[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 81 Hepatitis Caused by Other Viruses
Other viral diseases may sometimes involve the liver as part of a systemic infection. The agents of such infections include human immunodeficiency virus (HIV) (see Chapter 33), Epstein-Barr virus (EBV), cytomegalovirus (CMV), herpes simplex virus (HSV), varicella-zoster virus (VZV), and the virus that causes severe acute respiratory syndrome (SARS). Infection with any of these viruses may rarely lead to severe, sometimes fatal, liver disease.
HEPATITIS G AND GB AGENT INFECTION
During the long search for the cause of transfusion-associated non-A, non-B hepatitis (see Chapter 79), the GB agent (GBV) and hepatitis G virus (HGV) were discovered and later shown to be two isolates of the same virus. A 35-year-old surgeon with the initials GB developed an acute icteric hepatitis. When his serum was serially inoculated into healthy marmosets, they too developed hepatitis. Analysis of the marmosets infected with derivations of the GB serum led to the identification of two distinct viruses, labeled GBV-type A (GBV-A) and GBV-type B (GBV-B).1 A third virus closely related to the GB agents was subsequently identified by the same investigators from a human sample and was classified as GBV-C.2 At approximately the same time, another group independently identified a virus from the serum of a patient with cryptogenic non-A-to-E hepatitis, which they named HGV.3 Subsequent studies revealed 96% homology between the genomes of HGV and GBV-C, indicating that they were actually two strains of the same virus.4 Because large epidemiologic studies have not demonstrated any association between infection with GBV-C/HGV and acute or chronic hepatitis, the use of the term “hepatitis G virus” has been questioned. In fact, even the index patient (“GB”) was subsequently shown to be infected with hepatitis C virus (HCV) as the cause of his liver disease. For clarity, GBV-C/HGV will be referred to as GBV-C.
VIROLOGY
GBV-C is a positive-strand RNA virus with a genome of 9400 nucleotides encoding approximately 2900 amino acids and is classified as a member of the Flaviviridae family. GBV-C shares 44% and 28% nucleotide homology with GBV-A and GBV-B, respectively, and has five known genotypes with distinct global geographic distributions.4 Although similar in many respects to HCV, GBV-C shares only 27% nucleotide homology with HCV, and the two viruses are clearly distinct.
The GBV-C genome is organized like that of HCV. One long open reading frame encodes a single large polyprotein with structural proteins encoded at the 5′ aminoterminus and nonstructural proteins encoded at the 3′ carboxyterminus. A nontranslated region at the 5′ end serves as an internal ribosomal entry site (IRES), allowing translation of the uncapped messenger RNA.5
The structural proteins differ between HCV and GBV-C. Two glycoproteins—E1 and E2—predicted to compose the GBV-C viral envelope are cleaved from the polyprotein, likely by a host cell signal peptidase. Whereas HCV E1 and E2 have 5 and 11 N-linked glycosylation sites, GBV-C E1 and E2 have only 1 and 3 such sites, respectively.6 Perhaps of greater importance, amino acid polymorphisms do not cluster in the hypervariable region of GBC-C E2, as they do in HCV E2. The hypervariable region of HCV E2 is thought to account in part for the ability of HCV to evade immune attack and cause persistent infection. This difference in E2 polymorphisms may account for the 60% to 75% rate of chronic HCV infection, compared with only 25% in GBV-C infection.
The GBV-C genome does not encode a core protein, but biophysical and electron microscopic studies suggest that the virus does have a nucleocapsid structure, presumably with a core protein.7 Some GBV-C infected patients have antibodies that react with a synthetic peptide that corresponds to the region immediately proximal to the E1 coding region, suggesting that GBV-C may have a truncated core protein at the amino terminus of the genome.
In contrast with the structural differences, the GBV-C nonstructural proteins—designated NS1, NS2, and so on—are similar to those of HCV, including a zinc-dependent thiol protease that cleaves NS2 from NS3, a serine protease at the amino terminus of NS3 that probably cleaves all downstream proteolytic sites, and an RNA helicase (as in all positive-strand RNA viruses) that is also found in NS3 downstream from the protease region. NS4A likely serves as a cofactor for the NS3 protease. The NS5A protein is believed to interact with double-strand RNA protein kinase, and NS5B serves as the RNA-dependent RNA polymerase.8
Another important difference between GBV-C and HCV may be tissue tropism. Negative-strand RNA (indicating the presence of active viral replication) has been demonstrated in liver tissue during HCV infection, implying hepatotropism, but has not been clearly demonstrated during GBV-C infection. Negative-strand RNA, however, has been demonstrated in peripheral blood mononuclear cells (PBMCs), bone marrow, and spleens of patients with GBV-C infection, suggesting that GBV-C is a lymphotropic virus.9 The demonstration of replication of a GBV-C clone in CD4+ T cells confirms that GBV-C is able to replicate in lymphocytes and may help explain the interaction between GBV-C and HIV infection (see later).
GBV-C can be grown in cell culture and has been proposed as a model for studying HCV. Persistent GBV-C infection has been achieved in marmosets, which provide a small animal model of HCV infection in which a new serine protease inhibitor targeted to HCV has produced a marked reduction in GBV-C viremia.10 GBV-B has been proposed as a better model for HCV infection than GBV-C because GBV-B causes acute and rarely chronic hepatitis in tamarins and marmosets.11 Therefore, although the GB viruses do not cause overt human liver disease, they may be useful tools to study HCV infection.
EPIDEMIOLOGY
GBV-C is found worldwide. At least five genotypes have been identified, each with a specific geographic distribution: genotype 1 predominates in West Africa, genotype 2 in Europe and the United States, genotype 3 in parts of Asia, genotype 4 in Southeast Asia, and genotype 5 in South Africa.12
The development of GBV-C E2 antibodies correlates with loss of GBV-C viremia and suggests past exposure and clearance of GBV-C infection.13 Evidence of current and past GBV-C infection is found frequently in persons with parenteral risk factors and also is common among volunteer blood donors. Between 14% and 38% of persons with frequent exposures to blood are viremic with GBV-C, whereas 50% to 70% of such persons are seropositive for E2 antibodies.14 Up to 16% of healthy blood donors are positive for E2 antibodies, with much lower rates of active viremia.15 Past or current GBV-C viremia is found as often in blood donors with a normal serum alanine aminotransferase (ALT) level as in donors rejected because of an elevated serum ALT level.16 Consequently, GBV-C transmission is not prevented by exclusion of donors with a normal ALT value. GBV-C also has been shown to be transmitted sexually and vertically much more frequently than HCV, and the risk may vary with the GBV-C genotype.17 No evidence exists for hepatitis or other clinical sequelae in infected babies.18 Because both GBV-C and HCV are transmitted parenterally, GBV-C-HCV coinfection is common. GBV-C viremia is present in about 20% of HCV-infected persons, and 80% of the remaining subjects are seropositive for antibodies to E2.8 These findings suggest that the rate of natural clearance of GBV-C is higher (~75%) than that for HCV (~25%).
