chapter 5 Herb–drug interactions
INTRODUCTION
Complementary medicines such as herbal medicines are available through a variety of channels such as supermarkets, pharmacies, health-food stores, clinic rooms, internet sites and mail order companies. Many people self-select their products and do not receive professional advice about their safe and appropriate use.1,2 When using a complementary medicine, many do not discuss its use with their medical practitioner, either in the community or in the hospital setting.1,3–14 Importantly, people using complementary medicines tend to have poorer health than the general community15 and are not generally dissatisfied with conventional medicine but use complementary medicines as an adjunct to conventional medical care.1,4,16,17 This raises the possibility of dual care from both complementary and conventional practitioners and the concomitant use of herbs and pharmaceutical medicines. Widespread use, self-selection and poor disclosure suggest that many people feel sufficiently confident that over-the-counter (OTC) herbal medicines are beneficial and that their safety is assumed. This assumption is supported by several Australian studies.1,18
Australia’s risk-based regulatory process for therapeutic goods (including herbal medicines) provides some safeguard. Clinicians can feel reassured that OTC herbal medicines allocated an AUST L number have been produced according to the Code of Good Manufacturing Practice (GMP) and their ingredients assessed for safety. Alternatively, potentially unsafe herbal medicines have warning labels and may even be restricted from sale via the scheduling system, much like pharmaceutical medicines (more information about the regulation of herbal products can be found in Ch 4).
Despite this, it has become apparent over the past decade that some commonly used OTC herbal medicines are capable of causing significant drug interactions, which must be identified and managed to optimise patient safety. Open communication, familiarisation with the most commonly used complementary medicines and understanding the interaction mechanisms involved are vital first steps in promoting patient safety. This chapter provides an introduction to herb–drug interactions; detailed information about specific herb–drug interactions is beyond the scope of this chapter. Comprehensive drug–herb interaction charts and further information are available in Herbs and natural supplements—an evidence-based guide by Braun & Cohen.19
INTERACTIONS
Herbs contain a chemically complex cocktail of naturally occurring ingredients. In this way, they are very different from pharmaceutical medicines. The pharmacological effect of a herbal medicine is the end result of an interplay between various interactions. There are intra-herbal interactions between constituents, interactions between the herb and the vehicles it is mixed with during processing and manufacture, and then the final interaction between the product and person receiving it. Interactions can also occur between the foods, drugs and other herbal medicines being taken by that individual. As you can see, interactions are unavoidable and should not present any clinical problem unless they are unanticipated.
THE MAIN INTERACTION MECHANISMS: OVERVIEW
Considering the great variation in physical properties and pharmacological effects of the numerous substances used as medicines, together with the variable nature of herbal medicines, a virtually endless number of interactions is possible. Interaction mechanisms can be broadly categorised as pharmacodynamic or pharmacokinetic interactions (Fig 5.1). Regardless of the interaction mechanism at work, there are three possible outcomes:
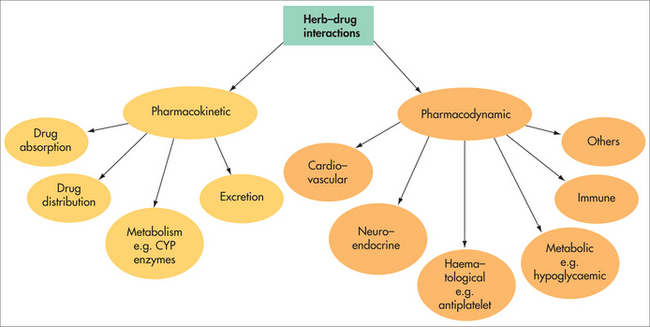
PHARMACOKINETIC INTERACTIONS
Absorption
A reduced rate of absorption can lead to a ‘sustained release’ effect, whereas a reduced extent of absorption is particularly problematic for drugs with a narrow therapeutic index (NTI). Gums and mucilages (such as guar gum and psyllium) are examples of substances known or thought to affect drug absorption. For example, a double-blind study found that guar gum slowed the absorption rate of digoxin but did not alter the extent of absorption, whereas penicillin absorption was both slowed and reduced.20 This brings into question the effects of other gums and highly mucilaginous herbal medicines such as Ulmus fulvus (slippery elm), Althea officinalis (marshmallow) and Plantago ovata (psyllium). Poorly lipid soluble, the mucilaginous content forms an additional physical barrier that needs to be traversed before the medicine can enter systemic circulation. Whether this will have clinically significant effects on the rate and/or extent of absorption of other medicines is uncertain and remains to be tested.
