CHAPTER 79 Hepatitis C
Hepatitis C virus (HCV) infects 170 million people worldwide and 1.6% of the population of the United States.1–4 Unfortunately, HCV successfully evades the host immune response in 55% to 85% of acutely infected persons, thus leading to chronic infection. The natural history of hepatitis C varies greatly; reasons for this heterogeneity remain incompletely understood but are related to both viral, host, and environmental factors. Chronic HCV infection can lead to cirrhosis and hepatocellular carcinoma. The incidence of these complications has risen dramatically in the 2000s but is expected to decline over the next 20 years.3 In fact, whereas HCV-related mortality increased dramatically after 1995, it has reached a plateau since 2002.5 Complications of HCV-related cirrhosis are currently the leading indication for liver transplantation in the United States and Europe.
Chronic hepatitis C is the only chronic viral infection that can be cured by antiviral therapy. Currently, 40% to 50% of patients infected with HCV genotype 1 who tolerate full-dose treatment with pegylated interferon and ribavirin achieve a sustained virologic response (SVR) to treatment, defined as absence of HCV RNA in serum six months after discontinuation of treatment; an SVR is almost always associated with a durable eradication of the virus.6,7 From 70% to 80% of genotype 2- and 3-infected patients achieve an SVR. Substantial progress in understanding the mechanisms of virus entry into the hepatocyte, replication, and the host immune response has led to the development of new therapeutic agents that target the steps in the viral life cycle. New agents under investigation promise to improve the SVR further when combined with interferons and ribavirin.
VIROLOGY
STRUCTURE
The HCV virion has been visualized by electron microscopy and is an enveloped virus 50 nm in diameter.8 The two envelope proteins, E1 and E2, heterodimerize and assemble into tetramers, which create a smooth outer layer. This layer has a “fishbone” configuration with icosahedral symmetry. The envelope proteins are anchored to a host cell–derived lipid bilayer envelope membrane that surrounds the nucleocapsid. The nucleocapsid is believed to be composed of multiple copies of the core protein and forms an internal icosahedral viral coat that encapsulates the genomic ribonucleic acid (RNA). HCV circulates in various forms in the serum of an infected host, including (1) virions that are bound to very-low-density and low-density lipoproteins and appear to represent the infectious fraction; (2) virions bound to immunoglobulins; and (3) free virions. In addition, viral particles that exhibit physicochemical, morphologic, and antigenic properties of nonenveloped HCV nucleocapsids have been detected in plasma.9
GENOMIC ORGANIZATION
HCV is a single-stranded positive-sense RNA virus that belongs to the Flaviviridae family and has been classified as the sole member of the genus Hepacivirus.10 The genome of HCV contains approximately 9600 nucleotides with an open reading frame (ORF) that encodes one large viral polypeptide precursor of 3008 to 3033 amino acids. The HCV ORF is flanked upstream by a 5′ untranslated region (UTR) that functions as an internal ribosome entry site (IRES) to direct cap-independent translation (i.e., without the addition of an extra ribonucleotide to the 5′ end of the viral messenger RNA) and downstream by a 3′ UTR that is critical for initiation of new RNA strand synthesis.11–13 The 5′ and portions of the 3′ UTR are the most conserved parts of the HCV genome.
VIRAL REPLICATION AND LIFE CYCLE
Although peripheral blood mononuclear cells, B cells, T cells, and dendritic cells have been reported to support HCV replication, hepatocytes are the major site of viral replication.14,15 Understanding of the mechanisms of viral replication comes from studies in chimpanzees, extrapolation of the mechanisms used by other flaviviruses, and infection of cells in vitro with subgenomic replicons (segments of HCV RNA that are capable of amplification and synthesis of viral proteins but that do not produce mature viruses).
Early events in viral binding to the hepatocyte surface are still not completely understood (Fig. 79-1). HCV entry involves the attachment of envelope proteins E1 and E2 to cell surface molecules. The expression and function of CD81, a member of the tetraspan superfamily, are essential for HCV entry into hepatocytes.16 In addition, human scavenger receptor class B type 1 (SR-B1), a selective importer of cholesteryl esters from high-density lipoproteins (HDL) into cells, has been shown to interact with E2 and is essential for HCV entry.17 Whereas CD81 and SR-B1 are required early in the process of viral entry, claudin-1 (CLDN1), a tight junction component that is highly expressed on hepatocytes, is required later in the cell entry process.18 Heparin sulfated proteoglycans have also been shown to be essential for HCV cell entry.19 Other receptors are also likely required for viral entry.18 There is some evidence that the low-density lipoprotein (LDL) receptor is involved during endocytosis of HCV.20
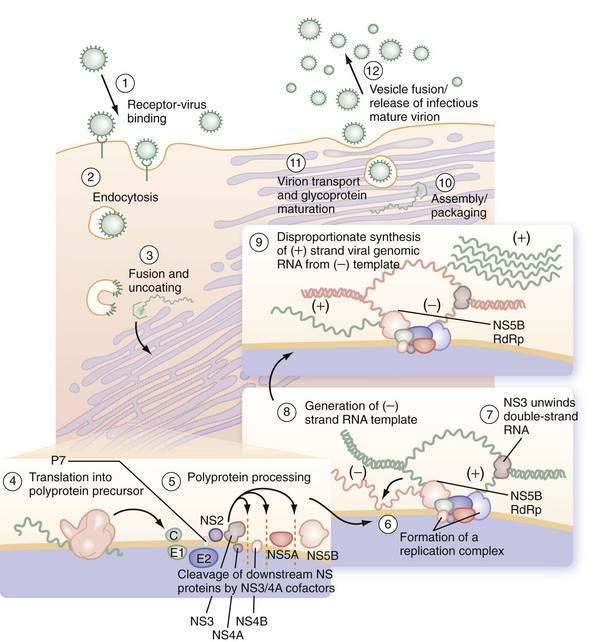
Figure 79-1. Putative life cycle of hepatitis C virus.
(Reproduced with permission from Pawlotsky JM, Chevaliez S, McHutchison JG. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology 2007; 132:1979-98.) (See text for details and Fig. 79-2 for functions of the hepatitis C virus proteins.) NS, nonstructural; RdRp, RNA-dependent RNA polymerase.
Not only do viral receptors on hepatocytes play a role in entry of HCV into the cell, but also other cofactors affect the efficiency of viral infectivity. C-type lectins DC-SIGN (dendritic cell–specific intercellular adhesion molecule-3-grabbing nonintegrin; CD209) and L-SIGN (DC-SIGNr, liver and lymph node specific; CD209L) are expressed on dendritic cells and liver sinusoidal endothelial cells, respectively.21 Both receptors bind E2 on the HCV virus and facilitate infection of adjacent hepatocytes by acting as a capture and delivery mechanism. In addition, HDL increases infectivity of hepatocytes ten-fold, although the mechanism is not understood.22
Once the HCV virus attaches to the cell, endocytosis of the bound virion is presumed to occur, as in other flaviviruses. A pH drop in the vesicle causes conformational changes in the glycoproteins that lead to fusion of the viral and cellular membranes23 and release of viral RNA into the cytoplasm. In the cytosol, the 5′ UTR contains several highly conserved and structured domains that function as an IRES, which directs the RNA to its docking site on the endoplasmic reticulum and mediates cap-independent internal initiation of HCV polyprotein translation by recruiting both cellular proteins, including eukaryotic initiation factors (eIF) 2 and 3, and viral proteins.24,25
The large polyprotein generated by translation of the HCV genome is co- and post-translationally processed proteolytically into at least 11 viral proteins, including both structural (nucleocapsid [C], or p21; envelope 1 [E1], or gp31; and envelope 2 [E2], or gp70) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins (Fig. 79-2).11,13 The functions of these specific proteins are described later in the chapter.
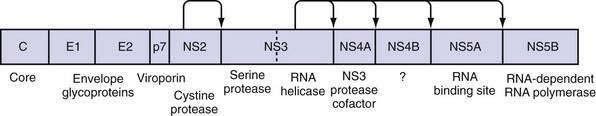
After polyprotein processing, NS4B expression causes the membrane alterations that are seen on electron microscopy as a membranous web.26,27 The replication complex associates viral proteins, cellular components, and nascent RNA strands and is essential for HCV replication, as demonstrated in replicon cell culture systems.28 HCV replication is catalyzed by the NS5B RNA-dependent RNA polymerase (RdRp). The positive-strand genomic RNA serves as a template for the synthesis of a negative-strand intermediate. Negative-strand RNA serves as a template for production of numerous strands of RNA of positive polarity that are used for polyprotein translation and synthesis of new intermediates of replication and are packaged into new virus particles.29
Finally, viral particle formation is initiated by the interaction of the core protein with genomic RNA in the endoplasmic reticulum, although the details of this process and subsequent export of mature virions from the hepatocyte are poorly understood.30,31 By analogy with pestiviruses, HCV packaging and release are likely to be inefficient because much of the virus remains in the cell. Following release, viral particles may infect adjacent hepatocytes or enter the circulation, where they are available for infection of another cell or host.
Virus Protein Function
The large polyprotein generated by translation of the HCV genome is cleaved by cellular and viral proteases to form structural and nonstructural proteins.11,13 The structural proteins are separated from the nonstructural proteins by the short membrane peptide p7, which is believed to be a viroporin, a protein that plays a role in viral particle maturation and release.32,33 At least one, and possibly three, alternative reading frame proteins (ARFP, or F for “frameshift”) exists.34–36 The exact number of alternative reading frames, the number of proteins that result, and the functions of the proteins are not known.36 One such protein is 17 kd in size; it can be expressed in vitro, and antibodies to it have been found in infected patients.
The core protein is first cleaved from the large polypeptide and then further processed by a host signal peptidase.11 In infectious HCV virions, core protein forms the viral nucleocapsid and binds RNA; it has many other functions as well. Core protein has been found attached to lipid rafts and the endoplasmic reticulum, and it translocates into the nucleus. When core protein attaches to lipid rafts, it recruits nonstructural proteins, resulting in the assembly of infectious virions. Core protein can also interact with the host immune system by inactivating the RNA silencing activity of Dicer, a cellular endoribonuclease that produces small interfering RNA to bind and target HCV RNA for destruction by the cell.37 Core protein can also bind to Janus kinase-1 (JAK1) and JAK2 and alter the activation of signal transducer activator of transcription (STAT) proteins, leading to impairment of interferon production.38 Extracellularly, core protein inhibits T-cell activation and proliferation, possibly by down-regulating co-stimulatory molecules on dendritic cells.39 Specific polymorphisms in core protein have also been associated with intracellular lipid accumulation40; this may be the result of facilitation of phosphorylation of insulin receptor substrate-1 (IRS-1), thereby leading to insulin resistance.41 Mutations in core protein have also been associated with an increased risk of hepatocellular carcinoma (HCC) in patients; core protein alone can cause HCC in transgenic mice.42
E1 and E2 proteins are cleaved from the polypeptide by host signal peptidase.11 The two proteins form highly glycosylated heterodimers and then tetramers that are essential for viral assembly (see earlier). They also mediate cell entry by binding to surface receptors.43 Subsequently, they are responsible for fusion between the host cell membrane and the viral envelope. Because E1 and E2 are expressed on the surface of the virion, they are targets of host antibodies. The first 27 amino acids of E2 form hypervariable region 1 (HVR1); alterations in HVR1 are believed to be an attempt by the virus at antibody-mediated immune evasion.
P7 is cleaved by the endoplasmic reticulum signal peptidase and forms an ion channel. This viroporin protein is essential for efficient assembly and release of infectious virions but not for cell entry.11,33 Because p7 is needed later in the viral life cycle, cleavage of the polypeptide is delayed.33
NS2 complexes with NS3 and zinc to form a cysteine protease, with two composite active sites, that autocatalytically cleaves NS2 from NS3.44 No other function of NS2 has been discovered to date. NS3 has several functions in addition to complexing with NS2 for autocatalytic cleavage of the NS2-NS3 site.44 Its function as a serine protease is markedly enhanced by its association with NS4A. The enzyme results in cleavage of the polyprotein at the NS3-NS4A, NS4A-NS4B, NS4B-NS5A, and NS5A-NS5B sites.11,45 The NS3 protease also cleaves and thereby destroys the function of Cardif and TRIF (Toll/interleukin receptor domain-containing adapter-inducing interferon-β), which are intermediates in two separate pathways of host-cell interferon secretion in response to viral infection.46–48 This property may have a significant effect in impairing the host response to HCV infection. Finally, a portion of the NS3 protein functions as a helicase that unwinds viral RNA as well as host DNA. The helicase function is dependent on adenosine triphosphate (ATP), may require dimerization of NS3, and progresses in discrete steps like an inchworm.12,49 NS4A complexes with NS3 and functions to stabilize the protease and helicase activities and anchor the complex to the endoplasmic reticulum membrane.11,12 It also regulates hyperphosphorylation of NS5A.50 The only known function of NS4B is to induce the formation of the “membranous web” on which HCV transcription occurs.26 NS5A binds zinc and forms homodimers that are bound to the endoplasmic reticulum membrane.12 NS5A is essential for viral replication and is believed to provide an RNA-binding site within the replication complex.51 In addition, NS5A inhibits apoptosis in infected cells,52,53 and some mutations confer improved sensitivity to interferon therapy.54 NS5B is the viral RNA-dependent RNA polymerase.11 The crystal structure elucidates the tunnel of the enzyme that directs single-stranded RNA into the active site.55 It can synthesize both negative-strand HCV RNA templates and positive-strand HCV RNA genomes.
GENOTYPES AND QUASISPECIES
HCV has an inherently high mutational rate that results in considerable heterogeneity throughout the genome.56 This high mutational rate is in part a consequence of the RNA-dependent RNA polymerase of HCV, which lacks 3′-to-5′-exonuclease proofreading ability that ordinarily would remove mismatched nucleotides incorporated during replication. An average of one error occurs for every 104 to 105 nucleotides copied. This phenomenon is favored by a high viral turnover rate; 1010 to 1012 virions are produced per day.57 A substantial proportion of newly synthesized viral genomes have alterations. Because of the functional differences in HCV proteins, genetic variation in some parts of the genome confers advantages by evading or inhibiting the host immune system, whereas other mutations may be lethal to the virus if they lead to defective replication machinery. Therefore, genetic variation is distributed irregularly along the genome. Each new genetic variant is produced in a single cell and may or may not spread through the liver and into the serum. The result is not only genetic diversity in the serum, but also compartmentalization of variant virions in different parts of the liver and perhaps in extrahepatic sites.
Because of the vast genetic variation, a classification scheme was devised whereby viral sequences are given a genotype and subtype. The first division used to describe the genetic heterogeneity of HCV is the viral genotype, which refers to genetically distinct groups of HCV isolates that have arisen during the evolution of the virus. Nucleotide sequencing has shown variation of up to 34% between genotypes.56 The most conserved region (5′ UTR) has a maximum nucleotide sequence divergence of 9% between genotypes, whereas the highly variable regions that encode the envelope proteins (E1 and E2) exhibit a nucleotide sequence divergence of 35% to 44% between genotypes. The sequences cluster into 6 major genotypes (designated by numbers), with sequence similarities of 60% to 70%, and more than 70 subtypes (designated by a lower case letter) within these major genotypes, with sequence similarities of 77% to 80%.56 In this scheme, the first variant, which was cloned by Choo and colleagues, is designated type 1a.58 The HCV genotype is an intrinsic characteristic of the infecting HCV strain and does not change over time; therefore, the genotype only needs to be determined once in an infected person. Mixed-genotype infections may be seen and reflect either coinfection with more than one HCV virus or methodologic problems in genotype testing. In addition, inter-genotypic HCV recombinants have been described59; these are thought to arise because of recombination between different genotypes in patients with repeated exposure. The recombination events have been reported to occur in or between NS2 and NS3.60
Global geographic differences exist in the distribution of HCV genotypes, as well as in the mode of acquisition. In the United States, genotype 1a is the most prevalent, accounting for approximately 57% of HCV infections, followed by genotype 1b in 17%, genotype 2 in 14%, genotype 3 in 7%, and genotype 4, 5, or 6 in less than 5%.61 Racial differences are seen in the prevalence of genotypes; approximately 90% of African Americans are infected with HCV genotype 1, whereas only 70% of whites and 71% of Hispanics are infected with genotype 1.62 In Europe, the most prevalent genotype is 1b (47%), followed by 1a (17%), 3 (16%), and 2 (13%).63 Genotype 4 is found mainly in Egypt, the Middle East, and Central Africa.64 In Egypt, approximately 15% of the population is infected with HCV, and more than 90% have HCV genotype 4. Because of the high prevalence rate in Egypt, genotype 4 represents 20% of the world’s HCV-infected population. Genotype 5, although originally isolated in South Africa, is also seen in specific regions of France, Belgium, and Spain.65 Genotype 6 is found predominantly in Asia. The distribution of genotypes is ever changing with immigration and alterations in the primary modes of viral transmission. Therefore, the frequencies of viral genotypes change over time.66
The second component of genetic heterogeneity is quasispecies generation.56 Quasispecies are closely related, yet heterogeneous, sequences of HCV RNA within a single infected person that result from mutations that occur during viral replication. The rate of nucleotide changes varies significantly among the different regions of the viral genome. The highest proportion of mutations is found in the E1 and E2 regions, particularly in HVR1. Even though this region represents only a minor part of the E2 region, it accounts for approximately 50% of the nucleotide changes and 60% of the amino acid substitutions within the envelope region.
The development of quasispecies may be one mechanism by which the virus escapes the host’s immune response and establishes persistent infection.67 During acute infection or during treatment, the lack of quasispecies is associated with viral clearance, and the development of numerous quasispecies is associated with viral persistence.68 In acute disease, patients in whom genetic variation in the HVR1 region develops after antibody seroconversion progress to chronic disease, whereas those in whom such genetic variation does not develop are more likely to achieve viral clearance.67 Genetic variation before seroconversion does not correlate with outcome, indicating that quasispecies formation results from antibody-mediated immune pressure. Interestingly, no intrinsically interferon-resistant variants of HCV have been defined, indicating that both viral and host factors play important roles in determining whether the virus persists or is cleared. An increased number of quasispecies has also been associated with more rapid progression to cirrhosis and the development of HCC.69
EPIDEMIOLOGY
INCIDENCE AND PREVALENCE
The worldwide seroprevalence of HCV infection, based on detection of antibody to HCV (anti-HCV), is estimated to be 3%, with more than 170 million people infected chronically. Marked geographic variation exists, with infection rates ranging from 1.3% to 1.6% in the United States to 15% in Egypt.1,64 Currently, between 3.2 and 5 million persons are infected with HCV in the United States.1 The prevalence rate is higher in persons 40 to 49 years old than in older or younger persons, in males (2.1%) than in females (1.1%), and in African Americans (3%) than in whites (1.5%).1 Other risk factors for HCV infection and the associated frequencies of infection are injection drug use (57.5%), blood transfusion before 1992 (5.8%), greater than 50 lifetime sexual partners (12%), and family income below the poverty level (3.2%). The current prevalence of HCV infection in the United States may be underestimated because the National Health and Nutrition Examination Survey (NHANES) data did not evaluate persons who are homeless, incarcerated, or in the military. Among incarcerated persons, 12% to 35% are positive for HCV RNA serum,70 whereas those in military service have a seroprevalence rate for anti-HCV of 0.5%.71
Worldwide, three different epidemiologic patterns of HCV infection have emerged: (1) previous exposure through health care with a peak prevalence in older persons; (2) exposure through intravenous drug use, the major risk factor since data first became available in about 1960, with a peak prevalence among middle-aged persons; and (3) ongoing high levels of infection in areas where high rates of infection occur in all age groups.63
Given the factors that influence viral diversity (see earlier), estimating the site of origin and age of HCV by phylogenetic analysis is difficult. The best estimate is that HCV originated in western and sub-Saharan Africa.72 Subsequent global spread probably occurred coincident with trade and human migration. Evolution of the virus led to a geographic distribution of genotypes, so that genotypes 1, 2, and 3 are most common in North America and Europe, genotype 4 is most common in the Middle East, and genotypes 5 and 6 are most common in Southeast Asia. In Japan, HCV transmission transitioned from constant to exponential growth in the 1920s, and the prevalence of HCV infection is highest in older persons.73 In Japan, and later in southern and Eastern Europe, health care–related procedures—particularly reuse of contaminated syringes—played a major role in viral spread. In the United States, Australia, and other developed countries, peak prevalence is in persons ages 40 to 49 years, and analysis of risk factors suggests that most HCV transmission occurred between the mid-1980s and the mid-1990s, through intravenous drug use. In Egypt, the spread of HCV increased exponentially from the 1930s to the 1980s because of mass vaccination campaigns with reuse of medical equipment.64 In Egypt and other developing countries, high rates of infection are observed in all age groups, suggesting that an ongoing risk of HCV acquisition exists.
