[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 77 Hepatitis A
The development of liver biopsy techniques in the 1930s allowed the recognition of hepatic necroinflammation that characterizes all forms of viral hepatitis. Subsequent experimental work in humans led to the clinical recognition that viruses are etiologic agents of hepatitis A (“infectious hepatitis”) and hepatitis B (“serum hepatitis”).1,2 Later, the existence of two hepatitis viruses was demonstrated—hepatitis A virus (HAV) and hepatitis B virus (HBV) (see Chapter 78).3 Additional viral causes of acute and chronic hepatitis were identified subsequently (see Chapters 79 to 81). HAV was first characterized in 1973, when scientists detected the virus in stools from human volunteers who were infected with HAV.4 The ensuing development of sensitive and specific serologic assays for the diagnosis of HAV infection and the isolation of HAV in cell culture5 were important advances that permitted understanding of the epidemiology of HAV infection and, ultimately, control of the disease.
VIROLOGY
In 1982, HAV was classified as an enterovirus type 72 belonging to the Picornaviridae family. Subsequent determination of the sequence of HAV nucleotides and amino acids led to questioning of this classification, and a new genus, Hepatovirus, was created for HAV.6
HAV has an icosahedral shape and lacks an envelope. It measures 27 to 28 nm in diameter, has a buoyant density of 1.33 to 1.34 g/cm3 in cesium chloride, and has a sedimentation coefficient of 156 to 160S on ultracentrifugation. HAV survives exposure to ether and an acid environment at pH 3. It also survives heat exposure of 60°C for 60 minutes but is inactivated at 85°C for 1 minute. HAV is capable of surviving in sea water (4% survival rate), in dried feces at room temperature for 4 weeks (17% survival), and in live oysters for 5 days (12% survival).7
HAV has only one known serotype, and no antigenic cross-reactivity with the hepatitis B, C, D, E, or G agents. The HAV genome consists of a positive-sense RNA that is 7.48 kb long, single-stranded, and linear (Fig. 77-1). HAV RNA has a sedimentation coefficient of 32 to 33S and a molecular weight of 2.8 × 10.4 The HAV RNA has a long open reading frame, consisting of 6681 nucleotides, and is covalently linked to a 5′ terminal protein and a 3′ terminal polyadenosine tract.
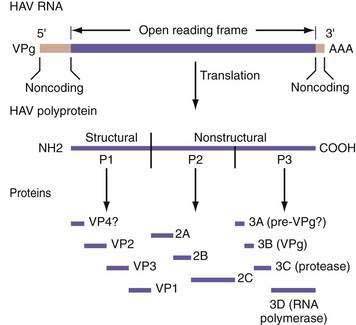
Figure 77-1. Genomic organization of hepatitis A virus (HAV). VP, viral protein; VPg, 5′ terminal protein.
(From Levine JE, Bull FG, Millward-Sadler GH, et al. Acute viral hepatitis. In: Millward-Sadler GH, Wright R, Arther MJP, editors. Wright’s Liver and Biliary Disease, 3rd ed. London: WB Saunders, 1992. p 679.)
The onset of HAV replication in cell culture systems takes from weeks to months. Primate cells, including African green monkey kidney cells, primary human fibroblasts, human diploid cells (MRC-5), and fetal rhesus kidney cells, are favored for cultivation of HAV in vitro. The virus is not cytopathic, and persistent infection in the cell cultures is the rule. Two conditions control the outcome of HAV replication in cell culture.8 The first condition is the genetic makeup of the virus; HAV strains mutate in distinct regions of the viral genome as they become adapted to cell culture. The second condition is the metabolic activity of the host cell at the time of infection. Cells in culture, although infected simultaneously, initiate HAV replication in an asynchronous manner. This asynchronicity may be caused by differences in the metabolic activity of individual cells, but definitive evidence of cell-cycle dependence of HAV replication is lacking.9
An initial step in the life cycle of a virus is its attachment to a cell surface receptor. The location and function of these receptors determine tissue tropism. Little is known about the mechanism of entry of HAV into cells. Some work has suggested that HAV could infect cells by a surrogate-receptor binding mechanism (involving a nonspecified serum protein). HAV infectivity in tissue culture has been shown to require calcium and to be inhibited by the treatment of the cells with trypsin, phospholipases, and β-galactosidase.10 A surface glycoprotein, named HAVcr-1, on African green monkey kidney cells has been identified as a receptor for HAV. Blocking of HAVcr-1 with specific monoclonal antibodies prevents infection of otherwise susceptible cells. Experimental data suggest that HAVcr-1 not only serves as an attachment receptor but also may facilitate uncoating of HAV and its entry into hepatocytes.11
Whatever the entry mechanism, once HAV enters a cell, the viral RNA is uncoated, cell host ribosomes bind to viral RNA, and polysomes are formed. HAV is translated into a large polyprotein of 2227 amino acids. This polyprotein is organized into three regions: P1, P2, and P3. The P1 region encodes structural proteins VP1, VP2, VP3, and a putative VP4. The P2 and P3 regions encode nonstructural proteins associated with viral replication (see Fig. 77-1).
The HAV RNA polymerase copies the plus RNA strand. The RNA transcript in turn is used for translation into proteins, which are used for assembly into mature virions. Down-regulation of HAV RNA synthesis appears to occur as defective HAV particles appear.12 In addition, a group of specific RNA-binding proteins has been observed during persistent infection.13 The origin and nature of these proteins is unknown, but they exert activity on the RNA template and are believed to play a regulatory role in the replication of HAV.14
Numerous strains of HAV exist, with considerable nucleotide sequence variability (15% to 25% difference within the P1 region of the genome). Human HAV strains can be grouped into four different genotypes (I, II, III, and VII), whereas simian strains of HAV belong to genotypes IV, V, and VI.15 Despite the nucleotide sequence heterogeneity, the antigenic structure of human HAV is highly conserved among strains.
The HAV VP1/2A and 2C genes are thought to be responsible for viral virulence. This conclusion is based on experiments in which the genotypes and phenotypes of viruses were compared after animals were infected with one of 14 chimeric virus genomes derived from two infectious cDNA clones that encoded a virulent HAV isolate and an attenuated HAV isolate (HM175 strain), respectively.16
Variations in the HAV genome are thought to play a role in the development of fulminant hepatic failure (FHF) during acute HAV infection. The 5′ untranslated region of the HAV genome was sequenced in serum samples from 84 patients with HAV infection, including 12 with FHF.17 The investigators observed fewer nucleotide substitutions in the HAV genome from patients with FHF than in those from patients without FHF (P < .001). The differences were most prominent between nucleotides 200 and 500, suggesting that nucleotide variations in the central portion of the 5′ untranslated region influence the clinical severity of HAV infection.
