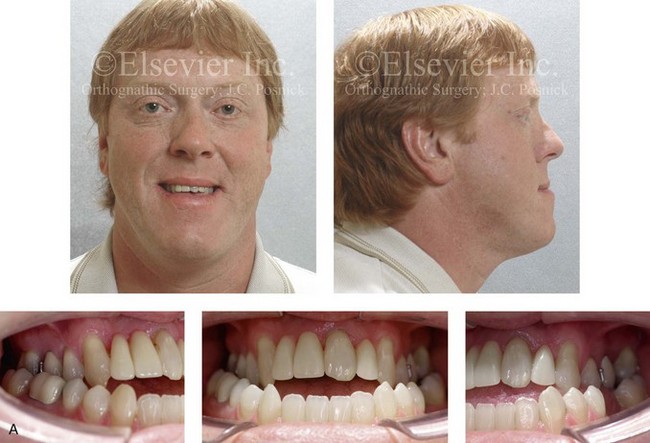
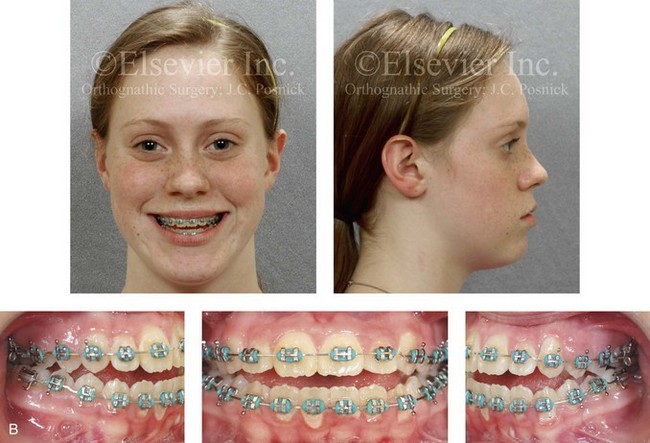
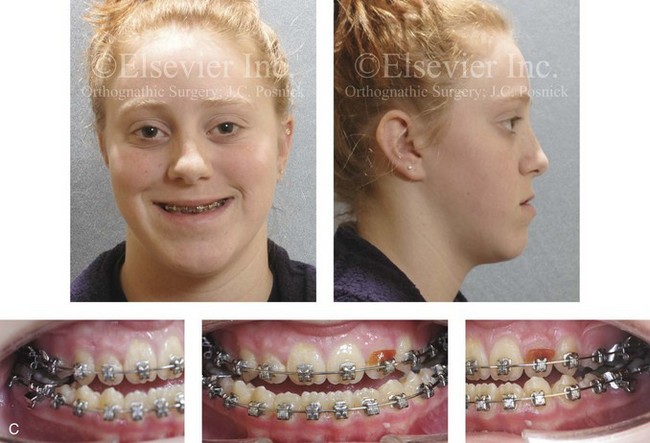
Figure 4-29 A father and his biologic daughters are shown during the daughters’ teenage years to confirm a hereditary tendency toward the occurrence of developmental jaw deformities with malocclusion. A, Facial and occlusal views of the father that demonstrate a Class III skeletal pattern. B, Daughter A with a similar class III skeletal pattern. C, Daughter B with a similar Class III skeletal pattern.
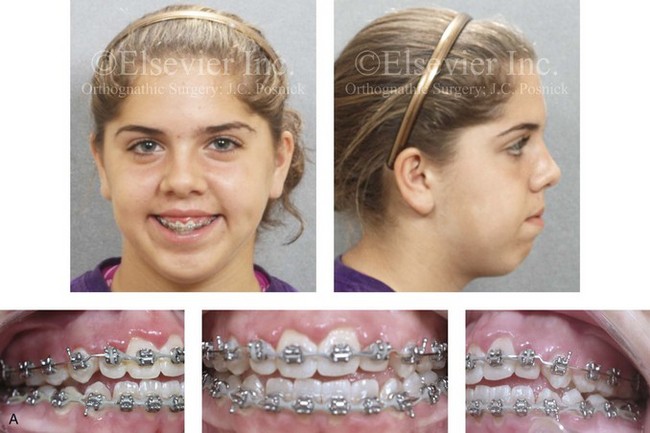
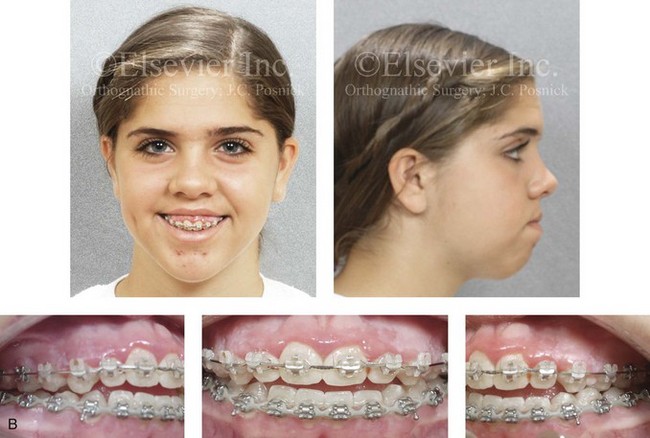
Figure 4-30 Biologic twin sisters are shown during their teenage years to confirm a hereditary tendency toward the occurrence of developmental jaw deformities with malocclusion. A, Facial and occlusal views of Twin A demonstrating a long face Class III growth pattern. B, Facial and occlusal views of Twin B demonstrating a similar long face Class III growth pattern.
The Class III skeletal pattern of maxillary deficiency with relative mandibular excess is known to be more frequent in certain races.2,73,84,123,139 Incidence rates of Class III malocclusion of as high as 13% have been reported in specific regions of Asia.136 Lew and colleagues examined occlusal parameters in 1050 Chinese school children between the ages of 12 and 14 years. They found an incidence of 12.6% for Class III malocclusion, 58.8% for Class I malocclusion, and 21.5% for Class II malocclusion.67 Samman and colleagues investigated 300 Chinese individuals residing in Hong Kong who were known to have a dentofacial deformity and who had been referred to an “orthognathic surgery clinic.” After excluding those individuals with cleft lip and palate, 222 study patients remained. Forty-seven percent of these individuals (n = 104) were found to have a Class III skeletal growth pattern.106
Several of the common maxillomandibular disharmonies are known to result from environmental influences (see Chapters 10 and 21). The dental position in each arch and the resulting occlusal variations are felt to have an even greater environmental component.3,11,29,41,64,92 For example, dento-alveolar morphology is commonly affected by thumb sucking and other para-oral habits (i.e., environmental influences; Figures 4-31 and 4-32). The more severe the dentofacial deformity, the more likely that inheritance plays at least a partial role. Maxillary deficiency with relative mandibular excess (i.e., a Class III skeletal pattern) and maxillomandibular deficiency (i.e., the short face growth pattern) are both thought to have a significant hereditary component.31,36,58,79,116,120,137
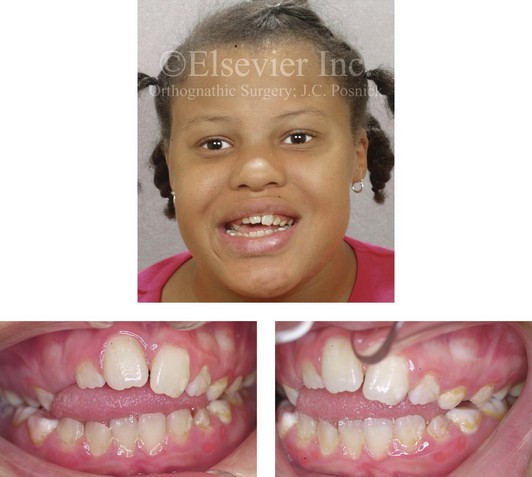
Figure 4-31 A 10-year-old girl with a severe anterior open bite. She demonstrates how a para-oral habit (thumb sucking) can exert the long duration of light force at the periodontal ligament that is required to move the teeth. This patient was a consistent and constant “thumb sucker” from soon after her birth and throughout the mixed dentition.
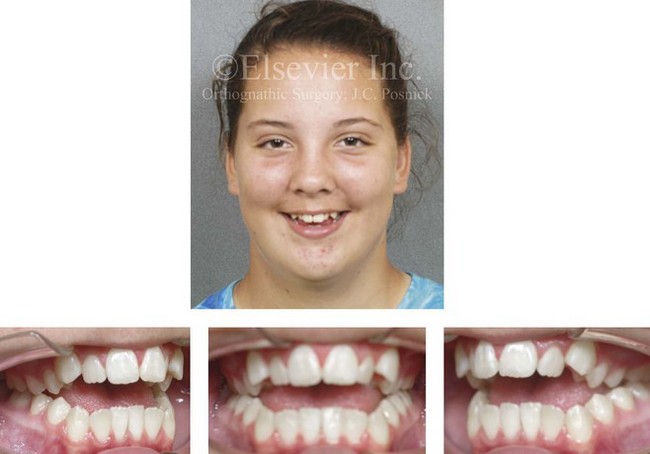
Figure 4-32 A 12-year-old girl was referred by an orthodontist for surgical evaluation. She had persistently sucked her thumb since infancy. This parafunctional habit resulted in the severe deformity of her maxilla and mandible. The upper jaw has a constricted arch width and an excessive curve of Spee, whereas the mandible has a Class III reversed curve of Spee. A severe anterior open-bite malocclusion is also present.
Environmental and Neuromotor Effects on Skeletal Growth
The mandible can be viewed as a somewhat malleable block of hard tissue that is adjacent to the skull base. It is separated from the skull by working temporomandibular joints. It houses the inferior alveolar neurovascular bundle, and it provides a matrix for the formation, eruption, and eventual alignment of the teeth. Muscular activity in the head and neck can directly affect skeletal growth through one of the following mechanisms: 1) hyperactivity of muscles at the point of attachment may cause excessive skeletal remodeling or contour deformities (as discussed later in this chapter) 2) hypoactivity of the masticatory muscles may cause distortions of the normal skeletal growth (i.e., long face growth pattern; Fig. 4-33) and 3) hyperactivity of specific muscles such as the sternocleidomastoid (i.e., torticollis) may result in the deformation of the growing bones (i.e., cranial vault, orbit, and zygoma; Fig. 4-34).71
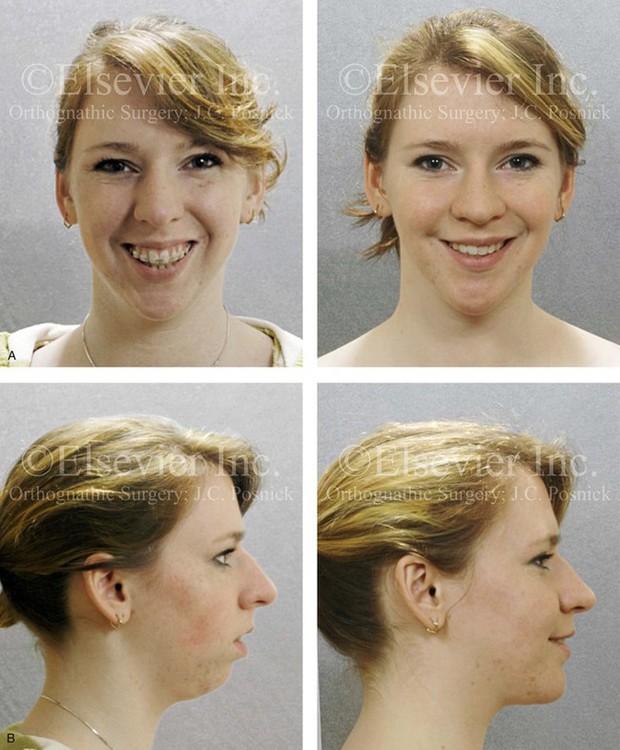
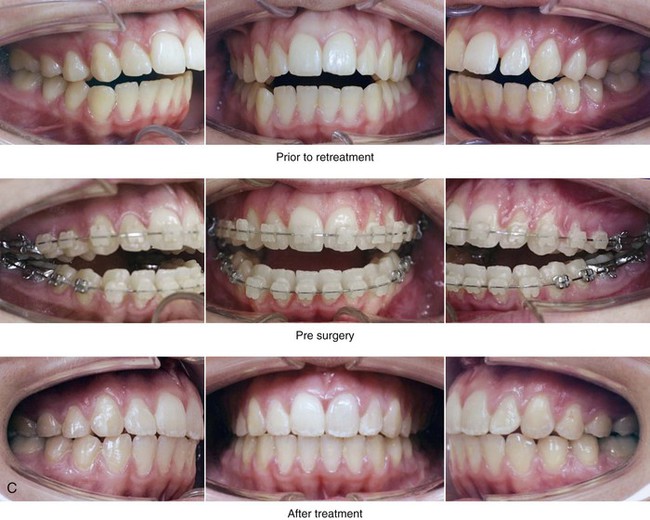
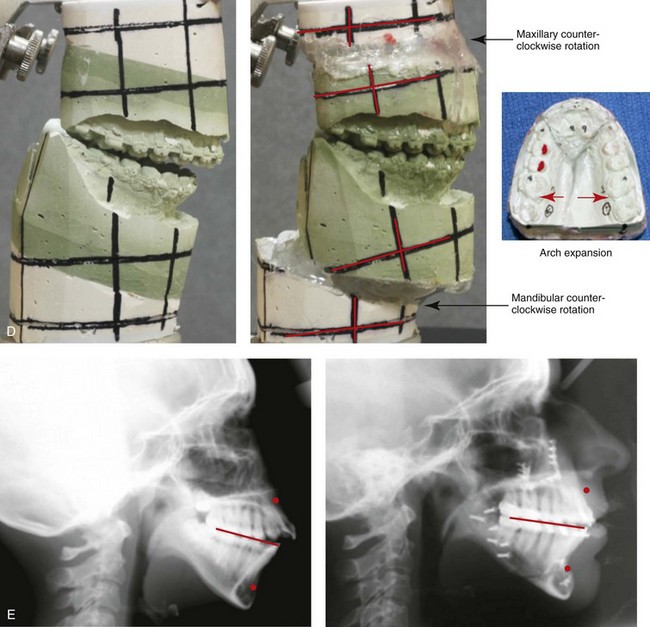
Figure 4-33 A teenage girl with a lifelong history of obstructed nasal breathing demonstrates the resulting long face growth pattern. The nasal obstruction resulted in forced mouth breathing with an open-mouth posture. This in turn is associated with maxillary dental hypereruption (i.e., hypereruption of the molars greater than the incisors), a steep mandibular plane, anterior open-bite Class II malocclusion, and increased lower anterior facial height. Clinically, there is a “gummy” smile, lip incompetence, and a flat, long face. A, Frontal views with smile before and after reconstruction. B, Profile views before and after reconstruction. C, Occlusal views before retreatment, with orthodontic decompensation in progress, and after the completion of treatment. D, Articulated dental casts that indicate analytic model planning. E, Cephalometric radiographs before and after surgery.