In the United States, the incidence of acute hepatitis C is falling. The peak incidence was estimated to be 180,000 cases per year in the mid-1980s, but the rate declined to approximately 19,000 new cases by 2006.74 Many factors have contributed to the falling incidence of acute hepatitis C. In the 1980s, when blood was purchased from donors, 2% to 10% of blood units were infected with HCV, leading to a high rate of transfusion-acquired HCV infection.75 The institution of volunteer blood donation, creation of recombinant clotting factors, and implementation of HCV blood testing (between 1990 and 1992) dramatically decreased transfusion-acquired HCV infection.66
TRANSMISSION
Percutaneous Transmission
Blood transfusion, before the introduction of screening, and injection drug use are the most clearly documented risk factors for HCV infection. Following the introduction of anti-HCV screening of blood donors between 1990 and 1992, the number of transfusion-related cases of HCV infection declined sharply, and currently less than 1 case occurs per 2,000,000 units transfused.76,77
Injection drug use has always been the major route of HCV acquisition in the United States and accounts for an increasingly large portion of cases, at least 68% of new cases of HCV infection, since the virus was essentially eliminated from the blood supply.74 The prevalence of HCV infection in injection drug users ranges from 57% to 90%.1,78 Although risk factors for hepatitis B virus (HBV) and human immunodeficiency virus (HIV) infection overlap with those for HCV infection, the prevalence of HCV infection in this population is the highest among the three viruses. The majority of injection drug users become anti-HCV positive within six months of initiating injection drug use with shared paraphernalia.
Chronic hemodialysis is also associated with increased rates of HCV infection. The frequency of anti-HCV in patients on hemodialysis ranges from 11.6% in the United States to 55% to 85% in Jordan, Saudi Arabia, and Iran.79 Serologic assays for anti-HCV may underestimate the frequency of HCV infection in this relatively immunocompromised population, and virologic assays may be necessary for accurate diagnosis.80
Transmission may occur from infected patients to health care workers. A serologic survey of emergency department patients found that 18% were infected with HCV.81 The proportion with HCV infection was even higher in patients with a history of intravenous drug use (83%), past blood transfusion (21%), or a male homosexual lifestyle (21%). Although all potential routes of transmission of HCV infection to hospital workers are not obvious, needlestick injuries probably account for a large proportion of cases. Anti-HCV seroconversion rates are approximately 0.3% to 4% in longitudinal studies of health care workers after percutaneous inoculation from anti–HCV-positive sources, although the risk is dependent on the type of needle (hollow versus solid, infusion versus withdrawal), volume of inoculum, depth of injury, time the body fluid has spent ex vivo, level of viremia (viral load), and HIV status of the inoculating body fluid.82,83 In one study, 99% of surgeons in training experienced at least one needlestick by their final year of residency. Fifty-three percent of these injuries involved a high-risk patient, and only 49% were reported to the employee health service.84 As a result, as many as 16,000 new cases of HCV are estimated to have been transmitted to health care workers worldwide in 2000.85 Although less common, transmission of HCV also may occur from health care workers to patients. Because acute HCV infection often is subclinical, nosocomial transmission may occur with greater frequency than has been recognized previously. Strict adherence to universal precautions to protect health care workers and patients is critically important. At this time, no treatment is effective for post-exposure prophylaxis, and no data support such treatment even if it were available.
Nonpercutaneous Transmission
Nonpercutaneous modes of HCV transmission include sexual practices and childbirth. Available evidence indicates that transmission by nonpercutaneous routes occurs but is inefficient. From 10% to 20% of patients with HCV infection report that their only risk factor is sexual exposure to a partner with HCV infection. Most seroepidemiologic studies, however, have demonstrated anti-HCV in only a small proportion of sexual contacts of infected persons. In a large prospective study of monogamous seronegative partners of HCV-infected patients who denied anal intercourse and intercourse during menstruation, no instances of HCV transmission of the same sequenced virus occurred over a 10-year period of time.86 Therefore, many of the cases presumed to be the result of sexual transmission are likely the result of other, perhaps unreported or unrecognized, exposures. If the index sexual partner is infected with HIV or the partners engage in high-risk sexual practices, such as anal intercourse, however, the transmissibility of HCV is likely increased.87 Furthermore, epidemiologic studies have shown that persons with multiple sex partners have a higher prevalence of HCV infection.1 Whether sexually transmitted diseases promote transmission of HCV through breakdown of mucosal or immune barriers is unclear.
HCV-infected persons commonly are counseled to notify sexual partners of their HCV status. The risk of sexual transmission is negligible in monogamous couples that do not engage in high-risk sexual practices.86 Barrier methods should be recommended, however, to persons in non-monogamous relationships or those engaging in high-risk sexual practices.
Compared with the high efficiency of perinatal transmission of HBV infection (see Chapter 78), the risk of perinatal transmission of HCV infection is low, averaging 5.1% to 6.7% for HCV-monoinfected patients and two to three times higher for HIV-HCV-coinfected patients.88,89 Mothers with a high viral load are more likely to transmit HCV to their infants, a finding that may explain why infants born to mothers with HIV-HCV coinfection are at higher risk of HCV infection. Interestingly, the use of highly active antiretroviral therapy (HAART) in HIV-HCV-coinfected mothers may decrease the risk of perinatal transmission of both HIV and HCV.89 Data regarding the risk associated with vaginal delivery as opposed to cesarean delivery are uncontrolled, but evidence for a higher risk of HCV transmission with vaginal delivery is unconvincing. This issue remains controversial, and some authorities recommend elective cesarean section before membrane rupture.88
Although little data exist, the risk of HCV transmission from breastfeeding is negligible to small. The Centers for Disease Control and Prevention have concluded that breastfeeding by HCV-infected mothers is generally safe. Some authorities have suggested, however, that mothers with a high viral load (greater than 108 copies/mL, see later) may pose a risk.90 Because anti-HCV can be acquired passively by the infant, molecular testing for HCV RNA is required if the diagnosis of HCV infection is suspected. Infants of infected mothers should not undergo serologic testing for anti-HCV before the age of 18 months because maternal antibodies may persist in the infant’s serum and lead to diagnostic confusion.
Sporadic HCV Infection
The source of transmission is unknown in 9% to 27% of cases of HCV infection.78 Such sporadic HCV infection probably results from an undisclosed or unrecognized percutaneous route of infection. This presumption is supported by the observation that intranasal cocaine use is not considered a risk factor for HCV transmission (although it was considered a risk factor in the past).91 HCV infection can be acquired from non-commercial tattooing and body piercing when equipment is reused, shared, or improperly sterilized. Commercial tattooing is now well controlled and probably conveys little risk of HCV infection. Iatrogenic transmission of HCV is well documented in a variety of circumstances, most notably via contaminated multi-use vials and inadequately sterilized multi-use instruments and syringes, as seen with schistosomal treatment campaigns in Egypt.92
PATHOGENESIS
Determinants of persistence of HCV include (1) the evasion of immune responses through several viral mechanisms, (2) inadequate induction of the innate immune response, (3) insufficient induction or maintenance of an adaptive immune response, (4) the production of viral quasispecies, and (5) the induction of immunologic tolerance.67,93,94 In 55% to 85% of acute HCV infections, the net result of the host-virus interplay is the inability to clear virus despite the development of antibodies against several viral proteins. In the minority of patients in whom acute HCV resolves, an early and multispecific CD4+ T-cell proliferative response occurs, with predominance of type 1 CD4+ helper T (Th1) cells in the peripheral blood,95 most of which produce interferon-α. This “protective” response is still detected 18 to 20 years after infection in a majority of asymptomatic recovered patients but in only a minority of patients in whom chronic HCV infection develops.96 Although the immune response is essential in preventing viral persistence after acute HCV infection in 15% to 45% of cases, in those without viral clearance, the immune response mediates hepatic cell destruction and fibrosis.
VIRAL MECHANISMS
In chronically infected patients, the pathogenesis of liver damage is largely immune mediated. In a small subset of immunocompromised HCV-infected patients among both HIV-infected patients and organ transplant recipients, however, a syndrome termed fibrosing cholestatic hepatitis develops.97,98 Such cases are thought to result from direct viral hepatotoxicity of infected cells because viral levels are typically greater than 30 million copies/mL and hepatocytes contain enormous concentrations of virus and viral proteins.99 Survival in such patients is quite poor.
IMMUNE-MEDIATED MECHANISMS
HCV infection elicits an immune response in the host that involves both an initial innate response as well as a subsequent adaptive response. The innate response is the first line of defense against the virus and includes several arms such as natural killer (NK) cell activation and cellular antiviral mechanisms triggered by pathogen-associated molecular patterns (PAMPs) recognized by the cell.100–102 These processes can lead to apoptosis of infected cells within the first few hours of infection. NK cells, as the effector cells of the innate immune system, also produce tumor necrosis factor (TNF)-β and interferon-α, cytokines that are critical for dendritic cell maturation and subsequent induction of adaptive immunity.94 After this, however, the virus initiates a number of mechanisms that undermine the ability of the host to control the infection.
Virus-related disruption of the innate, and later adaptive, immune response occurs at several levels.93 NK cell function is slowed possibly because NK cell–mediated cytotoxicity and production of cytokines are interrupted when the HCV E2 protein binds its cellular receptor CD81.103,104 PAMPs activate several cellular processes including the JAK-STAT pathway and Toll-like receptor-3 (TLR-3), activation of both of which ultimately results in production of cellular interferons and interferon-regulated factors that convey antiviral properties to the cell.93 NS3/4 protease degrades TRIF, an essential intermediate in this pathway, and cleaves interferon promoter stimulator-1 (IPS-1), an intermediate in the signaling cascade, to activate interferon when retinoic inducible gene-1 (RIG-1) binds viral intermediates.47,105,106 In addition, HCV core protein promotes STAT-1 degradation, inhibits STAT-1 phosphorylation, promotes suppressor of cytokine signaling (SOCS) induction (an inhibitor of JAK-STAT signaling), and impairs interferon-stimulated gene factor-3 (ISGF3), a heterotrimer of STAT-1, STAT-2, and interferon-β promoter stimulator (IRF-9) from binding to the promoter regions of interferon-stimulated response elements (ISRE), thereby inhibiting transcription of interferon-response genes. Even when interferon-response genes are activated, NS5A and E2 both can disrupt protein kinase R (PKR) function to suppress translation, thereby allowing viral replication to continue.93 In addition, NS5A inhibits 2′-5′-oligoadenylate synthetase (OAS). 2′-5′-OAS is expressed in response to HCV infection and leads to HCV RNA degradation.93 Taken together, HCV is able to disorient the innate immune response at several levels, and these strategies appear to be pivotal in establishing the chronicity of infection.
The ability of HCV to impair the innate immune response prevents development of a vigorous adaptive immune response to the infection. NK cells do not adequately activate dendritic cells, and as a result, the priming of CD8+ and CD4+ T cells in HCV-infected patients is inadequate.107–109 Dendritic cells from HCV-infected patients are more likely to produce IL-10, a cytokine that inhibits antigen-specific T-cell responses. In addition, CD4+ T cells primed from dendritic cells of HCV-infected patients are more likely to produce IL-10. Even if an adequate T-cell response is created, HCV-infected patients have a large number of regulatory T cells in their portal tracts110; intrahepatic immune regulation by these cells has not been demonstrated but is presumed.
HCV-specific T cells are enriched at the site of viral replication, with an increased number in the liver when compared with the peripheral blood.95,111 CD8+ lymphocytes predominate, suggesting that cytotoxic T lymphocytes are the main perpetrators of hepatocellular injury. The T-cell immune response in the liver may result in direct lysis of infected cells and inhibition of viral replication by secreted antiviral cytokines.95,111
Whereas the cellular immune response plays a pivotal role in the pathogenesis of HCV infection, the importance of the humoral immune response is less clear. Antibodies to viral proteins are produced in low levels and do not appear to correlate with the stage of infection or immune reactivity. Furthermore, administration of high-titer HCV-enriched or HCV-specific immunoglobulin has little effect on viral levels or persistence in humans.112
CLINICAL FEATURES
ACUTE AND CHRONIC HEPATITIS C
HCV accounted for an estimated 20% of cases of acute hepatitis in 2006.74 Acute hepatitis C is rarely seen in clinical practice because nearly all cases are asymptomatic.87,113 Jaundice probably occurs in about 10% of patients with acute HCV infection, whereas 20% to 30% of patients present with nonspecific symptoms such as fatigue, nausea, and vomiting. HCV RNA is detectable within 2 to 3 weeks of exposure, and anti-HCV seroconversion occurs between day 15 and month 3. Serum aminotransferase levels peak at about the first month after exposure (Fig. 79-3), exceed 1000 IU/L in 20% of cases, and generally follow a fluctuating pattern for the first few months. In patients in whom jaundice develops, peak serum bilirubin levels usually are less than 12 mg/dL, and jaundice typically resolves within one month. Severe impairment of liver function and liver failure are rare. The presentation may be more apparent and the clinical course more severe when acute HCV infection occurs in patients who drink large amounts of alcohol or have coinfection with HBV or HIV.
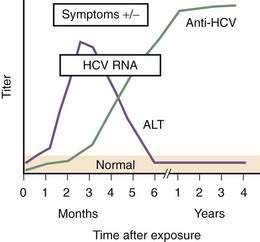
(Modified from the Centers for Disease Control and Prevention, www.cdc.gov/hepatitis/Resources/Professionals/Training/Serology/training.htm#one.)
The rate of viral persistence after acute infection varies, ranging from 45% to more than 90%. Age and gender clearly influence the risk of chronicity, with younger and female patients having the lowest rates of chronicity. Other factors that may play a role include the source of infection and size of inoculum (chronicity may be less common in injection drug users than in those who acquire HCV infection by blood transfusion), immune status of the host (chronicity rates are higher in persons with immunodeficiency states such as agammaglobulinemia and HIV infection), and the patient’s race (rates of viral persistence are higher in African Americans than in whites and Hispanic Americans in the United States). Finally, the rate of spontaneous clearance is higher in symptomatic patients in whom jaundice develops during acute infection than in those who remain asymptomatic.87,113,114 In patients with community-acquired hepatitis C in whom the infection resolves spontaneously, loss of HCV RNA from serum usually occurs within three to four months of the onset of clinical disease.114
Serum alanine aminotransferase (ALT) levels are usually elevated in patients with chronic HCV infection. Because levels commonly fluctuate, however, as many as one half of patients may have a normal ALT level at any given time.1 The ALT level may remain normal for prolonged periods of time in about 20% of cases,115 although transient elevations occur even in these cases.116 Persistently normal ALT levels are more common in women, and such cases typically are associated with lower serum HCV RNA levels and less inflammation and fibrosis on liver biopsy specimens.115
Most patients with chronic hepatitis C are asymptomatic before the onset of advanced hepatic fibrosis. Patients who have been diagnosed with chronic infection, however, often complain of fatigue or depression, and they consistently score lower than HCV-negative persons in all aspects of health-related quality of life (HRQL).117,118 Whether the decrease in HRQL is related to viral factors, social factors (e.g., intravenous drug use), social stigmatization, or worry related to the diagnosis itself is unclear. Nonetheless, HRQL scores improve if the patient achieves a sustained response to antiviral therapy (see later). Less common symptoms may include arthralgias, paresthesias, myalgias, sicca syndrome, nausea, anorexia, and difficulty with concentration. The severity of these symptoms may be, but is not necessarily, related to the severity of the underlying liver disease.
EXTRAHEPATIC MANIFESTATIONS
Patients with HCV infection may present with extrahepatic conditions, or these manifestations may occur in patients known to have chronic hepatitis C. Classification of the extrahepatic manifestations of HCV is shown in Table 79-1 and is based on the strength of available data to prove a correlation.119 Types 2 and 3 cryoglobulinemia, characterized by polyclonal immunoglobulin G (IgG) plus monoclonal IgM and polyclonal IgG plus polyclonal IgM, respectively, can both be caused by HCV infection. Among HCV-infected patients 19% to 50% have cryoglobulins in serum, but clinical manifestations of cryoglobulinemia are reported in only 5% to 10% of these patients and are more common in patients with cirrhosis. Symptoms and signs include fatigue, arthralgias, arthritis, purpura, Raynaud’s phenomenon, vasculitis, peripheral neuropathy, and nephropathy. The diagnosis is clear when a rheumatoid factor is detected, cryoglobulins are present, and complement levels are low in serum; however, the reliability of cryoglobulin measurements is dependent on proper handling and processing of the sample.
Table 79-1 Extrahepatic Manifestations of Hepatitis C Virus Infection
| Proven Associations |
| Autoimmune thyroiditis |
| B-cell non-Hodgkin’s lymphoma |
| Diabetes mellitus |
| Lichen planus |
| Mixed cryoglobulinemia |
| Monoclonal gammopathies |
| Porphyria cutanea tarda |
| Possible Associations |
| Chronic polyarthritis |
| Idiopathic pulmonary fibrosis |
| Non-cryoglobulinemic nephropathies |
| Sicca syndrome |
| Thyroid cancer |
| Renal cell carcinoma |
| Vitiligo |
Glomerular disease generally manifests as cryoglobulinemic nephropathy, membranoproliferative glomerulonephritis (MPGN), and membranous nephropathy.119,120 Cryoglobulinemic nephropathy manifests as hematuria, proteinuria, edema, and renal insufficiency of varying degrees, and on renal biopsy specimens it has features of MPGN. At diagnosis, 20% of patients with type 2 cryoglobulinemia have renal involvement, and renal involvement develops in another 35% to 60% over time. In about 15% of patients, cryoglobulinemic nephropathy progresses to end-stage kidney disease requiring dialysis.
Therapy should be considered in patients with symptomatic cryoglobulinemia. Cryoglobulinemia resolves in patients who achieve an SVR with pegylated interferon and ribavirin therapy (see later).119 Unfortunately, patients with significant renal involvement are at a disadvantage with respect to antiviral therapy because administration of ribavirin is generally contraindicated if the creatinine clearance is less than 50 mL/min. In these patients, treatment with rituximab should be considered.121,122 A durable clinical response to four doses of rituximab has been reported, although no prospective clinical trials have been completed to date. Prednisone, cyclophosphamide, other chemotherapeutic agents as well as plasmapheresis have been used with variable success; however, these approaches do not treat the underlying HCV infection.119 If cryoglobulinemia and renal disease improve with such treatment, then subsequent treatment of the HCV infection with pegylated interferon-α and ribavirin should be reconsidered.