EPIDEMIOLOGY
Acute hepatitis A is a reportable infectious disease in the United States. The incidence has declined by 90% since 1995. In 2006, 3579 cases of HAV infection were reported, corresponding to a rate of infection of 1.2 per 100,000, compared with 4 per 100,000 in 2001. With the underreporting of cases and the occurrence of asymptomatic infections taken into consideration, the true number of HAV infections in 2006 was calculated to be 32,000, compared with 93,000 in 2001. The greatest rate of decline has been among children from states where routine vaccination of children was recommended in 1999. The highest rate of reported disease historically has been among children ages 5 to 14 years. Because of the rapid rate of decline of disease in children, however, rates are now similar among age groups, with adults ages 20 to 44 having the highest rate of disease in 2006.18
The epidemiologic risk factors for HAV infection reported for the U.S. population in 2006 were as follows: unknown, 65%; international travel, 15%; contact with a patient who has hepatitis, 12%; sexual or household contact with a patient who has hepatitis A, 10%; men having sex with men, 9%; food or waterborne outbreak, 7%; child or employee in a daycare center, 4%; contact with a daycare child or employee, 4%; and injection drug use, 2%.18
HAV infection generally follows one of three epidemiologic patterns.19 In countries where sanitary conditions are poor, most children are infected at an early age. Although earlier seroepidemiologic studies routinely showed that 100% of preschool children in these countries had detectable antibody to HAV (anti-HAV) in serum, presumably reflecting previous subclinical infection, subsequent studies have shown that the average age of infection has risen rapidly to 5 years and older, when symptomatic infection is more likely. For example, 82% of 1393 Bolivian school children were shown to have detectable anti-HAV, but when they were stratified into two groups according to family income, a significant difference was found between the groups: 95% of children from low-income families, but only 56% of children from high-income families, had detectable anti-HAV.20
The second epidemiologic pattern is seen in industrialized countries, where the prevalence of HAV infection is low among children and young adults. In the United States, prior to universal HAV vaccination, the prevalence of anti-HAV was approximately 10% in children but 37% in adults.21
Whatever the epidemiologic pattern, the primary route of transmission of HAV is the fecal-oral route, by either person-to-person contact or ingestion of contaminated food or water. Although rare, transmission of HAV by a parenteral route has been documented after transfusion of blood22,23 or blood products.24 Cyclical outbreaks among users of injection and noninjection illicit drugs and among men who have sex with men (up to 10% may become infected in outbreak years) have been reported.25 Table 77-1 provides information about the detection of HAV and its infectivity in human body fluids.26–33
Table 77-1 Detection of Hepatitis A Virus (HAV) and Infectivity of Human Secretions or Excretions
| SECRETION/EXCRETION | COMMENT | REFERENCES |
|---|---|---|
| Stool | Main source of infection. HAV is detectable during the incubation period and for several weeks after the onset of disease. After the onset of symptoms, HAV is detectable in 45% and 11% of fecal specimens collected during the first and second weeks of illness, respectively, whereas HAV RNA (by polymerase chain reaction assay) is detectable for 4 to 5 months. | 26,27 |
| Blood | Viremia is present during the incubation period. Blood collected 3 and 11 days before the onset of symptoms has caused post-transfusion infection in recipients. Chronic viremia does not occur. | 28,29 |
| Bile | HAV has been detected in the bile of chimpanzees infected with HAV. | 30 |
| Urine | HAV is detected in low titer during the viremic phase. A urine sample was reported to be infectious after oral inoculation. Urine contaminated with blood was also infectious. | 31,32 |
| Nasopharyngeal secretions | Unknown in humans. HAV has been identified in the oropharynx of experimentally infected chimpanzees. | 33 |
| Semen, vaginal fluid | Uncertain. HAV may be detectable during the viremic phase. | — |
From 11% to 22% of patients with acute hepatitis A require hospitalization, with an average length of stay of 4.6 days, costing on average $7926 per patient in 2004. In one outbreak involving 43 persons, the total cost was approximately $800,000. On average, 27 workdays are lost per adult case of hepatitis A. In adolescents and adults, the combined direct and indirect costs associated with HAV infection in the United States totaled approximately $488.8 million in 1997, compared with $93 million in 2006.25,34,35 The decline in costs is a direct result of the dramatic reduction in the number of infections seen since the introduction of the HAV vaccine combined with changes in vaccination policies in the United States (see later).18
PATHOGENESIS
After HAV is ingested and survives gastric acid, it traverses the small intestinal mucosa and reaches the liver via the portal vein. The precise mechanism of hepatic uptake in humans is unknown (see earlier). In an experimental model using African green monkey kidney cells,11 the putative cellular receptor for HAV has been identified as a surface glycoprotein. Once the virus enters the hepatocyte, it starts replicating in the cytoplasm, where it is seen on electron microscopy as a fine granular pattern, but it is not present in the nucleus. HAV is distributed throughout the liver. Although HAV antigen has been detected in other organs (lymph nodes, spleen, kidney), the virus appears to replicate exclusively in hepatocytes. When the virus is mature, it reaches the systemic circulation via the hepatic sinusoids and is released into the biliary tree through bile canaliculi, passed into the small intestine, and eventually excreted in the feces.
CLINICAL FEATURES
Infection with HAV does not result in chronic infection, only in an acute self-limited episode of hepatitis. Rarely, acute hepatitis A can have a prolonged or a relapsing course, and occasionally profound cholestasis can occur.36 The incubation period is commonly 2 to 4 weeks, rarely up to 6 weeks. The mortality rate is low in previously healthy persons. Morbidity can be significant in adults and older children.
The clinical characteristics of cases of hepatitis A reported in 2002 were similar to those in previous years, with a preponderance of cases in men in all age groups. Overall, 72% of patients manifested jaundice, 25% required hospitalization, and 0.5 % died.37 The need for hospitalization rose with age, from 5% among children younger than 5 years to 34% among persons 60 years or older.37
Patients with HAV infection usually present with one of the following five clinical patterns: (1) asymptomatic without jaundice, (2) symptomatic with jaundice and self-limited after approximately 8 weeks, (3) cholestatic, with jaundice lasting 10 weeks or more,36 (4) relapsing, with two or more bouts of acute HAV infection occurring over a 6- to 10-week period, and (5) FHF.
Children younger than 2 years are usually asymptomatic; jaundice develops in only 20% of them, whereas symptoms develop in most children (80%) 5 years or older. The rate of symptoms is high in adolescents and adults. HAV infection with prolonged cholestasis is a rare variant but occasionally leads to invasive diagnostic procedures (inappropriately) because the diagnosis of acute hepatitis may not be readily accepted in patients who have jaundice for several months, even in the presence of detectable anti-HAV of the immunoglobulin M (IgM) class (see later).36 A relapsing course is observed in approximately 10% of patients with acute hepatitis A. Shedding of HAV in stool has been documented during the relapse phase.38 This variant is benign, and the infection ultimately resolves.38 Neither the cholestatic variant nor relapsing hepatitis A is associated with an increase in mortality. In all cases, treatment is symptomatic. Acute hepatitis A, unlike hepatitis E, is not associated with a higher mortality rate in pregnant women.