CLINICAL FEATURES
Although GBV-C is detected in many patients with non-A-to-E acute and chronic hepatitis and may persist for years, it does not appear to cause liver (or any other) disease, even in immunocompromised persons.16,19–21 Nor does GBV-C appear to modulate the course or response to treatment of chronic HCV or hepatitis B virus (HBV) infection.21 GBV-C infection does not affect the outcome of liver transplantation, even though liver transplant recipients have high rates of GBV-C viremia, likely because of their high transfusion requirements.22
The duration of GBV-C infection may depend on the immune status and age of the host. As with HBV infection, childhood acquisition of GBV-C appears to lead to chronic infection, whereas sexual transmission in immunocompetent adults typically leads to rapid clearance of viremia.23 HIV-infected persons are also more likely than non–HIV-infected persons to develop chronic GBV-C infection.24 In contrast to HCV infection, the development of antibodies to GBV-C E2 seems to protect against reinfection.25 No association between GBV-C and hepatocellular carcinoma,26 non-Hodgkin’s lymphoma,27 aplastic anemia, or lichen planus28 has been documented.
GB VIRUS TYPE C AND HUMAN IMMUNODEFICIENCY VIRUS
Once GBV-C was shown not to cause liver disease, interest in this virus diminished. In 1998, however, two independent groups of investigators observed that among a small number of HIV-infected persons, lower HIV viral loads as well as slower progression to acquired immunodeficiency syndrome (AIDS) and death correlated with the presence of GBV-C viremia (Fig. 81-1).29,30 Subsequently, larger studies have confirmed this finding in varied populations and have suggested that HIV–GBV-C–coinfected persons have a better prognosis than HIV-monoinfected persons.30–34 In addition, coinfected persons respond better to antiretroviral therapy, with a more rapid increase in CD4+ counts and suppression of HIV viral load.34,35 Children infected with GBV-C at birth also have a reduced risk of contracting HIV via vertical transmission, independent of the mother’s GBV-C status.36 Some studies, however, have not confirmed a protective effect of GBV-C infection in HIV-infected persons.37–41 The discrepant results may relate to differences in the HIV-inhibitory effect of different GBV-C genotypes and the duration of GBV-C infection42; the longer duration of GBV-C infection, the greater the benefit on HIV progression.33 The viral interaction may work in both directions. In a longitudinal study of coinfected persons, GBV-C viral titers increased during effective highly active antiretroviral therapy (HAART) and fell with interruptions in HAART, suggesting that HIV may also inhibit GBV-C.43 Although GBV-C does not affect the outcome of HCV monoinfection, a study of the effect of GBV-C infection on the progression of liver disease in HIV-HCV coinfected persons found that patients with current or past GBV-C infection had a lower risk of cirrhosis and hepatic complications but no difference in survival compared with patients without GBV-C infection.44
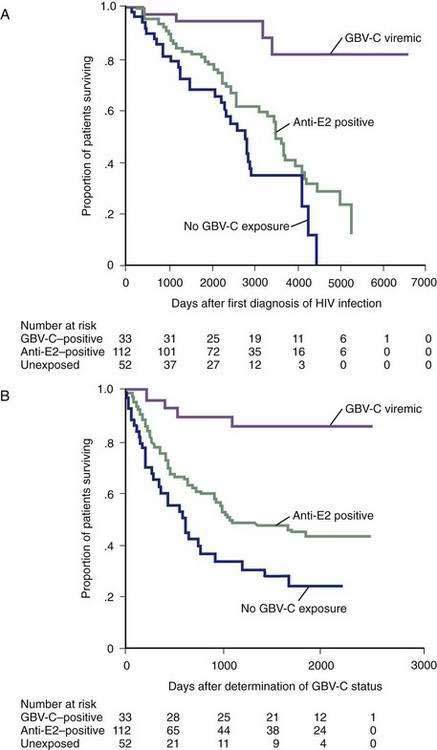
Why HIV-GBV-C coinfected persons have an improved response to HAART is unknown. HIV replication is diminished in PBMCs that are infected with both HIV and GBV-C, and a higher inoculum of HIV is required to infect cells already infected with GBV-C. The NS5A protein of GBV-C has been shown to down-regulate the expression of the HIV co-receptor CXCR4, and the E2 protein of GBV-C interferes with early steps in the HIV life cycle.45 Virologic inhibition may explain the interaction, but immune mechanisms, including GBV-C-induced stimulation of HIV-inhibitory cytokines, endogenous interferon production, and promotion of a T helper cell type 1 (Th1) immune profile, have also been proposed.46,47
TREATMENT
Because GBV-C infection is not associated with clinical liver disease, no treatments have targeted GBV-C specifically. In HIV–HCV–GBV-C coinfected persons, peginterferon and ribavirin treatment led to sustained GBV-C clearance in 31% of patients, with no observable subsequent effect on the course of HCV or HIV infection.48 In patients coinfected with GBV-C and HCV who were treated with interferon and ribavirin, GBV-C RNA disappeared from serum during therapy but reappeared in all patients following discontinuation of therapy.49 Importantly, no effect of GBV-C infection was observed on the response to treatment of HCV infection.50
TT VIRUS INFECTION
TTV was first identified in 1977 by the use of representational difference analysis in a patient (with the initials TT) in Japan who had acute post-transfusion non-A-to-G hepatitis.51 TTV is also referred to as the transfusion-transmitted and Torque-Teno virus.
VIROLOGY
The TTV genome is 3965 nucleotides long and contains at least three overlapping open reading frames. Three messenger RNAs (mRNAs) are expressed by TTV; each mRNA is translated from two start codons, leading to the production of six viral proteins. One protein product of the largest mRNA (3.0 kb) functions as the capsid protein and also serves as a replicase enzyme. The product of the second open reading frame interferes with host nuclear factor kappa B signaling, but its role in viral replication is unknown.52 TTV displays remarkable genomic sequence diversity, even among isolates found in an individual patient.53 At least 23 genotypes have been identified, with greater than 30% sequence divergence between isolates. Some genotypes differ in sequence by greater than 50%, and genotype prevalence rates vary geographically.54
TTV is believed to be hepatotropic on the basis of the observation that viral levels are higher in the liver than in the serum of infected patients. TTV has also been identified within hepatocytes, and shown to replicate, by in situ hybridization and PCR; however, no or only minor morphologic changes have been seen in cells with positive hybridization signals.55 TTV has also been shown to replicate in stimulated PBMCs and bone marrow cells.56
EPIDEMIOLOGY
TTV is found worldwide and is common. Initial studies documented infection in 1% to 40% of healthy blood donors.57 As more inclusive primers have been used to detect differing genotypes, the reported prevalence among blood donors has increased dramatically, approaching 100% in some studies.58 The prevalence of TTV infection increases with age but appears to reach a plateau by early childhood.59 TTV is also found in a variety of nonhuman primate species.