HCV infection is associated with the development of B-cell non-Hodgkin’s lymphoma and monoclonal gammopathy of uncertain significance.119,123,124 The relative risk of lymphoma is small (1.28) in the United States.123 The most prevalent forms of lymphoma found in patients infected with HCV are follicular lymphoma, chronic lymphocytic lymphoma, lymphoplasmacytic lymphoma, and marginal zone lymphoma. In addition, marginal splenic lymphoma has been reported to regress after therapy for HCV infection alone. An estimated 8% to 10% of patients with type 2 cryoglobulinemia evolve into lymphoma over time. Despite the known association of HCV infection with lymphoma, HCV RNA does not integrate into the host genome and cannot be considered a typical oncogenic virus. Rather, HCV shows lymphotropism and may facilitate the development and selection of abnormal B-cell clones by chronic stimulation of the immune system. In addition, genetic rearrangements in B cells, specifically the Bcl2/JH rearrangement and the t(14;18) translocation, have been found in HCV-infected patients.125,126 Patients with B-cell genetic alterations are less likely to respond to antiviral therapy.127
Other extrahepatic manifestations of HCV infection include porphyria cutanea tarda, lichen planus, and sicca syndrome.119 In addition, insulin resistance and diabetes mellitus are associated with HCV infection. The SVR to antiviral therapy for HCV infection is reduced in insulin-resistant patients; however, if HCV can be eradicated, insulin resistance often improves, an observation that further supports the relationship between HCV infection and insulin resistance.128 Although associations between HCV infection and both thyroid cancer and idiopathic pulmonary fibrosis have been described, data about the effect of HCV eradication on disease progression are lacking (see Table 79-1). A myriad of other conditions have been observed in association with HCV infection, but a true link has not been firmly established for these disorders.119
Although not associated with disease, seropositivity for autoantibodies is found in many HCV-infected persons (e.g., antinuclear antibodies with a titer greater than 1 : 40 in 9%, smooth muscle antibodies with a titer greater than 1 : 40 in 20%, anti-liver-kidney microsomal antibodies in 6%).129 In addition, anti-thyroid peroxidase is found in serum in 5% to 12% of HCV-infected patients, although associated thyroid disease is found in only 2% to 5% of patients.119 Therefore, the diagnosis of an autoimmune condition in a patient with HCV infection can never be based on serology alone.
DIAGNOSIS
Several immunologic and molecular assays are used to detect and monitor HCV infection.130 The presence of anti-HCV in high titer in serum (generally an enzyme immunoassay [EIA] ratio greater than 9) indicates exposure to the virus but does not differentiate among acute, chronic, and resolved infection. Anti-HCV usually persists for many years, and perhaps for life, in patients after spontaneous resolution of infection or an SVR following antiviral therapy. Serologic assays are used initially for diagnosis, whereas virologic assays are required for confirming infection, monitoring response to treatment, and evaluating immunocompromised patients.130,131
INDIRECT ASSAYS
EIAs detect antibodies against different HCV antigens. The time course of the development of symptoms, detection of anti-HCV, and appearance of HCV RNA after acute infection is shown in Figure 79-3. Three generations of EIAs have been developed. The latest, third-generation, EIAs detect antibodies against HCV core, NS3, NS4, and NS5 antigens as early as 7 to 8 weeks after infection, with sensitivity and specificity rates of 99%.131 Despite ongoing viral replication, serologic test results can be negative in patients who are on hemodialysis or are immunocompromised.80 Because the performance characteristics of third-generation EIAs are so good, confirmation with a recombinant immunoblot assay (RIBA) is no longer required. Instead, patients who are anti-HCV positive should undergo HCV RNA testing to determine if they have active viremia or have cleared the infection.
DIRECT ASSAYS
Two general types of direct assays exist: qualitative and quantitative HCV RNA tests.130 Qualitative HCV RNA nucleic acid tests (NAT) only report whether HCV RNA is found in serum or not and do not quantitate the amount of HCV RNA. These tests should be used only for screening purposes now (e.g., screening of blood donated to a blood bank) and should not be used in clinical practice. As a result of NAT technology, transfusion-related HCV infection has decreased to less than one case per two million units of blood transfused.76,77
Unlike NAT testing, quantitative HCV RNA tests are essential for monitoring the response to antiviral therapy (see later). Currently “real-time” tests, such as Taqman, are performed using polymerase chain reaction (PCR) methodology, with a lower limit of detection of 10 to 15 international units (IU)/mL.132 These assays have a linear dynamic range of 1 to 7 log10 IU/mL and are the preferred testing method in practice. Transcription-mediated amplification (TMA) is also extremely sensitive, but available assays are not quantitative in the lower dynamic range of the test. The advantages of these very sensitive tests include positivity within one to three weeks after acute infection and detection of low-level residual infection during antiviral therapy.
HCV Genotyping
Identifying the genotype of HCV can be accomplished by several methods. The most accurate method is PCR and direct sequencing of the NS5B or E1 region; however, this approach is not practical in clinical practice. HCV genotyping can be done by evaluating type-specific antibodies and has a 90% concordance in immunocompetent patients when results are compared with sequence analysis of the HCV genome.131 Testing can also be accomplished with reverse hybridization to genotype-specific probes, restriction fragment length polymorphism analysis, or PCR amplification of the 5′ noncoding region of the HCV genome. These tests have 92% to 96% concordance with the correct genotype; genotype 1 is identified with the highest accuracy. Because of mutations in the regions studied, regardless of the technique used, errors in subtype identification occur in 10% to 25% of cases. A line-probe assay (Inno-LIPA) using genotype-specific probes for reverse transcription of the 5′ portion of the HCV genome is currently the most popular commercial assay for HCV genotyping.133
SELECTION OF SEROLOGIC AND VIROLOGIC TESTS
Screening in Blood Banks
The risk of acquiring HCV infection from blood products has declined dramatically since blood donors have been screened routinely. Many blood banks have now switched from third-generation EIA testing to NAT testing, and this change has decreased the risk of transfusion-acquired HCV infection to one infection per two million units of blood transfused.76,77
Diagnosis Following Known Exposure
Following an occupational or recreational exposure or in the context of mother-to-infant transmission, the diagnosis of HCV infection is now based on HCV RNA testing by the most sensitive molecular method, usually a real-time PCR assay. If transmission has occurred, HCV RNA is detectable in serum 1 to 3 weeks following exposure, whereas anti-HCV may not be seen for 7 to 8 weeks following exposure. Treatment should be considered in adults with acute exposure because the risk of chronicity is high without treatment and clearance of HCV RNA with antiviral therapy is likely (see later). Therefore, early diagnosis of HCV infection is important in these settings.130 In patients with a continued risk of infection, as through injection drug use, periodic testing for HCV RNA should be done. Because spontaneous clearance of HCV RNA from serum is more likely in infants than in adults, an infant should not be tested until 18 months after birth if the mother is HCV RNA positive.
LIVER BIOPSY AND NONINVASIVE ASSESSMENT OF FIBROSIS
The risk of progressive hepatic injury from HCV infection varies considerably, with some patients showing little or no progression after decades of infection and others progressing rapidly to cirrhosis.134 Therefore, an assessment of the degree of liver injury is usually advisable. This assessment is usually done by percutaneous liver biopsy (Table 79-2), but indirect and noninvasive methods to assess liver injury and fibrosis are under study and becoming commercially available.
Table 79-2 Reasons to Perform a Liver Biopsy in a Patient with Hepatitis C
| Assessment of the need for surveillance for hepatocellular carcinoma |
| Evaluation for concomitant liver diseases |
| Guidance for decisions regarding treatment of hepatitis C |
| Staging of fibrosis |
Examination of liver biopsy specimens is used to quantify hepatic injury into discrete grades of inflammation and stages of fibrosis.134,135 Several scoring systems have been used and differ in range of possible scores and definitions (Fig. 79-4). The first system used was the Histology Activity Index (HAI) described by Knodell and colleagues. The components of this system include periportal inflammation and necrosis (graded as 0 to 10), lobular inflammation and necrosis (0 to 4), portal inflammation (0 to 4), and fibrosis (0 to 4). This scoring system combines inflammation and fibrosis into one score. Scheuer created a simplified scoring system that separates grade from stage: Portal inflammation and interface hepatitis (0 to 4), lobular activity (0 to 4), and fibrosis stage (0 to 4). The Ishak system is a modification of Knodell’s system but separates histologic grade from stage. Ishak’s fibrosis scores range from 0 to 6 (1 or 2, portal fibrotic expansion; 3 or 4, bridging fibrosis; 5 or 6, cirrhosis) (Fig. 79-5). The higher number of gradations of fibrosis has made the Ishak system popular for scoring progression of fibrosis in clinical trials. Currently, the METAVIR scoring system is the most popular; it is simpler than all the aforementioned systems. Inflammation is graded from 0 to 4 (none, mild, moderate, and severe), and fibrosis is staged from 0 to 4 (1, portal fibrotic expansion; 2, portal fibrosis with septa formation; 3, bridging fibrosis; 4, cirrhosis).
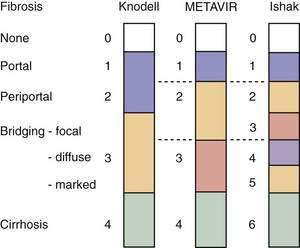
Figure 79-4. Comparison of the Knodell, METAVIR, and Ishak hepatic fibrosis staging systems. The METAVIR staging system is similar to the Scheuer system. Portal, periportal, bridging, and cirrhosis describe the degree (stage) of fibrosis (see also Fig. 79-5).
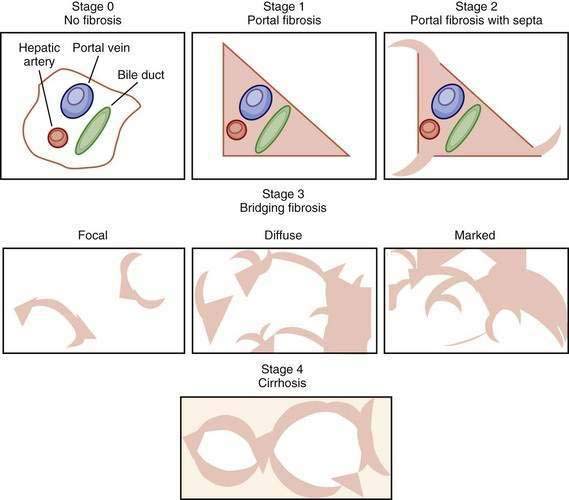
Figure 79-5. Visual depictions of fibrosis staging (METAVIR system) in patients with chronic hepatitis C. Stage 0 represents the absence of abnormal fibrosis, stage 1 shows portal fibrosis, and stage 2 shows portal fibrosis with septa. Bridging fibrosis (stage 3) is a continuous process ranging from focal periportal fibrosis with occasional bridging between portal structures to marked bridging without nodule formations, sometimes called early cirrhosis. Depending on the staging system (see Figure 79-4), bridging fibrosis may be stage 3, 4, or 5, emphasizing the importance of the reporting of the staging system used in pathology reports. A diagnosis of cirrhosis requires the presence of nodule formation.
Although examination of liver biopsy specimens is still considered the standard for establishing the grade of inflammation and stage of fibrosis, limitations of liver biopsy include (1) associated morbidity (pain occurs in as many as 30% in some series and hemorrhage or bile leak occurs in 0.3% of patients) and mortality (0.03%); (2) cost; (3) poor patient acceptance; (4) intraobserver and interobserver variability in the interpretation of findings (with current scoring systems, intraobserver and interobserver concordance for staging fibrosis among hepatopathologists is approximately 90% and 85%, respectively); (5) inaccuracy in interpretation of findings, particularly for the diagnosis of cirrhosis (with a false-negative rate of 15%); and (6) sampling error (a 33% difference in one stage of fibrosis and 2.4% difference in two stages of fibrosis is seen in simultaneously obtained biopsy specimens from the right and left hepatic lobes).136,137 Interobserver and intraobserver variability is increased when inexperienced pathologists use a complicated scoring system to evaluate liver tissue. Sampling error is especially common when small biopsy specimens are obtained. A biopsy should be done with at least a 16-gauge needle, be 15- to 20-mm or more in length, and contain at least 6 portal triads, although 11 or greater is considered optimal.138,139
Because of the limitations of liver biopsy, several noninvasive tests to estimate fibrosis have been developed (Table 79-3). Although many such tests have been evaluated, few have entered clinical practice. FibroSure (also known as FibroTest) is a noninvasive measure of fibrosis that creates a composite score, adjusted for gender and age, derived from the serum levels of α2-macroglobulin, haptoglobin, apolipoprotein A-1, gamma glutamyl transpeptidase, and total bilirubin.137 The test accurately categorizes patients with stage 0 and 1 fibrosis and those with cirrhosis; however, it is less useful in patients with intermediate scores. The AST-to-platelet ratio index (APRI) is used primarily to diagnose or exclude cirrhosis.140 In an initial evaluation, 81% of cirrhotic patients were accurately categorized with a score less than or equal to 0.5; however, the index does not discriminate among lower levels of fibrosis, and its reproducibility has been called into question.138,141 A new technology, transient elastography (Fibroscan), uses ultrasound to assess the elasticity of the liver, which correlates with the amount of hepatic fibrosis. In a meta-analysis, the area under the receiver operating curve (an estimate of accuracy) of Fibroscan for predicting cirrhosis was 0.94.142 When FibroSure and Fibroscan were evaluated, the areas under the curve (an estimate of accuracy) for predicting cirrhosis were 0.9 for FibroSure and 0.95 for Fibroscan.143
Table 79-3 Accuracy of Noninvasive Tests for Predicting Hepatic Fibrosis in Patients with Hepatitis C
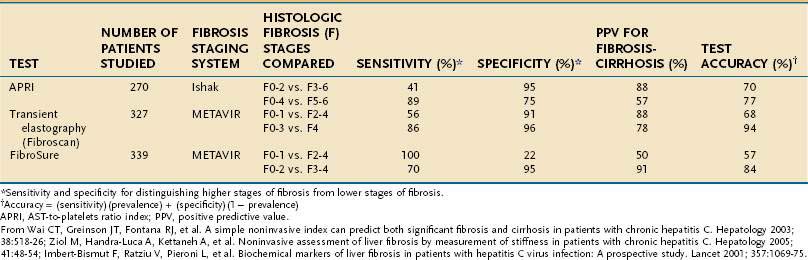
The following recommendations can be made regarding the role of liver biopsy in the management of patients with chronic hepatitis C (see Table 79-2). Regardless of the degree of serum aminotransferase elevations, an examination of a baseline liver biopsy specimen is recommended, but not mandatory, for initial assessment of patients with chronic hepatitis C, genotype 1 or 4, when examination of liver histology will influence treatment plans, and in patients infected with HCV genotype 2 or 3 who wish to defer treatment.144 Liver biopsy is not required when cirrhosis is already suggested by clinical findings (e.g., ascites, splenomegaly, spider angioma, low platelet count, or prolonged prothrombin time) or imaging (e.g., nodularity of the liver, evidence of portal hypertension). It is also not indicated following successful antiviral therapy, although histology generally improves significantly over time following eradication of HCV (see later).
In persons who do not achieve an SVR through treatment, a repeat assessment of liver fibrosis should be considered every four to five years.144–146 In patients who initially had stage 0 or 1 fibrosis, a noninvasive measurement may be adequate for following progression of fibrosis. In those with stage 2 or 3 fibrosis, however, liver biopsy is typically recommended because development of cirrhosis, if demonstrated, signifies the need for surveillance for hepatocellular carcinoma and varices. In the future, noninvasive testing for fibrosis will likely play a larger role in the management of patients with HCV infection.
NATURAL HISTORY
Progression of chronic hepatitis C is largely silent, as graphically depicted in Figure 79-6. The reported rate of progression of fibrosis depends on the cohort studied and the strategy used to collect data. Between 2% and 24% of HCV-infected patients progress to cirrhosis after 20 years of infection.147 Iatrogenic outbreaks of HCV infection have provided insight into the natural history of HCV infection. In 1977, young Irish women were infected with HCV genotype 1b when a contaminated lot of anti-D immunoglobulin was administered to them. After more than 17 years of follow-up, cirrhosis developed in only 2%, and complications of liver disease developed in none.148 These women continue to be followed, and 184 untreated patients with HCV infection had paired biopsy data over the subsequent decade, with cirrhosis developing in only an additional 2.1%.149 A similar study from Germany involved 1018 women infected through contaminated batches of anti-D immunoglobulin.150 Twenty years after exposure, 85% of patients tested positive for anti-HCV, and 55% tested positive for HCV RNA. An additional 3% had achieved an SVR with interferon therapy. Of the 220 who underwent liver biopsy, 3% had bridging fibrosis, and none had cirrhosis. These two studies demonstrate that the prognosis is excellent in women infected at a young age with a low viral inoculum. Longer-term studies are still needed to ensure that fibrosis progression does not accelerate later in life.
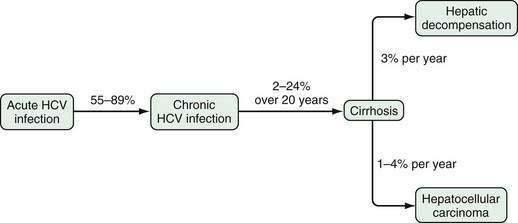
Most estimates of the rate of progression of fibrosis in HCV-infected patients are substantially higher than those reported in the aforementioned studies. The cohort followed appears to influence the observed rate of progression dramatically. The 20-year expected rate of progression to cirrhosis is 25% in patients referred to liver centers, 23.8% in patients followed after transfusion-associated hepatitis, 3.8% in healthy blood donors, and 4.7% in community-based studies.147 The factor that most distinguishes these different groups is the age at the time of infection. Fibrosis appears to progress more rapidly in older than in younger patients, although other factors such as alcohol intake may influence the rate of progression as well.
Among patients with HCV-induced cirrhosis, manifestations of liver failure (e.g., ascites, gastrointestinal bleeding, encephalopathy, hepatorenal syndrome, or synthetic dysfunction) develop in 3% per year, and HCC develops in 1% to 4% per year.151–154 The Hepatitis C Long-Term Treatment against Cirrhosis (HALT-C) trial, a prospective study of patients with bridging fibrosis or cirrhosis (see later), showed that over a period of three and one half years, the risk of death was 4.6% to 6.6%, the risk of decompensation was 13.2% to 14.3%, and the risk of HCC was 2.8% to 3.2%.155 The five-year survival rate is only about 50% after hepatic failure develops.151,152 An estimated 8000 to 10,000 deaths from HCV-related liver complications occur annually in the United States, although this number, which is based on death certificates, is likely an underestimate.156 The age-adjusted mortality rate from HCV infection has decreased slightly in the 2000s.5,157 Cirrhosis and HCC resulting from HCV infection are the leading indications for liver transplantation in the United States and Europe and account for approximately 40% of transplantations since 2000 in the United States. In patients who achieve an SVR with therapy and who have compensated cirrhosis from HCV, the risk of decompensation and HCC is reduced dramatically.155,158 Nevertheless, HCC can still occur, and surveillance for HCC is still needed in these patients.
FACTORS ASSOCIATED WITH PROGRESSION OF CHRONIC HEPATITIS C
Numerous host and environmental factors contribute to the presence or absence of histologic progression of HCV infection (Table 79-4). Identification of these factors is important because modifiable factors can be altered and high-risk patients can be treated promptly.