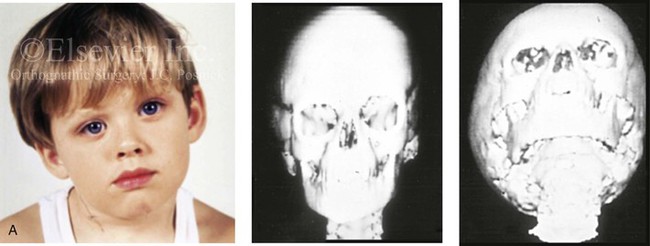
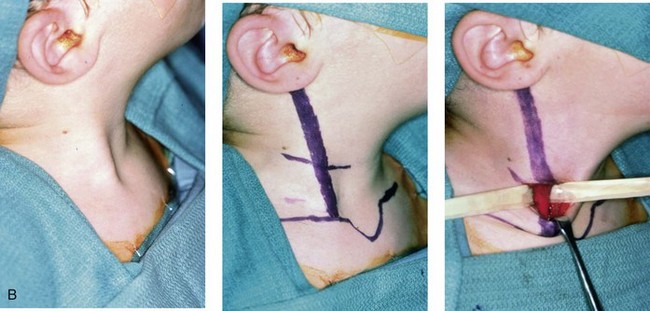
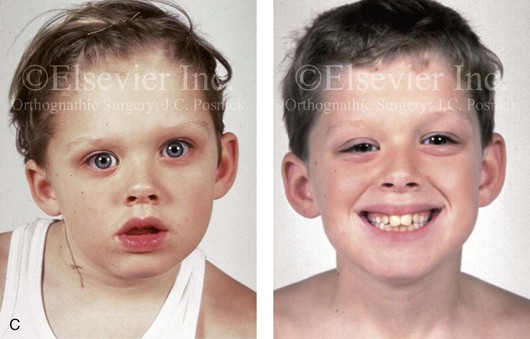
Figure 4-34 A child with right-sided congenital muscular torticollis and C1-C2 rotary subluxation. Ipsilateral right fronto-orbital flattening and contralateral left occipitoparietal flattening were noted. The right sternocleidomastoid muscle (SCM) remained tight when the patient was 2 years old. He underwent surgical release of the SCM, and this was followed by neck range-of-motion exercises with good maintenance of function. A degree of right fronto-orbital flattening (skull molding) remains. A, Frontal view with the neck in a relaxed position before treatment indicated a head tilt. Computed tomography scan views indicate fronto-orbitozygomatic asymmetry as a result of skull molding. B, Intraoperative view of the tight SCM and the proposed incision site, and a close-up view of the exposed SCM before release. C, Frontal views before and 2 years after neck muscle release. Residual fronto-orbital (skull molding) flattening remains. From Slate RK, Posnick JC, Armstrong DC, et al: Cervical spine subluxation associated with congenital muscular torticollis and craniofacial asymmetry. Plast Reconstr Surg 91:1187–1195, 1993.
Muscle and Soft-Tissue Effects on Skeletal Growth
Experimental animal research and clinical observational studies confirm that the influences of the mandibular rest posture, the neck rest posture, and the tongue and lip rest position of the affected individual’s eventual facial morphology are real and more important than the effects of intermittent muscle contracture (see Chapters 8 and 10).128–130 It is also known that functional stresses that are placed on the skeleton will increase bone density.113,114,118 A corollary is that, during periods of inactivity (e.g., mandibular and neck rest posture), skeletal demineralization occurs.
Erupting teeth carry alveolar bone with them. Therefore, forces that counteract the normal eruption of teeth can shape the dental arches and the alveolar processes of both the maxilla and the mandible.5,101–105,107,109,111,126,140 Experimental studies confirm that pressure against the active eruption of the teeth that is maintained for 4 to 6 hours can affect tooth movement and initiate bone remodeling (see Chapter 5). Habits such as finger or thumb sucking, which apply consistent pressure against the teeth, can have the same effect on the shaping of the dental arches (see Figs. 4-31 and 4-32). It seems as though the intensity (i.e., the amount of force) against the teeth is less important than the duration (i.e., the number of hours per day) with regard to causing this effect.
It has been confirmed that the postural position of the tongue at rest (e.g., the mandible open with the constant protrusion of the tongue) rather than where the tongue is placed during function (e.g., a tongue-thrust swallowing habit) is most important for determining the eventual position of the teeth and the morphology of the dento-alveolar complex.61,121 The intermittent forces that are generated against the teeth during chewing, swallowing, and speaking are documented to be more than heavy enough to produce tooth movement. However, during these normal functions, the force is not maintained long enough at any point in time to have an appreciable effect on the position of the teeth or the morphology of the bone.35 Even repetitive intermittent “abnormal” tongue and lip contraction with resulting pressure on the teeth during swallowing and speaking (i.e., tongue thrusting)—despite their frequency—do not sum up to the number of hours of force required to produce a pathologic effect. By contrast, even very light forces sustained for a long duration (i.e., the tonic contracture of masticatory muscles to maintain mandibular posture) can affect both the path of tooth eruption and jaw development.8,94
It is known that muscles of different size and force that attach to the mandible at specific locations will result in characteristic ridges and contours (e.g., gonial angles, coronoid process, genial tubercle).12,13 For patients with the condition known as masseteric muscle hypertrophy, there is excess bone proliferation at the masseter muscle’s point of attachment along the inferior border of the mandible (i.e., the gonial angle ridge) in response to greater than average muscle forces.43 By contrast, in patients with muscular dystrophies (i.e., hypotonic cerebral palsy, Moebius syndrome with cranial nerve palsy), the ramus may be short, and the gonial angles appear to be underdeveloped as a result of the limited muscle forces applied to these same regions.100
The lips, cheeks, and tongue contain muscles in their deep layers that exert varied pressures against the teeth and the alveolar process. They normally do so with a constant level of tone while at rest as well as during dynamic function. These light (tone) forces applied for a long duration are known to cause tooth movement and remodeling of the alveolar process. It is the “resting” postural pressures of these structures (i.e., lips, cheeks, tongue, and muscles), the presence of para-oral habits (i.e., thumb sucking, pencil biting; see Figs. 4-31 and 4-32), and the man-made and controlled orthodontic pressure that can exert the long duration of light force at the periodontal ligament that is required to move teeth.55 When the difference between applied orthodontic forces and pressures of the tongue, cheek, and lips that counteract those forces applied to the periodontal ligament becomes great enough for sustained periods of time, tooth movement will occur. Another example of dental adaptation is seen when the constant strong force of the tongue is juxtaposed with intrinsic weaker bilabial (i.e., upper and lower lip) muscle tone forces.135 This results in the predictable dental compensations (i.e., proclined maxillary and mandibular incisors) observed in patients with bimaxillary dento-alveolar protrusion (see Chapter 24). This also explains the consistent dental compensations that are typically observed in patients with specific developmental dentofacial deformities such as long face growth pattern (see Chapter 21 and Fig. 4-33) or isolated mandibular deficiency (see Chapter 19).122
Severe scar contracture of the anterior neck and lower lip soft tissues from a burn that occurred during childhood will also affect mandibular morphology. The soft-tissue contracture will distort ongoing skeletal growth and cause the mandible to be retrusive, with an obtuse angle. The chin region of the mandible will grow with increased vertical length and without sagittal prominence, and there will be an anterior open-bite malocclusion. This deformity and its surgical correction were first described by Simon Hullihen in 1849 (see Chapter 2 and Fig. 2-19).
With the congenital absence of part or most of the tongue, there will be a characteristic V-shaped deficiency or dysmorphology of the mandibular dental arch (e.g., hypoglossia–hypodactylia syndrome; Fig. 4-35).15 In the presence of a volumetrically enlarged tongue (e.g., vascular malformation, hemangioma, neurofibromatosis, Proteus syndrome, Beckwith-Wiedemann syndrome), there will be dento-alveolar effects that result in spaces between the teeth (Fig. 4-36). The lower anterior teeth will be flared, and the overall mandibular arch form will be enlarged. The important factor that causes these dental compensations and arch form deformities is the sustained and constant rest forces of the oversized tongue on the teeth and bone and not what the tongue does when it is in motion.
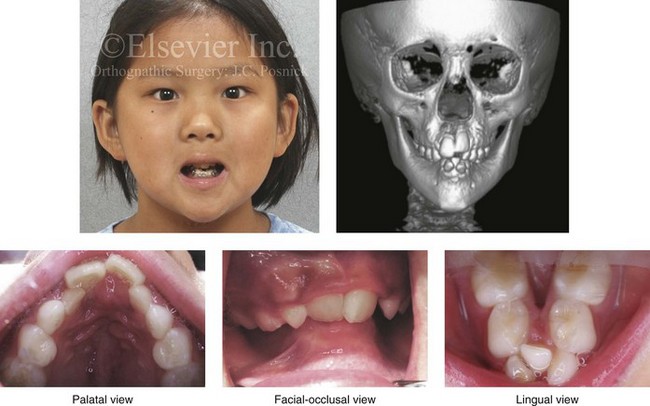
Figure 4-35 A 10-year-old girl who was eventually diagnosed with hypoglossia–hypodactylia syndrome. In this photo, she demonstrates how the congenital absence of part or most of the tongue will result in a characteristic V-shaped deficiency and dysmorphology of the mandibular dental arch and how this will negatively affect the maxillary arch form.
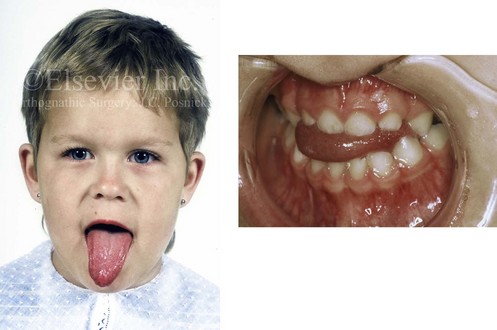
Figure 4-36 A 3-year-old child with a congenitally enlarged tongue is shown. The secondary effects on the oral cavity include a Class III anterior open-bite malocclusion and a lateral cross-bite malocclusion.
In the individual with a normal tongue volume and neuromotor capability, it is known that the rest position of the tongue in the mouth is primarily influenced by respiratory factors.73 It is the rest position of the tongue that influences the pattern of dentofacial development rather than the position of the tongue during function.
In the presence of chronic nasal obstruction, a typical long face growth pattern with anterior open-bite malocclusion is likely to occur. In these circumstances, a speech therapist may have success with teaching the individual altered movement of the tongue for the formation of more accurate voluntary independent speech sounds.90 However, unless the anterior open bite and the nasal obstruction are corrected, the myofunctional therapy will not allow the individual to reliably position his or her tongue, lips, teeth, and mandible for correct articulation during rapid conversational speech (see Chapter 8).
In the skeletal Class III adult patient (i.e., a retrusive maxilla with relative mandibular excess) who undergoes successful orthognathic correction, a normal oral cavity volume and a normal occlusion are established. Interestingly, in most cases, the tongue position and lip posture will then naturally adapt to the new morphology.80,81
Clinical observation confirms that, when an individual with a longstanding dentofacial deformity undergoes successful reconstruction, it is generally not necessary to retrain the tongue with active myofunctional therapy. This only needs to occur under the following conditions: 1) when the tongue volume is truly excessive (e.g., vascular malformation, neurofibromatosis) 2) when there is the presence of intrinsic neuromotor dysfunction (e.g., cerebral palsy) or 3) when the nasal airway remains obstructed and a failure of tongue adaptation is seen.63
Respiration Patterns and Their Effects on Skeletal Growth
Harvold conducted primate experiments that were designed to test hypotheses regarding the relationship between nasal obstruction, mouth breathing, mandibular rest posture, tongue rest position, jaw growth, and dental malocclusion.49–54 He created an experimental model in growing rhesus monkeys (Macaca mulatta) to study these relationships. To do this, he completely obstructed the nasal passages in the study subjects with silicone nose plugs. The experiments showed that the monkeys adapt to total nasal obstruction in two different ways. Predominantly, the experimental animals maintained an open-mouth posture with tongue protrusion. Interestingly, some adapted to a forced oral airway by holding the upper and lower teeth close together but with wide lip separation. Nevertheless, both adaptive mandibular, tongue, and lip positioning patterns gradually resulted in maxillomandibular dysmorphology and dental malocclusion that was notably different from that seen in the control animals.