Prodromal symptoms in patients with acute hepatitis A include fatigue, weakness, anorexia, nausea, vomiting, and abdominal pain. Less common symptoms are fever, headache, arthralgias, myalgias, and diarrhea. Dark urine precedes other symptoms in approximately 90% of infected persons; this symptom occurs within 1 to 2 weeks of the onset of prodromal symptoms. Symptoms of hepatitis may last from a few days to 2 weeks and usually decrease with the onset of clinical jaundice. Right upper quadrant tenderness and mild liver enlargement are found on physical examination in 85% of patients; splenomegaly and cervical lymphadenopathy are each present in 15%. Complete clinical recovery is achieved in 60% of affected persons within 2 months and in almost everyone by 6 months. The overall prognosis of acute hepatitis A in otherwise healthy adults is excellent. Potentially fatal complications (e.g., FHF) develop in a few patients (see later).39
Acute HAV infection must be differentiated by appropriate serologic testing from other causes of acute viral hepatitis, autoimmune hepatitis, and other causes of acute hepatitis (see Chapters 20 and 73). In some cases the diagnosis may be difficult to make because the patient may harbor another viral infection, such as chronic hepatitis B or chronic hepatitis C, with superimposed acute HAV infection.
FULMINANT HEPATITIS A
FHF due to HAV is rarely seen in children, adolescents, or young adults. The case-fatality rate in people older than 49 years with acute hepatitis A is reported to be 1.8%, compared with an overall rate of 0.3% in persons of all ages.37 Hepatic failure caused by hepatitis A becomes manifest in the first week of illness in about 55% of affected patients and during the first 4 weeks in 90%; the onset of FHF rarely occurs after 4 weeks of illness.39
The contribution of HAV to acute liver failure has been reported to be greater in populations classified as hyperendemic for HAV. In a report from India, where 276 patients with FHF were seen between 1994 and 1997, 10.6% of the cases among adults were caused by HAV. HAV had been responsible for only 3.5% of cases among 206 patients with FHF seen in the same community from 1978 to 1981.40
Certain populations have increased morbidity and a high risk of acute liver failure from HAV infection. Among these groups are the elderly41 and persons with chronic liver disease and HIV infection. A 1998 report described the clinical outcome of 256 persons hospitalized for acute hepatitis A in Tennessee from January 1994 through December 1995.42 On admission, 89% of the patients had experienced sustained nausea or vomiting, and 26% had a prolonged prothrombin time (>3 seconds); 39 had serious complications (19 hepatobiliary and 20 extrahepatic complications), and 5 (2%) died. Morbidity and mortality correlated with age. Twenty-five percent of patients 40 years and older had at least one complication, compared with 11% of patients younger than 40 years (P = .014).
Although two reports since the late 1990s have described a decline in the number of cases of acute viral hepatitis among patients with FHF in the United States,43,44 this decline is attributable principally to the control of hepatitis B. The contribution of HAV infection to FHF has remained unchanged since the 1970s despite the availability of highly efficacious vaccines (see also Chapter 90).
DIAGNOSIS
Acute hepatitis A is clinically indistinguishable from other forms of viral hepatitis. The diagnosis of infection is based on the detection of specific antibodies against HAV (anti-HAV) in serum (Fig. 77-2). A diagnosis of acute hepatitis A requires demonstration of IgM anti-HAV in serum. The test result is positive from the onset of symptoms47 and usually remains positive for approximately 4 months.48 Some patients may have low levels of detectable IgM anti-HAV for more than a year after the initial infection.48 IgG anti-HAV is also detectable at the onset of the disease, remains present usually for life, and, after clinical recovery, is interpreted as a marker of previous HAV infection (as demonstrated by a positive result on a commercial assay for total anti-HAV and negative result for IgM anti-HAV).
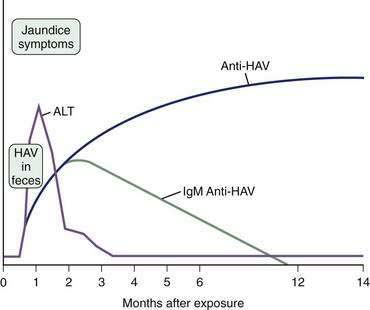
Testing for HAV RNA is limited to research laboratories. HAV RNA has been detected in serum, stool, and liver tissue. Viral RNA can be amplified by polymerase chain reaction (PCR) methodology.49 With a PCR assay, HAV RNA has been documented in human sera for up to 21 days after the onset of illness.50 The use of HCV RNA testing has been described in a report of 76 French patients with acute HAV infection seen between January 1987 and April 2000; 19 had FHF,51 10 of whom required liver transplantation and 1 of whom died while awaiting liver transplantation. The HAV RNA status was determined in 39 of the 50 patients in whom sera and clinical data were available, including the 19 with FHF. HAV RNA was detected in 36 of these 50 patients (72%). The likelihood that HAV RNA was undetectable was greater in patients with FHF than in those with nonfulminant hepatitis (P < .02). Of those in whom HAV RNA was detectable, titers were lower in patients with encephalopathy than in patients with nonfulminant hepatitis (3.6 log vs 4.4 log; P = .02). These data suggest that detection of IgM anti-HAV coupled with nondetection or finding of low-titer HAV RNA in patients with severe acute hepatitis may signal an ominous prognosis and the need for early referral for liver transplantation. As in other studies, HAV genotype did not seem to play a role in the severity of clinical manifestations.52
PREVENTION AND TREATMENT
Recommendations concerning immunoprophylaxis against HAV were published in December 1999 by the Advisory Committee on Immunization Practices (ACIP).25 The overall strategy was to protect persons from disease and to lower the incidence of HAV infection in the United States. The available monovalent vaccines were initially licensed for use in children greater than 2 years of age but are now licensed for use after 12 months of age.25,53 In accordance with the 1999 ACIP guidelines, high-risk populations were targeted for immunization. After achieving a dramatic reduction in incidence rates, a universal childhood vaccination policy was adopted in 2006, with the hope of eventually eliminating indigenous HAV transmission in the United States. The decline in incidence rates not surprisingly has been greater in children than in adults, effectively removing children as a high-risk population and potentially removing the primary reservoir for the virus in the United States.18,25 Table 77-2 lists the populations now considered to be at highest risk of HAV infection.