Table 79-4 Factors Associated with Progression of Hepatic Fibrosis in Patients with Chronic HCV Infection
| ESTABLISHED | POSSIBLE | NOT ASSOCIATED |
|---|---|---|
| Age >40 years Alcohol consumption | Increased hepatic iron concentration | Viral genotype Viral load |
| Hepatitis B virus coinfection | Male gender | |
| HIV coinfection Immunosuppressed state | Serum ALT level | |
| Insulin resistance | ||
| Marijuana use | ||
| Obesity | ||
| Schistosomiasis | ||
| Severe hepatic necroinflammation | ||
| Smoking | ||
| White race |
ALT, alanine aminotransferase; HIV, human immunodeficiency virus.
Age at infection is a strong predictor of outcome, as discussed earlier. Natural history studies also suggest that progression of fibrosis is not linear over time and that the progression is slower at younger ages and increases over time, with the greatest acceleration after the age of 50 years.159 This phenomenon likely explains some of the discrepancies seen in natural history studies. Accelerated fibrosis seen in older persons is independent of the age at acquisition. The mechanisms behind the accelerated rate of progression of fibrosis with aging are poorly understood.
Male gender is associated with more rapid progression to cirrhosis and HCC.157,159 The reason for this association is not clear, and hormonal effects on fibrogenesis have been suggested. Estrogen inhibits proliferation and activation of hepatic stellate cells in vitro.160 In addition, fibrosis appears to accelerate in postmenopausal women, and this acceleration may be attenuated with use of estrogen.157,161,162
Race also has been implicated in disease progression. African Americans have a higher prevalence of HCV infection than whites,1 but their risk of progression of the disease to cirrhosis is lower.163,164 Because African Americans have a higher prevalence of disease, decreased response to therapy, and possibly decreased access to health care and liver transplantation, they have higher rates of HCV-related inpatient mortality.165 Unlike African Americans, HCV-infected Hispanics have a higher rate of progression of fibrosis than do whites.166 Whether the higher risk is explained by a higher prevalence of alcohol abuse, fatty liver disease, or other factors is unclear.
Alcohol use of greater than 20 g/day clearly increases the risk of progression of fibrosis and development of HCC in patients who are chronically infected with HCV.167–171 Whether occasional or lesser amounts of alcohol also contribute to disease progression is less clear; however, in the absence of definitive evidence, most hepatologists recommend that HCV-infected patients avoid alcohol altogether.
In addition to alcohol, smoking cigarettes or cannabis leads to an increased rate of progression of fibrosis in HCV-infected patients.172,173 Daily users of cannabis also have a higher risk of liver steatosis. In experimental models, cannabinoid receptor antagonists have been shown to decrease hepatic steatosis.174
Several metabolic abnormalities and comorbid conditions, including insulin resistance or type 2 diabetes mellitus, obesity, and liver steatosis have been associated with accelerated progression of fibrosis.157 The frequencies of insulin resistance and of type 2 diabetes mellitus are markedly increased in patients infected with HCV genotypes 1 or 4.175–178 HCV infection results in down-regulation of IRS-1 and 2 through up-regulation of SOCS-3, an effect that is independent of other factors such as obesity that can also cause hepatic steatosis.179,180 Either insulin resistance or hepatic steatosis alone increases the risk of progression of fibrosis.179,180 Weight loss is associated with reductions in hepatic steatosis and the rate of fibrosis.181 Steatosis is also common in patients infected with HCV genotype 3, but this effect is virally induced and not associated with progression of fibrosis.182,183 Patients with insulin resistance have a lower SVR to HCV therapy.184
Mild-to-moderately increased hepatic iron stores are associated with more advanced fibrosis. A consistent relationship between C282Y or H63D heterozygosity and increased progression of fibrosis has not been established, however (see Chapter 74).185,186 Reduction of hepatic iron concentrations does not reduce the risk of progression of fibrosis or improve the response to antiviral treatment.187
Polymorphisms in genes implicated in the immune response and fibrogenesis have been studied extensively and may account for some of the variability in progression of fibrosis in patients who are otherwise similar, as reviewed elsewhere.157 The candidate genes are numerous, and most studies are limited by small sample sizes and lack of reproducibility; larger scale, genome-wide investigations are in progress.188
PATIENTS WITH PERSISTENTLY NORMAL SERUM AMINOTRANSFERASE LEVELS
Up to one half of anti-HCV-positive persons have a normal serum ALT level at any given time,1 and the ALT level will remain normal for at least six months in 8% to 20% of them.145,189 Serum HCV RNA levels are lower in these persons than in persons with elevated ALT levels, and they tend to have lesser degrees of hepatic inflammation and fibrosis. In one series, 86.5% of HCV-infected persons with a normal serum ALT level had METAVIR stage 0 or 1 fibrosis, 12% had stage 2 fibrosis, and just 1.5% had cirrhosis.189 In a study of 159 patients with persistently normal serum ALT levels, the mean Ishak fibrosis score was 0.87.116 Therefore, these patients tend to have less fibrosis, with the implication that fibrosis progresses more slowly in these patients than in those with an elevated ALT level.116,157,189 Progression to cirrhosis, although far less common in those with a normal ALT level, remains possible, and antiviral therapy may still be indicated.
IMMUNOCOMPROMISED PATIENTS
Studies of HCV-infected patients with humoral or cellular immune impairment have shown rates of progression to cirrhosis that are significantly higher than those observed in immunocompetent patients.98,190,191 Given the apparent predominance of cellular immunity in the pathogenesis of chronic hepatitis C (see earlier), it is fascinating that HCV-infected patients with common variable immune deficiency (CVID), which is characterized by a humoral immune defect, have an accelerated rate of progression of fibrosis. In patients with CVID who received pooled plasma products before the initiation of HCV screening, the frequency of HCV infection is high, and the rate of progression of liver disease is accelerated in these patients.190 During a follow-up period of approximately 10 years, 40% of 71 such patients progressed to decompensated cirrhosis.190
Cellular immune impairment is far more common than humoral immune impairment. The most common cause is iatrogenic as a result of exogenous immunosuppression following transplantation. Unfortunately, many patients with cellular immune impairment already have HCV infection before becoming immunosuppressed. Chronic HCV infection is the most common indication for liver transplantation and is present, either alone or in association with alcoholic liver disease, in 40% of patients who undergo liver transplantation in the United States. Recurrence of HCV infection is almost universal in patients who undergo transplantation and who are positive for HCV RNA in serum. Typically, levels of viremia increase at least one log following transplantation. The clinical course in patients with recurrent HCV infection after liver transplantation is variable; overall, however, the disease progresses more quickly to cirrhosis than in nontransplanted persons. Rapid recurrence of HCV infection and graft loss resulting from fibrosing cholestatic hepatitis occur in 1% to 10% of patients, and most of these persons die within one year.98 Of those without fibrosing cholestatic hepatitis, cirrhosis develops in 20% to 40% in five years.191 High serum HCV RNA levels before or early after liver transplantation, older age of the organ donor, severe and early recurrence of HCV infection after transplantation, and the use of high doses of prednisone or lymphocyte-depleting drugs to treat acute graft rejection are factors that most consistently are associated with a poor outcome.191 In patients in whom the disease progresses to cirrhosis after liver transplantation, decompensation occurs in 40% within one year, of whom 50% die in the following year.191,192 As a result, long-term survival of patients with HCV infection who undergo transplantation is inferior to long-term survival of patients who undergo transplantation for other indications.193 Fibrosing cholestatic hepatitis is generally accepted as a contraindication to retransplantation because of the rapid destruction of the new graft. For the same reason, most liver transplant centers are reluctant to consider retransplantation in patients with severe recurrent HCV infection within the first year. Because progression to cirrhosis is quicker after retransplantation, no consensus has been established as to whether retransplantation should be offered to a patient with recurrent HCV infection, even to a patient with slowly progressive disease (see also Chapter 95).194
Transmission of HCV to other solid-organ transplant recipients has been described. Before testing for HCV infection became widely available, such transmission was thought to occur infrequently in patients who underwent transplantation. In one large study of cardiac transplant recipients,195 survival of the 261 recipients of hearts from HCV-positive donors was significantly lower at 1, 5, and 10 years (83%, 53%, and 25%), respectively, than survival in recipients of hearts from HCV-negative donors (92%, 77%, and 53%, respectively). The additional risk of death was not affected by the recipients’ pretransplant HCV status or age. Excess mortality was attributable to liver disease and coronary vasculopathy.
Patients on hemodialysis and renal transplant recipients have a higher prevalence of HCV infection (up to 11% to 14% in some series in the United States) than that in the general population (1.6% in the United States).1,79,196 The higher prevalence is attributed to a higher frequency of risk factors, particularly prior blood transfusion, and possibly nosocomial spread within dialysis units. Survival is lower in HCV-infected patients on hemodialysis than in uninfected hemodialysis patients, with a relative risk of death of 1.57.197,198 Most mortality is related to HCC and complications of cirrhosis. Antiviral treatment is difficult in these patients, but the chance of response to treatment occurs in about 30% in genotype 1–infected patients, a surprisingly high rate considering that ribavirin is typically avoided in this group (see later).199 The impact of HCV infection on renal transplant recipients is controversial. HCV is the leading cause of liver disease in the postrenal transplant setting, and the rate of histologic progression is more rapid than in immunocompetent persons.200 Although the effect of HCV infection on short-term survival appears to be minimal, early studies found that the rate of late survival is reduced because of infectious complications rather than complications of liver disease mellitus. Moreover, HCV increases the risk of postrenal transplant glomerulopathy and diabetes mellitus. Long-term patient and graft survival rates are lower in renal transplant recipients for reasons that require further study. Despite the risks, renal transplantation still confers a survival benefit over hemodialysis in both HCV-positive and HCV-negative recipients.
Heart transplant recipients with preexisting chronic HCV infection appear to do well, although the experience is limited.201 On the other hand, as noted earlier, uninfected heart transplant recipients who receive a graft from an HCV-infected donor have poorer outcomes, with a reduction in the one-year survival rate from 92% to 83% and a reduction in the five-year survival rate from 77% to 55%.195,201 Fibrosing cholestatic hepatitis also has been reported.99 Few data are available on outcomes in lung and small bowel transplant recipients who are acutely or chronically infected with HCV.196
Interferon is generally contraindicated in recipients of solid organ transplants other than liver transplants because of the risk of graft rejection and loss.196 Therefore, treatment of HCV infection should be considered before solid organ transplantation is undertaken in transplant candidates with chronic hepatitis C (see later).
Liver disease is common in the first several weeks after allogeneic bone marrow transplantation (BMT), but most cases are the result of sinusoidal obstruction syndrome or graft-versus-host disease (see Chapter 34). Chronic hepatitis C is the most common cause of chronic liver disease after BMT. The cumulative frequency of cirrhosis has been estimated to be 11% after 15 years and 24% after 20 years.202 HCV is a major cause of cirrhosis and of liver-related mortality in this population. Unlike the case of solid-organ transplant recipients, however, antiviral treatment appears to be safe in BMT recipients.203 Graft-versus-host disease is a contraindication to interferon therapy, and cytopenias may limit the doses of interferon-α or ribavirin that can be used safely.
HCV infection is common in HIV-infected persons because the two infections share similar routes of transmission. Approximately 25% of HIV-infected persons are coinfected with HCV, and up to 8% of HCV-infected patients are coinfected with HIV.204 Differences in the efficiency of transmission of HCV and HIV by parenteral or sexual routes explain the wide variation in HCV seropositivity among HIV-infected persons. Higher rates of coinfection are seen among injection drug users and recipients of blood transfusions than among sexual contacts of HIV-infected persons. HIV infection decreases the spontaneous rate of HCV clearance during acute HCV infection and leads to a correspondingly higher rate of chronic HCV infection. Before the introduction of HAART, HCV had little impact on morbidity and mortality in HIV-infected persons. The reduction in the rate of mortality from the acquired immunodeficiency syndrome (AIDS) since the introduction of HAART, however, has allowed the sequelae of chronic HCV infection to become apparent. Indeed, complications of liver disease, mostly related to hepatitis C, are now almost as common as AIDS-related complications, and a major cause of mortality, in patients with HIV infection.204,205 Progression of hepatic fibrosis is accelerated in HIV-HCV-coinfected patients when compared with HCV-monoinfected patients. In a large study,206 10.6% of HIV-HCV coinfected patients with hemophilia progressed to decompensated cirrhosis over a median of 12.1 years as compared with 1.6% of HCV-monoinfected patients with hemophilia.206 Potential mechanisms of increased progression of fibrosis have been investigated. HIV increases the production of TGF-β1 in the liver and leads to increased HCV replication.207 HIV-positive patients with a low CD4+ count have greater gastrointestinal bacterial translocation, leading to increased levels of lipopolysaccharide in the portal circulation and greater progression of fibrosis.208 HIV proteins may also cause activation of collagen production by hepatic stellate cells.209 In addition, steatosis and steatohepatitis are common in HIV-HCV-coinfected patients, and both are associated with an increased risk of advanced fibrosis on liver biopsy specimens.210
PREVENTION
GENERAL MEASURES AND TREATMENT OF ACUTE INFECTION
Because neither an effective vaccine nor post-exposure prophylaxis against HCV infection is available, major efforts should be placed on preventing HCV infection. Needle exchange and methadone programs have decreased the rate of HCV transmission among injection drug users.211 In addition, universal precautions, disposable equipment, and rigorous sterilization of reusable medical and surgical equipment have reduced nosocomial HCV infections. Nonetheless, HCV infection is common among patients, and exposure to these patients is common in hospital workers. In one study, the frequency of hepatitis C was 15-fold higher in hospitalized patients than in the general population.212 Needlesticks are common in the health care setting and are under-reported. Almost all surgical house staff will sustain at least one needlestick, and in nearly one half the needlestick will be from a high-risk patient.84 Fortunately, the risk of transmission of HCV is about 0.3% when exposure occurs from hollow bore needles used to draw patients’ blood, although deep injuries increase the risk of transmission.83,213 Postexposure treatment with interferon-α has been used after occupational exposure to HCV, but the experience to date is uncontrolled and no benefit has been shown (not surprisingly given the low risk of transmission in this setting).214
Although postexposure prophylaxis is not effective, early treatment of acute HCV infection is effective.215–217 Treatment should be strongly considered in patients with acute HCV infection. Therapy with pegylated interferon alone should be started between weeks 8 and 12 after presentation if HCV RNA has not cleared spontaneously from serum by then.216,217 The addition of ribavirin to pegylated interferon does not increase the SVR in patients with acute HCV infection.215 Patients with acute HCV genotype 1 infection who are treated for 24 weeks have a greater than 80% chance of an SVR. A shorter duration of therapy may be acceptable for patients with acute HCV genotype 2 or 3 infection, but data are limited.
Because most patients with HCV infection are asymptomatic or minimally symptomatic, the vast majority of HCV-infected persons are not detected during acute infection. Therefore, screening of groups at high risk of HCV infection is recommended (see earlier). Persons at high risk include recipients of blood and blood products before 1992, past or present injection drug users (including those with a single exposure), persons with multiple sexual partners, patients on hemodialysis, infants of HCV-infected mothers, persons with occupational exposure to HCV-positive blood, and patients with persistently elevated serum ALT levels.1 HCV-infected patients should be instructed to avoid sharing razors. In addition, “safe sex” practices, such as the use of latex condoms, should be encouraged in persons with multiple sexual partners.87 Monogamous sexual partners who do not engage in high-risk sexual activity have a negligible risk of transmission.86 In view of the low rate of vertical transmission, pregnancy and breastfeeding are not contraindicated in HCV-infected women.88–90 HCV-infected patients are advised to undergo vaccination against hepatitis A and B because of the high risk of severe liver disease if superinfection with either of these viruses occurs.
IMMUNOPROPHYLAXIS
In contrast to hepatitis B, for which immunoprophylaxis or vaccination that results in a high level of envelope antibodies leads to protective immunity, the presence of anti-HCV in serum as a result of either administration of HCV-specific immunoglobulin or experimental vaccination does not prevent HCV infection effectively. HCV has an inherently high mutational rate that results in considerable heterogeneity in the envelope proteins and facilitates rapid escape from antibody recognition (see earlier). Therefore, traditional viral envelope or multiprotein vaccines have had limited success in preventing reinfection with homologous strains of HCV and no effect in preventing reinfection with heterologous strains of HCV. Monoclonal and polyclonal immunoglobulins have been used intra- and postoperatively in liver transplant recipients in the hope of avoiding reinfection of the graft, but this approach has been repeatedly unsuccessful.218,219
Given these difficulties, work on developing an HCV vaccine has focused instead on stimulating both humoral and cellular immune responses against the virus.220 If a vaccine were able to produce both neutralizing antibodies and a cellular immune response, it might have a role as both a prophylactic and therapeutic vaccine. Several strategies have been used to create a vaccine. Combining HCV proteins with an adjuvant such as the Toll-like receptor agonist dipalmitoyl-S-glyceryl cystine lipid moiety elicit CD8+ T-cell responses.221 Similarly, DNA vaccines lead to antibody production and cross-strain protective immunity by cytotoxic T lymphocytes in vivo. Dendritic cell vaccines have been created by transducing protein or transfecting viral constructs into dendritic cells. The source of the dendritic cells used and the mechanisms of maturation are crucial to their function. All of these approaches require further investigation to determine whether protective immunity is induced. Combinations of techniques are likely to be studied.
TREATMENT
Interferon-α monotherapy was approved for the treatment of chronic hepatitis C, then known as non-A, non-B hepatitis, before HCV was even identified. Substantial advances have been made since then with the introduction of prolonged treatment periods, longer-acting pegylated formulations of interferon, and the oral guanosine analog ribavirin. Treatment with the current standard of care—pegylated interferon and ribavirin—achieves an SVR (i.e., eradication of HCV) in about 42% to 52% of genotype 1–infected patients and 70% to 80% of genotype 2– or 3–infected patients.6,7,222 More recently, elucidation of the mechanisms of HCV replication has led to the development of the first-generation protease and polymerase inhibitors, some of which are in the final phases of clinical development and appear to improve response rates even further.
GOALS
The primary goal of therapy for HCV infection is eradication of the virus. A consequence of achieving this goal is prevention of liver-related deaths associated with the development of HCC and decompensated cirrhosis. SVR—the absence of detectable virus in blood 24 weeks after completion of therapy—is an excellent surrogate marker for the resolution of HCV infection. Seven retrospective follow-up studies that collectively included more than 500 patients, many of whom were immunosuppressed, followed for a mean of three and one half years found the risk of recurrent or new infection following SVR to be just 0.1% per year.223–229 SVR is also associated with a reduction in hepatic inflammation, regression of fibrosis, and improvement in HRQL.117,118 The risk of liver failure is essentially eliminated in patients with cirrhosis who achieve an SVR.158,230,231 Although the risk of HCC after an SVR in patients with cirrhosis is reduced by more than one half, the risk is not eliminated, and therefore, screening for HCC must continue.158
END POINTS
Although SVR is considered the endpoint of therapy, other indicators of response are often monitored during therapy because they may help guide and refine treatment (Figs. 79-7 and 79-8). A rapid virologic response (RVR), defined as undetectable HCV RNA in serum after the first 4 weeks of antiviral therapy, identifies those patients who are most sensitive to treatment and is associated with an 80% to 90% SVR rate in genotype 1–infected patients who complete 48 weeks of therapy or in genotype 2– or 3–infected patients who complete 24 weeks of therapy.232 An early virologic response (EVR), defined as a greater than 2-log drop in viral load at 12 weeks of therapy, is needed to continue therapy beyond 12 weeks because the absence of an EVR predicts failure of treatment in more than 98% of cases.233 An EVR can be subdivided further into a complete EVR (cEVR), defined as undetectable HCV RNA in serum at 12 weeks of therapy, and a partial EVR (pEVR), defined as a greater than 2-log decrease in the level of HCV RNA in serum despite residual detectable HCV RNA after 12 weeks of therapy. An end-of-treatment response (ETR) is defined as undetectable HCV RNA in serum at the end of treatment; a small and variable proportion of patients with an ETR will relapse when treatment is stopped. The absence of an ETR is considered nonresponse to treatment.