The observed morphologic and occlusal changes were most extreme when the mandible constantly remained open, with tongue protrusion (i.e., an open-mouth posture). The adaptive lowering of the mandible (with clockwise rotation) to achieve effective oral respiration was followed by secondary changes in the maxilla, including a downward displacement (i.e., vertical lengthening) of the alveolar process in conjunction with excess extrusion of the teeth. When molar (posterior) hypereruption was significantly greater than incisor hypereruption, an anterior open-bite malocclusion developed (Fig. 4-37).127
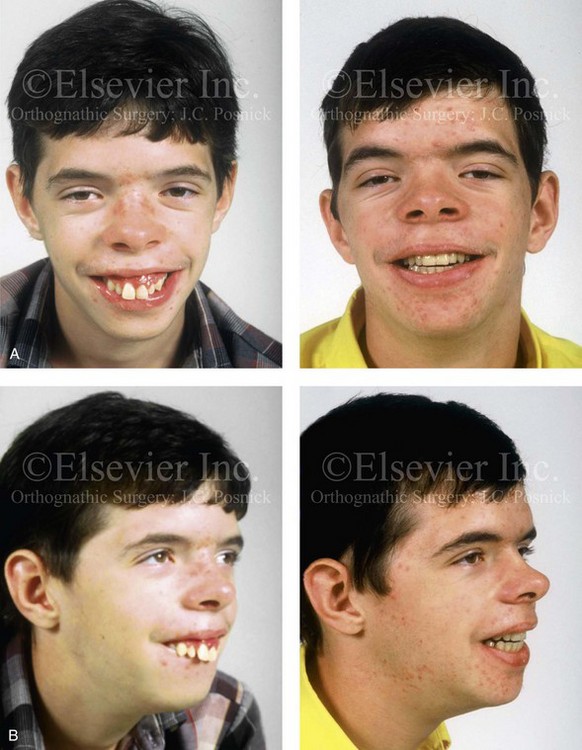
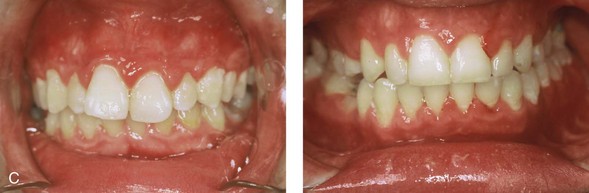
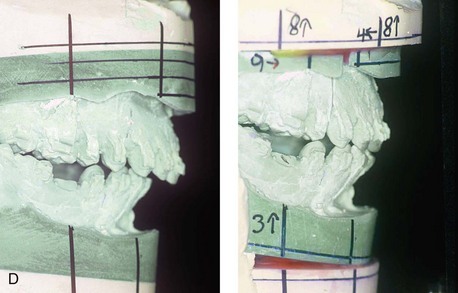
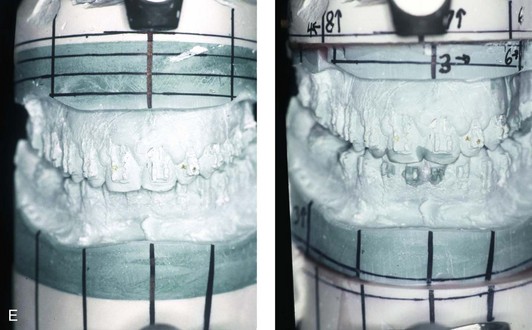
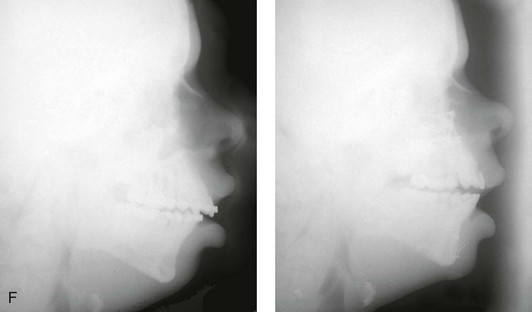
Figure 4-37 A 16-year-old with a nonprogressive congenital myopathy that resulted in a constant open-mouth posture. He developed a long face growth pattern that was characterized by severe maxillary vertical excess and protrusion, a retrusive mandible, and an Angle Class II malocclusion. He underwent a combined orthodontic and surgical approach. Orthodontic manipulation was limited as a result of hygiene control. The surgery included a maxillary Le Fort I osteotomy (vertical intrusion, first bicuspid extractions, and alveolar bone removal with premaxillary setback and cant correction) and sagittal split osteotomies of the mandible (clockwise rotation and asymmetric correction). A, Frontal views with smile before and after reconstruction. B, Oblique facial views before and after reconstruction. C, Occlusal views before and after reconstruction. D and E, Articulated dental casts that indicates analytic model planning. F, Lateral cephalometric radiographs before and after reconstruction.
It became apparent to Harvold that the changes in mandibular shape and the direction of mandibular growth (i.e., clockwise rotation) observed in a majority of the experimental monkeys were dependent on the adaptive activity of the facial, tongue, jaw, and neck muscles in response to respiratory need. Interestingly, in Harvold’s experimental animal model, there was no indication of abnormal bony apposition on the condyles in association with the forced oral respiratory pattern. The apposition of new bone on the condyles continued (in the usual way) independently of aberrant mandibular positioning or open-mouth posture.107
In review, the growing experimental animals studied by Harvold coped with nasal obstruction by changing neck, tongue, facial, and masticatory muscle tone, thereby resulting in alterations in mandibular posture and tongue position.44,45,55 The animals typically did this in one of two different ways but always with the goal of maintaining adequate respiration. This resulted in varied but consistent secondary changes in the maxillomandibular morphology (i.e., developmental jaw deformity) and dental compensations (i.e., malocclusion). The measured lower facial height increase that occurred in the presence of a constant open-mouth posture was primarily the result of maxillary molar—and, to a lesser extent, incisor dental—hypereruption (e.g., long face growth pattern).
Harvold also reviewed the human observation studies of Linder-Aronson and colleagues and found that they paralleled his experimental animal research.68–70,75–78 Their clinical research established that nasal obstruction with forced mouth breathing (i.e., open-mouth posture) in humans is frequently associated with maxillary dental extrusion, a steeper than normal mandibular plane, anterior open-bite malocclusion (i.e., the hypereruption of the molars is greater than that of the incisors), and increased lower anterior facial height. Similar to Harvold’s experimental monkeys, growing humans may develop different neuromotor adaptations to cope with nasal obstruction and the need to maintain adequate respiration. The secondary skeletal and dental morphologic deviations will therefore vary, but they tend to occur in a predictable long face growth pattern (see Chapter 21).39,125,133,134
Humans with severe hypoplasia of the maxilla, a retropositioned palate and a restricted nasal airway, as are seen in patients with Crouzon or Apert syndrome, also become dependent on oral respiration (see Chapter 30).4 With Crouzon or Apert syndrome, the individual’s neck is often maintained in hyperextension with an open-mouth posture, the protrusion of the tongue, and the clockwise rotation of the mandible; this head and neck posture is compensatory to support respiration. During the day, this compensatory posture may be sufficient, but it becomes inadequate at night and results in obstructive sleep apnea (see Chapter 26).4 When these compensatory adaptive changes occur during childhood, they contribute to additional secondary maxillomandibular skeletal dysmorphology with growth that further affects the occlusion (see Chapter 30).7,33,115 This is an example of a congenital midface deformity that is further influenced by the environment.
Congenital Myopathies and Effects on Skeletal Growth
Individuals with congenital myopathies constitute a heterogeneous subgroup of patients with congenital neuromuscular disorders in which the pathology is attributable to defects in the muscle fibers rather than in the neurons. Congenital fiber-type disproportions are rare congenital myopathies that are considered slow or nonprogressive. Patients with these conditions, like those with other types of primary myopathies, will generally exhibit facial features that often result in what is described as a “myopathic face.” The “myopathic face” has many similarities to the “adenoid face” that is seen in children with a blocked nasal airway due to enlarged adenoids and other intranasal obstructions, which result in an open-mouth posture. These abnormal human maxillomandibular growth patterns are also consistent with the findings reported by Harvold when rhesus monkeys were forced to maintain an open-mouth posture during jaw growth, as described previously in this chapter. The “myopathic face” findings described by Lehman and colleagues include characteristics of an increased lower anterior facial height, incompetent lips, a high-arched palate, and often anterior open-bite malocclusion (Figs. 4-37 and 4-38).66 Clinical human and animal model studies confirm that these features are to be expected in the presence of an open-mouth posture during jaw growth, no matter what the cause. Although blocked nasal breathing is the most common cause of an open-mouth posture during growth, clearly congenital myopathies that also result in an open-mouth posture will produce the same facial dysmorphology.
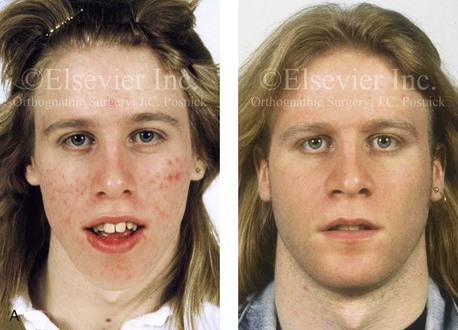
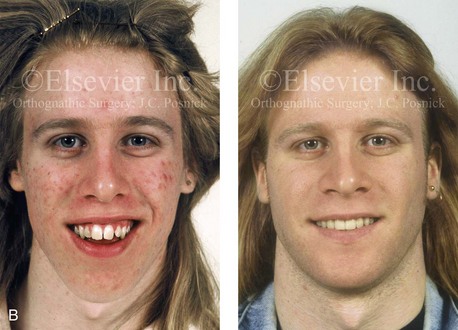
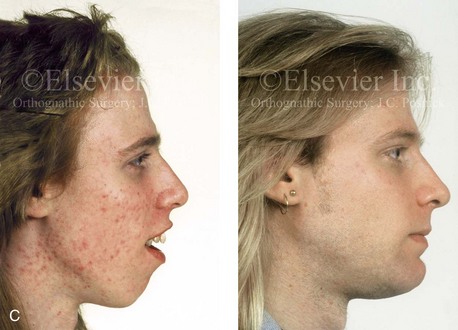
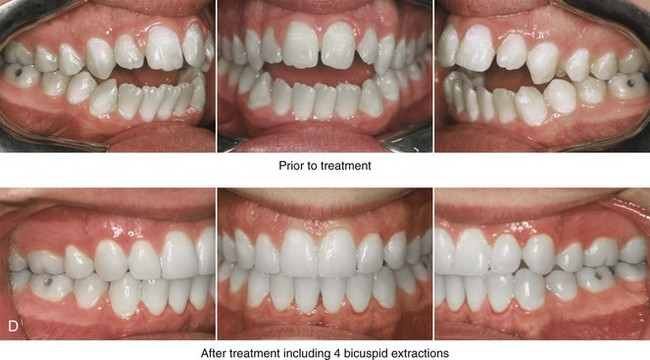
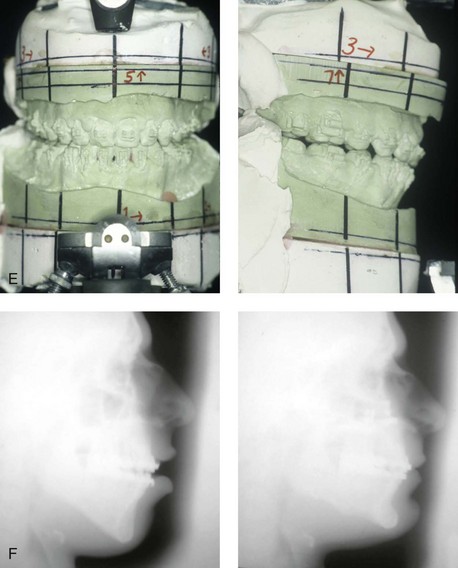
Figure 4-38 A 17-year-old boy who was born with a nonprogressive congenital myopathy was referred by an orthodontist for surgical evaluation. The lack of masticatory muscle strength resulted in a constant open-mouth posture. The long face growth pattern is characterized by vertical maxillary excess, a clockwise rotated retrusive mandible, a vertically long and retrusive chin, and an anterior open-bite malocclusion. The patient underwent a combined orthodontic and surgical approach. Four bicuspid extractions relieved dental crowding. This was followed by surgery that included a maxillary Le Fort I osteotomy (vertical intrusion and horizontal advancement); bilateral sagittal split osteotomies of the mandible (horizontal advancement); and osseous genioplasty (vertical reduction and horizontal advancement). A, Frontal views in repose before and after reconstruction. B, Frontal views with smile before and after reconstruction. C, Profile views before and after reconstruction. D, Occlusal views before and after reconstruction. E, Articulated dental casts that indicate analytic model planning. F, Lateral cephalometric radiographs before and after surgery.
Effects of Trauma on Skeletal Growth
Intrauterine compressive force against the developing face is a form of trauma that can lead to deformations of the craniofacial skeleton that are visually present at the time of birth. For example, when there is a decrease in the volume of amniotic fluid, the fetus’ head is typically flexed tightly against his or her chest in utero. The compressive forces to the chin will prevent the mandible from achieving its full prenatal growth potential. The normally developing tongue is then forced to rest high in the mouth, thereby preventing approximation and fusion of the lateral maxillary processes. At the time of birth, clefting of the secondary palate is recognized. A pathologic process continues after birth, and this includes retrognathia, a cleft of the secondary palate, and varying degrees of respiratory distress as a result of the otherwise normal tongue resting superior and posterior in the mouth. This intrauterine deformation is responsible for approximately two thirds of those individuals with observed Robin sequence at the time of birth. Significant but incomplete catch-up mandibular growth is expected. In general, there will be sufficient mandibular growth in combination with an expected degree of maxillary growth inhibition from cleft palate repair during childhood that results in maxillomandibular harmony but at least mild horizontal deficiency and the clockwise rotation of both jaws. Individuals with these features are typically managed with orthodontics to achieve an acceptable occlusion, but this does result in a degree of facial dysmorphology (see Fig. 4-20). Unless upper airway obstruction with obstructive sleep apnea is confirmed, the degree of maxillomandibular deficiency is such that a surgical consult is generally not requested.
An infant, child, or young adolescent who sustains injury to the craniofacial skeleton has the potential for growth disturbance of the injured bones. This is in addition to any skeletal deformity that remains if the fracture was not fully reduced with satisfactory initial healing (see Chapter 35). During some difficult deliveries through the vaginal canal, especially those that require forceps, damage to one or both of the temporomandibular joints or fracture of the mandible can occur. In modern times, with good prenatal care and skilled obstetrics support being available at the time of delivery, birth trauma to the mandible has become a rare event.
A childhood injury to the cartilage of the nasal septum may result in nasal deformity. It may also lead to deficient midface growth (see Fig. 35-2).38 This has been confirmed in experimental animals and documented through clinical observation in humans.28 In experimental animals, when the cartilage of the nasal septum is removed before growth is complete, there will be a dramatic decrease in the horizontal and vertical growth of the midface. Whether this is the result of a mechanical collapse with a secondary loss of growth velocity of the maxilla or an intrinsic growth restriction of the upper jaw remains open to question.
1. The mandibular condyle: Proliferating cartilage at the base of the fibrocartilage layer that covers the articular surface results in mandibular growth. At the completion of mandibular growth, the fibrocartilage layer is replaced by bone.
2. The outer surfaces of the ramus: Extensive remodeling of the mandible through the apposition of bone on the posterior and outer surfaces along with resorption on the anterior and inner surfaces is known to occur and is influenced by muscle forces and the compression of the associated soft tissues (e.g., tongue volume) within the mouth.