TTV is transmitted effectively by all parenteral routes; high prevalence rates have been documented in hemophiliacs, injection drug users, patients on hemodialysis, and organ transplant recipients.60 TTV has also been shown to be transmitted enterically, and high TTV DNA titers are present in the feces of viremic patients. Fecal-oral spread may account for the high prevalence in low-risk, healthy blood donor populations.58
CLINICAL FEATURES
Although TTV was associated with acute hepatitis in the patient in whom it was first identified, other studies have not supported a causal association between TTV and liver disease.61,62 In the original study,51 viremia was detected six weeks after exposure and two weeks before a rise in serum ALT levels. The viral DNA was cleared from serum, as documented by PCR testing, and serum ALT levels subsequently returned to normal. Viremia may persist for years in both immunocompetent and immunosuppressed persons. Most infected persons have no biochemical or histologic evidence of liver disease.61,63 Like GBV-C, TTV does not appear to alter the natural history or response to treatment of either chronic HCV or HBV infection.64 A report that TTV infection may increase the risk of hepatocellular carcinoma in patients with chronic HCV infection remains to be confirmed.65
TREATMENT
Formal studies of treatment of TTV infection have not been performed. A small study of HCV-TTV-coinfected patients showed that TTV infection had no effect on sustained virologic response of HCV to therapy with peginterferon and ribavirin. Although TTV viremia cleared after therapy in 6 of 10 patients, 4 of the 6 relapsed within six months.66
SANBAN, YONBAN, AND SEN VIRUSES AND TTV-LIKE MINI-VIRUS INFECTIONS
Since the discovery of TTV in 1997,51 several similar viruses with a small DNA genome have been isolated in Japan and named Sanban, Yonban, and TTV-like mini-virus (TLMV). These viruses have been divided into 29 genotypes, with sequence divergence of greater than 30%.67 Like TTV, they are readily transmitted parenterally and can also be passed by the fecal-oral route. None has been clearly associated with human liver disease to date.
In 1999, a novel virus was identified in an HIV-positive person (with the initials SEN) who had post-transfusion hepatitis of unknown etiology. This virus was found with the use of degenerative primers from the prototype TTV. The SEN virus is a small, nonenveloped, single-stranded DNA virus, but unlike that of TTV, the SEN genome is linear. Nucleotide sequencing has shown 50% homology with the prototype TTV, but only 30% of amino acids are homologous. Sequencing of multiple isolates has demonstrated sequence divergence of 15% to 50%.68
Like TTV, SEN virus is transmitted both parenterally and by the fecal-oral route.69 Vertical transmission occurs but in most cases does not lead to chronic infection. Natural clearance of both perinatally and parenterally acquired SEN virus does not appear to protect against reinfection.70 The prevalence varies markedly and is highest among patients with parenteral risk factors, particularly those coinfected with HCV.71 The prevalence among healthy blood donors is approximately 2% in the United States and 10% in Japan.72
The clinical significance of SEN infection remains controversial. One study of patients with post-transfusional non-A-to-E hepatitis suggested that SEN was the cause in a majority (11 of 12) of cases. SEN viremia persisted for more than one year in 45% of those infected; however, clinical hepatitis did not develop in a majority (86%) of transfused patients who acquired SEN infection. None of the patients with hepatitis had a fulminant course, nor did chronic liver disease or cirrhosis develop during follow up.73 Other reports and case series have identified SEN or TTV viremia in patients with both fulminant and chronic hepatitis, but causation has been difficult to establish. Most studies have shown neither an association between SEN or any of the other viruses in this group and human disease, nor an effect of these viruses on the course or response to treatment for chronic viral hepatitis.69,74
SYSTEMIC VIRAL INFECTIONS THAT MAY INVOLVE THE LIVER
EPSTEIN-BARR VIRUS
EBV infection is common and covers a wide spectrum of clinical presentations. Most infants and children are either asymptomatic or have mild, nonspecific complaints, whereas adolescents and adults typically present with the triad of pharyngitis, fever, and lymphadenopathy.75 Although usually subclinical, liver involvement is nearly universal in patients with EBV mononucleosis and ranges from serum aminotransferase elevations to rare cases of fulminant and even fatal hepatitis.76
Up to 90% of patients with acute mononucleosis have serum aminotransferase and lactate dehydrogenase elevations two to three times the upper limit of normal. The enzyme levels typically rise over a one- to two-week period, and peak levels are usually much lower than those normally seen in patients with acute hepatitis A, B, D, or E. Elevated levels of alkaline phosphatase are common, and mild hyperbilirubinemia is observed in as many as 45% of cases.77 In most patients, liver test levels normalize within one month, often before complete resolution of clinical symptoms.78 As with infectious mononucleosis, EBV hepatitis tends to be more severe in adults older than age 30 than in younger adults and children.79 Elderly patients occasionally present with jaundice, fever, and right upper quadrant pain that may suggest a diagnosis of extrahepatic biliary obstruction.80 Although jaundice may be caused by viral-induced cholestasis, autoimmune hemolytic anemia should be excluded in all hyperbilirubinemic patients. Cholestatic jaundice with pruritus may be observed in young women with EBV infection who continue taking oral contraceptive pills.
Rarely, EBV may cause persistent chronic infection with involvement of the liver, lungs, and other organs. In the largest series of fatal mononucleosis, hepatic involvement was universal and was the cause of death in 13 of 30 patients.81 Granulomatous hepatitis also has been attributed to chronic EBV infection on occasion.82 EBV infection detected on a liver biopsy specimen may account for some cases of otherwise unexplained hepatitis. Fatal, fulminant EBV hepatitis has been described in both immunocompetent and immunocompromised persons and appears to be associated with a greater than usual EBV viral burden, particularly in T cells as opposed to B cells.83 A hemophagocytic syndrome characterized by fever, hepatosplenomegaly, hepatic synthetic dysfunction, cytopenias, and marked hyperferritinemia may develop in patients with EBV infection. The syndrome is caused by natural killer T cell dysregulation, leading to lymphoctye proliferation and activation with uncontrolled hemophagocytosis and cytokine production. Although rare, the syndrome is usually severe and may be fatal.84
The diagnosis of EBV hepatitis is based on clinical features of mononucleosis and laboratory data suggestive of acute EBV infection. Most patients (70%) have a leukocytosis with a predominance of lymphocytes and monocytes, and up to 50% have mild thrombocytopenia.85 The Monospot test is sensitive for the detection of heterophile antibodies but is not a specific test for EBV infection. Levels of EBV-specific immunoglobulin M (IgM) antibodies peak early in serum and may persist for many months, at which point IgG antibodies appear. Findings on abdominal ultrasonography are usually nonspecific and may include hepatosplenomegaly, lymphadenopathy, and possibly gallbladder thickening, which has been reported to portend more severe liver disease.86 Liver biopsy is rarely necessary for diagnosis but, if done, shows portal and sinusoidal mononuclear cell infiltration with no disruption of hepatic architecture; multinucleated giant cells are not a feature. In more severe cases, focal hepatic necrosis may be evident. In situ hybridization or PCR testing of biopsy samples may be used to confirm the diagnosis, but immunohistochemistry for EBV proteins is rarely positive.87
No specific treatment for EBV hepatitis exists. Acyclovir inhibits EBV replication and reduces viral shedding from the nasopharynx but has no effect on clinical symptoms or outcome.88 Improvement in acute and chronic EBV hepatitis has been reported with gancyclovir treatment, but this approach has not been well studied.89 Liver transplantation has been performed for fulminant EBV hepatitis. EBV rarely causes hepatitis after liver transplantation but has been associated with post-transplantation lymphoproliferative disease (see Chapter 95). In a patient with chronic EBV hepatitis after kidney transplantation, treatment with rituximab was reported to lead to improvement in serum liver enzyme levels and hepatic pathology.90
CYTOMEGALOVIRUS
CMV is the largest member of the Herpesviridae family and, like other herpesviruses, persists lifelong in a latent, nonreplicative state after resolution of primary infection. Consequently, clinical disease caused by CMV may occur as a primary infection or, more commonly, as reactivation of latent infection,91 particularly in immunocompromised persons.