Table 77-2 Groups at High Risk of Hepatitis A Virus Infection
No specific medications are available to treat acute hepatitis A; symptomatic treatment is the rule. Attention to sanitation and administration of serum immune globulin (IG) have been the mainstays of preventing HAV infection. The availability of excellent HAV vaccines has rendered use of IG for pre-exposure prophylaxis unnecessary. Furthermore, in June 2007, the HAV vaccine was approved for use in postexposure prophylaxis of immunocompetent persons, ages 12 months to 40 years, without chronic liver disease.54 This new indication for the HAV vaccine was based on the results of a study that compared the efficacy of the HAV vaccine with that of IG for postexposure prophylaxis against HAV infection. The rates of clinical infection were low in both groups, with clinical hepatitis A developing in 4.4% of subjects in the vaccine group compared with 3.3% of those in the IG group.55 This study, however, likely excluded persons with asymptomatic infection. In the vaccine group, 162 persons with IgM HAV in serum were excluded, compared with 50 persons in the IG group because of either a lack of symptoms or absence of an elevated serum alanine aminotransferase level of at least two times the upper limit of normal. The possibility exists that a number of persons with asymptomatic hepatitis A still posed an infectious risk to others.
Taking into account data from Canada and the United Kingdom, where the HAV vaccine has been used for postexposure prophylaxis since the early 2000s, the ACIP concluded the HAV vaccine is safe and comparable to IG in protecting recipients against clinical hepatitis A. The ACIP guidelines allow persons who have recently been exposed to HAV and who have not been vaccinated previously to be given a single dose of single-antigen HAV vaccine or IG (0.02 mL/kg) as soon as possible, within 2 weeks of exposure. Some of the benefits of the vaccine include long-term immunity if a second dose of the vaccine is administered in accordance with the standard vaccine schedule (see Table 77-3), as well as cost savings and wide availability compared with IG.54 Although IG is considered safe, the perception is widespread that it poses a risk because it is a blood-derived product. IG can cause fever and myalgias just as the vaccine can, but pain at the injection site is usually more pronounced with IG than with the vaccine. Postexposure prophylaxis with IG can be administered at the same time as initiation of active immunization with the vaccine.56
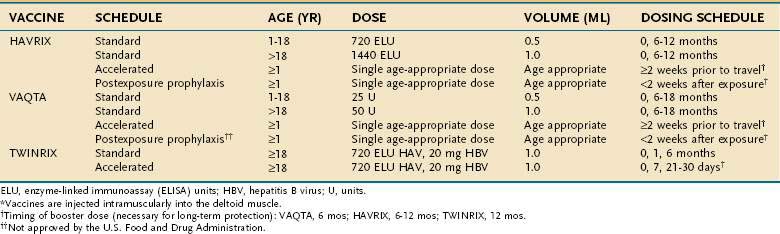
The HAV vaccine was first licensed in the United States in 1995; two inactivated HAV vaccines are commercially available. Extensive use of the vaccines in clinical trials and postmarketing surveillance support the safety and efficacy of these products. HAVRIX is manufactured by SmithKline Biologicals, Rixensart, Belgium, and VAQTA by Merck Sharp & Dohme, West Point, Pennsylvania. Both vaccines are derived from HAV grown in cell culture. The final products are purified and formalin-inactivated; they contain alum as an adjuvant. The basic difference between the two commercially available vaccines is the HAV strain used for preparation. HAVRIX was prepared with the HM175 strain, whereas VAQTA was prepared with the CR326 strain.57,58 The difference is of little practical importance because both vaccines are safe and immunogenic. The doses and schedule of immunization are shown in Table 77-3. After vaccination with HAVRIX, anti-HAV is estimated to remain detectable in serum for approximately 20 years; immunity may last longer.59 From the time the HAV vaccine was licensed in the United States through 2005, more than 50 million doses were administered. Worldwide, more than 188 million doses of HAV vaccine were administered through 2005. Among adults, the most common local side effects have been soreness at the injection site (56%), headache (14%), and malaise (7%). In children, the most common side effects have been soreness at the injection site (15%), feeding problems (8%), headache (4%), and induration at the injection site (4%).25
In the United States, through 2005, the Vaccine Adverse Event Reporting System received 6136 reports of unexplained adverse events after immunization with the HAV vaccine alone or in combination with other vaccines. Of the 6136 reports, 871 were considered serious and included Guillian-Barré syndrome, immune thrombocytopenic purpura, elevated serum aminotransferase levels, and seizures in children.25 No reported serious event, however, could be attributed definitively to the HAV vaccine, and the reported rates did not exceed the expected background rates. For example, the general population incidence of Guillain-Barré syndrome ranges from 0.5 to 2.4 cases per 100,000 person-years, and among adult HAV vaccine recipients the incidence of Guillain-Barré was 0.2 cases per 100,000 person-years.25
A combined formulation of hepatitis A and B vaccines (TWINRIX) is available and has an excellent record of efficacy and safety.60 Although some long-term studies have shown persistence of anti-HAV in children and adolescents, seroconversion rates for TWINRIX are apparently lower in children ages 1 to 6 years than those for standard monovalent vaccines.61 Currently, therefore, TWINRIX is approved only for persons 18 years of age and older.
As a result of the reduction in endemic cases of hepatitis A in the United States, the largest proportion of patients who now become infected with HAV are non-immune adults traveling to endemic areas. Even if medical advice is sought before travel, the time is usually insufficient for completing the standard immunization schedule. HAVRIX and VAQTA are approved by the U.S. Food and Drug Administration (FDA) for use in an accelerated vaccination schedule before planned travel. If given at least 2 weeks before travel, a single dose of either monovalent vaccine results in protective anti-HAV titers.54 In 2008, the FDA also approved an accelerated vaccination schedule for TWINRIX that can be completed within 30 days, with a booster at 12 months, after studies showed equivalent protection when TWINRIX was compared with standard and alternative schedules of the individual monovalent vaccines. After 1 year, HAV seroconversion rates were 100%, and HBV seroconversion rates were 96.4% to 100% with TWINRIX.62,63 The TWINRIX accelerated schedule is being considered for use in new inmates at U.S. correctional facilities, where high-risk activities place the inmates at risk for both HAV and HBV infections.64 The dosing schedules are shown in Table 77-3.