3. The alveolar process: New alveolar bone is added only as individual teeth develop and erupt into the arch.
In individuals who sustain a displaced condylar neck fracture, a sequence of events generally occurs that includes the contraction of the lateral pterygoid muscle with the displacement of the condylar fragment anteriorly and medially (Fig. 4-39). The displaced fragment (i.e., the medial pole of the condyle or the total condyle) has a tendency toward a degree of resorption and malunion. This may then result in a loss of posterior facial height, with a shift of the ipsilateral mandible toward the affected side. In these cases, an asymmetric retrusive lower jaw and predictable malocclusion can be documented when the jaw is placed in centric relation. Experimental and clinical studies confirm that the ability of the individual’s mandible and masticatory muscles to adapt will determine the degree of eventual jaw deformity and residual malocclusion as well as what treatment is needed (see Chapter 35).
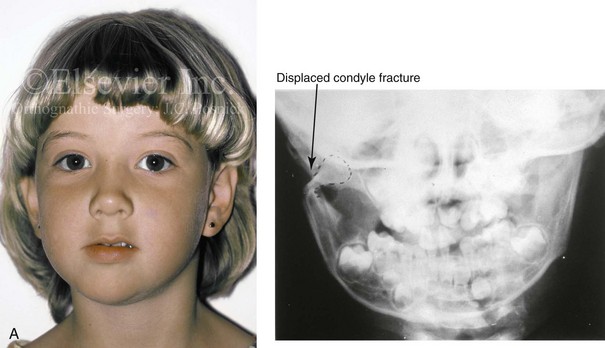
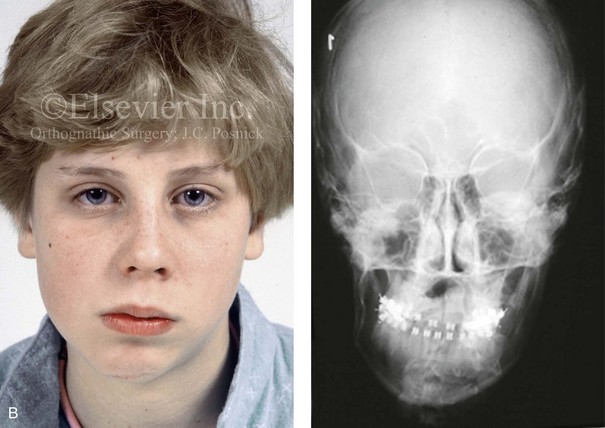
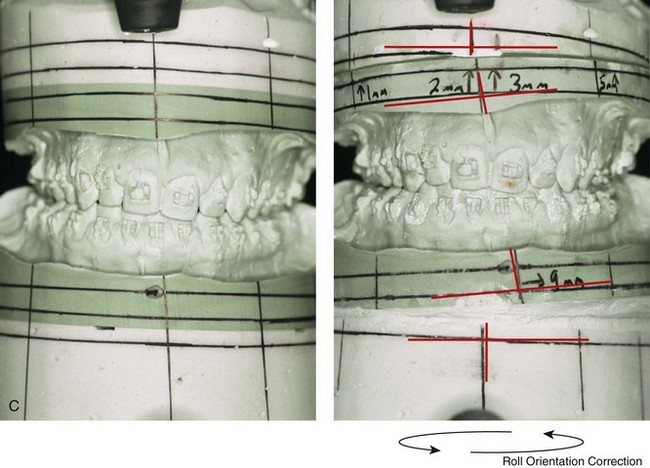
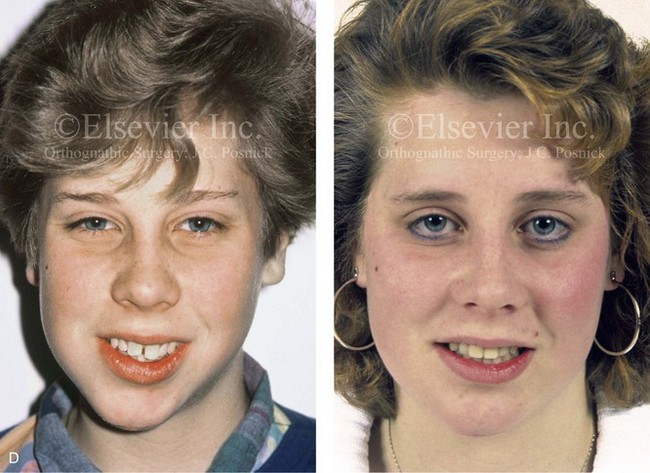
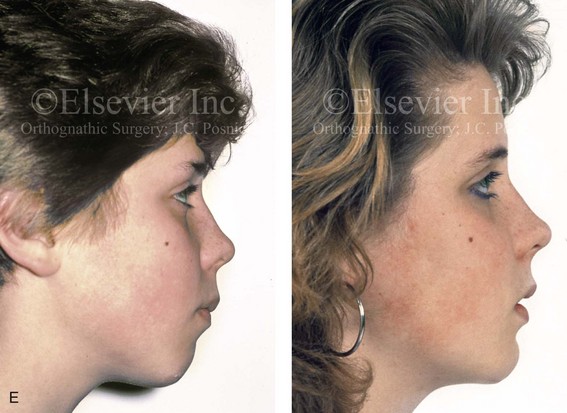
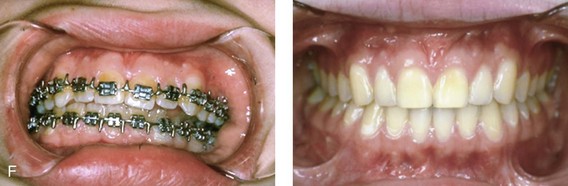
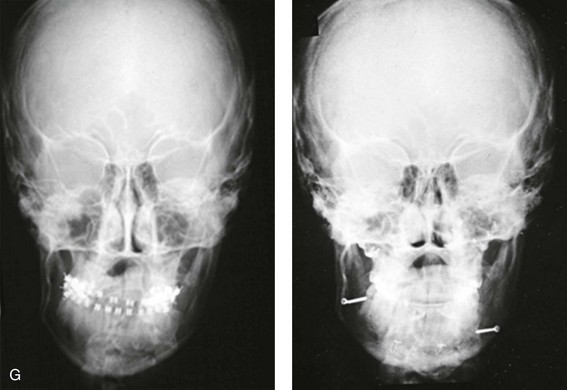
Figure 4-39 A 5-year-old girl sustained a right condyle fracture of the mandible and then presented as a teenager with resulting facial asymmetry involving the maxilla and the mandible. She had full painless mandibular mobility. She then underwent a combined orthodontic and orthognathic approach that included a Le Fort I osteotomy (cant correction), bilateral sagittal split ramus osteotomies (asymmetry correction), an osseous genioplasty (horizontal advancement), and intranasal procedures (septoplasty, inferior turbinate reduction, and nasal recontouring) during her teenage years (see Chapter 35). A, Frontal view when the patient was 6 years old, which was 1 year after the right condyle fracture. The anteroposterior facial radiograph indicates a medially displaced right condyle fracture. B, Frontal view when the patient was 15 years old. An anteroposterior cephalometric radiograph confirms the resulting facial asymmetry that primarily involved the mandible but that now also involves the secondary deformity of the maxilla and the chin. C, Articulated dental casts that indicate analytic model planning. D, Frontal views before and after reconstruction. E, Profile views before and after reconstruction. F, Occlusal views before and after reconstruction. G, Posteroanterior cephalometric radiographs before and after reconstruction.
When significant condylar trauma occurs during childhood, with or without fracture, there will have been injury to the fibrocartilage, with a potential effect on growth. If in fact the “condylar cartilage–push theory” of mandibular growth is all important, a devastating growth distortion should occur over time in all affected individuals. Walker tested this theory by deliberately producing condylar fractures in young monkeys and then observing the effect of these fracture on subsequent growth.131,132 He was unable to document a consistent progressive mandibular deficiency or deformity with malocclusion. Mandibular growth that approximated normal occurred in the majority of experimental animals. Late postmortem gross and histologic evaluation confirmed that there was regeneration of the condylar process with a new layer of cartilage over the articular surface that was identical to the contralateral (noninjured) side. Clinical studies on a series of human children who sustained similar condylar neck fractures confirmed the potential to regenerate bone that approximated the lost (injured) condyle adverse effects on growth in many of the cases.42,74 It also became evident from these studies that a notable minority (approximately 25%) of affected individuals do sustain significant growth disturbances (i.e., mandibular hypoplasia or asymmetry) after a condylar injury, often with secondary deformity in the growing maxilla (see Fig. 4-39). The explanation for this variation in human response (i.e., repair versus deformity) is likely dependent on the following: 1) the extent of initial injury; 2) the extent of resulting scarring or the lack of regenerative potential; 3) the variable importance of the condyle as a growth center in each individual child; and 4) the variable adaptive ability of the muscles of mastication (see Chapter 35).
Over the decades, clinicians have confirmed the work of Gilhuus-Moe 42 and Lund74 by documenting a spectrum of possible outcomes after condylar fracture. Reports confirm instances in which the fracture dislocation of a condylar process has occurred and is followed by a continued normal downward and forward translation of the mandibular body with regeneration of a neocondyle back to baseline. Other clinical reports show a complete loss of the condylar morphology without regeneration followed by decreased posterior facial height, ipsilateral mandibular hypoplasia, and secondary deformity of the contralateral side of the mandible, the maxilla, and the chin region. This sequence of events results in significant maxillomandibular asymmetry and disproportion (see Fig. 35-8). There are also numerous reported examples of bony ankylosis of the condylar stump to the glenoid fossa after the fracture of the condyle (see Fig. 35-1).
Effects of Tumors and Growths on Skeletal Growth
The presence of a sustained expansile mass (e.g., cyst, cellular tumor, skeletal mass) will distort the normal growth and morphology of the bones that are being compressed or expanded.1,6,9,20,24,26,34,46,87–89,95–99 Frequent examples of this occurrence in the maxillofacial region include the effects of neurofibromatosis, vascular malformations, Proteus syndrome, fibrous dysplasia, and odontogenic tumors and cysts (Fig. 4-40). In the adult, the compressive forces of a tumor mass may cause resorption and even result in pathologic fracture. The tumor mass may also compress visceral structures and cavities, thereby causing an impairment of function. Depending on the tumor’s location, volume, and velocity of expansion, there may be alterations in cognitive function, vision, speech, breathing, swallowing, chewing, lip control, hearing, neck range of motion, or mandibular range of motion. When radiation therapy is required for the management of a malignancy, damage to the adjacent soft tissues and skeletal structures frequently occurs. The arrest of growth in irradiated immature bones may result in orbital dysplasia, zygomatic hypoplasia jaw deformity with malocclusion, and damage to the developing teeth and periodontium (Fig. 4-41).
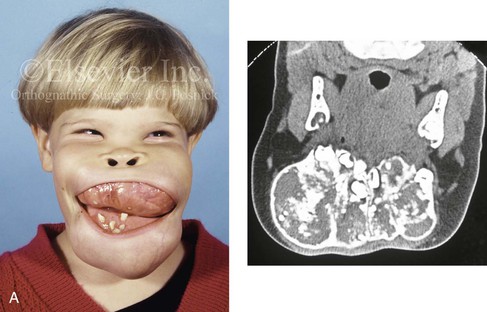
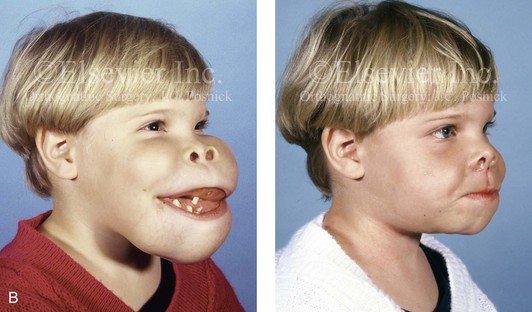
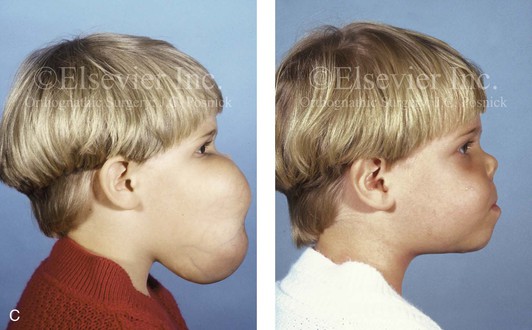
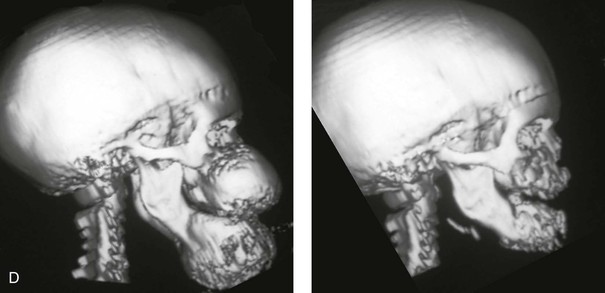
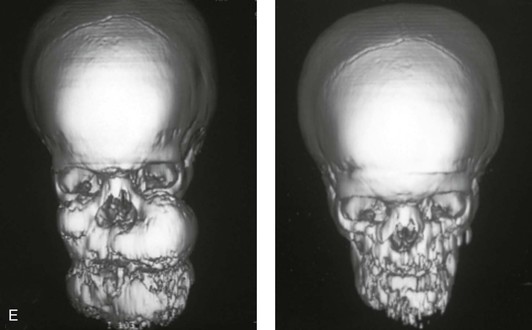
Figure 4-40 A 5-year-old girl with polyostotic fibrous dysplasia with massive involvement of the maxilla and mandible bilaterally. An asymmetric swelling of the mandible (left greater than right) was first recognized when the child was 18 months old; it rapidly enlarged, and maxillary involvement became evident with the deterioration of breathing, chewing, speech, and swallowing. Pathologic fractures were sustained in all four extremities as a result of the involvement of fibrous dysplasia. A, Frontal and computed tomography scan views before surgery. B, Oblique facial views before and after reconstruction. C, Profile facial views before and after reconstruction. D and E, Computed tomography views before and after a debulking procedure. From Posnick JC, Hughes CA, Milmoe G, et al: Polyostotic fibrous dysplasia: an unusual presentation in childhood. J Oral Maxillofac Surg 54:1458–1464, 1996.