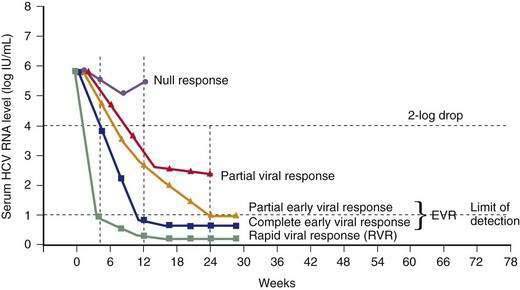
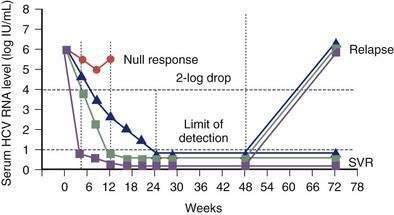
DRUGS
Ribavirin is an oral guanosine analog with activity against DNA and RNA viruses. When ribavirin is used in combination with interferon, the ETR improves and the relapse rate decreases. Several mechanisms to explain the synergistic effect of ribavirin when administered in combination with interferon have been proposed, including (1) alterations of the cytokine milieu leading to a change from a type 2 T-helper cell (Th2) to a Th1 immune response; (2) depletion of intracellular guanosine triphosphate through inhibition of the host enzyme inosine monophosphate dehydrogenase (IMPDH); (3) inhibition of the action of the HCV RNA-dependent RNA polymerase; and (4) induction of lethal mutagenesis during HCV RNA replication.234 Ribavirin generally is well tolerated, although it results in a dose-dependent hemolytic anemia. The dose administered is based on the patient’s weight, and the patient’s hemoglobin level must be monitored during treatment. Furthermore, in patients with a history of cardiopulmonary disease who cannot tolerate a sudden fall in the hemoglobin level, ribavirin must be used with caution, if at all. In addition, ribavirin is teratogenic; patients taking ribavirin and their partners are required to avoid pregnancy during therapy and for six months after cessation of the drug. Ribavirin has a long cumulative half-life in serum and is excreted by the kidneys; as a result it can lead to severe side effects, particularly hemolysis, in patients with kidney disease. The dose of ribavirin must be adjusted for renal function, and the drug should be administered with extreme caution to patients with a creatinine clearance less than 50 mL/min. Ribavirin is not removed by hemodialysis.
EFFICACY
The current standard of care for the treatment of HCV infection is the combination of a pegylated interferon administered subcutaneously once per week and ribavirin taken orally every day. A treatment algorithm is depicted in Figure 79-9. The dose of interferon is the same for all HCV genotypes, but the dose of ribavirin is based on the patient’s weight for genotypes 1 and 4 (13.3 mg/kg/day divided twice daily) and is fixed at 800 mg per day (in two divided doses) for genotypes 2 and 3. Treatment is administered for 48 weeks in patients with genotype 1 or 4 infection and 24 weeks in those with genotype 2 or 3 infection. In genotype 1–infected patients, treatment should be stopped if an EVR is not achieved. The SVR in genotype 1–infected patients is 42% to 52%.6,7,222 In the Individualized Dosing Efficacy versus Fixed Dosing to Assess Optimal Pegylated Interferon Therapy (IDEAL) trial, 3070 genotype 1–infected patients were randomized to one of the two pegylated interferons, and no difference in SVR was noted between the two formulations.235
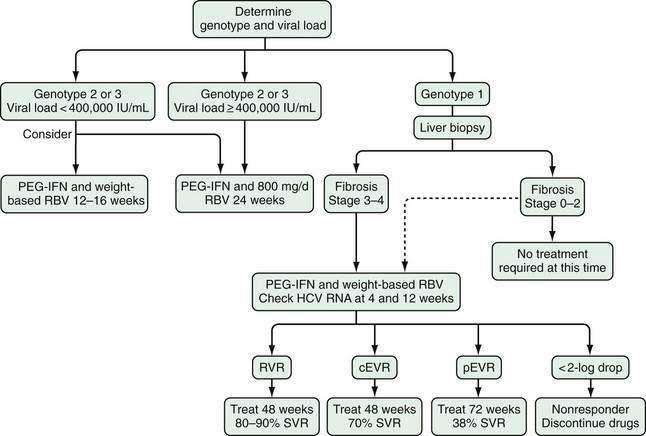
Figure 79-9. Algorithm for the treatment of hepatitis C virus (HCV) infection. Patients infected with HCV genotype 2 or 3 and with a low viral load (level of viremia <400,000 IU/mL) may be treated with pegylated interferon (PEG-IFN) and weight-based ribavirin (RBV) for 12 to 16 weeks, with sustained virologic response (SVR) rates similar to those for patients treated for 24 weeks with PEG-IFN and 800 mg/day of RBV; either regimen is acceptable for this group. Patients at higher risk of relapse or nonresponse (those with genotype 1 or with cirrhosis or a high viral load) should not be considered for a shortened course of therapy. Patients with HCV genotype 1 infection and advanced fibrosis (stage 3-4 according to the METAVIR system) should be treated, but patients infected with genotype 1 who have stage 0-2 fibrosis may not require immediate therapy. (Dotted arrow indicates that treatment is an option in these patients.) The viral load should be checked at weeks 4 and 12 of treatment because the likelihood of response is determined by the viral load at these time points (see Figure 79-7). cEVR, complete early virologic response; pEVR, partial early virologic response; RVR, rapid virologic response; IU, international units.
(Based on Mangia A, Minerva N, Bacca D, et al. Individualized treatment duration for hepatitis C genotype 1 patients: A randomized controlled trial. Hepatology 2008; 47:43-50; Pearlman B, Ehleben G, Saifee S, et al. Treatment extension to 72 weeks of peginterferon and ribavirin in hepatitis C genotype 1–infected slow responders. Hepatology 2007; 46:1671-4; Shiffman ML, Suter F, Bacon BR, et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med 2007; 357:124-34.)
Higher doses of pegylated interferon or ribavirin may be helpful in some patients. In one study,236 patients with HCV genotype 1 infection, high viral load, and a weight greater than 85 kg—all factors that predict a poor response to treatment—had a higher SVR when the dose of peginterferon alfa-2a or the dose of ribavirin was increased. In this difficult-to-treat patient population, peginterferon alfa-2a in a dose of 270 µg/week improved the SVR rate when compared with a standard dose of 180 µg/week. When higher doses of ribavirin are used (15.2 mg/kg/day) in genotype 1–infected patients, the relapse rate decreases (from 38% for standard-dose to 8% for high-dose ribavirin); however, hemolysis can limit treatment with high doses of ribavirin.236,237
Some patients may not require the standard duration of treatment to achieve an SVR. Patients with HCV genotypes 2 or 3 infection may achieve an SVR in some cases with just 12 to 16 weeks of treatment.238–240 If a truncated course of treatment is anticipated, the patient should be given a ribavirin dose based on weight rather than a fixed dose of 800 mg/day and should only be treated with a shortened course if an RVR is achieved.241 In general, patients with HCV genotype 1 infection should not have the treatment duration shortened, even if they achieve an RVR because SVR rates are about 10% less than those that follow a full year of therapy. Occasionally, however, drug intolerance may make a shorter course of treatment preferable.242,243
A longer than standard course of therapy appears to be helpful in genotype 1–infected patients who have a slow response to antiviral therapy. In patients with a pEVR, extending the duration of therapy from 48 to 72 weeks improves the SVR rate from 30% to 45.5% (with use of weight-based ribavirin).244 Furthermore, if genotype 1–infected patients are unable to tolerate the full dose of ribavirin, extending treatment to 72 weeks increases the SVR rate from 32% to 45%, a rate similar to that for 48 weeks of treatment with a weight-based dose of ribavirin and pegylated interferon.245
Mangia and colleagues examined the effect of tailoring the duration of treatment–either longer or shorter than usual—to the time when HCV RNA first became undetectable in serum. They randomized patients to 48 weeks of therapy or a variable length of therapy of 24, 48, or 72 weeks based on a first negative HCV RNA result at weeks 4, 8, and 12, respectively.246 If HCV RNA was negative in serum at week 4, the SVR rate was 87% with 48 weeks and 77% with 24 weeks of therapy. When HCV RNA became negative after eight weeks, all patients received 48 weeks of treatment, and the SVR rate was 70% to 72%. When the HCV RNA first became undetectable at week 12, the SVR rate was 38% with 48 weeks and 64% with 72 weeks of therapy.
FACTORS THAT PREDICT A SUSTAINED VIROLOGIC RESPONSE
The best predictor of response to pegylated interferon and ribavirin is the rate of the initial fall in serum HCV RNA levels during treatment. The highest SVRs occur in patients with an RVR, followed respectively by those with a cEVR, those with a pEVR, and those without an EVR. Patients are usually interested in an estimate of their chances of a response before making a decision to proceed with therapy. Pretreatment factors associated with a greater chance of an SVR include infection with non-genotype 1 HCV, a low baseline serum HCV RNA level, absence of bridging fibrosis or cirrhosis on a liver biopsy specimen, age younger than 40 years, absence of obesity, lack of hepatic steatosis or insulin resistance, absence of HIV infection, and white race (Fig. 79-10). Although the likelihood of an SVR is marginally lower in patients without these favorable factors, patients should not be discouraged and treatment should not be withheld because of the presence of any or all of these factors.
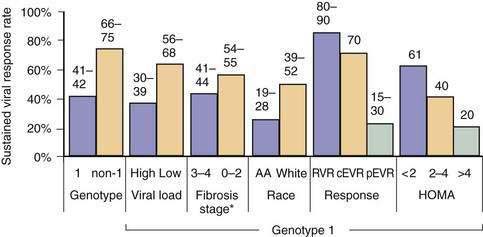
Of known pretreatment variables, the most powerful predictor of a response to treatment is the viral genotype. Genotype 2 is the most responsive to therapy. Response rates for genotype 3 infections are close to those of genotype 2, although genotype 3–infected patients with a high viral load have been shown to have lower response rates (86% for those with a serum HCV RNA level less than 600,000 IU/mL and 70% for those with a HCV RNA level of 600,000 IU/mL or greater).247 Genotype 4 infection is associated with an SVR similar to that for genotype 3 infection but requires drug doses and a duration of therapy similar to those for genotype 1 infection. Of the common HCV genotypes, HCV genotype 1 is the least responsive to therapy.
Pretreatment viral load also predicts the SVR. Studies have used different cutoff values to define a “low” viral load, with a range of <400,000 IU/mL to <800,000 IU/mL. A low viral load has consistently been associated with a higher SVR, independent of HCV genotype.241–243246
Obesity, insulin resistance, and hepatic steatosis all decrease the chance of SVR.184 These factors appear to affect the SVR independently. Weight reduction leads to an improved SVR to HCV therapy.
African Americans respond poorly to standard therapy for HCV infection, with SVR rates ranging from 19% to 28%.248–250 African Americans have a higher proportion of genotype 1 infections than do whites, but this difference does not explain the difference in SVR rates. Some evidence suggests that African Americans have improved response to higher doses of interferon.251
Advanced hepatic fibrosis is a negative predictor of SVR to therapy.252 Patients with advanced fibrosis, however, are most in need of antiviral therapy and should be treated. Patients who have advanced fibrosis should expect a 10% reduction in SVR compared with patients who do not have advanced fibrosis.158 Thrombocytopenia and neutropenia occur more frequently in these patients and may necessitate a reduction in the dose of pegylated interferon or discontinuation of therapy, further limiting the success of therapy in cirrhotic patients (see later).
Finally, strict adherence by the patient to the prescribed regimen improves the likelihood of an SVR, and compliance must be emphasized at each visit. Adherence to therapy appears to be particularly relevant in the first months of therapy; the impact of a reduction in dose is less if it occurs after an RVR has been achieved.253,254
INDICATIONS AND CONTRAINDICATIONS
The best time to treat a patient with chronic hepatitis C is before the development of cirrhosis because response rates are better and the risk of later complications of cirrhosis can be eliminated if treatment is successful. Treatment of patients with decompensated cirrhosis can still be successful but should only be undertaken by an experienced clinician. Managing patients with decompensated cirrhosis who are treated for HCV infection is labor intensive and requires reductions in dose because of cytopenias in 50% to 75% of cases as well as administration of growth factors in some cases; however, the treatment is generally poorly tolerated. Many prescribers prefer to begin with low doses of both drugs and increase the dose as tolerated by the patient.224 The chance of an SVR is low in patients with decompensated cirrhosis (13% in those with genotype 1 and 50% in those with genotype 2 and 3 infections).224 Nonetheless, the risk-benefit ratio clearly favors treatment in most of these cases. If these patients subsequently require liver transplantation, those who have achieved an SVR will avoid recurrence of HCV infection.224
Relative and absolute contraindications to interferon and ribavirin therapy are listed in Table 79-5. In general, more patients with relative contraindications to treatment have been treated successfully as practitioners have gained experience and familiarity with the treatment. Many relative contraindications to therapy may resolve over time or as a result of a specific intervention. A few absolute contraindications remain. Because ribavirin is a teratogen, unwillingness of the patient and his or her partner to practice adequate contraception and avoid pregnancy during treatment or for six months after the discontinuation of therapy is an absolute contraindication to starting or continuing treatment. Any severe or uncontrolled psychiatric condition is considered an absolute contraindication to therapy. If a patient’s psychiatric disorder is treated appropriately and is stable, however, the patient may become a candidate for HCV therapy. Severe cardiac or pulmonary disease is an absolute contraindication to HCV therapy because of the risk of worsening tissue hypoxia if severe hemolytic anemia occurs because of ribavirin. Moreover, in most patients with severe comorbid disease, successful antiviral therapy would offer no survival benefit. Patients with an underlying autoimmune condition are at risk of experiencing an exacerbation of the condition on pegylated interferon. Although previously considered an absolute contraindication, an autoimmune disorder should probably be considered a strong relative contraindication, and the risk of an exacerbation of the underlying autoimmune disease must be weighed against the potential benefit of clearing the HCV infection.
Table 79-5 Contraindications to Therapy with Pegylated Interferon and Ribavirin
| Absolute | Acute pancreatitis |
| Autoimmune hepatitis | |
| Comorbid conditions that markedly limit life expectancy | |
| History of hypersensitivity to one of the drugs | |
| Pregnancy or unwillingness to use birth control during and for six months after treatment | |
| Severe cardiac disease | |
| Severe pulmonary disease | |
| Uncontrolled psychiatric condition | |
| Uncontrolled seizure disorder | |
| Relative | Active alcohol or drug abuse |
| Active infection | |
| Baseline hemoglobin level <10 g/dL (for ribavirin) | |
| Baseline neutrophil count <1500/mm3 | |
| Baseline platelet count <90,000/mm3 | |
| Creatinine clearance <50 mL/min (use ribavirin with extreme caution) | |
| Decompensated cirrhosis | |
| Hemoglobinopathy | |
| Ophthalmologic disorders (may worsen during therapy) | |
| Other autoimmune conditions | |
| Uncontrolled hyperthyroidism or hypothyroidism |
MONITORING AND SAFETY
Drugs are discontinued consequent to an adverse event or laboratory test abnormality in approximately 5% and 16% of patients who receive a combination of pegylated interferon and ribavirin for 24 and 48 weeks, respectively.6,7,222,255 A reduction in the dose of pegylated interferon is required in 26% to 36% of patients and of ribavirin in 19% to 38%, depending on the initial dose and anticipated duration of therapy. Neutropenia, anemia, and thrombocytopenia are the most frequent reasons for reductions in dose. To ensure maximal response rates, dose reductions should be avoided, and the patient’s adherence to therapy should be encouraged.
Administration of hematopoietic growth factors (e.g., erythropoietin, filgrastim) may enable a patient to continue full-dose pegylated interferon and ribavirin. Use of growth factors may improve the patient’s subjective well-being but has not been shown to have an effect on response to antiviral treatment.256
RETREATMENT
The decision to retreat a patient who failed to respond to an earler course of antiviral therapy should take into consideration the previous regimen used, the appropriateness of the doses given throughout the course, the patient’s ability to tolerate therapy, and the response to treatment, especially whether and when HCV RNA became undetectable. Generally, retreatment with the same regimen, even with a different brand of interferon, is not justified. In nonresponders to an initial course of therapy consisting of either standard interferon monotherapy or standard interferon and ribavirin, the response rate to retreatment with pegylated interferon and ribavirin is 21% to 28% and 12% to 29%, respectively (Fig. 79-11).257–259 Persons who relapsed after a course of standard interferon alone or a combination of interferon with ribavirin achieve an SVR rate with pegylated interferon and ribavirin of 40% to 58% and 42%, respectively.257–259 Response rates are higher in patients who are infected with genotypes 2 and 3, have lower baseline levels of serum HCV RNA, and have lesser degrees of hepatic fibrosis.
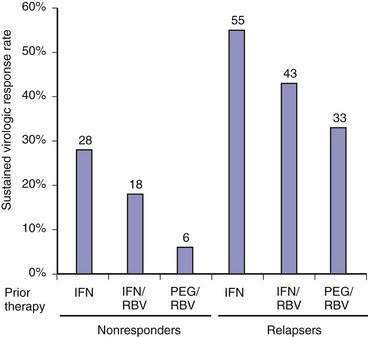
Other strategies for retreating nonresponders include use of higher doses of drugs or maintenance therapy. In the prospective Daily-Dose Consensus Interferon and Ribavirin: Efficacy of Combined Therapy (DIRECT) trial, 343 patients, who were previous nonresponders to pegylated interferon and ribavirin, were randomized to interferon alfacon-1, a non-pegylated recombinant interferon, 9 mg or 15 mg daily, and ribavirin for 48 weeks.260 The SVR rates were 5% and 9%, respectively.260 In the Retreatment with Pegasys Plus Ribavirin in Patients Not Responding to Prior Peg-Intron/Ribavirin Combination Therapy (REPEAT) trial, 942 nonresponders to pegylated interferon and ribavirin were randomized to pegylated interferon and ribavirin for 48 or 72 weeks. A subset of each group also received a higher (induction) dose of interferon for the first 12 weeks. The SVR rate was 7% to 9% in the patients in the 48-week arms and 14% to 16% for those in the 72-week arms.261 The use of an induction dose made no difference.
Maintenance therapy for nonresponders to pegylated interferon and ribavirin with low-dose (e.g., half of the usual dose) pegylated interferon has been studied in two large randomized prospective trials.155,262 In the Colchicine versus Peg-Intron Long-Term (COPILOT) trial, a decrease in the frequency of variceal hemorrhage was observed in treated patients in the subset with portal hypertension, but no differences in the frequency of liver failure, liver transplantation, HCC, or liver-related death after four years were observed.262 In the HALT-C trial (see earlier), no differences in these end points were observed after three and one half years.155 Although neither study was adequately powered to detect a benefit in the subset of patients in whom HCV RNA was suppressed throughout therapy, the conclusion can be drawn that maintenance therapy with low-dose peginterferon confers no benefit in nonresponders to full-dose therapy.
HIV-HCV COINFECTED PATIENTS
Patients with HIV-HCV coinfection probably progress more rapidly to cirrhosis than do HCV-monoinfected patients, and therefore treatment of HCV infection should always be considered in this group.263 Patients are at even higher risk of progression of fibrosis if they are female, older than age 33 years, have an increase in the CD4+ count of less than 100/mm3 with HAART, continue to have a detectable HIV viral load during antiretroviral therapy, or have untreated HCV infection.