IMMUNIZATION AGAINST HEPATITIS A VIRUS IN PATIENTS WITH CHRONIC ILLNESSES
Persons with chronic liver disease are at increased risk of HAV-related morbidity and mortality if they acquire the infection. Therefore, pre-exposure prophylaxis with the HAV vaccine has been recommended for patients with chronic liver disease who are susceptible to HAV.65 This recommendation should be extended to patients awaiting liver transplantation as well as those who have already undergone liver transplantation, although the immunogenicity of the HAV vaccine is reduced in such persons.66
An episode of acute hepatitis in a patient with underlying chronic liver disease poses the risk of considerable morbidity and mortality. Although the current guidelines recommend immunization against HAV for all patients with chronic liver disease,25 the results of several cost-effectiveness analyses have been conflicting. A report published in 2000 found that saving the life of one patient with HCV infection through HAV vaccination would cost 23 million Canadian dollars,67 although some of the assumptions in this report have been challenged.68 Two other studies of patients with chronic hepatitis C showed a decided benefit to immunization against HAV.69,70 The methods used in these studies were dissimilar, and some analyses may have been insensitive to the incidence of HAV or may have underestimated the economic and societal costs of a case of FHF. Universal immunization against HAV during childhood, before the possible occurrence of chronic liver disease, offers the greatest promise of preventing HAV infection.71
Patients infected with the human immunodeficiency virus (HIV) should be vaccinated against HAV. The response to vaccination, however, may be reduced because of a blunted immune system. Earlier studies suggested seropositivity rates above 97% in HIV-infected children on antiretroviral therapy72; however, a more recent study found that a CD4+ count of less than 25/mm3 and an HIV viral load of more than 400 copies/mL predicted a reduced seroconversion rate.73 Although the discrepancies among studies can be explained in part by different sensitivities of assays for anti-HAV, it appears that the more immunosuppressed a person is, the less likely the person is to respond to vaccination. In this population, consideration should be given to checking post-vaccination IgG anti-HAV titers to assess immunity. Small studies have shown limited additional benefit to a third dose of HAV vaccine in persons who fail to respond to the standard vaccine schedule.73
American Academy of Pediatrics, Committee on Infectious Disease. Hepatitis A vaccine recommendations. Pediatrics. 2007;120:189-99. (Ref 53.)
Centers for Disease Control and Prevention. Hepatitis Surveillance Report No. 59. Atlanta: CDC; 2004. (Ref 37.)
Centers for Disease Control and Prevention. Prevention of hepatitis A thorough active or passive immunization. MMWR. 2006;55(No. RR07):1-23. (Ref 25.)
Centers of Disease Control and Prevention. Prevention of hepatitis A after exposure to hepatitis A virus and in international travelers. MMWR. 2007;56:1080-4. (Ref 54.)
Centers for Disease Control and Prevention. Surveillance for Acute Viral Hepatitis, United States-2006. MMWR. 2008;57(SS02):1-24. (Ref 18.)
Connor BA, Blatter MM, Beran J, et al. Rapid and sustained immune response against hepatitis A and B achieved with combined vaccine using an accelerated administration schedule. J Travel Med. 2007;14:9-15. (Ref 63.)
FDA approval for a combined hepatitis A and B vaccine. MMWR. 2001;50:806. (Ref 60.)
Feinstone SM, Kapikian AZ, Purcell RH. Hepatitis A: Detection by immune electron microscopy of a viruslike antigen associated with acute illness. Science. 1973;182:1026-8. (Ref 4.)
Krugman S, Ward R, Giles JP, et al. Infectious hepatitis: Detection of virus during the incubation period and in clinically inapparent infections. N Engl J Med. 1959;261:729-34. (Ref 3.)
Mathiesen LR, Feinstone SM, Purcell RH, Wagner JA. Detection of hepatitis A antigen by immunofluorescence. Infect Immun. 1977;18:524-30. (Ref 15.)
Nothdurft HD, Dietrich M, Zuckerman JN, et al. A new accelerated vaccination schedule for rapid protection against hepatitis A and B. Vaccine. 2002;20:1157-62. (Ref 62.)
Rezende G, Roque-Alsonso M, Samuel D, et al. Viral and clinical factors associated with fulminant course of hepatitis A infection. Hepatology. 2003;38:613-18. (Ref 51.)
Rosenthal P. Cost-effectiveness of hepatitis A vaccination in children, adolescents and adults. Hepatology. 2003;37:44-51. (Ref 71.)
Victor JC, Monto AS, Surdina TY, et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med. 2007;357:1685-94. (Ref 55.)
Zhou F, Shefer A, Weinbaum C, et al. Impact of hepatitis A vaccination on health care utilization in the United States, 1996-2004. Vaccine. 2007;25:3581-7. (Ref 34.)
1. MacCallum FO, McFarlan AM, Miles JAR, et al, editors. Infective Hepatitis: Studies in East Anglia during the period 1943-1947 Medical Research Council, Special Report No. 273. 1951. HMSO, London: 1
2. Havens WP, Ward R, Drill VA, et al. Experimental production of hepatitis by feeding icterogenic materials. Proc Soc Exp Biol Med. 1944;57:206.
3. Krugman S, Ward R, Giles JP, et al. Infectious hepatitis: Detection of virus during the incubation period and in clinically inapparent infections. N Engl J Med. 1959;261:729-34.
4. Feinstone SM, Kapikian AZ, Purcell RH. Hepatitis A: Detection by immune electron microscopy of a viruslike antigen associated with acute illness. Science. 1973;182:1026-8.
5. Provost PJ, Hilleman MR. An inactivated hepatitis A virus vaccine prepared from infected marmoset liver. Proc Soc Exp Biol Med. 1978;159:201-3.
6. Minor PD. Picornaviridae: Classification and nomenclature of viruses: Fifth Report of the International Committee on Taxonomy of Viruses. Arch Virol Suppl. 1991;2:320.
7. Sobsey MD, Shields PA, Hauchman FS, et al. Survival and persistence of hepatitis A virus in environmental samples. In: Zuckerman AJ, editor. Viral Hepatitis and Liver Disease. New York: Alan R. Liss; 1988:121-6.
8. Siegl G. Replication of hepatitis A virus and processing of proteins. Vaccine. 1992;10:S32.
9. Harmon SA, Summers DF, Ehrenfeld E. Detection of hepatitis A virus RNA and capsid antigen in individual cells. Virus Res. 1989;12:361-9.
10. Seganti L, Superti F, Orsi N, et al. Study of the chemical nature of Frp/3 cell recognition units for hepatitis A virus. Med Microbiol Immunol. 1987;176:21-6.
11. Kaplan G, Totsuka A, Thompson P, et al. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 1996;15:4282-96.
12. Siegl G, Nüesch JPF, de Chastonay J. DI-particles of hepatitis A virus in cell culture and clinical specimens. In: Brinton MA, Heinz FX, editors. New Aspects of Positive Strand RNA Viruses. Washington, DC: American Society for Microbiology; 1990:102-7.
13. Nüesch JPF, Weitz M, Siegl G. Proteins specifically binding to the 3′ untranslated region of hepatitis A virus RNA in persistently infected cells. Arch Virol. 1993;128:65-79.
14. Robertson BH, Jansen RW, Khanna B, et al. Genetic relatedness of hepatitis A virus strains recovered from different geographic regions. J Gen Virol. 1992;73:1365-77.