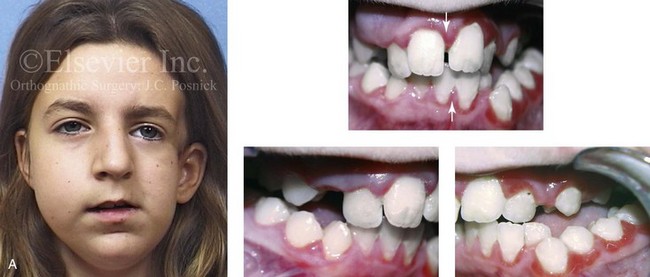
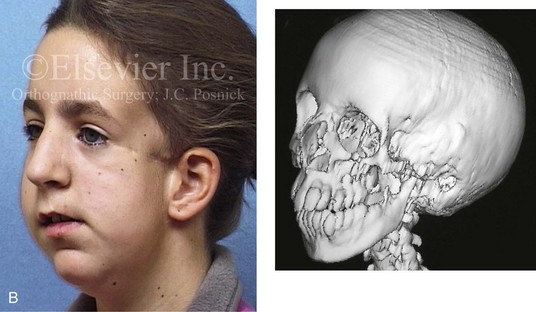
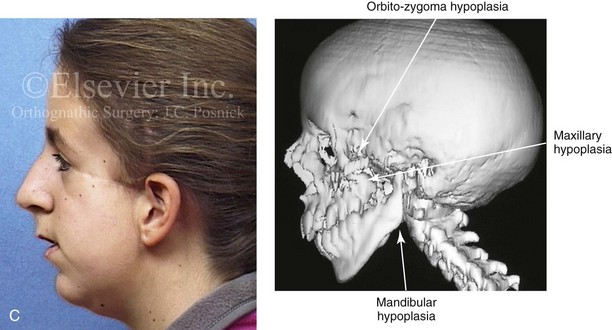
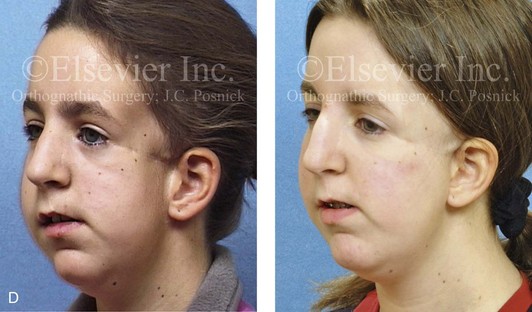
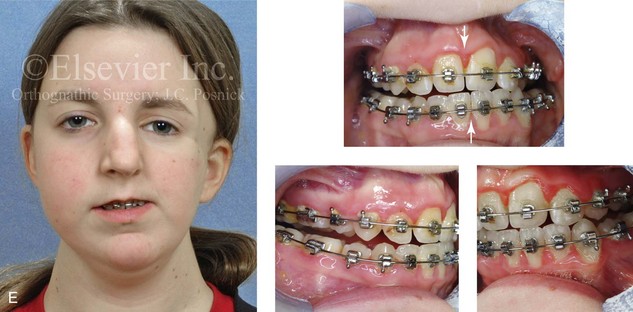
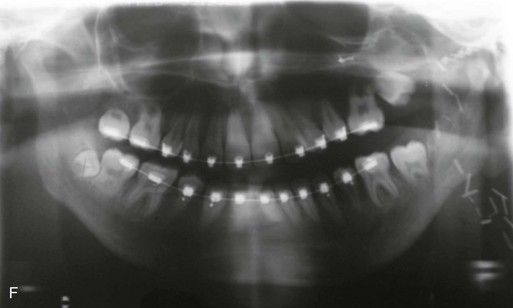
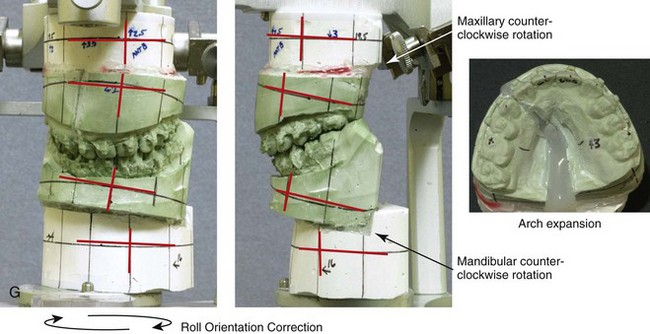
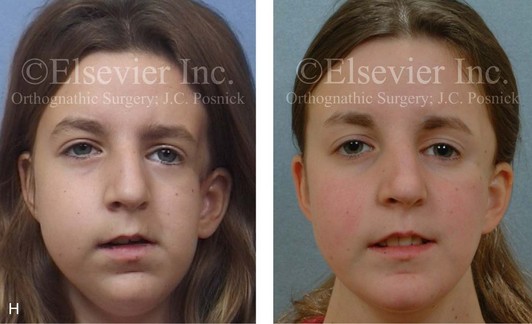
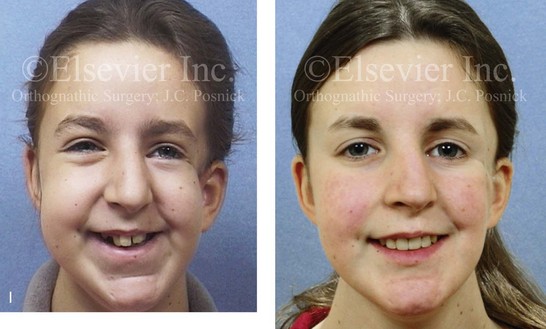
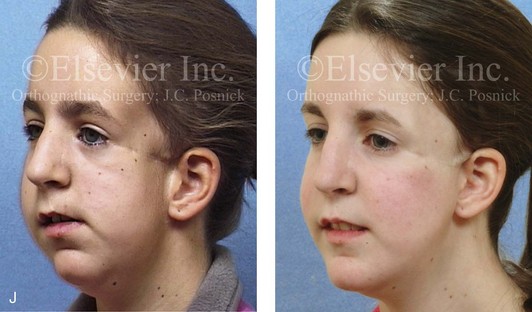
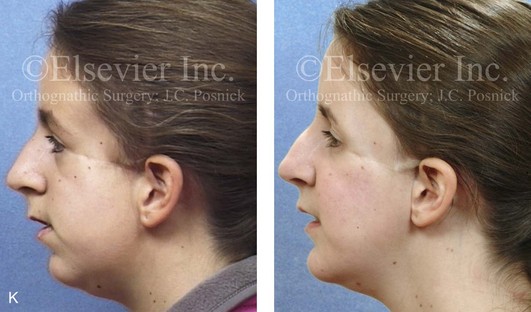
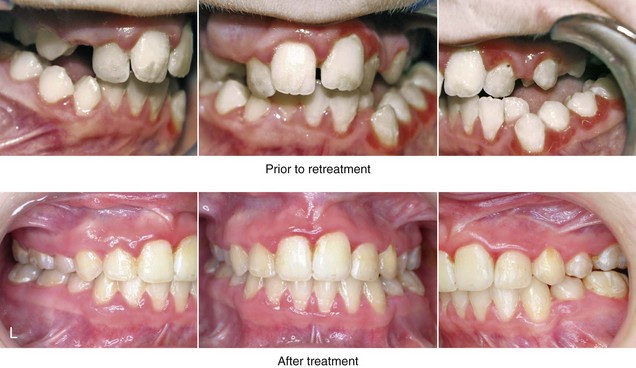
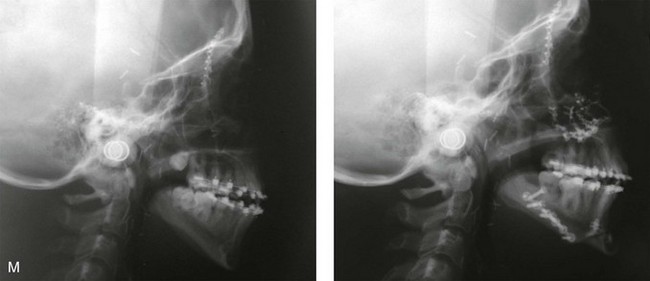
Figure 4-41 A teenage girl who at 9 years of age was found to have a malignancy of the parameningeal region. Biopsy confirmed the diagnosis of rhabdomyosarcoma. She was treated with surgical debulking, chemotherapy, and radiation according to intergroup protocols. There was resulting radiation growth disturbance of the skeletal and soft-tissue structures. The patient underwent staged reconstruction, including stage I serratus muscle microvascular transfer to the left temporal region in combination with full-thickness autogenous cranial graft reconstruction of the left zygomatic orbital complex when she was 13 years old. This was followed when she was 16 years old by further staged orthognathic reconstruction to manage the jaw deformity and the malocclusion. The orthodontic portion of her treatment was carried out cautiously, because there was radiation injury to her left posterior maxillary and mandibular dentition. The procedures included maxillary Le Fort I osteotomy in two segments with interpositional autogenous iliac (hip) grafting, bilateral sagittal split ramus osteotomies of the mandible, and an osseous genioplasty. Septoplasty, inferior turbinate reduction, and recontouring of the floor of the nose were also carried out to open the airway. A, Facial and occlusal views are shown when the patient was 12 years old, before reconstruction. B, Oblique facial and computed tomography scan views when the patient was 12 years old, before reconstruction. C, Profile facial and computed tomography scan views when the patient was 12 years old, before reconstruction. D, Before and 1 year after left zygomatic and orbital cranial graft reconstruction and serratus muscle free-tissue transfer reconstruction. E, Frontal and occlusal views when the patient was 13 years old, with orthodontics in progress in preparation for jaw reconstruction. F, Panorex radiograph when the patient was 13 years old, with preoperative orthodontics complete in preparation for jaw reconstruction. Note the short dental roots on the left side. G, Articulated dental casts that indicate analytic model planning. H, Frontal facial views in repose before and after reconstruction. I, Frontal views with smile before and after reconstruction. J, Oblique facial views before and after reconstruction. K, Profile facial views before and after reconstruction. L, Occlusal views before and after orthodontics and orthognathic surgery. M, Lateral cephalometric radiographs before and after reconstruction.
References
1. Adamides, IP, Spyropoulos, MN. The effects of lymphadenoid hypertrophy on the position of the tongue, the mandible and the hyoid bone. Eur J Orthod. 1983; 5:287–294.
2. Allwright, WC, Bundred, WH. A survey of handicapping dentofacial anomalies among Chinese in Hong Kong. Int Dent J. 1964; 14:505–519.
3. Bachrach, FH, Young, M. A comparison of the degree of resemblance in dental characteristics shown in pairs of twins of identical and fraternal types. Br Dent J. 1927; 21:1293–1304.
4. Bacon, WH, Turlet, JC, Krieger, J, Stierle, JL. Craniofacial characteristics in patients with obstructive sleep apnea syndrome. Cleft Palate J. 1988; 25:374–378.
5. Behrents, RG. The biological basis for understanding craniofacial growth during adulthood. Prog Clin Biol Res. 1985; 187:307–319.
6. Bradley, JP, Zide, BM, Berenstein, A, Longaker, MT. Large arteriovenous malformations of the face: aesthetic results with recurrence control. Plast Reconstr Surg. 1999; 103:351–361.
7. Bresolin, D, Shapiro, PA, Shapiro, GG, et al. Mouth breathing in allergic children: its relationship to dentofacial development. Am J Orthod. 1983; 84:334–340.
8. Brudevold, F. A basic study of the chewing forces of a denture wearer. J Am Dent Assoc. 1951; 43:45–51.
9. Burns, AJ, Kaplan, LC, Mulliken, JB. Is there association between hemangioma and syndromes with dysmorphic features? Pediatrics. 1991; 88:1257–1267.
10. Burton, PR, Tobin, MD, Hopper, JL. Key concepts in genetic epidemiology. Lancet. 2005; 366:941–951.
11. Chung, CS, Runck, DW, Niswander, JD, et al. Genetic and epidemiologic studies of oral characteristics in Hawaii’s schoolchildren. I. Caries and periodontal disease. J Dent Res. 1970; 49:1374–1385.
12. Cleall, JF. Deglutition: a study of form and function. Am J Orthod. 1965; 51:566–594.
13. Close, RJ. Dynamic properties of mammalian skeletal muscle. Physiol Rev. 1972; 52:129–197.
14. Cohen, MM, Jr., Rollnick, BR, Kaye, CL. Oculoauriculovertebral spectrum: an updated critique. Cleft Palate J. 1989; 26:276–286.
15. Cohen, MM, Jr. The child with multiple birth defects, ed 2. New York: Oxford University Press; 1997.
16. Cohen, MM, Jr. Holoprosencephaly survival and performance. Am J Med Genet (Semin Med Genet). 1999; 89:116–120.
17. Cohen, MM, Jr., MacClean, RE. Craniosynostosis: diagnosis, evaluation, and management. New York: Oxford University Press; 2000.
18. Cohen, MM, Jr. Problems in the definition of holoprosencephaly. Am J Med Genet. 2001; 103:183–187.
19. Cohen, MM, Jr. Perspectives on craniofacial anomalies, syndromes, and other disorders. In: Lin KY, Ogle RC, Jane JA, eds. Craniofacial surgery: science and surgical technique. Philadelphia: W. B. Saunders Company; 2002:3–38.
20. Cohen, MM, Jr. Vasculogenesis, angiogenesis, hemangiomas, and vascular malformations. Am J Med Genet. 2002; 108:265–274.
21. Cohen, MM, Jr., Neri, G, Weksberg, R. Overgrowth syndromes. New York: Oxford University Press; 2002.
22. Cohen, MM, Jr. FGFs/FGFRs and associated disorders. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, eds. Inborn errors of development. New York: Oxford University Press; 2004:380–400.
23. Cohen, MM, Jr. Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res A Clin Mol Teratol. 2006; 76:658–673.
24. Cohen, MM, Jr. Vascular update: morphogenesis, tumors, malformations, and molecular dimensions. Am J Med Genet. 2006; 140A:2013–2038.
25. Cohen, MM, Jr. Craniofacial disorders. In: Rimoin DL, Conner JM, Pyeritz RE, Korf BR, eds. Emory and Rimoin’s principles and practice of medical genetics. ed 5. Philadelphia: Elsevier; 2007:3303–3348.