SVR rates were lower in HIV-HCV coinfected patients in clinical trials than in HCV-monoinfected historical controls.264–266 SVR rates with pegylated interferon and ribavirin (800 mg/day) for 48 weeks are 44% to 73% in genotype 2– or 3–infected patients and 14% to 29% in genotype 1– and 4–infected patients.264–266 One reason for a lower SVR in coinfected patients is the use of lower doses of ribavirin in most of the studies. In one study,267 however, standard weight-based dosing of ribavirin was used, and the SVR rate in genotype 1–infected patients was 35%. No doubt other factors played a role in the lower response rates, including high dose-reduction and drug-discontinuation rates and the presence among study subjects of factors that predict a poor response to treatment, including a high viral load, preponderance of genotype 1, a high rate of advanced hepatic fibrosis, concurrent alcohol use, and a high proportion of African Americans. Despite a lower SVR among HIV-HCV coinfected patients, however, an RVR is still associated with a high SVR, and failure to achieve an EVR signifies nonresponse.268 The safety and efficacy of pegylated interferon and ribavirin have not been established in patients with a CD4+ count less than 200/mm3.269 Ideally, therapy for HCV infection should be started before antiretroviral therapy for HIV is initiated. When HCV therapy cannot be started first, zidovudine, stavudine, and didanosine should not be used in combination with ribavirin because of the additive risk of mitochondrial toxicity. Contraindications to antiviral therapy for HCV infection in HIV-HCV-coinfected patients do not differ from those for monoinfected patients.
LIVER TRANSPLANT RECIPIENTS
Complications of chronic hepatitis C are the most common indication for liver transplantation (see Chapter 95). Patients who have detectable HCV RNA in serum at the time of liver transplantation almost universally experience reinfection of the allograft. Therefore, therapy with interferon and ribavirin should be considered in cirrhotic patients, preferably before liver failure or HCC occurs. Treatment is difficult to tolerate in this setting, and response rates are poor when cirrhosis is decompensated (see earlier). Another option in these patients is to initiate antiviral treatment after transplantation. Preemptive therapy of all HCV-infected patients within the first few weeks after transplantation is associated with substantial toxicity and a low chance of an SVR; it is not recommended.270 Still another option is to follow the patient expectantly post-transplantation and begin antiviral therapy when surveillance liver biopsy specimens demonstrate progression of disease, usually to grade 3 inflammation or stage 2 fibrosis. Often, this degree of liver injury occurs at least one year post-transplantation, but initiating treatment before then is not contraindicated. Treatment is better tolerated more than one year post-transplantation, but reductions in drug doses are required in most patients. Few patients tolerate full doses of ribavirin because of the renal dysfunction associated with calcineurin inhibitors. Nonetheless, 23% to 26% of genotype 1–infected patients achieve an SVR with 48 weeks of pegylated interferon and ribavirin. The SVR rate may be higher than 80% in patients infected with HCV genotype 2 or 3.270–272 Although patients tolerate therapy poorly and SVR rates are low, a survival advantage has been demonstrated for patients who achieve an SVR compared with those who remain viremic.273
FUTURE THERAPIES
Several inhibitors of the NS3/4A serine protease are currently in clinical trials. Telaprevir (VX-950) and boceprevir (SCH503034) are the two that are furthest along in development. Initial monotherapy studies demonstrated early selection of preexisting drug-resistant strains of the HCV virus, particularly if the dose of the drug was insufficient or the dosing interval was greater than eight hours.274 The combination of peginterferon alfa-2a and ribavirin with telaprevir, given orally three times a day for 12 weeks, followed by 12 weeks of peginterferon alfa-2a and ribavirin, in previously untreated genotype 1–infected patients led to a rapid reduction in serum HCV RNA levels that prevented the emergence of resistance and resulted in an SVR rate of 61%, compared with 41% in those treated with pegylated interferon and ribavirin alone for 48 weeks (standard treatment).275 A second trial in previously untreated genotype 1-infected patients showed an SVR rate of 68% with the 24-week triple-drug regimen and an SVR rate of 48% with standard treatment.276 Ribavirin remains a critical component of the regimen because telaprevir and pegylated interferon dual therapy produce an inferior SVR compared with triple-drug therapy. Side effects of telaprevir include rash, gastrointestinal complaints, and anemia, although most are mild to moderate and do not require a dose reduction or discontinuation.
Boceprevir, a second protease inhibitor, is also given orally three times a day, and, in combination with peginterferon alfa-2b and ribavirin, led to an SVR rate of 55% in previously untreated genotype 1–infected patients after 28 weeks of therapy.277 Because of concerns about resistance, a second group of patients was treated with a 4-week lead-in phase of pegylated interferon and ribavirin to reduce HCV RNA levels before the introduction of boceprevir. The SVR rate was 57% after completion of an additional 24 weeks of triple-drug therapy.
A second target of HCV replication, the RNA-dependent RNA polymerase, was inhibited by R1626, a nucleoside analog, given orally twice daily, in combination with peginterferon alfa-2a and ribavirin.278 Triple-drug therapy in previously untreated genotype 1–infected patients resulted in undetectable HCV RNA levels in serum at four weeks in 74% of patients, compared with 5% of patients treated with standard therapy. Another orally bioavailable nucleoside analog inhibitor of the RNA-dependent RNA polymerase, R7128, reduced serum HCV RNA levels in patients by 5.1 log when used in combination with pegylated interferon and ribavirin, compared with a 2.5-log reduction with standard treatment, after 28 days.279 Other non-nucleoside inhibitors target allosteric sites on the RNA-dependent RNA polymerase and lead to modest reductions in serum HCV RNA levels when given as monotherapy; studies of combination therapy with pegylated interferon and ribavirin have not yet been undertaken.
Another strategy for impairing viral polymerase function is to inhibit cyclophylin B, which facilitates attachment of HCV RNA to the replicase complex. The cyclophilin inhibitor DEBIO-025, given daily in combination with pegylated interferon, caused a 4.75-log drop in serum HCV RNA levels in 29 days, compared with a 2.49-log decline in HCV RNA levels with pegylated interferon alone and a 2.2-log decline in HCV RNA levels with DEBIO-025 alone.280 Other approaches to antiviral therapy include targeting the HCV helicase and viral assembly; these approaches to antiviral therapy are in the early stages of development.
Armstrong GL, Wasley A, Simard EP, et al. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705-14. (Ref 1.)
Bruno S, Stroffolini T, Colombo M, et al. Sustained virological response to interferon-alpha is associated with improved outcome in HCV-related cirrhosis: A retrospective study. Hepatology. 2007;45:579-87. (Ref 158.)
Everson GT, Trotter J, Forman L, et al. Treatment of advanced hepatitis C with a low accelerating dosage regimen of antiviral therapy. Hepatology. 2005;42:255-62. (Ref 224.)
Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975-82. (Ref 6.)
Mangia A, Minerva N, Bacca D, et al. Individualized treatment duration for hepatitis C genotype 1 patients: A randomized controlled trial. Hepatology. 2008;47:43-50. (Ref 246.)
Manning DS, Afdhal NH. Diagnosis and quantitation of fibrosis. Gastroenterology. 2008;134:1670-81. (Ref 138.)
Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet. 2001;358:958-65. (Ref 7.)
Missiha SB, Ostrowski M, Heathcote EJ. Disease progression in chronic hepatitis C: Modifiable and nonmodifiable factors. Gastroenterology. 2008;134:1699-714. (Ref 157.)
Poordad F, Reddy KR, Martin P. Rapid virologic response: A new milestone in the management of chronic hepatitis C. Clin Infect Dis. 2008;46:78-84. (Ref 232.)
Powell EE, Jonsson JR, Clouston AD. Steatosis: Co-factor in other liver diseases. Hepatology. 2005;42:5-13. (Ref 178.)
Sangiovanni A, Prati GM, Fasani P, et al. The natural history of compensated cirrhosis due to hepatitis C virus: A 17-year cohort study of 214 patients. Hepatology. 2006;43:1303-10. (Ref 154.)
Shiffman ML, Salvatore J, Hubbard S, et al. Treatment of chronic hepatitis C virus genotype 1 with peginterferon, ribavirin, and epoetin alpha. Hepatology. 2007;46:371-9. (Ref 237.)
Shiffman ML, Suter F, Bacon BR, et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med. 2007;357:124-34. (Ref 241.)
Strader DB, Wright T, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C. Hepatology. 2004;39:1147-71. (Ref 144.)
Wiesner RH, Sorrell M, Villamil F. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl. 2003;9:S1-9. (Ref 191.)
1. Armstrong GL, Wasley A, Simard EP, et al. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144:705-14.
2. Alter MJ, Kruszon-Moran D, Nainan OV, et al. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341:556-62.
3. Davis GL, Albright JE, Cook SF, Rosenberg DM. Projecting future complications of chronic hepatitis C in the United States. Liver Transpl. 2003;9:331-8.
4. Global surveillance and control of hepatitis C. Report of a WHO Consultation organized in collaboration with the Viral Hepatitis Prevention Board, Antwerp, Belgium. J Viral Hepat. 1999;6:35-47.
5. Wise M, Bialek S, Finelli L, et al. Changing trends in hepatitis C-related mortality in the United States, 1995-2004. Hepatology. 2008;47:1128-35.
6. Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975-82.
7. Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet. 2001;358:958-65.
8. Yu X, Qiao M, Atanasov I, et al. Cryo-electron microscopy and three-dimensional reconstructions of hepatitis C virus particles. Virology. 2007;367:126-34.
9. Maillard P, Krawczynski K, Nitkiewicz J, et al. Nonenveloped nucleocapsids of hepatitis C virus in the serum of infected patients. J Virol. 2001;75:8240-50.
10. Robertson B, Myers G, Howard C, et al. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch Virol. 1998;143:2493-503.
11. Penin F, Dubuisson J, Rey FA, et al. Structural biology of hepatitis C virus. Hepatology. 2004;39:5-19.
12. Dubuisson J. Hepatitis C virus proteins. World J Gastroenterol. 2007;13:2406-15.
13. Reed KE, Rice CM. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr Top Microbiol Immunol. 2000;242:55-84.
14. Lerat H, Hollinger FB. Hepatitis C virus (HCV) occult infection or occult HCV RNA detection? J Infect Dis. 2004;189:3-6.
15. Pham TN, King D, Macparland SA, et al. Hepatitis C virus replicates in the same immune cell subsets in chronic hepatitis C and occult infection. Gastroenterology. 2008;134:812-22.
16. Cormier EG, Tsamis F, Kajumo F, et al. CD81 is an entry coreceptor for hepatitis C virus. Proc Natl Acad Sci U S A. 2004;101:7270-4.
17. Zeisel MB, Koutsoudakis G, Schnober EK, et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology. 2007;46:1722-31.
18. Evans MJ, von Hahn T, Tscherne DM, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801-5.
19. Barth H, Schafer C, Adah MI, et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol Chem. 2003;278:41003-12.
20. Agnello V, Abel G, Elfahal M, et al. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1999;96:12766-71.
21. Cormier EG, Durso RJ, Tsamis F, et al. L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc Natl Acad Sci U S A. 2004;101:14067-72.
22. Bartosch B, Verney G, Dreux M, et al. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J Virol. 2005;79:8217-29.
23. Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197:633-42.
24. Honda M, Ping LH, Rijnbrand RC, et al. Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology. 1996;222:31-42.
25. Ji H, Fraser CS, Yu Y, et al. Coordinated assembly of human translation initiation complexes by the hepatitis C virus internal ribosome entry site RNA. Proc Natl Acad Sci U S A. 2004;101:16990-5.
26. Egger D, Wolk B, Gosert R, et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol. 2002;76:5974-84.
27. Elazar M, Liu P, Rice CM, Glenn JS. An N-terminal amphipathic helix in hepatitis C virus (HCV) NS4B mediates membrane association, correct localization of replication complex proteins, and HCV RNA replication. J Virol. 2004;78:11393-400.
28. Gosert R, Egger D, Lohmann V, et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol. 2003;77:5487-92.
29. Bartenschlager R, Frese M, Pietschmann T. Novel insights into hepatitis C virus replication and persistence. Adv Virus Res. 2004;63:71-180.
30. Tanaka Y, Shimoike T, Ishii K, et al. Selective binding of hepatitis C virus core protein to synthetic oligonucleotides corresponding to the 5′ untranslated region of the viral genome. Virology. 2000;270:229-36.
31. Mizuno M, Yamada G, Tanaka T, et al. Virion-like structures in HeLa G cells transfected with the full-length sequence of the hepatitis C virus genome. Gastroenterology. 1995;109:1933-40.
32. Sakai A, Claire MS, Faulk K, et al. The p7 polypeptide of hepatitis C virus is critical for infectivity and contains functionally important genotype-specific sequences. Proc Natl Acad Sci U S A. 2003;100:11646-51.
33. Steinmann E, Penin F, Kallis S, et al. Hepatitis C Virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007;3:e103.
34. Walewski JL, Keller TR, Stump DD, Branch AD. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA. 2001;7:710-21.
35. Xu Z, Choi J, Yen TS, et al. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J. 2001;20:3840-8.
36. Branch AD, Stump DD, Gutierrez JA, et al. The hepatitis C virus alternate reading frame (ARF) and its family of novel products: The alternate reading frame protein/F-protein, the double-frameshift protein, and others. Semin Liver Dis. 2005;25:105-17.
37. Wang Y, Kato N, Jazag A, et al. Hepatitis C virus core protein is a potent inhibitor of RNA silencing-based antiviral response. Gastroenterology. 2006;130:883-92.
38. Hosui A, Ohkawa K, Ishida H, et al. Hepatitis C virus core protein differently regulates the JAK-STAT signaling pathway under interleukin-6 and interferon-gamma stimuli. J Biol Chem. 2003;278:28562-71.
39. Zimmermann M, Flechsig C, La Monica N, et al. Hepatitis C virus core protein impairs in vitro priming of specific T cell responses by dendritic cells and hepatocytes. J Hepatol. 2008;48:51-60.
40. Jhaveri R, McHutchison J, Patel K, et al. Specific polymorphisms in hepatitis C virus genotype 3 core protein associated with intracellular lipid accumulation. J Infect Dis. 2008;197:283-91.
41. Banerjee S, Saito K, Ait-Goughoulte M, et al. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J Virol. 2008;82:2606-12.
42. Akuta N, Suzuki F, Kawamura Y, et al. Amino acid substitutions in the hepatitis C virus core region are the important predictor of hepatocarcinogenesis. Hepatology. 2007;46:1357-64.
43. Bartosch B, Cosset FL. Cell entry of hepatitis C virus. Virology. 2006;348:1-12.
44. Lorenz IC, Marcotrigiano J, Dentzer TG, Rice CM. Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature. 2006;442:831-5.
45. Kim JL, Morgenstern KA, Lin C, et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996;87:343-55.
46. Binder M, Kochs G, Bartenschlager R, Lohmann V. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy. Hepatology. 2007;46:1365-74.
47. Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992-7.
48. Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167-72.
49. Myong S, Bruno MM, Pyle AM, Ha T. Spring-loaded mechanism of DNA unwinding by hepatitis C virus NS3 helicase. Science. 2007;317:513-16.
50. Lindenbach BD, Pragai BM, Montserret R, et al. The C terminus of hepatitis C virus NS4A encodes an electrostatic switch that regulates NS5A hyperphosphorylation and viral replication. J Virol. 2007;81:8905-18.
51. Tellinghuisen TL, Marcotrigiano J, Rice CM. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature. 2005;435:374-9.
52. Nanda SK, Herion D, Liang TJ. The SH3 binding motif of HCV [corrected] NS5A protein interacts with Bin1 and is important for apoptosis and infectivity. Gastroenterology. 2006;130:794-809.
53. Wang J, Tong W, Zhang X, et al. Hepatitis C virus non-structural protein NS5A interacts with FKBP38 and inhibits apoptosis in Huh7 hepatoma cells. FEBS Lett. 2006;580:4392-400.
54. Enomoto N, Sakuma I, Asahina Y, et al. Mutations in the nonstructural protein 5A gene and response to interferon in patients with chronic hepatitis C virus 1b infection. N Engl J Med. 1996;334:77-81.
55. Bressanelli S, Tomei L, Roussel A, et al. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci U S A. 1999;96:13034-9.
56. Pawlotsky JM. Hepatitis C virus genetic variability: pathogenic and clinical implications. Clin Liver Dis. 2003;7:45-66.
57. Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282:103-7.
58. Choo QL, Kuo G, Weiner AJ, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359-62.
59. Kalinina O, Norder H, Mukomolov S, Magnius LO. A natural intergenotypic recombinant of hepatitis C virus identified in St. Petersburg. J Virol. 2002;76:4034-43.
60. Noppornpanth S, Lien TX, Poovorawan Y, et al. Identification of a naturally occurring recombinant genotype 2/6 hepatitis C virus. J Virol. 2006;80:7569-77.
61. Blatt LM, Mutchnick MG, Tong MJ, et al. Assessment of hepatitis C virus RNA and genotype from 6807 patients with chronic hepatitis C in the United States. J Viral Hepat. 2000;7:196-202.
62. Nainan OV, Alter MJ, Kruszon-Moran D, et al. Hepatitis C virus genotypes and viral concentrations in participants of a general population survey in the United States. Gastroenterology. 2006;131:478-84.
63. Wasley A, Alter MJ. Epidemiology of hepatitis C: Geographic differences and temporal trends. Semin Liver Dis. 2000;20:1-16.
64. Kamal SM, Nasser IA. Hepatitis C genotype 4: What we know and what we don’t yet know. Hepatology. 2008;47:1371-83.
65. Verbeeck J, Peigue-Lafeuille H, Ross RS, et al. HCV genotype 5: Epidemiology and spread of an uncommon genotype. J Clin Virol. 2008;41:170-1.
66. Esteban JI, Sauleda S, Quer J. The changing epidemiology of hepatitis C virus infection in Europe. J Hepatol. 2008;48:148-62.
67. Farci P, Shimoda A, Coiana A, et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288:339-44.
68. Sobesky R, Feray C, Rimlinger F, et al. Distinct hepatitis C virus core and F protein quasispecies in tumoral and nontumoral hepatocytes isolated via microdissection. Hepatology. 2007;46:1704-12.
69. Sheridan I, Pybus OG, Holmes EC, Klenerman P. High-resolution phylogenetic analysis of hepatitis C virus adaptation and its relationship to disease progression. J Virol. 2004;78:3447-54.
70. Weinbaum CM, Sabin KM, Santibanez SS. Hepatitis B, hepatitis C, and HIV in correctional populations: A review of epidemiology and prevention. AIDS. 2005;19(Suppl 3):S41-6.
71. Hyams KC, Riddle J, Rubertone M, et al. Prevalence and incidence of hepatitis C virus infection in the US military: A seroepidemiologic survey of 21,000 troops. Am J Epidemiol. 2001;153:764-70.
72. Simmonds P. The origin and evolution of hepatitis viruses in humans. J Gen Virol. 2001;82:693-712.
73. Tanaka Y, Kurbanov F, Mano S, et al. Molecular tracing of the global hepatitis C virus epidemic predicts regional patterns of hepatocellular carcinoma mortality. Gastroenterology. 2006;130:703-14.
74. Wasley A, Grytdal S, Gallagher K. Surveillance for acute viral hepatitis—United States, 2006. MMWR Surveill Summ. 2008;57:1-24.