15. Mathiesen LR, Feinstone SM, Purcell RH, Wagner JA. Detection of hepatitis A antigen by immunofluorescence. Infect Immun. 1977;18:524-30.
16. Emerson SU, Huang YK, Nguyen H, et al. Identification of VP1/2A and 2C as a virulence genes of hepatitis A virus and demonstration of genetic instability of 2C. J Virol. 2002;76:8551-9.
17. Fujiwara K, Yokosuka O, Ehata T, et al. Association between severity of type A hepatitis and nucleotide variations in the 5′ non-translated region of hepatitis A virus RNA: Strains from fulminant hepatitis have fewer nucleotide substitutions. Gut. 2002;51:82-8.
18. Centers for Disease Control and Prevention. Surveillance for Acute Viral Hepatitis, United States-2006. MMWR. 57(SS02), 2008.
19. Gust ID. Epidemiological patterns of hepatitis A in different parts of the world. Vaccine. 1992;10:S56-8.
20. Gandolfo GM, Ferri GM, Conti L, et al. Prevalence of infections by hepatitis A, B, C and E viruses in two different socio-economic groups of children from Santa Cruz, Bolivia. Med Clin (Barc). 2003;120:725-7.
21. Prevention of hepatitis A through active or passive immunization: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR. 1996;45(RR-15):1.
22. Skidmore SJ, Boxall EH, Ala F. A case report of post-transfusion hepatitis A. J Med Virol. 1982;10:223.
23. Hollinger FB, Khan NC, Oefinger PE, et al. Posttransfusion hepatitis type A. JAMA. 1983;250:2313-17.
24. Mannucci PM, Gdovin S, Gringeri A, et al. Transmission of hepatitis A to patients with hemophilia by factor VIII concentrates treated with organic solvent and detergent to inactivate viruses. Ann Intern Med. 1994;120:1-7.
25. Centers for Disease Control and Prevention. Prevention of hepatitis A through active or passive immunization. MMWR. 2006;55(No. RR07):1-23.
26. Coulepis AG, Locarnini SA, Lehmann NI, Gust ID. Detection of HAV in feces. J Infect Dis. 1980;141:151-6.
27. Rosenblum LS, Villarino ME, Nainan OV, et al. Hepatitis A outbreak in a neonatal intensive care unit: Risk factors for transmission and evidence of prolonged viral excretion among preterm infants. J Infect Dis. 1991;164:476-82.
28. Francis Jr T, Frisch AW, Quilligan JJ. Demonstration of infectious hepatitis virus in presymptomatic period after transfer by transfusion. Proc Soc Exp Biol Med. 1946;61:276.
29. Harden AG, Barondess JA, Parker B. Transmission of infectious hepatitis by transfusion of whole blood. N Engl J Med. 1955;253:923-5.
30. Schulman AN, Dienstag JL, Jackson DR, et al. Hepatitis A antigen particles in liver, bile and stool of chimpanzees. J Infect Dis. 1976;134:80-4.
31. Giles JP, Liebhaber H, Krugman S, Lattimer C. Early viremia and viruria in infectious hepatitis. Virology. 1964;24:107-8.
32. Findlay GM. Infective hepatitis in West Africa: 1. Mon Bull Ministry Health. 1948;7:2.
33. Cohen JI, Feinstone S, Purcell RH. Hepatitis A virus infection in a chimpanzee: Duration of viremia and detection of virus in saliva and throat swabs. J Infect Dis. 1989;160:887-9.
34. Zhou F, Shefer A, Weinbaum C, et al. Impact of hepatitis A vaccination on health care utilization in the United States, 1996-2004. Vaccine. 2007;25:3581-7.
35. Berge JJ, Drennan D, Jacobs J, et al. The cost of hepatitis A infections in American adolescents and adults in 1997. Hepatology. 2000;31:469-73.
36. Gordon SC, Reddy KR, Schiff ER. Prolonged intrahepatic cholestasis secondary to acute hepatitis A. Ann Intern Med. 1984;101:635-7.
37. Centers for Disease Control and Prevention. Hepatitis Surveillance Report No. 59. Atlanta: CDC; 2004.
38. Sjögren MH, Tanno H, Fay O, et al. Hepatitis A virus in stool during clinical relapse. Ann Intern Med. 1987;106:221-6.
39. William R. Classification, etiology and considerations of outcome in acute liver failure. Semin Liver Dis. 1996;16:343.
40. Chadha MS, Walimbe AM, Chobe LP, Arankalle VA. Comparison of etiology of sporadic acute and fulminant viral hepatitis in hospitalized patients in Pune, India during 1978-81 and 1994-97. Indian J Gastroenterol. 2003;22:11-15.
41. Brown GR, Persley K. Hepatitis A epidemic in the elderly. South Med J. 2002;95:826-33.
42. Willner IR, Uhl MD, Howard SC, et al. Serious hepatitis A: An analysis of patients hospitalized during an urban epidemic in the United States. Ann Intern Med. 1998;128:111-14.
43. Ostapowicz G, Fontana R, Schiødt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947-54.
44. Schiødt FV, Atillasoy E, Shakill AO, et al. Etiology and outcome for 295 patients with acute liver failure in the United States. Liver Transpl Surg. 1999;5:29-34.
45. Vento S, Cainelli F. Is there a role for viruses triggering auto-immune hepatitis? Autoimmun Rev. 2004;3:61-9.
46. Tagle Arrospide M, Leon Barua R. Viral hepatitis A as a triggering agent of autoimmune hepatitis report of a case and review of literature. Rev Gastroenterol Peru. 2003;23:134-7.
47. Liaw YF, Yang CY, Chu CM, Huang MJ. Appearance and persistence of hepatitis A IgM antibody in acute clinical hepatitis A observed in an outbreak. Infection. 1986;14:156-8.
48. Kao HW, Ashcavai M, Redeker AG. The persistence of hepatitis A IgM antibody after acute clinical hepatitis A. Hepatology. 1984;4:933-6.
49. Yotsuyanagi H, Iino S, Koike K, et al. Duration of viremia in human hepatitis A viral infection as determined by polymerase chain reaction. J Med Virol. 1993;40:35-8.
50. Mannucci PM, Gdovin S, Gringeri A, et al. Transmission of hepatitis A to patients with hemophilia by factor VIII concentrates treated with organic solvent and detergent to inactivate viruses. Ann Intern Med. 1994;120:1-7.
51. Rezende G, Roque-Alsonso M, Samuel D, et al. Viral and clinical factors associated with fulminant course of hepatitis A infection. Hepatology. 2003;38:613-18.
52. Fujiwara K, Yokosuka O, Imazeki F, et al. Analysis of the genotype-determining region of hepatitis A viral RNA in relation to disease severities. Hepatol Res. 2003;25:124-34.