26. Cohen, MM, Jr., Craniofacial abnormalities. Potter’s pathology of the fetus, infant, and child. ed 2. Gilbert-Barness, E, eds. Potter’s pathology of the fetus, infant, and child; vol 1. Mosby/Elsevier, Philadelphia, 2007:885–918.
27. Cohen, MM, Jr. Holoprosencephaly: a mythologic and teratologic distillate. Am J Med Genet Part C Semin Med Genet. 2010; 154C:8–12.
28. D’Ascanio, L, Lancione, C, Pompa, G, et al. Craniofacial growth in children with nasal septum deviation: a cephalometric comparative study. Int J Pediatr Otorhinolaryngol. 2010; 74:1180–1183.
29. Downs, WG. Studies in the causes of dental anomalies. J Dent Res. 1928; 16:367–379.
30. Edwards, SJ, Gladwin, AJ, Dixon, MJ. The mutational spectrum in Treacher Collins syndrome reveals a predominance of mutations that create a premature termination codon. Am J Hum Genet. 1997; 60:515–524.
31. El-Gheriani, AA, Maher, BS, El-Gheriani, AS, et al. Segregation analysis of mandibular prognathism in Libya. J Dent Res. 2003; 82:523–527.
32. Emrich, RE, Brodie, AG, Blayney, JR. Prevalence of class I, class II, and class III malocclusions (Angle) in an urban population: an epidemiological study. J Dent Res. 1965; 44:947–953.
33. Enache, AM, Nimigean, VR, Mihaltan, F, et al. Assessment of sagittal and vertical skeletal patterns in Romanian patients with obstructive sleep apnea. Rom J Morphol Embryol. 2010; 51:505–508.
34. Enjolras, O, Mulliken, JB. Vascular tumors and vascular malformations. Adv Dermatol. 1998; 13:375–423.
35. Finn, RA. Relationship of vertical maxillary dysplasias, bite force, and integrated EMG. In: Abstracts of Conference on Craniofacial Research. Ann Arbor, Mich: University of Michigan Center for Human Growth and Development; 1978.
36. Fox, RR, Crary, DD. Mandibular prognathism in the rabbit. J Hered. 1971; 62:23–27.
37. Fraser, FC. Liability, thresholds, malformations, and syndromes. Am J Med Genet. 1996; 66:75–76.
38. Freng, A, Kvam, E. Facial sagittal growth following partial basal resection of the nasal septum: a retrospective study in man. Eur J Orthod. 1979; 1:89–96.
39. Freng, A. Dentofacial development in the long-lasting nasal stenosis. Scand J Dent Res. 1979; 89:260–267.
40. Garner, LD, Butt, MH. Malocclusion in black American and Nyeri Kenyans, an epidemiologic study. Angle Orthod. 1985; 55:139–146.
41. Gass, JR, Valiathan, M, Tiwari, HK, et al. Familial correlations and heritability of maxillary midline diastema. Am J Orthod Dentofacial Orthop. 2003; 123:35–39.
42. Gilhus-Moe, O. Fractures of the mandibular condyle in the growth period. Stockholm: Scandinavian University Books, Universitatsforlaget; 1969.
43. Graf, H, Grassl, J, Aeberhard, H. A method for measurement of occlusal force in three directions. Helv Odont Acta. 1974; 18:7–11.
44. Grewe, JM, Cervenka, J, Shapiro, BL, et al. Prevalence of malocclusion in Chippewa Indian children. J Dent Res. 1968; 47:302–305.
45. Handelman, CS, Osborne, G. Growth on nasopharynx and adenoid development from one to eighteen years. Angle Orthod. 1976; 46:243–258.
46. Hannuksela, A. The effects of moderate and severe atopy on the facial skeleton. Eur J Orthod. 1981; 3:187–193.
47. Harding, CO, Green, CG, Perloff, WH. Respiratory complications in children with spondyloepiphyseal dysplasia congenital. Pediatr Pulmonol. 1990; 9:49–54.
48. Harris, EF, Johnson, MG. Heritability of crainiometric and occlusal variables: a longitudinal sib analysis. Am J Orthod Dentofac Orthop. 1991; 99:258–268.
49. Harvold, EP. The role of function in the etiology and treatment of malocclusion. Am J Orthod. 1968; 54:883–898.
50. Harvold, EP, Chierici, G, Varervik, K. Experiments on the development of dental malocclusion. Am J Orthod. 1972; 61:38–44.
51. Harvold, EP, Vargervik, K, Chierici, G. Primate experiments on oral sensation and dental malocclusion. Am J Orthod Dentofacial Orthop. 1973; 63:494–508.
52. Harvold, EP. Centric relation: a study of pressure and tension systems in bone modeling and mandibular positioning. Dent Clin North Am. 1975; 19:473–484.
53. Harvold, EP, Tomer, BS, Varervik, K, Chierici, G. Primate experiments on oral respiration. Am J Orthod. 1981; 79:359–372.
54. Harvold EP, Chierici G, Vargervik K, eds. Treatment of hemifacial microsomia. New York: Alan R. Liss, 1983.
55. Haryett, RD, Hansen, FC, Davidson, PO. Chronic thumb sucking: the psychological effects and relative effectiveness of various methods of treatment. Am J Orthod Dentofacial Orthop. 1967; 53:569–585.
56. Holmberg, H, Linder-Aronson, S. Cephalometric radiographs as a means of evaluating the capacity of the nasal and nasopharyngeal airway. Am J Orthod. 1979; 76:479–490.
57. Horowitz, SL, Osborne, RH, Degeorge, FV. A cephalometric study of craniofacial variation in adult twins. Angle Orthod. 1960; 30:1–5.
58. Huang, CM, Mi, MP, Vogt, DW. Mandibular prognathism in the rabbit: discrimination between single-locus and multifactorial models of inheritance. J Hered. 1981; 72:296–298.
59. Hunt, EE. The continuing evolution of modern man. Cold Spring Harb Symp Quant Biol. 1959; 24:245–254.
60. Iwagake, H. Hereditary influence of malocclusion. Am J Orthod Oral Surg. 1938; 24:328–336.
61. Johnson, AL. The constitutional factor in skull form and dental occlusion. Am J Orthod Oral Surg. 1940; 26:627–663.
62. Kraus, BS, Wise, WJ, Frie, RA. Heredity and the craniofacial complex. Am J Orthod. 1959; 45:172–217.
63. Kreiborg, S, Jensen, BL, Møller, E, Björk, A. Craniofacial growth in a case of congenital muscular dystrophy. Am J Orthod. 1978; 74:207–215.
64. Krogman, WM. The role of urbanization in the dentition of various population groups. Zf Rassenkunde. 1938; 9:41–72.
65. Lear, CSC, Moorrees, CFA. Bucco-lingual muscle force and dental arch form. Am J Orthod Dentofacial Orthop. 1969; 56:379.
66. Lehman, H, Harari, D, Tarazi, E, et al. Orthognathic surgery in primary myopathies: severe case of congenital fiber type disproportion with long-term follow-up and review of the literature. J Oral Maxillofac Surg. 2012; 70:1636–1642.
67. Lew, KK, Foong, WC, Loh, E. Malocclusion prevalence in an ethnic Chinese population. Aust Dent J. 1993; 38:442–449.
68. Linder Aronson, S, Aschan, G. Nasal resistance to breathing and palatal height before and after expansion of the median palatine suture. Odontol Rev. 1963; 14:254–270.
69. Linder-Aronson, S. Adenoids: their effect on mode of breathing and nasal airflow and their relationship to characteristics of the facial skeleton and the dentition: a biometric, rhino-manometric and cephalometro-radiographic study on children with and without adenoids. Acta Otolaryngol Suppl. 1970; 265:1–132.
70. Linder-Aronson, S, Lindgren, J. The skeletal and dental effects of rapid maxillary expansion. Br J Orthod. 1979; 6:25–29.
71. Linderholm, H, Wennstrom, A. Isometric bite force and its relation to general force and body build. Acta Odontol Scand. 1970; 28:679–689.
72. Linderholm, H, Lindquist, B, Ringquist, M, Wennstrom, A. Isometric bite force in children and its relation to body build and general muscle force. Acta Odontol Scand. 1971; 29:563–568.
73. Litton, SF, Ackerman, LV, Issacson, RJ, Shapiro, BL. A genetic study of Class III malocclusion. Am J Orthod. 1970; 58:565–577.
74. Lund, K. Mandibular growth and remodeling processes after mandibular fractures. Acta Odontol Scand Suppl. 1974; 32:64.
75. Lundström, A. The importance of genetic and non-genetic factors in the facial skeleton studies in 100 pairs of twins. Trans Eur Orthod Soc. 1954.
76. Lundström, A. The significance of genetic and non-genetic factors in the profile of the facial skeleton. Am J Orthod. 1955; 41:910–916.
77. Lundström, A, Woodside, DG. Individual variation in growth directions expressed at the chin and the midface. Eur J Orthod. 1980; 2:65–79.
78. Lundström, A. Nature versus nature. Eur J Orthod. 1986.
79. Mah, J. Genetics of the “Habsburg jaw,”. Eur Orthod Soc Res. 2001; 1:1–8.
80. Mason, RM, Proffit, WR. The tongue thrust controversy: background and recommendations. J Speech Hear Disord. 1974; 39:115–132.
81. McNamara, JA, Jr. Neuromuscular and skeletal adaptation to altered function in the orofacial region. Am J Orthod. 1973; 64:578–606.
82. Mills, JRE. Occlusion and malocclusion of the teeth of primates. In: Brothwell DR, ed. Dental anthropology. New York: Macmillan; 1963:29–51.
83. Mills, LF. Epidemiologic studies of occlusion: IV. The prevalence of malocclusion in a population of 1,455 school children. J Dent Res. 1966; 45:332–336.
84. Miotti, A. Rassegna bibliografica sull’eziologia ed epidemiologia del prognatismo mandibolare. Dent Cadmos. 1982; 50:29–32.
85. Morton, NE, Chung, CS, Mi, MP. Genetics of interracial crosses in Hawaii. Basel: Karger; 1967.
86. Mossey, PA. The heritability of malocclusion: Part 1–Genetics, principles and terminology. Br J Orthod. 1999; 26:103–113.
87. Mulliken, JB, Glowacki, J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. 1982; 69:412–422.
88. Mulliken, JB. Cutaneous vascular anomalies. Semin Vasc Surg. 1993; 6:204–218.
89. Mulliken, JB. Vascular anomalies. In: Aston SJ, Beasley RW, Thorne CHM, eds. Grabb and Smith’s plastic surgery. ed 5. Philadelphia: Lippincott-Raven; 1997:191.
90. Nagan, P, Fields, HW. Open bite: a review of etiology and management. Pediatr Dent. 1997; 19:91–98.
91. Newman, GV. The Eskimo’s dentofacial complex; effects of environmental and genetic factors. U S Armed Forces Med J. 1952; 3:1653–1662.
92. Niswander, JD, Sujaku, C. Congenital anomalies of teeth in Japanese children. Am J Phys Anthropol. 1963; 21:569–574.
93. Niswander, JD. Further studies on the Xavante Indians: VII. The oral status of the Xavante of Simões Lopes. Am J Hum Genet. 1967; 19:543–553.
94. Nordstrom, SW, Yemm, R. The relationship between jaw position and isometric active tension produced by direct stimulation of the rat masseter muscle. Arch Oral Biol. 1974; 19:353–359.
95. Posnick, JC, Cleland, HJ, Zuker, RM, Mock, D. Recurrent giant cell lesion of the nasomaxillary region in a child: excision and immediate reconstruction with a free rectus abdominis muscle transfer. J Oral Maxillofac Surg. 1992; 50:1009–1015.
96. Posnick, JC, Al-Qattan, MM, Zuker, RM. Large vascular malformation of the face undergoing resection with facial nerve preservation in infancy. Ann Plast Surg. 1993; 30(1):67–70.
97. Posnick, JC, Chen, P, Zuker, R, et al. Extensive malignant melanoma of the uvea in childhood: resection and immediate reconstruction with microsurgical and craniofacial techniques. Ann Plast Surg. 1993; 31(3):265–270.
98. Posnick, JC, Wells, MD, Drake, JM, et al. Childhood fibrous dysplasia presenting as blindness: a skull base approach for resection and immediate reconstruction. Pediatr Neurosurg. 1993; 19(5):260–266.
99. Posnick, JC. Pediatric craniomaxillofacial tumors. Oral Maxillofac Surg Clin North Am. 1994; 197–210.
100. Proffit, WR, Gamble, JW, Christiansen, RL. Generalized muscular weakness with severe anterior open bite. Am J Orthod. 1968; 54:104–110.
101. Proffit, WR, Norton, LA. The tongue and oral morphology: influences of tongue activity during speech and swallowing. J Speech Hear Res. 1970; 5:106.
102. Proffit, WR. Muscle pressures and tooth position: North American whites and Australian aborigines. Angle Orthod. 1975; 45:1–11.
103. Proffit, WR, Sellers, KT. The effect of intermittent forces on eruption of the rabbit incisor. J Dent Res. 1986; 65:118–122.
104. Proffit, WR, Phillips, C. Adaptations in lip posture and pressure following orthognathic surgery. Am J Orthod. 1988; 93:294–304.
105. Proffit, WR, Phillips, C, Fields, HW, Turvey, TA. The effect of orthognathic surgery on occlusal force. J Oral Maxillofac Surg. 1989; 47:457–463.
106. Samman, N, Tong, AC, Cheung, LK, et al. Analysis of 300 dentofacial deformities in Hong Kong. Int J Adult Orthodon Orthognath Surg. 1992; 7:181–185.
107. Schudy, FF. The rotation of the mandible resulting from growth: its implications in orthodontic treatment. Angle Orthod. 1965; 35:36–50.
108. Schull, WJ, Neel, JV. The effects of inbreeding on Japanese children. New York: Harper & Row; 1965.
109. Seigel, ML. Mechanisms of early maxillary growth: implications for surgery. J Oral Surg. 1976; 34:106–112.
110. Shprintzen, RJ. Pierre Robin, micrognathia, and airway obstruction: the dependency of treatment on accurate diagnosis. Int Anesthesiol Clin. 1988; 26:64–71.