In immunocompetent children and adults, primary CMV infection is usually subclinical but may cause a mononucleosis-like illness. Liver involvement is common and is characterized by mild to moderate aminotransferase (88%) and alkaline phosphatase (64%) elevations with or without hepatosplenomegaly.92 Although the clinical course is mild in most patients, rare instances of granulomatous, cholestatic CMV hepatitis, with or without jaundice, and even massive, fatal hepatic necrosis have been described.93 In addition to the congenital CMV syndrome (jaundice, hepatosplenomegaly, thrombocytopenic purpura, and severe neurologic impairment), CMV is a common cause of neonatal hepatitis.94 A number of case reports also suggest a possible association between CMV infection and acute portal vein thrombosis; however, the mechanism is unclear.95
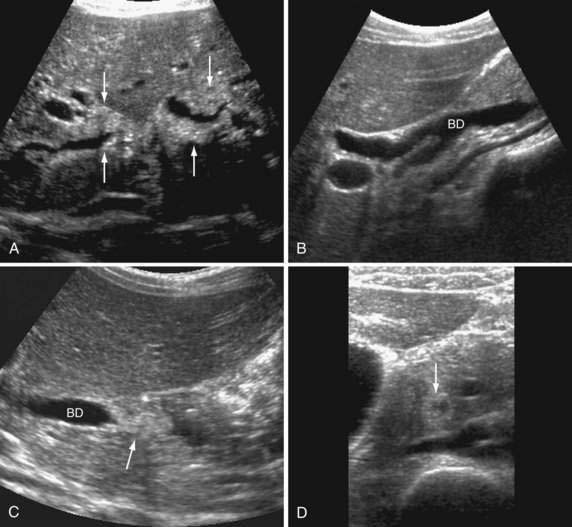
Disseminated, life-threatening CMV infection with multiorgan involvement may develop in patients with impaired cell-mediated immunity (see Chapters 33 and 34). Hepatobiliary involvement by CMV is common in patients with AIDS and may manifest as hepatitis, pancreatitis, or acalculous gangrenous cholecystitits.96 CMV also may cause AIDS-associated cholangiopathy, which manifests with chronic cholestasis and mimics primary sclerosing cholangitis (PSC) clinically and radiographically.97 Patients may have papillary stenosis alone or in combination with intra- or extrahepatic (or both) biliary stricturing and dilatation (Fig. 81-2; see also Fig. 33-7). Antiviral therapy has no effect on this syndrome, but papillotomy, with or without placement of a biliary stent, may lead to symptomatic improvement.96 Organ transplant recipients are also at risk for aggressive CMV hepatitis, including fibrosing cholestatic hepatitis (see Chapter 95), but for unclear reasons cholangiopathy does not develop in these patients.98 CMV hepatitis can be difficult to distinguish from graft rejection in liver transplant recipients.99
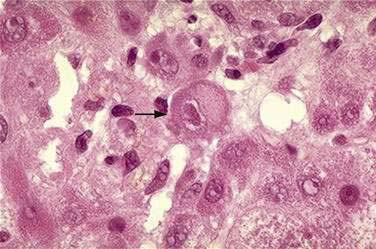
The diagnosis of CMV infection is based on results of serologic studies, liver biopsy, or both. In acute primary CMV infection, IgM antibodies to CMV are present. For patients with reactivation of latent CMV, “shell vial” assays, which use monoclonal antibodies to detect CMV antigens, or direct CMV antigenemia testing are necessary. Because CMV viremia precedes organ involvement, testing for CMV antigenemia or CMV PCR testing of blood are useful screening tools in immunocompromised patients.100 Multinucleated giant cells with mononuclear portal and parenchymal inflammatory infiltrates and cholestasis are commonly seen on liver biopsy specimens. Large nuclear inclusions, sometimes referred to as “owl’s eye” inclusions may be seen in hepatocytes or biliary epithelial cells (Fig. 81-3).
With mild CMV disease in an immunocompetent adult, treatment is unnecessary. In immunocompromised patients, antiviral therapy is indicated. Ganciclovir, a guanosine nucleoside analog with a much longer intracellular half-life than that of acyclovir, has proved to be most effective. The major toxicity is bone marrow suppression, particularly granulocytopenia. Because viremia correlates with disease outcome, ganciclovir should be continued until CMV antigenemia is undetectable.101 For patients resistant to or intolerant of ganciclovir, alternative agents include foscarnet and cidofovir.