More research has been conducted on the way in which nutrients interact and alter the absorption of other medicines. The interactions between iron and mineral-based antacids are a useful example. Separating the intake of iron and the last antacid dose by at least 2 hours reduces the risk of interaction.19
Metabolism
Enzyme inhibition is an immediate response, with effects seen rapidly.21 It can be reversible, quasi-reversible or irreversible. In practice, most inhibition is reversible, ceasing when use of the inhibitor agent is discontinued. The result of CYP enzyme inhibition is elevated serum levels of those drugs chiefly metabolised by the affected enzyme. Medicines with narrow therapeutic margins, such as digoxin, are of particular concern as small elevations in serum levels have the potential to produce toxic effects. In practice, enzyme inhibition is not always harmful and has been manipulated to raise serum drug levels without the need to increase the dose administered. The result has obvious cost advantages when expensive drugs are involved and has been used in some hospitals for medicines such as cyclosporin. Grapefruit is one example of a natural product having significant enzyme inhibition effects.
Unlike enzyme inhibition, enzyme induction is a relatively slow process and results in reduced serum levels of the drugs chiefly metabolised by the affected CYP enzyme. Many different medicines and everyday substances have been found to be capable of inducing CYP enzymes—examples are broccoli, brussel sprouts, chargrilled meat, high-protein diets and alcohol.19
St John’s wort is a good example of a herbal substance capable of interacting with a variety of drugs through this mechanism. Clinical studies have confirmed that long-term administration of St John’s wort has significant CYP inducer activity, particularly CYP3A4.22–25 This is significant because CYP3A4 is responsible for the metabolism of many pharmaceutical drugs. Studies have isolated the hyperforin component as a potent ligand for the pregnane X receptor, which regulates expression of CYP3A4 mono-oxygenase. In this way, hyperforin increases the availability of CYP3A4, resulting in enzyme induction.26 Examples of CYP3A4 substrates are alprazolam, codeine, erythromycin and simvastatin. Importantly, low-hyperforin St John’s wort extracts such as Ze 117 do not significantly induce CYP3A4, 2D6, 2C9, 1A2 or 2C19, according to human pharmacokinetic studies.27–29 As such, the Ze 117 extract may present a safer treatment option for patients taking multiple drugs at the same time.
PHARMACODYNAMIC INTERACTIONS
A pharmacodynamic interaction occurs when one substance alters the sensitivity or responsiveness of tissues to another substance. This type of interaction results in additive, synergistic or antagonistic drug effects. Pharmacodynamic interactions can also occur when therapeutic substances with overlapping side effects or toxicities are used together, leading to more serious side effects than when either substance is used alone. A good understanding of the pharmacological activity of the therapeutic substances involved can help to theoretically predict potential interactions and optimise therapy.
EVALUATING HERB INTERACTIONS
Most studies investigating herb–drug interactions have used in vitro testing of herbal constituents in microsomal systems, supersomes, cytosols, expressed enzymes or cell culture systems such as transfected cell lines, primary cultures of human hepatocytes and tumour-derived cells. There has also been some in vivo investigation in normal, transgenic and humanised animals and, increasingly, in humans. Although these studies are useful, they have limitations. Table 5.1 presents a summary of the limitations of different types of research. Most interaction studies conducted to date have focused on herbal constituents and their effects on cytochrome (CYP) enzymes and, increasingly, P-glycoprotein (P-gp), with few studies investigating effects on drug transport or phase 2 metabolism.