111. Sillman, JH. Dimensional changes in the dental arches: longitudinal study from birth to 25 years. Am J Orthod Dentofacial Orthop. 1964; 50:824.
112. Singh, GD, McNamara, JA, Jr., Lozanoff, S. Procrustes, Euclidean and cephalometric analysis of the morphology of the mandible in human class III malocclusions. Arch Oral Biol. 1998; 43:535–543.
113. Singh, GD. Morphologic determinants in the etiology of class III malocclusion: a review. Clin Anat. 1999; 12:382–405.
114. Siriwat, PP, Jarabak, JR. Malocclusion and facial morphology: is there a relationship? Angle Orthod. 1985; 55:127–138.
115. Sousa, JB, Anselmo-Lima, WT, Valera, FC, et al. Cephalometric assessment of the mandibular growth pattern in mouth-breathing children. Int J Pediatr Otorhinolaryngol. 2005; 69:311–317.
116. Stiles, KA, Luke, JE. The inheritance of malocclusion due to mandibular prognathism. J Hered. 1953; 44:241–245.
117. Stockard, CR. The genetic and endocrine basis for differences in form and behavior as elucidated by studies of contrasted pure-line dog breeds and their hybrids. Philadelphia: Wistar Institute; 1941.
118. Subtelny, JD. Oral respiration: facial maldevelopment and corrective dentofacial orthopedics. Angle Orthod. 1980; 50:147–164.
119. Tenczar, P, Bader, RS. Maternal effect in dental traits of the house mouse. Science. 1966; 15:1398–1400.
120. Thompson, EM, Winter, RM. Another family with the “Habsburg jaw,”. J Med Genet. 1988; 25:838–842.
121. Thompson, JR, Broadie, AG. Factors in the position of the mandible. J Am Dent Assoc. 1942; 29:925–941.
122. Throckmorton, G, Finn, R, Bell, W. Biomechanics of differences in lower face height. Am J Orthod. 1980; 77:410–420.
123. Tollaro, I, Baccetti, T, Franchi, L. Class III malocclusion in the deciduous dentition: a morphological and correlation study. Eur J Orthod. 1994; 16:401–408.
124. Treacher Collins Syndrome Collaborative Group. Positional cloning of a gene involved in pathogenesis of Treacher Collins syndrome. Nat Genet. 1996; 12:130–136.
125. Turvey, TA, Hall, DJ, Warren, DW. Alterations in nasal airway resistance following superior repositioning of the maxilla. Am J Orthod. 1984; 85:109–115.
126. Tweed, CH. Clinical orthodontics. St. Louis: Mosby; 1966.
127. Valera, FC, Travitzki, LV, Mattar, SE, et al. Muscular, functional and orthodontic changes in preschool children with enlarged adenoids and tonsils. Int J Pediatr Otorhinolaryngol. 2003; 67:761–770.
128. Vargervik, K, Miller, AJ, Chierici, G, et al. Morphologic response to changes in neuromuscular patterns experimentally induced by altered mode of respiration. Am J Orthod. 1984; 85:115–124.
129. Vargervik, K. Relationship between bone and muscles of mastication in hemifacial microsomia [discussion]. Plast Reconstr Surg. 1997; 99:998–999.
130. Vargervik, K, Mill, AJ, Chierici, G, et al. Morphologic response to changes in neuromuscular patterns experimentally induce by altered mode of respiration. Am J Orthod. 1984; 85:115–124.
131. Walker, RV. Traumatic mandibular condyle fracture dislocations. Effect on growth in the Macaca rhesus monkey. Am J Surg. 1960; 100:850–863.
132. Walker, RV. Condylar abnormalities. In: Transactions of the 2nd International Conference on Oral Surgery. Copenhagen: Munksgaard Copenhagen; 1965:81–96.
133. Warren, DW, Hershey, HG, Turvey, TA, et al. The nasal airway following maxillary expansion. Am J Orthod. 1987; 91:111–116.
134. Wenzel, A, Henriksen, J, Melsen, B. Nasal respiratory resistance and head posture: effect of intra-nasal corticosteroid (Budesonide) in children with asthma and perennial rhinitis. Am J Orthod. 1983; 84:422–426.
135. Wickwire, NA, White, RP, Proffit, WR. The effect of mandibular osteotomy on tongue position. J Oral Surg. 1972; 30:184–190.
136. Williams, MD, Sarver, DM, Sadowsky, PL, Bradley, E. Combined rapid maxillary expansion and protraction facemask in the treatment of Class III malocclusion in growing children: a prospective long-term study. Semin Orthod. 1997; 3:265–274.
137. Wollf, G, Wienker, TF, Sander, H. On the genetics of mandibular prognathism: analysis of large European noble families. J Med Genet. 1993; 30:112–116.
138. Wyszynski, DF, Beaty, TH, Maestri, NE. Genetics of nonsyndromic oral clefts revisited. Cleft Palate Craniofac J. 1996; 33:406–417.
139. Yamaguchi, T, Park, SB, Narita, A, et al. Genome-wide linkage analysis of mandibular prognathism in Korean and Japanese patients. J Dent Res. 2005; 84:255–259.
140. Yurkstas, A, Curby, W. Force analysis of prosthetic appliance during function. J Prosthet Dent. 1953; 3:82–87.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]4
Hereditary, Developmental, and Environmental Influences on the Formation of Dentofacial Deformities
• Development of the Head and Neck
• Syndromes and Anomalies that Include Dentofacial Deformities
• Hereditary Tendencies in the Development of Dentofacial Deformities
• Environmental and Neuromotor Effects on Skeletal Growth
• Effects of Trauma on Skeletal Growth
The term dentofacial deformity is generally used to describe a significant disproportion of the jaws in association with malocclusion. It is estimated that, at a minimum, 5% of the population will have a discrepancy that falls into the developmental dentofacial deformity category. Another 45% of the population will fall into a much larger group of those with malocclusion but without what is considered a “notable” jaw discrepancy component.2,3,5,56,65,72 After jaw growth is complete (i.e., 14 to 16 years of age in girls and 16 to 18 years of age in boys), the definitive reconstruction of a dentofacial deformity can go forward.
Development of the Head and Neck
Skull
The skull is formed from the lateral plate mesoderm (the neck region), the paraxial mesoderm, and the neural crest (Fig. 4-1). The bony skull is formed by one of two mechanisms: intramembranous ossification or endochondral ossification. Skull development is divided into two parts: the viscerocranium and the neurocranium. The viscerocranium forms the bones of the face, whereas the neurocranium forms the bones of the cranial base and the cranial vault. The neurocranium can be divided into the membranous neurocranium and the cartilaginous neurocranium. The anterior fontanel (bregma) has a wide range of expected closure between 4 and 26 months of age. The posterior fontanel (lambda) generally closes between 1 and 2 months of age.
Face
The face is formed mainly from the neural crest, which makes three “swellings” that surround the stomodeum (Fig. 4-2). The three swellings are the frontonasal prominence, the maxillary prominence (from the first pharyngeal arch), and the mandibular prominence (from the first pharyngeal arch).
Palate
The palate is formed by the primary palate (intermaxillary segment) and the secondary palate (protrusions from the lateral prominences) (Fig. 4-3). The intermaxillary segment (primary palate) is the initial portion of the palate to develop. It contains the central and lateral incisors. Swellings of the maxillary prominence form shelves that project medially but that are separated by the tongue. When the tongue no longer occupies the space between the palatal shelves, these processes fuse together to form the secondary palate. The primary and secondary palatal tissues all meet at the incisal foramen. The primary and secondary palates and the nasal septum fuse to form the definitive palate.
Syndromes and Anomalies that Include Dentofacial Deformities
Many of the syndromes and anomalies that involve the cranio-orbito-zygomatic (upper face) region also affect the maxillomandibular (lower face) location.1,20,124 These conditions may involve any of the tissue components, including the skin, the muscles, the nerves, the fat, the vessels, the bone, the teeth, the cartilage, and the associated viscera (i.e., brain, ears, eyes, salivary glands, sinuses). Known syndromes and anomalies comprise only a small proportion of the dentofacial deformity group (see Chapters 27 through 31).19 These conditions can be further subdivided according to etiology or tissue of origin.
Deficiencies of Midline Tissue Structures
Holoprosencephalies involve variable deficiencies of midline tissues. These structural deficiencies have been documented to occur early during embryologic development.16,18,23,27 They may range from severe brain anomalies to simple absence or hypoplasia of the corpus callosum. Severe facial anomalies such as cyclopia, ethmocephaly, and cebocephaly are always associated with severe holoprosencephaly, and they are incompatible with life. This can also be true of premaxillary agenesis; however, in some instances, patients may survive for several months or even years. There are reported cases of premaxillary agenesis with normal head circumference and no brain abnormalities (i.e., without holoprosencephaly), although these patients sometimes have mild mental deficiencies. Syntelencephaly (the absence or hypoplasia of the corpus callosum or absent or hypoplastic olfactory tracts and bulbs) is compatible with life and should thus be treated. Associated facial anomalies include retinal colobomas, cleft lip, cleft palate, single maxillary central incisor, and midface deficiencies. Holoprosencephaly has multiple causes and includes many chromosomal types, many monogenetic disorders, and at least three teratogens.
Deficiencies and Anomalies of Neural Crest Origin
Most of the tissues of the face, including the muscular and skeletal components, are derived from the mesoderm. Interestingly, throughout the rest of the body, these components (i.e., muscle and skeleton) are derived from an ectodermal origin.19 The facial mesodermal elements develop from the neural crest and then migrate downward beside the neural tube and laterally under the surface ectoderm. When the neural crest cells have completed migration to their specific location, then facial development is dominated by specific growth centers, the formation of visceral structures (e.g., brain, eyes, sinuses), and the differentiation of the tissue layers. Facial anomalies of neural crest origin are thought to arise when the neuroepithelium has apoptosis (i.e., the cells die).25 This results in less neuroepithelium to migrate through into the facial locations and to ultimately differentiate. Current knowledge of Treacher Collins syndrome indicates that this is not a migration problem but simply a lack of cells to migrate (Figs. 4-4 and 4-5).
Figure 4-4 An 11-year-old boy who was born with Treacher Collins syndrome has typical malformations of the soft tissues and the skeletal structures within the first and second branchial arches on both sides. There is significant hypoplasia of each zygomatic complex, the orbits, and the maxillomandibular structures (Kaban type II-A mandible). The soft tissues of the adnexal region are deficient. The external ears are malformed, but all parts are present. The central part of the face is fully formed. A, Frontal facial and computed tomography scan views. B, Profile facial and computed tomography scan views. C, Oblique facial and computed tomography scan views.
Figure 4-5 This mother and daughter demonstrate the extent of variation of expression of Treacher Collins syndrome within a family. The mother was not aware that she carried the Treacher Collins gene until after the birth of her daughter. From Posnick JC: Treacher Collins syndrome: perspectives in evaluation and treatment. J Oral Maxillofac Surg 55:1120–1133, 1997.
Teratogens that are known to result in head and neck (neural crest) congenital anomalies include cytomegalovirus (microcephaly, hydrocephaly, microophthalmia); Dilantin (cleft lip and palate); vitamin D excess (premature suture closure); Valium (cleft lip and palate); Rubella virus (microophthalmia, cataracts, deafness); and thalidomide (variations of hemifacial microsomia and Treacher Collins syndrome). Despite these known relationships, teratogenic agents are not the most frequent causes of these syndromes.1
The branchial arch syndromes are neural crest anomalies that comprise an etiologically heterogeneous group of disorders.14 They account for only a small percentage of the patients who present to the surgeon or orthodontist in need of jaw reconstruction. The best known of these are hemifacial microsomia, its variant Goldenhar syndrome, and Treacher Collins syndrome.14 Hemifacial microsomia affects aural, zygomatic, and mandibular growth at a minimum (Fig. 4-6). The disorder may be mild or severe, and it is generally limited to one side of the face. However, bilateral involvement, with more severe expression on one side, is also known to occur. Goldenhar syndrome, which is a variant of hemifacial microsomia, also includes epibulbar dermoids and vertebral anomalies. Involvement is often but not always limited to the face. There may be cardiac, renal, skeletal, and central nervous system anomalies (see Chapter 28). At a minimum, patients with Treacher Collins syndrome demonstrate the physical findings of bilateral zygomatic hypoplasia, down-slanting palpebral fissures, malformed ears, and micrognathia (see Chapter 27). Inheritance occurs in an autosomal-dominant fashion, and expressivity varies (see Figs. 4-4 and 4-5). The syndrome maps to 5q32-q33.1, and mutations in the TCOF1 gene are of the nonsense, insertion, deletion, or splice-site types.14,30,139
Figure 4-6 A 7-year-old boy born with hemifacial microsomia and unilateral cleft lip and palate (left side) has typical malformations of the soft tissues and skeletal structures within the first and second branchial arches on the left side. There is an absence of the left zygomatic complex and hypoplasia of the orbit, the anterior cranial vault, and the maxilla. The mandibular malformation is a Kaban type III. The soft tissues of the left external ear, the ear canal adnexal structures, and the cheek region are markedly deficient. A, Frontal facial and computed tomography scan views. B, Oblique facial and computed tomography scan views.
Clefting of the Lip and Palate
The most prevalent congenital defect of dentofacial development is clefting of the lip, the palate, or both (Figures 4-7 through 4-19). This condition occurs in approximately 1 in 1000 whites, 1 in 500 Asians, and 1 in 2000 blacks.27 The etiology of clefts is often complex and multifactorial. Evidence supports the view that genetic factors are associated with orofacial clefting.37 In twins with cleft lips and palates, concordance is far greater among monozygotic twins (40%) as compared with dizygotic twins (4.2%). In twins with isolated cleft palate, concordance is also higher among monozygotic twins (35%) as compared with dizygotic twins (7.8%).138 Nevertheless, orofacial clefting is heterogeneous and variable, and it is likely determined by a number of major genes, minor genes, environmental factors, and a developmental threshold.37 Some syndromes associated with clefting in which specific gene mutations have been identified include van der Woude syndrome (IRF6), popliteal pterygium syndrome (IRF6), autosomal-recessive cleft palate/ectodermal dysplasia (PVRL1), hypodontia/clefting (MSX1), Hay-Wells syndrome (TP63), and cleft palate/ankyloglossia (TBX22). These mutations explain only about 5% of clefting cases, with an additional 10% to 15% of cases explained by variations involving the IRF6 gene.110 Although orofacial clefting is caused by a malformation, the development of secondary deformities of the jaws after birth is well known (see Chapters 32, 33, and 34). For the most part, maxillary hypoplasia in patients with orofacial clefts is felt to be the result of palate surgical interventions carried out during infancy and early childhood and not from the primary congenital anomaly. Although van der Woude and Stickler are just 2 of more than 100 clefting syndromes, they demonstrate the importance of having an awareness of associated anomalies to assist with family counseling and to achieve effective reconstruction.