53. American Academy of Pediatrics, Committee on Infectious Disease. Hepatitis A vaccine recommendations. Pediatrics. 2007;120:189-99.
54. Centers of Disease Control and Prevention. Prevention of hepatitis A after exposure to hepatitis A virus and in international travelers. MMWR. 2007;56:1080-4.
55. Victor JC, Monto AS, Surdina TY, et al. Hepatitis A vaccine versus immune globulin for postexposure prophylaxis. N Engl J Med. 2007;357:1685-94.
56. Leentvaar-Kuijpers A, Coutinho RA, Brulein V, Safary A. Simultaneous passive and active immunization against hepatitis A. Vaccine. 1992;10:S138.
57. Andre FE, D’Hondt E, Delem A, Safary A. Clinical assessment of the safety and efficacy of an inactivated hepatitis A vaccine. Vaccine. 1992;10(Suppl 1):S160.
58. Provost PJ, Hughes JN, Miller WJ, et al. An inactivated hepatitis A viral vaccine of cell culture origin. J Med Virol. 1986;19:23-31.
59. Van Damme P, Thoelen S, Cramm K, et al. Inactivated hepatitis A vaccine: Reactogenicity, immunogenicity, and long-term antibody persistence. J Med Virol. 1994;44:446-51.
60. FDA approval for a combined hepatitis A and B vaccine. MMWR. 2001;50:806.
61. Diaz-Mitoma F, Law B, Subramanya A, Hoet B. Long-term antibody persistence induced by a combined hepatitis A and B vaccine in children and adolescents. Vaccine. 2008;26:1759-63.
62. Nothdurft HD, Dietrich M, Zuckerman JN, et al. A new accelerated vaccination schedule for rapid protection against hepatitis A and B. Vaccine. 2002;20:1157-62.
63. Connor BA, Blatter MM, Beran J, et al. Rapid and sustained immune response against hepatitis A and B achieved with combined vaccine using an accelerated administration schedule. J Travel Med. 2007;14:9-15.
64. Centers of Disease Control and Prevention. Prevention and Control of Infections with Hepatitis Viruses in Correctional Settings. MMWR. 2003;52(RRO1):1-33.
65. Reiss G, Keeffe EB. Review article: Hepatitis vaccination in patients with chronic liver disease. Aliment Pharmacol Ther. 2004;19:715-27.
66. Aeslan M, Wiesner RH, Poterucha JJ, Zein NN. Safety and efficacy of hepatitis A vaccination in liver transplantation recipients. Transplantation. 2001;72:272-6.
67. Myers RP, Gregor JC, Marotta P. The cost-effectiveness of hepatitis A vaccination in patients with chronic hepatitis C. Hepatology. 2000;31:834-9.
68. Jacobs RJ, Koff RS. Cost-effectiveness of hepatitis A vaccination in patients with chronic hepatitis C. Hepatology. 2000;32:873-4.
69. Jacobs RJ, Koff RS, Meyerhoff AS. The cost-effectiveness of vaccinating chronic hepatitis C patients against hepatitis A. Am J Gastroenterol. 2002;97:427-34.
70. Arguedas MR, Heudebert GR, Fallon MB, Stinnett AA. The cost-effectiveness of hepatitis A vaccination in patients with chronic hepatitis C viral infection in the United States. Am J Gastroenterol. 2002;97:721-8.
71. Rosenthal P. Cost-effectiveness of hepatitis A vaccination in children, adolescents and adults. Hepatology. 2003;37:44-51.
72. Weinberg A, Gona P, Nachman SA, et al. Antibody responses to hepatitis A virus vaccine in HIV-infected children with evidence of immunologic reconstitution while receiving highly active antiretroviral therapy. J Infect Dis. 2006;193:302-11.
73. Siberry G, Coller R, Henkle E, et al. Antibody response to hepatitis A immunization among human immunodeficiency virus-infected children and adolescents. Pediatr Infect Dis J. 2008;27:465-8.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 77 Hepatitis A
The development of liver biopsy techniques in the 1930s allowed the recognition of hepatic necroinflammation that characterizes all forms of viral hepatitis. Subsequent experimental work in humans led to the clinical recognition that viruses are etiologic agents of hepatitis A (“infectious hepatitis”) and hepatitis B (“serum hepatitis”).1,2 Later, the existence of two hepatitis viruses was demonstrated—hepatitis A virus (HAV) and hepatitis B virus (HBV) (see Chapter 78).3 Additional viral causes of acute and chronic hepatitis were identified subsequently (see Chapters 79 to 81). HAV was first characterized in 1973, when scientists detected the virus in stools from human volunteers who were infected with HAV.4 The ensuing development of sensitive and specific serologic assays for the diagnosis of HAV infection and the isolation of HAV in cell culture5 were important advances that permitted understanding of the epidemiology of HAV infection and, ultimately, control of the disease.
VIROLOGY
In 1982, HAV was classified as an enterovirus type 72 belonging to the Picornaviridae family. Subsequent determination of the sequence of HAV nucleotides and amino acids led to questioning of this classification, and a new genus, Hepatovirus, was created for HAV.6
HAV has an icosahedral shape and lacks an envelope. It measures 27 to 28 nm in diameter, has a buoyant density of 1.33 to 1.34 g/cm3 in cesium chloride, and has a sedimentation coefficient of 156 to 160S on ultracentrifugation. HAV survives exposure to ether and an acid environment at pH 3. It also survives heat exposure of 60°C for 60 minutes but is inactivated at 85°C for 1 minute. HAV is capable of surviving in sea water (4% survival rate), in dried feces at room temperature for 4 weeks (17% survival), and in live oysters for 5 days (12% survival).7
HAV has only one known serotype, and no antigenic cross-reactivity with the hepatitis B, C, D, E, or G agents. The HAV genome consists of a positive-sense RNA that is 7.48 kb long, single-stranded, and linear (Fig. 77-1). HAV RNA has a sedimentation coefficient of 32 to 33S and a molecular weight of 2.8 × 10.4 The HAV RNA has a long open reading frame, consisting of 6681 nucleotides, and is covalently linked to a 5′ terminal protein and a 3′ terminal polyadenosine tract.
Figure 77-1. Genomic organization of hepatitis A virus (HAV). VP, viral protein; VPg, 5′ terminal protein.
(From Levine JE, Bull FG, Millward-Sadler GH, et al. Acute viral hepatitis. In: Millward-Sadler GH, Wright R, Arther MJP, editors. Wright’s Liver and Biliary Disease, 3rd ed. London: WB Saunders, 1992. p 679.)