75. Prati D. Transmission of hepatitis C virus by blood transfusions and other medical procedures: A global review. J Hepatol. 2006;45:607-16.
76. Alter HJ, Houghton M. Clinical Medical Research Award. Hepatitis C virus and eliminating post-transfusion hepatitis. Nat Med. 2000;6:1082-6.
77. Schreiber GB, Busch MP, Kleinman SH, Korelitz JJ. The risk of transfusion-transmitted viral infections. The Retrovirus Epidemiology Donor Study. N Engl J Med. 1996;334:1685-90.
78. Alter MJ. Prevention of spread of hepatitis C. Hepatology. 2002;36:S93-8.
79. Rahnavardi M, Hosseini Moghaddam SM, Alavian SM. Hepatitis C in hemodialysis patients: Current global magnitude, natural history, diagnostic difficulties, and preventive measures. Am J Nephrol. 2008;28:628-40.
80. Lau JY, Davis GL, Brunson ME, et al. Hepatitis C virus infection in kidney transplant recipients. Hepatology. 1993;18:1027-31.
81. Kelen GD, Green GB, Purcell RH, et al. Hepatitis B and hepatitis C in emergency department patients. N Engl J Med. 1992;326:1399-404.
82. Sulkowski MS, Ray SC, Thomas DL. Needlestick transmission of hepatitis C. JAMA. 2002;287:2406-13.
83. De Carli G, Puro V, Ippolito G. Risk of hepatitis C virus transmission following percutaneous exposure in healthcare workers. Infection. 2003;31(Suppl 2):22-7.
84. Makary MA, Al-Attar A, Holzmueller CG, et al. Needlestick injuries among surgeons in training. N Engl J Med. 2007;356:2693-9.
85. Pruss-Ustun A, Rapiti E, Hutin Y. Estimation of the global burden of disease attributable to contaminated sharps injuries among health-care workers. Am J Ind Med. 2005;48:482-90.
86. Vandelli C, Renzo F, Romano L, et al. Lack of evidence of sexual transmission of hepatitis C among monogamous couples: Results of a 10-year prospective follow-up study. Am J Gastroenterol. 2004;99:855-9.
87. Blackard JT, Shata MT, Shire NJ, Sherman KE. Acute hepatitis C virus infection: A chronic problem. Hepatology. 2008;47:321-31.
88. Gibb DM, Goodall RL, Dunn DT, et al. Mother-to-child transmission of hepatitis C virus: evidence for preventable peripartum transmission. Lancet. 2000;356:904-7.
89. Conte D, Fraquelli M, Prati D, et al. Prevalence and clinical course of chronic hepatitis C virus (HCV) infection and rate of HCV vertical transmission in a cohort of 15,250 pregnant women. Hepatology. 2000;31:751-5.
90. Kumar RM, Shahul S. Role of breast-feeding in transmission of hepatitis C virus to infants of HCV-infected mothers. J Hepatol. 1998;29:191-7.
91. Hwang LY, Kramer JR, Troisi C, et al. Relationship of cosmetic procedures and drug use to hepatitis C and hepatitis B virus infections in a low-risk population. Hepatology. 2006;44:341-51.
92. Frank C, Mohamed MK, Strickland GT, et al. The role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet. 2000;355:887-91.
93. Saito T, Gale MJr. Regulation of innate immunity against hepatitis C virus infection. Hepatol Res. 2008;38:115-22.
94. Golden-Mason L, Rosen HR. Natural killer cells: Primary target for hepatitis C virus immune evasion strategies? Liver Transpl. 2006;12:363-72.
95. Racanelli V, Rehermann B. Hepatitis C virus infection: When silence is deception. Trends Immunol. 2003;24:456-64.
96. Takaki A, Wiese M, Maertens G, et al. Cellular immune responses persist and humoral responses decrease two decades after recovery from a single-source outbreak of hepatitis C. Nat Med. 2000;6:578-82.
97. Dixon LR, Crawford JM. Early histologic changes in fibrosing cholestatic hepatitis C. Liver Transpl. 2007;13:219-26.
98. Gane E. The natural history and outcome of liver transplantation in hepatitis C virus-infected recipients. Liver Transpl. 2003;9:S28-34.
99. Lim HL, Lau GK, Davis GL, et al. Cholestatic hepatitis leading to hepatic failure in a patient with organ-transmitted hepatitis C virus infection. Gastroenterology. 1994;106:248-51.
100. Guo JT, Sohn JA, Zhu Q, Seeger C. Mechanism of the interferon alpha response against hepatitis C virus replicons. Virology. 2004;325:71-81.
101. Guo JT, Zhu Q, Seeger C. Cytopathic and noncytopathic interferon responses in cells expressing hepatitis C virus subgenomic replicons. J Virol. 2003;77:10769-79.
102. Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623-8.
103. Tseng CT, Klimpel GR. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J Exp Med. 2002;195:43-9.
104. Crotta S, Stilla A, Wack A, et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med. 2002;195:35-41.
105. Foy E, Li K, Sumpter RJr., et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci U S A. 2005;102:2986-91.
106. Zhu H, Dong H, Eksioglu E, Hemming A, et al. Hepatitis C virus triggers apoptosis of a newly developed hepatoma cell line through antiviral defense system. Gastroenterology. 2007;133:1649-59.
107. Della Bella S, Crosignani A, Riva A, et al. Decrease and dysfunction of dendritic cells correlate with impaired hepatitis C virus-specific CD4+ T-cell proliferation in patients with hepatitis C virus infection. Immunology. 2007;121:283-92.
108. Pachiadakis I, Pollara G, Chain BM, Naoumov NV. Is hepatitis C virus infection of dendritic cells a mechanism facilitating viral persistence? Lancet Infect Dis. 2005;5:296-304.
109. Bain C, Fatmi A, Zoulim F, et al. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120:512-24.
110. Ward SM, Fox BC, Brown PJ, et al. Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J Hepatol. 2007;47:316-24.
111. Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946-52.
112. Davis GL. Hepatitis C immune globulin to prevent HCV recurrence after liver transplantation: Chasing windmills? Liver Transpl. 2006;12:1317-19.
113. Orland JR, Wright TL, Cooper S. Acute hepatitis C. Hepatology. 2001;33:321-7.
114. Santantonio T, Medda E, Ferrari C, et al. Risk factors and outcome among a large patient cohort with community-acquired acute hepatitis C in Italy. Clin Infect Dis. 2006;43:1154-9.
115. Shiffman ML, Diago M, Tran A, et al. Chronic hepatitis C in patients with persistently normal alanine transaminase levels. Clin Gastroenterol Hepatol. 2006;4:645-52.
116. Puoti C, Castellacci R, Montagnese F, et al. Histological and virological features and follow-up of hepatitis C virus carriers with normal aminotransferase levels: The Italian prospective study of the asymptomatic C carriers (ISACC). J Hepatol. 2002;37:117-23.
117. Kallman J, O’Neil MM, Larive B, et al. Fatigue and health-related quality of life (HRQL) in chronic hepatitis C virus infection. Dig Dis Sci. 2007;52:2531-9.
118. Younossi Z, Kallman J, Kincaid J. The effects of HCV infection and management on health-related quality of life. Hepatology. 2007;45:806-16.
119. Zignego AL, Ferri C, Pileri SA, et al. Extrahepatic manifestations of Hepatitis C Virus infection: A general overview and guidelines for a clinical approach. Dig Liver Dis. 2007;39:2-17.
120. Kamar N, Izopet J, Alric L, et al. Hepatitis C virus-related kidney disease: An overview. Clin Nephrol. 2008;69:149-60.
121. Saadoun D, Rosenzwajg M, Landau D, et al. Restoration of peripheral immune homeostasis after Rituximab in mixed cryoglobulinemia vasculitis. Blood. 2008;111:5334-41.
122. Ahmed MS, Wong CF. Should rituximab be the rescue therapy for refractory mixed cryoglobulinemia associated with hepatitis C? J Nephrol. 2007;20:350-6.
123. Giordano TP, Henderson L, Landgren O, et al. Risk of non-Hodgkin lymphoma and lymphoproliferative precursor diseases in US veterans with hepatitis C virus. JAMA. 2007;297:2010-7.
124. de Sanjose S, Benavente Y, Vajdic CM, et al. Hepatitis C and non-Hodgkin lymphoma among 4784 cases and 6269 controls from the International Lymphoma Epidemiology Consortium. Clin Gastroenterol Hepatol. 2008;6:451-8.
125. Zuckerman E, Zuckerman T, Sahar D, et al. The effect of antiviral therapy on t(14;18) translocation and immunoglobulin gene rearrangement in patients with chronic hepatitis C virus infection. Blood. 2001;97:1555-9.
126. Zignego AL, Ferri C, Giannelli F, et al. Prevalence of bcl-2 rearrangement in patients with hepatitis C virus-related mixed cryoglobulinemia with or without B-cell lymphomas. Ann Intern Med. 2002;137:571-80.
127. Giannelli F, Moscarella S, Giannini C, et al. Effect of antiviral treatment in patients with chronic HCV infection and t(14;18) translocation. Blood. 2003;102:1196-201.
128. Romero-Gomez M, Fernandez-Rodriguez CM, Andrade RJ, et al. Effect of sustained virological response to treatment on the incidence of abnormal glucose values in chronic hepatitis C. J Hepatol. 2008;48:721-7.
129. Cassani F, Cataleta M, Valentini P, et al. Serum autoantibodies in chronic hepatitis C: Comparison with autoimmune hepatitis and impact on the disease profile. Hepatology. 1997;26:561-6.
130. Scott JD, Gretch DR. Molecular diagnostics of hepatitis C virus infection: A systematic review. JAMA. 2007;297:724-32.
131. Pawlotsky JM. Use and interpretation of virological tests for hepatitis C. Hepatology. 2002;36:S65-73.
132. Sabato MF, Shiffman ML, Langley MR, et al. Comparison of performance characteristics of three real-time reverse transcription-PCR test systems for detection and quantification of hepatitis C virus. J Clin Microbiol. 2007;45:2529-36.
133. van Doorn LJ, Kleter B, Stuyver L, et al. Analysis of hepatitis C virus genotypes by a line probe assay and correlation with antibody profiles. J Hepatol. 1994;21:122-9.
134. Scheuer PJ, Standish RA, Dhillon AP. Scoring of chronic hepatitis. Clin Liver Dis. 2002;6:335-47. v-vi
135. Lefkowitch JH. Liver biopsy assessment in chronic hepatitis. Arch Med Res. 2007;38:634-43.
136. Regev A, Berho M, Jeffers LJ, et al. Sampling error and intraobserver variation in liver biopsy in patients with chronic HCV infection. Am J Gastroenterol. 2002;97:2614-18.
137. Imbert-Bismut F, Ratziu V, Pieroni L, et al. Biochemical markers of liver fibrosis in patients with hepatitis C virus infection: A prospective study. Lancet. 2001;357:1069-75.
138. Manning DS, Afdhal NH. Diagnosis and quantitation of fibrosis. Gastroenterology. 2008;134:1670-81.
139. Schiano TD, Azeem S, Bodian CA, et al. Importance of specimen size in accurate needle liver biopsy evaluation of patients with chronic hepatitis C. Clin Gastroenterol Hepatol. 2005;3:930-5.
140. Wai CT, Greenson JK, Fontana RJ, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology. 2003;38:518-26.
141. Le Calvez S, Thabut D, Messous D, et al. The predictive value of Fibrotest vs. APRI for the diagnosis of fibrosis in chronic hepatitis C. Hepatology. 2004;39:862-3.
142. Friedrich-Rust M, Ong MF, Martens S, et al. Performance of transient elastography for the staging of liver fibrosis: A meta-analysis. Gastroenterology. 2008;134:960-74.
143. Shaheen AA, Wan AF, Myers RP. FibroTest and FibroScan for the prediction of hepatitis C-related fibrosis: A systematic review of diagnostic test accuracy. Am J Gastroenterol. 2007;102:2589-600.
144. Strader DB, Wright T, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C. Hepatology. 2004;39:1147-71.
145. Ryder SD, Irving WL, Jones DA, et al. Progression of hepatic fibrosis in patients with hepatitis C: A prospective repeat liver biopsy study. Gut. 2004;53:451-5.
146. Ghany MG, Kleiner DE, Alter H, et al. Progression of fibrosis in chronic hepatitis C. Gastroenterology. 2003;124:97-104.
147. Freeman AJ, Dore GJ, Law MG, et al. Estimating progression to cirrhosis in chronic hepatitis C virus infection. Hepatology. 2001;34:809-16.
148. Kenny-Walsh E. Clinical outcomes after hepatitis C infection from contaminated anti-D immune globulin. Irish Hepatology Research Group. N Engl J Med. 1999;340:1228-33.
149. Levine RA, Sanderson SO, Ploutz-Snyder R, et al. Assessment of fibrosis progression in untreated Irish women with chronic hepatitis C contracted from immunoglobulin anti-D. Clin Gastroenterol Hepatol. 2006;4:1271-7.
150. Wiese M, Berr F, Lafrenz M, et al. Low frequency of cirrhosis in a hepatitis C (genotype 1b) single-source outbreak in Germany: A 20-year multicenter study. Hepatology. 2000;32:91-6.
151. El-Serag HB. Hepatocellular carcinoma: Recent trends in the United States. Gastroenterology. 2004;127:S27-34.
152. Fattovich G, Giustina G, Degos F, et al. Morbidity and mortality in compensated cirrhosis type C: A retrospective follow-up study of 384 patients. Gastroenterology. 1997;112:463-72.
153. Serfaty L, Aumaitre H, Chazouilleres O, et al. Determinants of outcome of compensated hepatitis C virus-related cirrhosis. Hepatology. 1998;27:1435-40.
154. Sangiovanni A, Prati GM, Fasani P, et al. The natural history of compensated cirrhosis due to hepatitis C virus: A 17-year cohort study of 214 patients. Hepatology. 2006;43:1303-10.
155. Di Bisceglie AM, Everson GT, Linsay KL, et al. Prolonged antiviral therapy with peginterferon to prevent complications of advanced liver disease associated with hepatitis C: Results of the hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) trial [abstract]. Hepatology. 2007;46:ALB1.
156. Manos MM, Leyden WA, Murphy RC, et al. Limitations of conventionally derived chronic liver disease mortality rates: Results of a comprehensive assessment. Hepatology. 2008;47:1150-7.
157. Missiha SB, Ostrowski M, Heathcote EJ. Disease progression in chronic hepatitis C: Modifiable and nonmodifiable factors. Gastroenterology. 2008;134:1699-714.
158. Bruno S, Stroffolini T, Colombo M, et al. Sustained virological response to interferon-alpha is associated with improved outcome in HCV-related cirrhosis: A retrospective study. Hepatology. 2007;45:579-87.
159. Poynard T, Ratziu V, Charlotte F, et al. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis c. J Hepatol. 2001;34:730-9.
160. Bissell DM. Sex and hepatic fibrosis. Hepatology. 1999;29:988-9.
161. Di Martino V, Lebray P, Myers RP, et al. Progression of liver fibrosis in women infected with hepatitis C: Long-term benefit of estrogen exposure. Hepatology. 2004;40:1426-33.
162. Codes L, Asselah T, Cazals-Hatem D, et al. Liver fibrosis in women with chronic hepatitis C: Evidence for the negative role of the menopause and steatosis and the potential benefit of hormone replacement therapy. Gut. 2007;56:390-5.
163. Wiley TE, Brown J, Chan J. Hepatitis C infection in African Americans: Its natural history and histological progression. Am J Gastroenterol. 2002;97:700-6.
164. Sterling RK, Stravitz RT, Luketic VA, et al. A comparison of the spectrum of chronic hepatitis C virus between Caucasians and African Americans. Clin Gastroenterol Hepatol. 2004;2:469-73.
165. Kim WR, Gross JB, Poterucha JJ, et al. Outcome of hospital care of liver disease associated with hepatitis C in the United States. Hepatology. 2001;33:201-6.
166. Rodriguez-Torres M. Latinos and chronic hepatitis C: A singular population. Clin Gastroenterol Hepatol. 2008;6:484-90.
167. Donato F, Tagger A, Gelatti U, et al. Alcohol and hepatocellular carcinoma: The effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol. 2002;155:323-31.
168. Anand BS, Currie S, Dieperink E, et al. Alcohol use and treatment of hepatitis C virus: Results of a national multicenter study. Gastroenterology. 2006;130:1607-16.
169. Harris DR, Gonin R, Alter HJ, et al. The relationship of acute transfusion-associated hepatitis to the development of cirrhosis in the presence of alcohol abuse. Ann Intern Med. 2001;134:120-4.
170. Hutchinson SJ, Bird SM, Goldberg DJ. Influence of alcohol on the progression of hepatitis C virus infection: A meta-analysis. Clin Gastroenterol Hepatol. 2005;3:1150-9.
171. Monto A, Patel K, Bostrom A, et al. Risks of a range of alcohol intake on hepatitis C-related fibrosis. Hepatology. 2004;39:826-34.
172. Pessione F, Ramond MJ, Njapoum C, et al. Cigarette smoking and hepatic lesions in patients with chronic hepatitis C. Hepatology. 2001;34:121-5.
173. Hezode C, Zafrani ES, Roudot-Thoraval F, et al. Daily cannabis use: A novel risk factor of steatosis severity in patients with chronic hepatitis C. Gastroenterology. 2008;134:432-9.
174. Gary-Bobo M, Elachouri G, Gallas JF, et al. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology. 2007;46:122-9.
175. Moucari R, Asselah T, Cazals-Hatem D, et al. Insulin resistance in chronic hepatitis C: Association with genotypes 1 and 4, serum HCV RNA level, and liver fibrosis. Gastroenterology. 2008;134:416-23.
176. Sheikh MY, Choi J, Qadri I, et al. Hepatitis C virus infection: Molecular pathways to metabolic syndrome. Hepatology. 2008;47:2127-33.
177. Lonardo A, Adinolfi LE, Loria P, et al. Steatosis and hepatitis C virus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. 2004;126:586-97.
178. Powell EE, Jonsson JR, Clouston AD. Steatosis: Co-factor in other liver diseases. Hepatology. 2005;42:5-13.
179. Kawaguchi T, Yoshida T, Harada M, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165:1499-508.
180. Hu KQ, Currie SL, Shen H, et al. Clinical implications of hepatic steatosis in patients with chronic hepatitis C: A multicenter study of U.S. veterans. Dig Dis Sci. 2007;52:570-8.
181. Hickman IJ, Clouston AD, Macdonald GA, et al. Effect of weight reduction on liver histology and biochemistry in patients with chronic hepatitis C. Gut. 2002;51:89-94.
182. Sharma P, Balan V, Hernandez J, et al. Hepatic steatosis in hepatitis C virus genotype 3 infection: Does it correlate with body mass index, fibrosis, and HCV risk factors? Dig Dis Sci. 2004;49:25-9.
183. Patton HM, Patel K, Behling C, et al. The impact of steatosis on disease progression and early and sustained treatment response in chronic hepatitis C patients. J Hepatol. 2004;40:484-90.
184. Hanouneh IA, Feldstein AE, Lopez R, et al. Clinical significance of metabolic syndrome in the setting of chronic hepatitis C virus infection. Clin Gastroenterol Hepatol. 2008;6:584-9.
185. Smith BC, Gorve J, Guzail MA, et al. Heterozygosity for hereditary hemochromatosis is associated with more fibrosis in chronic hepatitis C. Hepatology. 1998;27:1695-9.
186. Kazemi-Shirazi L, Datz C, Maier-Dobersberger T, et al. The relation of iron status and hemochromatosis gene mutations in patients with chronic hepatitis C. Gastroenterology. 1999;116:127-34.