HERPES SIMPLEX VIRUS
HSV typically causes mucocutaneous vesicular oral or genital lesions; visceral involvement may occur in certain clinical settings. HSV hepatitis is seen in neonates, pregnant women, and immunocompromised persons and can be aggressive and life-threatening.102 Cases of severe HSV hepatitis in immunocompetent persons have been reported as well.103 Severe hepatitis with multiorgan involvement and, often, adrenal insufficiency, may develop in neonates exposed to infected maternal genital secretions at the time of delivery.104 In pregnant women, HSV hepatitis usually has a fulminant course. The disease is most common in late gestation, typically (in 65% of patients) in the third trimester. Mucocutaneous lesions are present in only 50% of cases, and a high index of clinical suspicion is important to ensure timely diagnosis.105 Maternal and perinatal mortality rates approach 40%, and in the largest series, 25% of patients were diagnosed only at autopsy. Early diagnosis and initiation of antiviral therapy are critical.106
Mild, asymptomatic liver enzyme elevations may be seen in 14% of immunocompetent patients with acute genital HSV infection. By contrast, immunocompromised patients may present with fulminant hepatitis.107 Hepatitis is more common with acute infection than reactivation and presents with fever, leukopenia, and markedly elevated serum aminotransferase levels. Coagulopathy, including disseminated intravascular coagulation, and jaundice may be seen. Of reported cases, only 50% had a rash at presentation, and 58% were diagnosed at autopsy.108
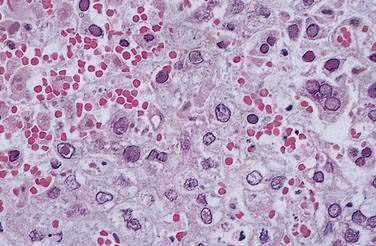
Liver biopsy is essential for diagnosis, particularly in pregnancy. The transjugular route may be required because liver failure may develop rapidly, precluding percutaneous biopsy. Focal or extensive hemorrhagic or coagulative necrosis, with few inflammatory infiltrates, is seen. Intranuclear (Cowdry A type) inclusions may be identified in hepatocytes at the margins of the necrosis. In addition, some periportal multinucleated hepatocytes show a ground-glass appearance suggestive of viral inclusions (Fig. 81-4).109 Electron microscopy, immunohistochemical staining, and PCR techniques can be used to confirm the diagnosis.110
HSV hepatitis constitutes an emergency, and empirical therapy should be instituted pending diagnostic confirmation. High-dose intravenous acyclovir (at least 10 mg/kg every eight hours) is effective and appears to be safe in pregnancy.111 Prolonged therapy may be required because severe relapse has been reported.103 Successful liver transplantation has also been reported.112
VARICELLA-ZOSTER VIRUS
Like HSV infection, VZV infection occasionally can be complicated by hepatitis. Serum liver enzyme levels may be elevated in up to 3.4% of children with chickenpox; however, clinically significant hepatitis has been reported only rarely.113 Although VZV reactivation in adults usually is limited to the skin, dissemination with liver, lung, and pancreatic involvement may occur.114 Rarely, visceral involvement has been reported to develop before cutaneous manifestations in bone marrow or solid organ transplant recipients. If visceral involvement is suspected, treatment with high-dose intravenous acyclovir should be instituted.
CORONAVIRUS CAUSING SEVERE ACUTE RESPIRATORY SYNDROME
During the 2003 outbreak of SARS, a rapidly progressive respiratory illness, elevated serum aminotransferase levels were commonly observed during the acute illness. Subsequently, cases of SARS hepatitis were reported in which the coronavirus that causes SARS was demonstrated in the liver by reverse transcriptase–PCR techniques in three patients; no viral particles were seen on electron microscopy. All three cases fulfilled the World Health Organization criteria for SARS. Examination of liver tissue revealed marked apoptosis, ballooning of hepatocytes, and moderate lobular lymphocytic infiltration.115
Alter HJ, Nakatsuji Y, Melpolder J, et al. The incidence of transfusion-associated hepatitis G virus infection and its relation to liver disease. N Engl J Med. 1997;336:747-54. (Ref 16.)
Alter MJ, Gallagher M, Morris TT, et al. Acute non-A-E hepatitis in the United States and the role of hepatitis G virus infection. Sentinel Counties Viral Hepatitis Study Team. N Engl J Med. 1997;336:741-6. (Ref 20.)
Berzsenyi MD, Bowden DS, Kelly HA, et al. Reduction in hepatitis C-related liver disease associated with GB virus C in human immunodeficiency virus coinfection. Gastroenterology. 2007;133:1821-30. (Ref 44.)
Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology. 1998;28:839-42. (Ref 63.)
Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Intern Med. 1993;119:924-35. (Ref 91.)
Linnen J, Wages JJr, Zhang-Keck ZY, et al. Molecular cloning and disease association of hepatitis G virus: A transfusion-transmissible agent. Science. 1996;271:505-8. (Ref 3.)
Markin RS. Manifestations of Epstein-Barr virus-associated disorders in liver. Liver. 1994;14:1-13. (Ref 81.)
Pellise M, Miquel R. Liver failure due to herpes simplex virus. J Hepatol. 2000;32:170. (Ref 110.)
Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-22. (Ref 84.)
Sheng WH, Hung CC, Wu RJ, et al. Clinical impact of GB virus C viremia on patients with HIV type 1 infection in the era of highly active antiretroviral therapy. Clin Infect Dis. 2007;44:584-90. (Ref 37.)
Stapleton JT. GB virus type C/Hepatitis G virus. Semin Liver Dis. 2003;23:137-48. (Ref 8.)
Tanaka E, Alter HJ, Nakatsuji Y, et al. Effect of hepatitis G virus infection on chronic hepatitis C. Ann Intern Med. 1996;125:740-3. (Ref 21.)
Tillmann HL, Heiken H, Knapik-Botor A, et al. Infection with GB virus C and reduced mortality among HIV-infected patients. N Engl J Med. 2001;345:715-24. (Ref 32.)
Williams CF, Klinzman D, Yamashita TE, et al. Persistent GB virus C infection and survival in HIV-infected men. N Engl J Med. 2004;350:981-90. (Ref 33.)
Xiang J, McLinden JH, Chang Q, et al. Characterization of a peptide domain within the GB virus C NS5A phosphoprotein that inhibits HIV replication. PLoS ONE. 2008;3:e2580. (Ref 45.)
1. Simons JN, Pilot-Matias TJ, Leary TP, et al. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc Natl Acad Sci U S A. 1995;92:3401-5.
2. Simons JN, Leary TP, Dawson GJ, et al. Isolation of novel virus-like sequences associated with human hepatitis. Nat Med. 1995;1:564-9.
3. Linnen J, Wages JJr, Zhang-Keck ZY, et al. Molecular cloning and disease association of hepatitis G virus: A transfusion-transmissible agent. Science. 1996;271:505-8.
4. Smith DB, Cuceanu N, Davidson F, et al. Discrimination of hepatitis G virus/GBV-C geographical variants by analysis of the 5′ non-coding region. J Gen Virol. 1997;78(Pt 7):1533-42.
5. Muerhoff AS, Leary TP, Simons JN, et al. Genomic organization of GB viruses A and B: Two new members of the Flaviviridae associated with GB agent hepatitis. J Virol. 1995;69:5621-30.
6. Xiang J, Klinzman D, McLinden J, et al. Characterization of hepatitis G virus (GB-C virus) particles: Evidence for a nucleocapsid and expression of sequences upstream of the E1 protein. J Virol. 1998;72:2738-44.
7. Xiang J, Daniels KJ, Soll DR, et al. Visualization and characterization of GB virus-C particles: Evidence for a nucleocapsid. J Viral Hepat. 1999;6(Suppl 1):16-22.
8. Stapleton JT. GB virus type C/Hepatitis G virus. Semin Liver Dis. 2003;23:137-48.
9. George SL, Varmaz D, Stapleton JT. GB virus C replicates in primary T and B lymphocytes. J Infect Dis. 2006;193:451-4.
10. Bright H, Carroll AR, Watts PA, Fenton RJ. Development of a GB virus B marmoset model and its validation with a novel series of hepatitis C virus NS3 protease inhibitors. J Virol. 2004;78:2062-71.