TABLE 5.1 Advantages and limitations of herb–drug interaction studies
| Study type | Advantages | Limitations |
|---|---|---|
| In vitro |
PROBLEMS IN EXTRAPOLATING DATA
In vitro tests and/or tests with animal models have shown both CYP induction and inhibition for Ginkgo biloba.30–44 In contrast, four clinical studies have failed to identify a clinically significant effect on a variety of cytochromes. In one clinical study, Gurley and colleagues demonstrated that Ginkgo biloba had no significant effect on CYP1A2, CYP2D6 or CYP3A4 activity.45 Markowitz and colleagues also conducted a human study and found no significant effects on CYP2D6 or CYP3A4 activity.46 Two further clinical studies found no significant effect for Ginkgo biloba on CYP2C9 activity.47,48
In the case of saw palmetto, the herb has shown potent inhibition of CYP3A4, CYP2D6 and CYP2C9 in vitro,49 but no significant effect was observed on CYP2D6 or CYP3A4 activity in a clinical study by Markowitz et al.50 Gurley et al. also found no significant effect for saw palmetto on CYP1A2, CYP2D6, CYP2E1 or CYP3A4 activity in healthy subjects.51
To add to the complexity of the problem, in some instances researchers have tested individual herbal components or different forms of a herb, and found different effects on CYPs. For example, one study of different constituents of garlic used animal models and showed that diallyl sulfide (100 μmol/kg) slightly but significantly increased cytochrome CYP2E1 activity (1.6-fold vs control), whereas diallyl disulfide and diallyl trisulfide did not affect CYP2E1 activity or the hepatic total CYP level or CYP1A1/2 activity.52 The significance of these results in clinical practice is difficult to determine, as the overall effect on CYP activity will depend on the concentrations of these various constituents present in a garlic product. The example also highlights the general difficulty of extrapolating results for one herbal extract to another, as there may be significant chemical variations between batches of the same herbal product and between different products of the same herb produced by various manufacturers.
PUTTING THEORY INTO PRACTICE: CLINICAL CONSIDERATIONS
METOPIA ALGORITHM
The METOPIA algorithm provides clinicians with a framework for making rational decisions about the possibility of a herb–drug interaction.19 The acronym METOPIA refers to:
If you suspect an interaction, what are the next steps?




CONCLUSION
The evidence on the safety of herbal medicines is incomplete, complex and confusing. As with pharmaceutical medicines, their use is associated with both risks and benefits. As more people take herbal medicines, the pressure increases on healthcare professionals such as doctors, pharmacists, naturopaths and herbalists to be well informed about the subject, and on researchers to fill the many and somewhat embarrassing gaps in our current knowledge. It could be argued that failing to do (and fund) this work constitutes the true risk associated with herbal medicines.
Adverse Drug Reactions Advisory Committee (ADRAC). http://www.tga.gov.au.
Braun L, Cohen M. Herbs and natural supplements: an evidence-based guide. 3rd edn. Sydney: Elsevier, 2009.
Brinker F. Herb contraindications and drug interactions. 3rd edn. USA: Eclectic Medical Publications, 2001.
Bone K, Mills S. The essential guide to herbal safety. Sydney: Elsevier, 2005.
Natural Standard. http://www.naturalstandard.com/.
Natural Medicines Comprehensive Database. http://www.naturaldatabase.com/(S(frcdl52p5bn02uqyb2buvg33))/home.aspx.
1 MacLennan AH, Wilson DH, Taylor AW. The escalating cost and prevalence of alternative medicine. Prev Med. 2002;35(2):166-173.
2 Jamison JR. Herbal and nutrient supplementation practices of chiropractic patients: an Australian case study. J Manipulative Physiol Ther. 2003;26(4):242.
3 Barraco D, Valencia G, Riba AL, et al. Complementary and alternative medicine (CAM) use patterns and disclosure to physicians in acute coronary syndrome patients. Complement Ther Med. 2005;13(1):34-40.
4 Eisenberg DM, Davis RB, Ettner SL, et al. Trends in alternative medicine use in the United States, 1990–1997: results of a follow-up national survey. JAMA. 1998;280(18):1569-1575.
5 Norred CL, Zamudio S, Palmer SK. Use of complementary and alternative medicines by surgical patients. AANA J. 2000;68(1):13-18.
6 Kaye AD, Clarke RC, Sabar R, et al. Herbal medicines: current trends in anesthesiology practice—a hospital survey. J Clin Anesth. 2000;12(6):468-471.
7 Leung JM, Dzankic S, Manku K, et al. The prevalence and predictors of the use of alternative medicine in presurgical patients in five California hospitals. Anesth Analg. 2001;93(4):1062-1068.
8 Norred CL. Complementary and alternative medicine use by surgical patients. AORN J. 2002;76(6):1013-1021.
9 Adusumilli PS, Ben Porat L, Pereira M, et al. The prevalence and predictors of herbal medicine use in surgical patients. J Am Coll Surg. 2004;198(4):583-590.
10 Norred CL. A follow-up survey of the use of complementary and alternative medicines by surgical patients. AANA J. 2002;70(2):119-125.
11 Tsen LC, Segal S, Pothier M, et al. Alternative medicine use in presurgical patients. Anesthesiology. 2000;93(1):148-151.