187. Di Bisceglie AM, Bonkovsky HL, Chopra S, et al. Iron reduction as an adjuvant to interferon therapy in patients with chronic hepatitis C who have previously not responded to interferon: A multicenter, prospective, randomized, controlled trial. Hepatology. 2000;32:135-8.
188. Huang H, Shiffman ML, Cheung RC, et al. Identification of two gene variants associated with risk of advanced fibrosis in patients with chronic hepatitis C. Gastroenterology. 2006;130:1679-87.
189. Pradat P, Alberti A, Poynard T, et al. Predictive value of ALT levels for histologic findings in chronic hepatitis C: A European collaborative study. Hepatology. 2002;36:973-7.
190. Quinti I, Pierdominici M, Marziali M, et al. European surveillance of immunoglobulin safety—results of initial survey of 1243 patients with primary immunodeficiencies in 16 countries. Clin Immunol. 2002;104:231-6.
191. Wiesner RH, Sorrell M, Villamil F. Report of the first International Liver Transplantation Society expert panel consensus conference on liver transplantation and hepatitis C. Liver Transpl. 2003;9:S1-9.
192. Berenguer M, Prieto M, Rayon JM, et al. Natural history of clinically compensated hepatitis C virus-related graft cirrhosis after liver transplantation. Hepatology. 2000;32:852-8.
193. Thuluvath PJ, Krok KL, Segev DL, Yoo HY. Trends in post-liver transplant survival in patients with hepatitis C between 1991 and 2001 in the United States. Liver Transpl. 2007;13:719-24.
194. McCashland T, Watt K, Lyden E, et al. Retransplantation for hepatitis C: Results of a U.S. multicenter retransplant study. Liver Transpl. 2007;13:1246-53.
195. Gasink LB, Blumberg EA, Localio AR, et al. Hepatitis C virus seropositivity in organ donors and survival in heart transplant recipients. JAMA. 2006;296:1843-50.
196. Wells JT, Lucey MR, Said A. Hepatitis C in transplant recipients of solid organs, other than liver. Clin Liver Dis. 2006;10:901-17.
197. Kalia H, Lopez PM, Martin P. Treatment of HCV in patients with renal failure. Arch Med Res. 2007;38:628-33.
198. Fabrizi F, Martin P, Dixit V, et al. Meta-analysis: Effect of hepatitis C virus infection on mortality in dialysis. Aliment Pharmacol Ther. 2004;20:1271-7.
199. Fabrizi F, Dulai G, Dixit V, et al. Meta-analysis: Interferon for the treatment of chronic hepatitis C in dialysis patients. Aliment Pharmacol Ther. 2003;18:1071-81.
200. Fabrizi F, Martin P, Ponticelli C. Hepatitis C virus infection and renal transplantation. Am J Kidney Dis. 2001;38:919-34.
201. Cano O, Almenar L, Martinez-Dolz L, et al. Course of patients with chronic hepatitis C virus infection undergoing heart transplantation. Transplant Proc. 2007;39:2353-4.
202. Peffault de Latour R, Levy V, Asselah T, et al. Long-term outcome of hepatitis C infection after bone marrow transplantation. Blood. 2004;103:1618-24.
203. Peffault de Latour R, Asselah T, Levy V, et al. Treatment of chronic hepatitis C virus in allogeneic bone marrow transplant recipients. Bone Marrow Transplant. 2005;36:709-13.
204. Thomas DL. The challenge of hepatitis C in the HIV-infected person. Annu Rev Med. 2008;59:473-85.
205. Salmon-Ceron D, Lewden C, Morlat P, et al. Liver disease as a major cause of death among HIV infected patients: Role of hepatitis C and B viruses and alcohol. J Hepatol. 2005;42:799-805.
206. Goedert JJ, Eyster ME, Lederman MM, et al. End-stage liver disease in persons with hemophilia and transfusion-associated infections. Blood. 2002;100:1584-9.
207. Lin W, Weinberg EM, Tai AW, et al. HIV increases HCV replication in a TGF-beta-1-dependent manner. Gastroenterology. 2008;134:803-11.
208. Balagopal A, Philp FH, Astemborski J, et al. Human immunodeficiency virus-related microbial translocation and progression of hepatitis C. Gastroenterology. 2008;135:226-33.
209. Tuyama A, Hong F, Schecter AD, et al. HIV entry and replication in stellate cells promotes cellular activation and fibrogenesis: Implications for hepatic fibrosis in HIV/HCV co-infection [abstract]. Hepatology. 2007;46:ALB3.
210. Sterling RK, Contos MJ, Smith PG, et al. Steatohepatitis: Risk factors and impact on disease severity in human immunodeficiency virus/hepatitis C virus coinfection. Hepatology. 2008;47:1118-27.
211. Van Den Berg C, Smit C, Van Brussel G, et al. Full participation in harm reduction programs is associated with decreased risk for human immunodeficiency virus and hepatitis C virus: Evidence from the Amsterdam Cohort Studies among drug users. Addiction. 2007;102:1454-62.
212. Wicker S, Cinatl J, Berger A, et al. Determination of risk of infection with blood-borne pathogens following a needlestick injury in hospital workers. Ann Occup Hyg. 2008;52:615-22.
213. Yazdanpanah Y, De Carli G, Migueres B, et al. Risk factors for hepatitis C virus transmission to health care workers after occupational exposure: A European case-control study. Clin Infect Dis. 2005;41:1423-30.
214. Chung H, Kudo M, Kumada T, et al. Risk of HCV transmission after needlestick injury, and the efficacy of short-duration interferon administration to prevent HCV transmission to medical personnel. J Gastroenterol. 2003;38:877-9.
215. Kamal SM, Ismail A, Graham CS, et al. Pegylated interferon alpha therapy in acute hepatitis C: Relation to hepatitis C virus-specific T cell response kinetics. Hepatology. 2004;39:1721-31.
216. Kamal SM, Moustafa KN, Chen J, et al. Duration of peginterferon therapy in acute hepatitis C: A randomized trial. Hepatology. 2006;43:923-31.
217. Kamal SM, Fouly AE, Kamel RR, et al. Peginterferon alfa-2b therapy in acute hepatitis C: Impact of onset of therapy on sustained virologic response. Gastroenterology. 2006;130:632-8.
218. Davis GL, Nelson DR, Terrault N, et al. A randomized, open-label study to evaluate the safety and pharmacokinetics of human hepatitis C immune globulin (Civacir) in liver transplant recipients. Liver Transpl. 2005;11:941-9.
219. Schiano TD, Charlton M, Younossi Z, et al. Monoclonal antibody HCV-AbXTL68 in patients undergoing liver transplantation for HCV: Results of a phase 2 randomized study. Liver Transpl. 2006;12:1381-9.
220. Wintermeyer P, Wands JR. Vaccines to prevent chronic hepatitis C virus infection: Current experimental and preclinical developments. J Gastroenterol. 2007;42:424-32.
221. Chua BY, Eriksson EM, Brown LE, et al. A self-adjuvanting lipopeptide-based vaccine candidate for the treatment of hepatitis C virus infection. Vaccine. 2008;26:4866-75.
222. Hadziyannis SJ, Sette H, Morgan TR, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140:346-55.
223. Soriano V, Maida I, Nunez M, et al. Long-term follow-up of HIV-infected patients with chronic hepatitis C virus infection treated with interferon-based therapies. Antivir Ther. 2004;9:987-92.
224. Everson GT, Trotter J, Forman L, et al. Treatment of advanced hepatitis C with a low accelerating dosage regimen of antiviral therapy. Hepatology. 2005;42:255-62.
225. Formann E, Steindl-Munda P, Hofer H, et al. Long-term follow-up of chronic hepatitis C patients with sustained virological response to various forms of interferon-based anti-viral therapy. Aliment Pharmacol Ther. 2006;23:507-11.
226. Desmond CP, Roberts SK, Dudley F, et al. Sustained virological response rates and durability of the response to interferon-based therapies in hepatitis C patients treated in the clinical setting. J Viral Hepat. 2006;13:311-15.
227. Yu ML, Dai CY, Chen SC, et al. High versus standard doses interferon-alpha in the treatment of naive chronic hepatitis C patients in Taiwan: A 10-year cohort study. BMC Infect Dis. 2005;5:27.
228. Tsuda N, Yuki N, Mochizuki K, et al. Long-term clinical and virological outcomes of chronic hepatitis C after successful interferon therapy. J Med Virol. 2004;74:406-13.
229. Abdelmalek MF, Firpi RJ, Soldevila-Pico C, et al. Sustained viral response to interferon and ribavirin in liver transplant recipients with recurrent hepatitis C. Liver Transpl. 2004;10:199-207.
230. Maylin S M-PM, Moucari R, Boyer N, et al. Eradication of hepatitis C virus in patients successfully treated for chronic hepatitis C. Gastroenterology. 2008;135:821-9.
231. Veldt BJ, Heathcote EJ, Wedemeyer H, et al. Sustained virologic response and clinical outcomes in patients with chronic hepatitis C and advanced fibrosis. Ann Intern Med. 2007;147:677-84.
232. Poordad F, Reddy KR, Martin P. Rapid virologic response: A new milestone in the management of chronic hepatitis C. Clin Infect Dis. 2008;46:78-84.
233. Davis GL. Monitoring of viral levels during therapy of hepatitis C. Hepatology. 2002;36:S145-51.
234. Chung RT, Gale MJr., Polyak SJ, et al. Mechanisms of action of interferon and ribavirin in chronic hepatitis C: Summary of a workshop. Hepatology. 2008;47:306-20.
235. Sulkowski M, Lawitz E, Shiffman ML, et al. Final results of the IDEAL (individualized dosing efficacy versus fixed dosing to assess optimal pegylated interferon therapy) phase IIIb study [abstract]. J Hepatol. 2008;48:A991.
236. Fried MW, Jensen DM, Rodriguez-Torres M, et al. Improved outcomes in patients with hepatitis C with difficult-to-treat characteristics: Randomized study of higher doses of peginterferon alpha-2a and ribavirin. Hepatology. 2008;48:1033-43.
237. Shiffman ML, Salvatore J, Hubbard S, et al. Treatment of chronic hepatitis C virus genotype 1 with peginterferon, ribavirin, and epoetin alpha. Hepatology. 2007;46:371-9.
238. von Wagner M, Huber M, Berg T, et al. Peginterferon-alpha-2a (40KD) and ribavirin for 16 or 24 weeks in patients with genotype 2 or 3 chronic hepatitis C. Gastroenterology. 2005;129:522-7.
239. Mangia A, Santoro R, Minerva N, et al. Peginterferon alfa-2b and ribavirin for 12 vs. 24 weeks in HCV genotype 2 or 3. N Engl J Med. 2005;352:2609-17.
240. Dalgard O, Mangia A. Short-term therapy for patients with hepatitis C virus genotype 2 or 3 infection. Drugs. 2006;66:1807-15.
241. Shiffman ML, Suter F, Bacon BR, et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med. 2007;357:124-34.
242. Yu ML, Dai CY, Huang JF, et al. Rapid virological response and treatment duration for chronic hepatitis C genotype 1 patients: A randomized trial. Hepatology. 2008;47:1884-93.
243. Ferenci P, Laferl H, Scherzer TM, et al. Peginterferon alfa-2a and ribavirin for 24 weeks in hepatitis C type 1 and 4 patients with rapid virological response. Gastroenterology. 2008;135:451-8.
244. Pearlman B, Ehleben C, Saifee S. Treatment extension to 72 weeks of peginterferon and ribavirin in hepatitis c genotype 1-infected slow responders. Hepatology. 2007;46:1671-4.
245. Sanchez-Tapias JM, Diago M, Escartin P, et al. Peginterferon-alfa2a plus ribavirin for 48 versus 72 weeks in patients with detectable hepatitis C virus RNA at week 4 of treatment. Gastroenterology. 2006;131:451-60.
246. Mangia A, Minerva N, Bacca D, et al. Individualized treatment duration for hepatitis C genotype 1 patients: A randomized controlled trial. Hepatology. 2008;47:43-50.
247. Zeuzem S, Hultcrantz R, Bourliere M, et al. Peginterferon alfa-2b plus ribavirin for treatment of chronic hepatitis C in previously untreated patients infected with HCV genotypes 2 or 3. J Hepatol. 2004;40:993-9.
248. Conjeevaram HS, Fried MW, Jeffers LJ, et al. Peginterferon and ribavirin treatment in African American and Caucasian American patients with hepatitis C genotype 1. Gastroenterology. 2006;131:470-7.
249. Jeffers LJ, Cassidy W, Howell CD, et al. Peginterferon alfa-2a (40 kd) and ribavirin for black American patients with chronic HCV genotype 1. Hepatology. 2004;39:1702-8.
250. Muir AJ, Bornstein JD, Killenberg PG. Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-Hispanic whites. N Engl J Med. 2004;350:2265-71.
251. Gross J, Johnson S, Kwo P, et al. Double-dose peginterferon alfa-2b with weight based ribavirin improves response for interferon/ribavirin non-responders with hepatitis C: Final results of “renew” [abstract]. Hepatology. 2005;42:A60.
252. Everson GT, Hoefs JC, Seeff LB, et al. Impact of disease severity on outcome of antiviral therapy for chronic hepatitis C: Lessons from the HALT-C trial. Hepatology. 2006;44:1675-84.
253. Reddy KR, Shiffman ML, Morgan TR, et al. Impact of ribavirin dose reductions in hepatitis C virus genotype 1 patients completing peginterferon alfa-2a/ribavirin treatment. Clin Gastroenterol Hepatol. 2007;5:124-9.
254. Shiffman ML, Ghany MG, Morgan TR, et al. Impact of reducing peginterferon alfa-2a and ribavirin dose during retreatment in patients with chronic hepatitis C. Gastroenterology. 2007;132:103-12.
255. Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology. 2002;36:S237-44.
256. Afdhal NH, Dieterich DT, Pockros PJ, et al. Epoetin alfa maintains ribavirin dose in HCV-infected patients: A prospective, double-blind, randomized controlled study. Gastroenterology. 2004;126:1302-11.
257. Jacobson IM, Gonzalez SA, Ahmed F, et al. A randomized trial of pegylated interferon alpha-2b plus ribavirin in the retreatment of chronic hepatitis C. Am J Gastroenterol. 2005;100:2453-62.
258. Shiffman ML, Di Bisceglie AM, Lindsay KL, et al. Peginterferon alfa-2a and ribavirin in patients with chronic hepatitis C who have failed prior treatment. Gastroenterology. 2004;126:1015-23.
259. Taliani G, Gemignani G, Ferrari C, et al. Pegylated interferon alfa-2b plus ribavirin in the retreatment of interferon-ribavirin nonresponder patients. Gastroenterology. 2006;130:1098-106.
260. Bacon B, Regev A, Ghalib R, et al. The DIRECT trial (daily-dose consensus interferon and ribavirin: Efficacy of combined therapy): Treatment of non-responders to previous pegylated interfereon plus ribavirin: Sustained virologic response data [abstract]. Hepatology. 2007;46:A168.
261. Jensen DM, Freilich B, Andreone P, et al. Pegylated interferon alfa-2a (40KD) plus ribavirin (RBV) in prior non-responders to pegylated interferon alfa-2a (12KD)/RBV): Final efficacy and safety outcomes of the repeat study [abstract]. Hepatology. 2007;46:ALB4.
262. Afdhal N, Levine R, Brown R, et al. Colchicine versus Peg-Interferon alpha 2B long term therapy: Results of the 4 year COPILOT trial [abstract]. J Hepatol. 2008;48:A3.
263. Pineda JA, Garcia-Garcia JA, Aguilar-Guisado M, et al. Clinical progression of hepatitis C virus-related chronic liver disease in human immunodeficiency virus-infected patients undergoing highly active antiretroviral therapy. Hepatology. 2007;46:622-30.
264. Chung RT, Andersen J, Volberding P, et al. Peginterferon Alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. N Engl J Med. 2004;351:451-9.
265. Torriani FJ, Rodriguez-Torres M, Rockstroh JK, et al. Peginterferon Alfa-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. N Engl J Med. 2004;351:438-50.
266. Carrat F, Bani-Sadr F, Pol S, et al. Pegylated interferon alfa-2b vs standard interferon alfa-2b, plus ribavirin, for chronic hepatitis C in HIV-infected patients: A randomized controlled trial. JAMA. 2004;292:2839-48.
267. Nunez M, Miralles C, Berdun MA, et al. Role of weight-based ribavirin dosing and extended duration of therapy in chronic hepatitis C in HIV-infected patients: The PRESCO trial. AIDS Res Hum Retroviruses. 2007;23:972-82.
268. Shea DO, Tuite H, Farrell G, et al. Role of rapid virological response in prediction of sustained virological response to Peg-IFN plus ribavirin in HCV/HIV co-infected individuals. J Viral Hepat. 2008;15:482-9.
269. Sulkowski MS. Viral hepatitis and HIV coinfection. J Hepatol. 2008;48:353-67.
270. Terrault NA, Berenguer M. Treating hepatitis C infection in liver transplant recipients. Liver Transpl. 2006;12:1192-204.
271. Lodato F, Berardi S, Gramenzi A, et al. Peg-interferon alfa-2b and ribavirin for the treatment of genotype 1 hepatitis C recurrence after liver transplantation. Aliment Pharmacol Ther. 2008;28:450-7.
272. Hanouneh IA, Miller C, Aucejo F, et al. Recurrent hepatitis C after liver transplantation: On-treatment prediction of response to peginterferon/ribavirin therapy. Liver Transpl. 2008;14:53-8.
273. Berenguer M, Palau A, Aguilera V, et al. Clinical benefits of antiviral therapy in patients with recurrent hepatitis C following liver transplantation. Am J Transplant. 2008;8:679-87.
274. Kieffer TL, Sarrazin C, Miller JS, et al. Telaprevir and pegylated interferon-alpha-2a inhibit wild-type and resistant genotype 1 hepatitis C virus replication in patients. Hepatology. 2007;46:631-9.
275. McHutchison JG, Everson GT, Gordon SC, et al. PROVE1: Results from a phase 2 study of telaprevir with peginterferon alpha-2a and ribavirin in treatment-naive subjects with hepatitis C [abstract]. J Hepatol. 2008;48:A4.
276. Dusheiko GM, Hezode C, Pol S, et al. Treatment of chronic hepatitis C with telaprevir (TVR) in combination with peginterferon-alpha-2a with or without ribavirin: Further interim analysis results of the PROVE2 study [abstract]. J Hepatol. 2008;48:A58.
277. Kwo P, Lawitz KP, McCone J, et al. Interim resutls from HCV SPRINT-1: RVR/EVR from phase 2 study of boceprevir plus pegintron (peginterferon alpha-2b)/ribavirin in treatment-naive subjects with genotype-1 CHC [abstract]. J Hepatol. 2008;48:A995.
278. Pockros PJ, Nelson D, Godofsky E, et al. R1626 plus peginterferon Alfa-2a provides potent suppression of hepatitis C virus RNA and significant antiviral synergy in combination with ribavirin. Hepatology. 2008;48:385-97.
279. Lalezari J, Gane E, Rodriguez-Torres M, et al. Potent antiviral activity of the HCV nucleoside polymerase inhibitor R7128 500 mg BID for 28 days [abstract]. J Hepatol. 2008;48:A66.
280. Flisiak R, Feinman SV, Jablkowski M, et al. Efficacy and safety of increasing doses of the cyclophilin inhibitor DEBIO 025 in combination with pegylated interferon alpha-1a in treatment naive chronic HCV patients [abstract]. J Hepatol. 2008;48:A143.