11. Martin A, Bodola F, Sangar DV, et al. Chronic hepatitis associated with GB virus B persistence in a tamarin after intrahepatic inoculation of synthetic viral RNA. Proc Natl Acad Sci U S A. 2003;100:9962-7.
12. Muerhoff AS, Leary TP, Sathar MA, et al. African origin of GB virus C determined by phylogenetic analysis of a complete genotype 5 genome from South Africa. J Gen Virol. 2005;86(Pt 6):1729-35.
13. Schaluder GG, Dawson GJ, Simons JN, et al. Molecular and serologic analysis in the transmission of the GB hepatitis agents. J Med Virol. 1995;46:81-90.
14. Jarvis LM, Davidson F, Hanley JP, et al. Infection with hepatitis G virus among recipients of plasma products. Lancet. 1996;348:1352-5.
15. Tacke M, Schmolke S, Schlueter V, et al. Humoral immune response to the E2 protein of hepatitis G virus is associated with long-term recovery from infection and reveals a high frequency of hepatitis G virus exposure among healthy blood donors. Hepatology. 1997;26:1626-33.
16. Alter HJ, Nakatsuji Y, Melpolder J, et al. The incidence of transfusion-associated hepatitis G virus infection and its relation to liver disease. N Engl J Med. 1997;336:747-54.
17. Li C, Danso K, Addo-Yobo E, et al. GB virus C genotype 1 is rarely transmitted vertically but acquired during infancy in West Africa. AIDS. 2006;20:1458-60.
18. Wejstal R, Manson AS, Widell A, Norkrans G. Perinatal transmission of hepatitis G virus (GB virus type C) and hepatitis C virus infections—a comparison. Clin Infect Dis. 1999;28:816-21.
19. Tan D, Matsumoto A, Conry-Cantilena C, et al. Analysis of hepatitis G virus (HGV) RNA, antibody to HGV envelope protein, and risk factors for blood donors coinfected with HGV and hepatitis C virus. J Infect Dis. 1999;179:1055-61.
20. Alter MJ, Gallagher M, Morris TT, et al. Acute non-A-E hepatitis in the United States and the role of hepatitis G virus infection. Sentinel Counties Viral Hepatitis Study Team. N Engl J Med. 1997;336:741-6.
21. Tanaka E, Alter HJ, Nakatsuji Y, et al. Effect of hepatitis G virus infection on chronic hepatitis C. Ann Intern Med. 1996;125:740-3.
22. Fried MW, Khudyakov YE, Smallwood GA, et al. Hepatitis G virus co-infection in liver transplantation recipients with chronic hepatitis C and nonviral chronic liver disease. Hepatology. 1997;25:1271-5.
23. Christensen PB, Fisker N, Mygind LH, et al. GB virus C epidemiology in Denmark: Different routes of transmission in children and low- and high-risk adults. J Med Virol. 2003;70:156-62.
24. Clevenberg P, Durant J, Halfon P, et al. High prevalence of GB virus C/hepatitis G virus infection in different risk groups of HIV-infected patients. Clin Microbiol Infect. 1998;4:644-7.
25. Tillmann HL, Heringlake S, Trautwein C, et al. Antibodies against the GB virus C envelope 2 protein before liver transplantation protect against GB virus C de novo infection. Hepatology. 1998;28:379-84.
26. Kanda T, Yokosuka O, Imazeki F, et al. GB virus-C RNA in Japanese patients with hepatocellular carcinoma and cirrhosis. J Hepatol. 1997;27:464-9.
27. Collier JD, Zanke B, Moore M, et al. No association between hepatitis C and B-cell lymphoma. Hepatology. 1999;29:1259-61.
28. Nagao Y, Sata M, Noguchi S, et al. GB virus infection in patients with oral cancer and oral lichen planus. J Oral Pathol Med. 1997;26:138-41.
29. Heringlake S, Ockenga J, Tillmann HL, et al. GB virus C/hepatitis G virus infection: A favorable prognostic factor in human immunodeficiency virus-infected patients? J Infect Dis. 1998;177:1723-6.
30. Toyoda H, Fukuda Y, Hayakawa T, et al. Effect of GB virus C/hepatitis G virus coinfection on the course of HIV infection in hemophilia patients in Japan. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;17:209-13.
31. Yeo AE, Matsumoto A, Hisada M, et al. Effect of hepatitis G virus infection on progression of HIV infection in patients with hemophilia. Multicenter Hemophilia Cohort Study. Ann Intern Med. 2000;132:959-63.
32. Tillmann HL, Heiken H, Knapik-Botor A, et al. Infection with GB virus C and reduced mortality among HIV-infected patients. N Engl J Med. 2001;345:715-24.
33. Williams CF, Klinzman D, Yamashita TE, et al. Persistent GB virus C infection and survival in HIV-infected men. N Engl J Med. 2004;350:981-90.
34. Mosam A, Sathar MA, Dawood H, et al. Effect of GB virus C co-infection on response to generic HAART in African patients with HIV-1 clade C infection. AIDS. 2007;21:1377-9.
35. Rodriguez B, Woolley I, Lederman MM, et al. Effect of GB virus C coinfection on response to antiretroviral treatment in human immunodeficiency virus-infected patients. J Infect Dis. 2003;187:504-7.
36. Supapol WB, Remis RS, Raboud J, et al. Reduced mother-to-child transmission of HIV associated with infant but not maternal GB virus C infection. J Infect Dis. 2008;197:1369-77.
37. Sheng WH, Hung CC, Wu RJ, et al. Clinical impact of GB virus C viremia on patients with HIV type 1 infection in the era of highly active antiretroviral therapy. Clin Infect Dis. 2007;44:584-90.
38. Descamps D, Damond F, Benard A, et al. No association between GB virus C infection and disease progression in HIV-2-infected patients from the French ANRS HIV-2 cohort. AIDS. 2006;20:1076-9.
39. Zhang W, Chaloner K, Tillmann HL, et al. Effect of early and late GB virus C viraemia on survival of HIV-infected individuals: A meta-analysis. HIV Med. 2006;7:173-80.
40. Kaye S, Howard M, Alabi A, et al. No observed effect of GB virus C coinfection on disease progression in a cohort of African woman infected with HIV-1 or HIV-2. Clin Infect Dis. 2005;40:876-8.
41. Oxenius A, Price DA, Hersberger M, et al. HIV-specific cellular immune response is inversely correlated with disease progression as defined by decline of CD4+ T cells in relation to HIV RNA load. J Infect Dis. 2004;189:1199-208.
42. Kaiser T, Tillmann HL. GB virus C infection: Is there a clinical relevance for patients infected with the human immunodeficiency virus? AIDS Rev. 2005;7:3-12.
43. Bjorkman P, Flamholc L, Molnegren V, et al. Enhanced and resumed GB virus C replication in HIV-1-infected individuals receiving HAART. AIDS. 2007;21:1641-3.