12 Wang SM, Caldwell-Andrews AA, Kain ZN. The use of complementary and alternative medicines by surgical patients: a follow-up survey study. Anesth Analg. 2003;97(4):1010-1015.
13 Skinner CM, Rangasami J. Preoperative use of herbal medicines: a patient survey. Br J Anaesth. 2002;89(5):792-795.
14 Vaabengaard P, Clausen LM. Surgery patients’ intake of herbal preparations and dietary supplements. Ugeskr Laeger. 2003;165(35):3320-3323.
15 MacLennan AH, Myers SP, Taylor AW. The continuing use of complementary and alternative medicine in South Australia: costs and beliefs in 2004. Med J Aust. 2006;184(1):27-31.
16 Astin JA. Why patients use alternative medicine: results of a national study. JAMA. 1998;279(19):1548-1553.
17 Eisenberg DM, Kessler RC, Van Rompay MI, et al. Perceptions about complementary therapies relative to conventional therapies among adults who use both: results from a national survey. Ann Intern Med. 2001;135(5):344-351.
18 Siahpush M. Why do people favour alternative medicine? Aust NZ J Public Health. 1999;23(3):266-271.
19 Braun L, Cohen MM. Herbs and natural supplements—an evidence-based guide. 3rd edn. Sydney: Elsevier, 2009.
20 Huupponen R, Seppala P, Iisalo E. Effect of guar gum, a fibre preparation, on digoxin and penicillin absorption in man. Eur J Clin Pharmacol. 1984;26(2):279-281.
21 Rodrigues AD, Lin JH. Screening of drug candidates for their drug–drug interaction potential. Curr Opin Chem Biol. 2001;5(4):396-401.
22 Roby CA, Anderson GD, Kantor E, et al. St John’s Wort: effect on CYP3A4 activity. Clin Pharmacol Ther. 2000;67(5):451-457.
23 Ruschitzka F, Meier PJ, Turina M, et al. Acute heart transplant rejection due to Saint John’s wort. Lancet. 2000;355(9203):548-549.
24 Durr D, Stieger B, Kullak-Ublick GA, et al. St John’s wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin Pharmacol Ther. 2000;68(6):598-604.
25 Wang Z, Gorski JC, Hamman MA, et al. The effects of St John’s wort (Hypericum perforatum) on human cytochrome P450 activity. Clin Pharmacol Ther. 2001;70(4):317-326.
26 Moore LB, Goodwin B, Jones SA, et al. St John’s wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci USA. 2000;97(13):7500-7502.
27 Madabushi R, Frank B, Drewelow B, et al. Hyperforin in St John’s wort drug interactions. Eur J Clin Pharmacol. 2006;62(3):225-233.
28 Mueller SC, Uehleke B, Woehling H, et al. Effect of St John’s wort dose and preparations on the pharmacokinetics of digoxin. Clin Pharmacol Ther. 2004;75(6):546-557.
29 Arold G, Donath F, Maurer A, et al. No relevant interaction with alprazolam, caffeine, tolbutamide, and digoxin by treatment with a low-hyperforin St John’s wort extract. Planta Med. 2005;71(4):331-337.
30 Zhao LZ, Huang M, Chen J, et al. Induction of propranolol metabolism by Ginkgo biloba extract EGb 761 in rats. Curr Drug Metab. 2006;7(6):577-587.
31 Mohutsky MA, Anderson GD, Miller JW, et al. Ginkgo biloba: evaluation of CYP2C9 drug interactions in vitro and in vivo. Am J Ther. 2006;13(1):24-31.
32 Chang TK, Chen J, Teng XW. Distinct role of bilobalide and ginkgolide A in the modulation of rat CYP2B1 and CYP3A23 gene expression by Ginkgo biloba extract in cultured hepatocytes. Drug Metab Dispos. 2006;34(2):234-242.
33 Chang TK, Chen J, Yeung EY. Effect of Ginkgo biloba extract on procarcinogen-bioactivating human CYP1 enzymes: identification of isorhamnetin, kaempferol, and quercetin as potent inhibitors of CYP1B1. Toxicol Appl Pharmacol. 2006;213(1):18-26.
34 Chatterjee SS, Doelman CJ, Noldner M, et al. Influence of the Ginkgo extract EGb 761 on rat liver cytochrome P450 and steroid metabolism and excretion in rats and man. J Pharm Pharmacol. 2005;57(5):641-650.