Figure 4-7 Cleft lip and palate represents a spectrum of morphologic findings that are initially dependent on the individual’s specific anomalies but that are then affected by surgical intervention during growth. A newborn with a complete (left side) unilateral cleft lip and palate is shown.
Figure 4-8 A Hispanic child was born with complete clefting of the left primary lip and palate. He was adopted when he was approximately 1 year old, before repair. He then underwent single-stage primary lip, nasal, and palate repair. He is shown before repair and then at the age of 2 years after single-stage repair.
Figure 4-9 A child was born with an incomplete cleft of the left lip that also involved the alveolar ridge back to the incisal foramen. She is shown before primary lip and nasal reconstruction and then at the age of 5 years before alveolar cleft grafting.
Figure 4-10 A child was born with a complete (left side) unilateral cleft lip and palate. He is shown before and at intervals after primary lip and palate repair. A, He is shown before primary lip and nasal reconstruction and B, at the age of 10 months, just before cleft palate repair. C, The same child at the age of 3 years and D, at the age of 8 years just before mixed dentition bone grafting.
Figure 4-11 Three newborns with bilateral cleft lip and palate (BCLP) are shown. A, A newborn with complete BCLP. B, A newborn with BCLP but with partial attachment of the lip and the nasal floor on the left side. C, A 3-month-old infant with complete BCLP is shown. The premaxilla is attached to the septum of the nose; it is forwardly projecting without attachment to the lateral lip segments. The wide separation of the palatal shelves is also demonstrated by the intraoral view. D and E, A series of computed tomography scan views of the maxillofacial complex of the infant shown in C demonstrate the skeletal anatomy associated with BCLP.
Figure 4-12 A child born with complete bilateral cleft lip and palate. He is shown before and then 7 years after primary lip and palate repair, just before mixed dentition bone grafting.
Figure 4-13 A child born with incomplete bilateral cleft lip and palate. He is shown before and then 8 years after primary lip and palate repair, just before mixed dentition bone grafting.
Figure 4-14 A child born with a complete cleft of the right lip and palate and incomplete clefting of the left lip. He is shown A, before and then B, at 2 and C, 8 years after primary lip and palate repair. The last photo was taken just before mixed dentition bone grafting.
Figure 4-15 A, A child born with complete clefting of the secondary palate (i.e., incisal foramen through the uvula) is shown. B, An adult born with a complete unilateral cleft lip and palate is also shown. He underwent lip repair during childhood, but the cleft palate was neglected. Note the severe septal deviation that obstructs the right nasal valve.
Figure 4-16 A teenage boy from Jamaica who was born with a bilateral cleft lip and a unilateral cleft of the alveolar ridge and palate. He underwent rudimentary lip repair as a child. The unrepaired clefted alveolus and palatal anatomy are shown. Note the normal growth parameters of the upper jaw when the palate is unrepaired. With a lack of continuity of the alveolar ridge, there has been typical distorted growth of the maxilla.
Figure 4-17 A woman who was born with bilateral cleft lip and palate and pitting of the lower (central) lip. At the time of her pregnancy, an ultrasound confirmed twins, one of which was suspected of having bilateral cleft lip and palate and the other of having unilateral cleft lip and palate. This was documented at the time of delivery. A, A family with van der Woude syndrome, including a mother with a repaired bilateral cleft lip and palate and newborn twins. B, Twin A with bilateral cleft lip and palate. C, Twin B with unilateral cleft lip and palate.
Figure 4-18 A 16-year-old boy who was born with Stickler syndrome (type II collagen mutation). At the time of birth, Pierre Robin sequence was appreciated. The patient underwent repair of the cleft palate before he was 1 year old. He had positive eye findings, and he required the treatment of a retinal detachment during his teenage years. A small cataract is being followed. He arrived for the evaluation of a jaw deformity and malocclusion characterized by maxillomandibular deficiency with anterior open-bite malocclusion. This was combined with chronic obstructive nasal breathing and a long face growth pattern. Attempted growth modification and camouflage orthodontics earlier during life were ineffective. There was generalized root deficiency throughout the maxillary and mandibular dentition, likely as a result of the collagenopathy. A, Frontal views in repose before and after treatment. B, Frontal views with smile before and after treatment. The patient agreed to an orthodontic and surgical approach. Further orthodontic (dental) decompensation was cautiously carried out as a result of the compromised periodontal apparatus. The procedures included maxillary Le Fort I osteotomy (vertical intrusion, horizontal advancement, counterclockwise rotation, and arch expansion); bilateral sagittal split ramus osteotomies (horizontal advancement and counterclockwise rotation); osseous genioplasty (vertical shortening and horizontal advancement); and septoplasty, inferior turbinate reduction, and nasal floor recontouring. C, Oblique facial views before and after treatment. D, Profile views before and after treatment. E, Occlusal views before retreatment, with orthodontics in progress, and after treatment. F, Articulated dental casts that indicate analytic model planning. G, Lateral cephalometric radiographs before and after treatment. H, Panorex radiographs before and after treatment that demonstrate generalized limited root formation. The right maxillary first molar was lost 5 years earlier as a result of limited roots. The right maxillary first molar was lost 1½ years after surgery as a result of limited root support; the right mandibular first molar will likely be lost to similar pathology. Periodontal evaluation and treatment are ongoing. Future plans are for the placement of dental implants in the right posterior maxilla and mandible.
Figure 4-19 A child born with Robin sequence, which consists of retrognathia with glossoptosis, clefting of the secondary palate, and a degree of respiratory distress. A, Frontal facial and intraoral views demonstrating clefting of the secondary palate. B, Profile facial view indicating retrognathia. Computed tomography scan demonstrating retrognathia but with all of the components of the jaw present. For this child, the small mandible is the result of deforming forces during fetal development rather than the result of a malformation. Catch-up growth of the mandible is anticipated during childhood. C, Profile and occlusal views when the patient was 9 years old, with expected catch-up growth.
Van der Woude Syndrome
In 1845, Demarquay was the first to report the occurrence of congenital sinuses of the lower lip in combination with cleft lip and palate; this condition became known as van der Woude syndrome. This syndrome occurs in roughly 1% to 2% of patients with lip and palatal clefts. Van der Woude syndrome has an autosomal-dominant inheritance pattern; it has approximately 90% penetrance, and it is variably expressed (see Fig. 4-17). Manifestations of the syndrome outside of the oral and facial regions are unusual. Generally, the pits are characterized as depressions that are observed on the vermilion border of the lower lip; they are usually bilateral and symmetrically placed, although an asymmetrical pit or single central pit may occur. Roughly 33% of patients with van der Woude syndrome have pits without clefting, 33% have pits with cleft lip and palate, and 33% have lip pits with isolated cleft palate or submucous cleft palate. Approximately 10% of those individuals with van der Woude syndrome do not exhibit lip pits.
Stickler Syndrome
In 1967, Stickler and Pugh were the first to describe a series of patients who had a combination of eye findings, hearing loss, isolated cleft palate, a Marfanoid habitus, and long-bone changes.1 It is now known that Stickler syndrome is associated with basic defects in collagen. These collagenopathies include mutations on type II collagen and mutations in type XI collagen.25 The inheritance pattern with this syndrome is autosomal dominant with variable expressivity. Cohen and others have reviewed the relationship between high myopia and clefting of the secondary palate and the other features of Stickler syndrome.19 High myopia (i.e., 8 to 18 diopters) is found in 75% of patients by the time they are 5 years old. The eye findings are progressive, with vitreous and chorioretinal degeneration and retinal detachment observed in 70% of patients by the time they are 20 years old. Additional eye findings include astigmatism, cataracts, strabismus, and glaucoma. Individuals with Stickler syndrome may manifest any or all of a range of craniofacial findings, including mild to moderate midface hypoplasia, shallow orbits with eye proptosis, epicanthal folds, a flat nasal bridge, and submucous or full clefting of the secondary palate. Some affected individuals will have progressive degeneration of the dental roots and the associated periodontium. This is primarily due to the inborn error in collagen production, but it may be exacerbated by malocclusion (secondary trauma) and orthodontic manipulations (see Fig. 4-18). Progressive neurosensory high-tone hearing loss has been reported in 80% of patients. Some patients have a Marfanoid body habitus, whereas others have short stature. There may be progressive early joint degeneration by the mid-adult years. Any newborn who is found to have Robin sequence should undergo an initial genetic and ophthalmologic assessment to rule out Stickler syndrome.
Robin Sequence
Robin sequence is commonly defined as cleft palate, micrognathia, and glossoptosis (see Fig. 4-19). This sequence of events can occur as a result of a variety of etiologic and pathogenetic conditions, and it involves a wide spectrum of phenotypes. The distinction must be made between a malformed mandible (micrognathia) and a deformed mandible that is simply retrognathic. The malformed mandible (i.e., Treacher Collins syndrome) may also be missing parts or sections of the jaw (i.e., Kaban types IIB and III) and cannot be expected to “grow out” and self-correct. This is different from a deformed mandible (i.e., retrognathic at birth) caused by fetal malpositioning, which can be expected to “grow out” and become relatively normal. The newborn’s respiratory compromise that occurs with Robin sequence will vary according to the child’s physical findings, and treatment should proceed accordingly. For example, with spondyloepiphyseal dysplasia congenita, the causes of respiratory compromise include a small and mechanically abnormal chest, tracheobronchial malacia, and central apnea as a result of cervical or medullary compression caused by cervical instability.47 This will require a different level of airway management as compared with an isolated retrognathic mandible, which is simply deformed as a result of intrauterine compression. If the deformed (retrognathic) mandible is present at birth, it is expected to “catch up” during growth and basically to self-correct. For these infants, mandibular osteotomies with advancement carried out during childhood should be avoided.
Achondroplasia
Achondroplasia is the most common condition associated with severe disproportionate short stature.22 The cause of this condition is the failure during development of the primary growth cartilages of the limbs and the cranial base. This results in short arms and legs as well as a characteristic midface deficiency or deformity that is most visually notable at the nasofrontal process.
Most affected individuals do well but attain a final height of only approximately 4 feet. The condition is known to be caused by mutations on the FGFR3 (fibroblast growth factor receptor 3) gene.22 Eighty percent of reported cases are sporadic, whereas 20% are familial. Although craniosynostosis is not a typical feature, three cases with craniosynostosis have been reported.
Premature Suture Closure of the Cranial Vault and Skull Base
The flat bones of the cranial vault develop from mesenchymal condensations over the developing brain (see Fig. 4-1). These bones grow primarily via the apposition of bone at their edges. The regions in which the cranial bony edges collide are called sutures. In addition to appositional growth at the edges (i.e., suture growth), cranial vault development also occurs through the process of remodeling (i.e., resorption and deposition).17 Continued separation of the sutures during the process of the apposition of bone at the edges is also important to the normal growth of the bones of the cranial base and the midface.
Synostosis is a condition of premature fusion (i.e., the arrest of appositional growth at the bone edges) that occurs before the associated visceral structures (e.g., brain, eyes) complete growth.17 When this happens, there is distortion or deformity of the shape of the affected bones and possible compression of the underlying visceral structures (i.e., the brain). Premature closure of the cranial vault sutures (craniosynostosis) that do not extend into the cranial base will have minimal effects on the maxillofacial form (e.g., metopic, unilateral coronal, and sagittal suture synostosis; Figures 4-20, 4-21, and 4-22). However, prematurely fused cranial vault sutures that extend into the cranial base will also affect midfacial growth and development (e.g., Crouzon, Apert, and Pfeiffer syndrome; Figures 4-23 through 4-26; see Chapter 30).
Figure 4-20 A child born with metopic synostosis that resulted in trigonocephaly. He underwent anterior cranial vault and three-quarter orbital osteotomies with reshaping when he was 10 months old. A, Facial and computed tomography scan views before surgery. B, Illustrations of the craniofacial skeleton in a child with metopic synostosis that resulted in trigonocephaly before and after anterior cranial vault and three-quarter orbital osteotomies. C, Bird’s-eye view of removed orbital osteotomy unit before and after reshaping. Part B modified from an original illustration by Bill Winn. D, Frontal views before and 1 year after reconstruction. E, Facial views of patient at 5 years old. F and G, Computed tomography scan views before and after reconstruction.
Figure 4-21 A child born with right unilateral coronal synostosis that resulted in anterior plagiocephaly. He underwent anterior cranial vault and three-quarter orbital osteotomies with reshaping when he was 9 months old. A, Facial and computed tomography scan views before reconstruction. B, Illustrations of the craniofacial skeleton in a child with unilateral coronal synostosis that resulted in anterior plagiocephaly before and after anterior cranial vault and three-quarter orbital osteotomies. Part B modified from an original illustration by Bill Winn.C, Bird’s-eye view of removed orbital osteotomy units before and after reshaping. D, Frontal views before and 5 years after reconstruction. E, Oblique views 5 years after reconstruction. F and G, Computed tomography scan views of the cranial vault before and just after reconstruction.
Figure 4-22 A child born with sagittal synostosis that resulted in a scaphocephalic shape of the cranial vault that was characterized by increased anteroposterior length and decreased bitemporal and biparietal width. It was not until the child was 2 years old that he was diagnosed with sagittal synostosis. After a comprehensive evaluation, he underwent total cranial vault reshaping through a coronal (scalp) incision. He is shown before and after a single-stage reconstruction. A, Frontal facial views before and after reconstruction. B, Profile facial views before and after reconstruction. C, Illustrations of the craniofacial skeleton in a child with sagittal synostosis that resulted in scaphocephaly before and after total cranial vault and upper orbital osteotomies with reconstruction. Part C modified from an original illustration by Bill Winn.
Figure 4-23 A 6-month-old girl who was born with Apert syndrome and bilateral coronal synostosis. A,
[/not-level-membership-for-surgery-category]