44. Berzsenyi MD, Bowden DS, Kelly HA, et al. Reduction in hepatitis C-related liver disease associated with GB virus C in human immunodeficiency virus coinfection. Gastroenterology. 2007;133:1821-30.
45. Xiang J, McLinden JH, Chang Q, et al. Characterization of a peptide domain within the GB virus C NS5A phosphoprotein that inhibits HIV replication. PLoS ONE. 2008;3:e2580.
46. Capobianchi MR, Lalle E, Martini F, et al. Influence of GBV-C infection on the endogenous activation of the IFN system in HIV-1 co-infected patients. Cell Mol Biol (Noisy-le-grand). 2006;52:3-8.
47. Xiang J, George SL, Wunschmann S, et al. Inhibition of HIV-1 replication by GB virus C infection through increases in RANTES, MIP-1alpha, MIP-1beta, and SDF-1. Lancet. 2004;363:2040-6.
48. Schwarze-Zander C, Blackard JT, Zheng H, et al. GB virus C (GBV-C) infection in hepatitis C virus (HCV)/HIV-coinfected patients receiving HCV treatment: Importance of the GBV-C genotype. J Infect Dis. 2006;194:410-19.
49. Cheng PN, Jen CM, Young KC, et al. High-dose interferon-alpha 2b plus ribavirin combination therapy for GB virus-C/hepatitis G virus infection—a study in patients with chronic hepatitis C. Hepatogastroenterology. 2003;50:449-52.
50. Piroth L, Carrat F, Larrat S, et al. Prevalence and impact of GBV-C, SEN-V and HBV occult infections in HIV-HCV co-infected patients on HCV therapy. J Hepatol. 2008;49:892-8.
51. Nishizawa T, Okamoto H, Konishi K, et al. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem Biophys Res Commun. 1997;241:92-7.
52. Zheng H, Ye L, Fang X, et al. Torque teno virus (SANBAN isolate) ORF2 protein suppresses NF-kappaB pathways via interaction with IkappaB kinases. J Virol. 2007;81:11917-24.
53. Jelcic I, Hotz-Wagenblatt A, Hunziker A, et al. Isolation of multiple TT virus genotypes from spleen biopsy tissue from a Hodgkin’s disease patient: Genome reorganization and diversity in the hypervariable region. J Virol. 2004;78:7498-507.
54. Khudyakov YE, Cong ME, Nichols B, et al. Sequence heterogeneity of TT virus and closely related viruses. J Virol. 2000;74:2990-3000.
55. Rodriguez-Inigo E, Casqueiro M, Bartolome J, et al. Detection of TT virus DNA in liver biopsies by in situ hybridization. Am J Pathol. 2000;156:1227-34.
56. Desai M, Pal R, Deshmukh R, Banker D. Replication of TT virus in hepatocyte and leucocyte cell lines. J Med Virol. 2005;77:136-43.
57. Simmonds P, Davidson F, Lycett C, et al. Detection of a novel DNA virus (TTV) in blood donors and blood products. Lancet. 1998;352:191-5.
58. Hsu HY, Ni YH, Chen HL, et al. TT virus infection in healthy children, children after blood transfusion, and children with non-A to E hepatitis or other liver diseases in Taiwan. J Med Virol. 2003;69:66-71.
59. Naganuma M, Tominaga N, Miyamura T, et al. TT virus prevalence, viral loads and genotypic variability in saliva from healthy Japanese children. Acta Paediatr. 2008;97:1686-90.
60. Desai SM, Muerhoff AS, Leary TP, et al. Prevalence of TT virus infection in U.S. blood donors and populations at risk for acquiring parenterally transmitted viruses. J Infect Dis. 1999;179:1242-4.
61. Gimenez-Barcons M, Forns X, Ampurdanes S, et al. Infection with a novel human DNA virus (TTV) has no pathogenic significance in patients with liver diseases. J Hepatol. 1999;30:1028-34.
62. Irshad M, Sharma Y, Dhar I, et al. Transfusion-transmitted virus in association with hepatitis A-E viral infections in various forms of liver diseases in India. World J Gastroenterol. 2006;12:2432-6.
63. Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology. 1998;28:839-42.
64. Maggi F, Pistello M, Vatteroni M, et al. Dynamics of persistent TT virus infection, as determined in patients treated with alpha interferon for concomitant hepatitis C virus infection. J Virol. 2001;75:11999-2004.
65. Tokita H, Murai S, Kamitsukasa H, et al. High TT virus load as an independent factor associated with the occurrence of hepatocellular carcinoma among patients with hepatitis C virus-related chronic liver disease. J Med Virol. 2002;67:501-9.
66. Moreno J, Moraleda G, Barcena R, et al. Response of TT virus to IFN plus ribavirin treatment in patients with chronic hepatitis C. World J Gastroenterol. 2004;10:143-6.
67. Hijikata M, Takahashi K, Mishiro S. Complete circular DNA genome of a TT virus variant (isolate name SANBAN) and 44 partial ORF2 sequences implicating a great degree of diversity beyond genotypes. Virology. 1999;260:17-22.
68. Tanaka Y, Primi D, Wang RY, et al. Genomic and molecular evolutionary analysis of a newly identified infectious agent (SEN virus) and its relationship to the TT virus family. J Infect Dis. 2001;183:359-67.
69. Akiba J, Umemura T, Alter HJ, et al. SEN virus: Epidemiology and characteristics of a transfusion-transmitted virus. Transfusion. 2005;45:1084-8.
70. Wilson LE, Umemura T, Astemborski J, et al. Dynamics of SEN virus infection among injection drug users. J Infect Dis. 2001;184:1315-19.
71. Pfeiffer RM, Tanaka Y, Yeo AE, et al. Prevalence of SEN viruses among injection drug users in the San Francisco Bay area. J Infect Dis. 2003;188:13-18.
72. Shibata M, Wang RY, Yoshiba M, et al. The presence of a newly identified infectious agent (SEN virus) in patients with liver diseases and in blood donors in Japan. J Infect Dis. 2001;184:400-4.
73. Umemura T, Yeo AE, Sottini A, et al. SEN virus infection and its relationship to transfusion-associated hepatitis. Hepatology. 2001;33:1303-11.
74. Umemura T, Tanaka E, Ostapowicz G, et al. Investigation of SEN virus infection in patients with cryptogenic acute liver failure, hepatitis-associated aplastic anemia, or acute and chronic non-A-E hepatitis. J Infect Dis. 2003;188:1545-52.
75. Hoagland RJ. The clinical manifestations of infectious mononucleosis: A report of two hundred cases. Am J Med Sci. 1960;240:55-63.
76. Grotto I, Mimouni D, Huerta M, et al. Clinical and laboratory presentation of EBV positive infectious mononucleosis in young adults. Epidemiol Infect. 2003;131:683-9.
77. Shaukat A, Tsai HT, Rutherford R, Anania FA. Epstein-Barr virus induced hepatitis: An important cause of cholestasis. Hepatol Res. 2005;33:24-6.