35 von Moltke LL, Weemhoff JL, Bedir E, et al. Inhibition of human cytochrome P450 by components of Ginkgo biloba. J Pharm Pharmacol. 2004;56(8):1039-1044.
36 Sugiyama T, Kubota Y, Shinozuka K, et al. Ginkgo biloba extract modifies hypoglycemic action of tolbutamide via hepatic cytochrome P450 mediated mechanism in aged rats. Life Sci. 2004;75(9):1113-1122.
37 Gaudineau C, Beckerman R, Welbourn S, et al. Inhibition of human P450 enzymes by multiple constituents of the Ginkgo biloba extract. Biochem Biophys Res Commun. 2004;318(4):1072-1078.
38 He N, Edeki T. The inhibitory effects of herbal components on CYP2C9 and CYP3A4 catalytic activities in human liver microsomes. Am J Ther. 2004;11(3):206-212.
39 Sugiyama T, Kubota Y, Shinozuka K, et al. Induction and recovery of hepatic drug metabolizing enzymes in rats treated with Ginkgo biloba extract. Food Chem Toxicol. 2004;42(6):953-957.
40 Kuo I, Chen J, Chang TK. Effect of Ginkgo biloba extract on rat hepatic microsomal CYP1A activity: role of ginkgolides, bilobalide, and flavonols. Can J Physiol Pharmacol. 2004;82(1):57-64.
41 Kubota Y, Kobayashi K, Tanaka N, et al. Pretreatment with Ginkgo biloba extract weakens the hypnosis action of phenobarbital and its plasma concentration in rats. J Pharm Pharmacol. 2004;56(3):401-405.
42 Ohnishi N, Kusuhara M, Yoshioka M, et al. Studies on interactions between functional foods or dietary supplements and medicines. I. Effects of Ginkgo biloba leaf extract on the pharmacokinetics of diltiazem in rats. Biol Pharm Bull. 2003;26(9):1315-1320.
43 Ryu SD, Chung WG. Induction of the procarcinogen-activating CYP1A2 by a herbal dietary supplement in rats and humans. Food Chem Toxicol. 2003;41(6):861-866.
44 Shinozuka K, Umegaki K, Kubota Y, et al. Feeding of Ginkgo biloba extract (GBE) enhances gene expression of hepatic cytochrome P-450 and attenuates the hypotensive effect of nicardipine in rats. Life Sci. 2002;70(23):2783-2792.
45 Gurley BJ, Gardner SF, Hubbard MA, et al. Cytochrome P450 phenotypic ratios for predicting herb–drug interactions in humans. Clin Pharmacol Ther. 2002;72(3):276-287.
46 Markowitz JS, Donovan JL, Lindsay DC, et al. Multiple-dose administration of Ginkgo biloba did not affect cytochrome P-450 2D6 or 3A4 activity in normal volunteers. J Clin Psychopharmacol. 2003;23(6):576-581.
47 Greenblatt DJ, von Moltke LL, Luo Y, et al. Ginkgo biloba does not alter clearance of flurbiprofen, a cytochrome P450-2C9 substrate. J Clin Pharmacol. 2006;46(2):214-221.
48 Mohutsky MA, Anderson GD, Miller JW, et al. Ginkgo biloba: evaluation of CYP2C9 drug interactions in vitro and in vivo. Am J Ther. 2006;13(1):24-31.
49 Yale SH, Glurich I. Analysis of the inhibitory potential of Ginkgo biloba, Echinacea purpurea, and Serenoa repens on the metabolic activity of cytochrome P450 3A4, 2D6, and 2C9. J Altern Complement Med. 2005;11(3):433-439.
50 Markowitz JS, Donovan JL, Devane CL, et al. Multiple doses of saw palmetto (Serenoa repens) did not alter cytochrome P450 2D6 and 3A4 activity in normal volunteers. Clin Pharmacol Ther. 2003;74(6):536-542.
51 Gurley BJ, Gardner SF, Hubbard MA, et al. In vivo assessment of botanical supplementation on human cytochrome P450 phenotypes: Citrus aurantium, Echinacea purpurea, milk thistle, and saw palmetto. Clin Pharmacol Ther. 2004;76(5):428-440.
52 Fukao T, Hosono T, Misawa S, et al. The effects of allyl sulfides on the induction of phase II detoxification enzymes and liver injury by carbon tetrachloride. Food Chem Toxicol. 2004;42(5):743-749.