78. Baron DN, Bell JL, Dunnet WN. Biochemical studies on hepatic involvement in infectious mononucleosis. J Clin Pathol. 1965;18:209-11.
79. Cheng CC, Chang LY, Shao PL, et al. Clinical manifestations and quantitative analysis of virus load in Taiwanese children with Epstein-Barr virus-associated infectious mononucleosis. J Microbiol Immunol Infect. 2007;40:216-21.
80. Edoute Y, Baruch Y, Lachter J, et al. Severe cholestatic jaundice induced by Epstein-Barr virus infection in the elderly. J Gastroenterol Hepatol. 1998;13:821-4.
81. Markin RS. Manifestations of Epstein-Barr virus-associated disorders in liver. Liver. 1994;14:1-13.
82. Biest S, Schubert TT. Chronic Epstein-Barr virus infection: A cause of granulomatous hepatitis? J Clin Gastroenterol. 1989;11:343-6.
83. Hara S, Hoshino Y, Naitou T, et al. Association of virus infected-T cell in severe hepatitis caused by primary Epstein-Barr virus infection. J Clin Virol. 2006;35:250-6.
84. Rouphael NG, Talati NJ, Vaughan C, et al. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7:814-22.
85. Edwards JM, Vandervelde EM, Cohen BJ, McSwiggan DA. Laboratory diagnosis of EB virus infection in some cases presenting as hepatitis. J Clin Pathol. 1978;31:179-82.
86. Kilpatrick ZM. Structural and functional abnormalities of liver in infectious mononucleosis. Arch Intern Med. 1966;117:47-53.
87. Suh N, Liapis H, Misdraji J, et al. Epstein-Barr virus hepatitis: Diagnostic value of in situ hybridization, polymerase chain reaction, and immunohistochemistry on liver biopsy from immunocompetent patients. Am J Surg Pathol. 2007;31:1403-9.
88. van der Horst C, Joncas J, Ahronheim G, et al. Lack of effect of peroral acyclovir for the treatment of acute infectious mononucleosis. J Infect Dis. 1991;164:788-92.
89. Adams LA, Deboer B, Jeffrey G, et al. Ganciclovir and the treatment of Epstein-Barr virus hepatitis. J Gastroenterol Hepatol. 2006;21:1758-60.
90. Ohta K, Shimizu M, Nakai A, et al. Rituximab therapy for Epstein-Barr virus-related chronic hepatitis following living donor kidney transplantation. Am J Kidney Dis. 2006;48:986-9.
91. Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Intern Med. 1993;119:924-35.
92. Wreghitt TG, Teare EL, Sule O, et al. Cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 2003;37:1603-6.
93. Eddleston M, Peacock S, Juniper M, Warrell DA. Severe cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 1997;24:52-6.
94. Domiati-Saad R, Dawson DB, Margraf LR, et al. Cytomegalovirus and human herpesvirus 6, but not human papillomavirus, are present in neonatal giant cell hepatitis and extrahepatic biliary atresia. Pediatr Dev Pathol. 2000;3:367-73.
95. Squizzato A, Ageno W, Cattaneo A, Brumana N. A case report and literature review of portal vein thrombosis associated with cytomegalovirus infection in immunocompetent patients. Clin Infect Dis. 2007;44:e13-16.
96. Benhamou Y, Caumes E, Gerosa Y, et al. AIDS-related cholangiopathy. Critical analysis of a prospective series of 26 patients. Dig Dis Sci. 1993;38:1113-18.
97. Mahajani RV, Uzer MF. Cholangiopathy in HIV-infected patients. Clin Liver Dis. 1999;3:669-84.
98. Agarwal SK, Kalra V, Dinda A, et al. Fibrosing cholestatic hepatitis in renal transplant recipient with CMV infection: A case report. Int Urol Nephrol. 2004;36:433-5.
99. Sampathkumar P, Paya CV. Management of cytomegalovirus infection after liver transplantation. Liver Transpl. 2000;6:144-56.
100. St George K, Rinaldo CRJr. Comparison of cytomegalovirus antigenemia and culture assays in patients on and off antiviral therapy. J Med Virol. 1999;59:91-7.
101. Nigro G, Krzysztofiak A, Bartmann U, et al. Ganciclovir therapy for cytomegalovirus-associated liver disease in immunocompetent or immunocompromised children. Arch Virol. 1997;142:573-80.
102. Corey L, Spear PG. Infections with herpes simplex viruses (2). N Engl J Med. 1986;314:749-57.
103. Czartoski T, Liu C, Koelle DM, et al. Fulminant, acyclovir-resistant, herpes simplex virus type 2 hepatitis in an immunocompetent woman. J Clin Microbiol. 2006;44:1584-6.
104. Overall JCJr. Herpes simplex virus infection of the fetus and newborn. Pediatr Ann. 1994;23:131-6.
105. Yaziji H, Hill T, Pitman TC, et al. Gestational herpes simplex virus hepatitis. South Med J. 1997;90:347-51.
106. Chatelain S, Neumann DE, Alexander SM. Fatal herpetic hepatitis in pregnancy. Infect Dis Obstet Gynecol. 1994;1:246-8.
107. Bissig KD, Zimmermann A, Bernasch D, et al. Herpes simplex virus hepatitis 4 years after liver transplantation. J Gastroenterol. 2003;38:1005-8.
108. Norvell JP, Blei AT, Jovanovic BD, Levitsky J. Herpes simplex virus hepatitis: An analysis of the published literature and institutional cases. Liver Transpl. 2007;13:1428-34.
109. Fink CG, Read SJ, Hopkin J, et al. Acute herpes hepatitis in pregnancy. J Clin Pathol. 1993;46:968-71.
110. Pellise M, Miquel R. Liver failure due to herpes simplex virus. J Hepatol. 2000;32:170.
111. Greenspoon JS, Wilcox JG, McHutchison LB, Rosen DJ. Acyclovir for disseminated herpes simplex virus in pregnancy. A case report. J Reprod Med. 1994;39:311-17.
112. Twagira M, Hadzic N, Smith M, et al. Disseminated neonatal herpes simplex virus (HSV) type 2 infection diagnosed by HSV DNA detection in blood and successfully managed by liver transplantation. Eur J Pediatr. 2004;163:166-9.
113. Feldman S, Crout JD, Andrew ME. Incidence and natural history of chemically defined varicella-zoster virus hepatitis in children and adolescents. Scand J Infect Dis. 1997;29:33-6.
114. Fehr T, Bossart W, Wahl C, Binswanger U. Disseminated varicella infection in adult renal allograft recipients: Four cases and a review of the literature. Transplantation. 2002;73:608-11.
115. Chau TN, Lee KC, Yao H, et al. SARS-associated viral hepatitis caused by a novel coronavirus: Report of three cases. Hepatology.. 2004;39:302-10.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]<
[/not-level-membership-for-gastroenterology-and-hepatology-category]