The onset of HAV replication in cell culture systems takes from weeks to months. Primate cells, including African green monkey kidney cells, primary human fibroblasts, human diploid cells (MRC-5), and fetal rhesus kidney cells, are favored for cultivation of HAV in vitro. The virus is not cytopathic, and persistent infection in the cell cultures is the rule. Two conditions control the outcome of HAV replication in cell culture.8 The first condition is the genetic makeup of the virus; HAV strains mutate in distinct regions of the viral genome as they become adapted to cell culture. The second condition is the metabolic activity of the host cell at the time of infection. Cells in culture, although infected simultaneously, initiate HAV replication in an asynchronous manner. This asynchronicity may be caused by differences in the metabolic activity of individual cells, but definitive evidence of cell-cycle dependence of HAV replication is lacking.9
An initial step in the life cycle of a virus is its attachment to a cell surface receptor. The location and function of these receptors determine tissue tropism. Little is known about the mechanism of entry of HAV into cells. Some work has suggested that HAV could infect cells by a surrogate-receptor binding mechanism (involving a nonspecified serum protein). HAV infectivity in tissue culture has been shown to require calcium and to be inhibited by the treatment of the cells with trypsin, phospholipases, and β-galactosidase.10 A surface glycoprotein, named HAVcr-1, on African green monkey kidney cells has been identified as a receptor for HAV. Blocking of HAVcr-1 with specific monoclonal antibodies prevents infection of otherwise susceptible cells. Experimental data suggest that HAVcr-1 not only serves as an attachment receptor but also may facilitate uncoating of HAV and its entry into hepatocytes.11
Whatever the entry mechanism, once HAV enters a cell, the viral RNA is uncoated, cell host ribosomes bind to viral RNA, and polysomes are formed. HAV is translated into a large polyprotein of 2227 amino acids. This polyprotein is organized into three regions: P1, P2, and P3. The P1 region encodes structural proteins VP1, VP2, VP3, and a putative VP4. The P2 and P3 regions encode nonstructural proteins associated with viral replication (see Fig. 77-1).
The HAV RNA polymerase copies the plus RNA strand. The RNA transcript in turn is used for translation into proteins, which are used for assembly into mature virions. Down-regulation of HAV RNA synthesis appears to occur as defective HAV particles appear.12 In addition, a group of specific RNA-binding proteins has been observed during persistent infection.13 The origin and nature of these proteins is unknown, but they exert activity on the RNA template and are believed to play a regulatory role in the replication of HAV.14
Numerous strains of HAV exist, with considerable nucleotide sequence variability (15% to 25% difference within the P1 region of the genome). Human HAV strains can be grouped into four different genotypes (I, II, III, and VII), whereas simian strains of HAV belong to genotypes IV, V, and VI.15 Despite the nucleotide sequence heterogeneity, the antigenic structure of human HAV is highly conserved among strains.
The HAV VP1/2A and 2C genes are thought to be responsible for viral virulence. This conclusion is based on experiments in which the genotypes and phenotypes of viruses were compared after animals were infected with one of 14 chimeric virus genomes derived from two infectious cDNA clones that encoded a virulent HAV isolate and an attenuated HAV isolate (HM175 strain), respectively.16
Variations in the HAV genome are thought to play a role in the development of fulminant hepatic failure (FHF) during acute HAV infection. The 5′ untranslated region of the HAV genome was sequenced in serum samples from 84 patients with HAV infection, including 12 with FHF.17 The investigators observed fewer nucleotide substitutions in the HAV genome from patients with FHF than in those from patients without FHF (P < .001). The differences were most prominent between nucleotides 200 and 500, suggesting that nucleotide variations in the central portion of the 5′ untranslated region influence the clinical severity of HAV infection.
EPIDEMIOLOGY
Acute hepatitis A is a reportable infectious disease in the United States. The incidence has declined by 90% since 1995. In 2006, 3579 cases of HAV infection were reported, corresponding to a rate of infection of 1.2 per 100,000, compared with 4 per 100,000 in 2001. With the underreporting of cases and the occurrence of asymptomatic infections taken into consideration, the true number of HAV infections in 2006 was calculated to be 32,000, compared with 93,000 in 2001. The greatest rate of decline has been among children from states where routine vaccination of children was recommended in 1999. The highest rate of reported disease historically has been among children ages 5 to 14 years. Because of the rapid rate of decline of disease in children, however, rates are now similar among age groups, with adults ages 20 to 44 having the highest rate of disease in 2006.18
The epidemiologic risk factors for HAV infection reported for the U.S. population in 2006 were as follows: unknown, 65%; international travel, 15%; contact with a patient who has hepatitis, 12%; sexual or household contact with a patient who has hepatitis A, 10%; men having sex with men, 9%; food or waterborne outbreak, 7%; child or employee in a daycare center, 4%; contact with a daycare child or employee, 4%; and injection drug use, 2%.18
HAV infection generally follows one of three epidemiologic patterns.19 In countries where sanitary conditions are poor, most children are infected at an early age. Although earlier seroepidemiologic studies routinely showed that 100% of preschool children in these countries had detectable antibody to HAV (anti-HAV) in serum, presumably reflecting previous subclinical infection, subsequent studies have shown that the average age of infection has risen rapidly to 5 years and older, when symptomatic infection is more likely. For example, 82% of 1393 Bolivian school children were shown to have detectable anti-HAV, but when they were stratified into two groups according to family income, a significant difference was found between the groups: 95% of children from low-income families, but only 56% of children from high-income families, had detectable anti-HAV.20
The second epidemiologic pattern is seen in industrialized countries, where the prevalence of HAV infection is low among children and young adults. In the United States, prior to universal HAV vaccination, the prevalence of anti-HAV was approximately 10% in children but 37% in adults.21
Whatever the epidemiologic pattern, the primary route of transmission of HAV is the fecal-oral route, by either person-to-person contact or ingestion of contaminated food or water. Although rare, transmission of HAV by a parenteral route has been documented after transfusion of blood22,23 or blood products.24 Cyclical outbreaks among users of injection and noninjection illicit drugs and among men who have sex with men (up to 10% may become infected in outbreak years) have been reported.25 Table 77-1 provides information about the detection of HAV and its infectivity in human body fluids.26–33
Table 77-1 Detection of Hepatitis A Virus (HAV) and Infectivity of Human Secretions or Excretions
| SECRETION/EXCRETION | COMMENT | REFERENCES |
|---|---|---|
| Stool | Main source of infection. HAV is detectable during the incubation period and for several weeks after the onset of disease. After the onset of symptoms, HAV is detectable in 45% and 11% of fecal specimens collected during the first and second weeks of illness, respectively, whereas HAV RNA (by polymerase chain reaction assay) is detectable for 4 to 5 months. | 26,27 |
| Blood | Viremia is present during the incubation period. Blood collected 3 and 11 days before the onset of symptoms has caused post-transfusion infection in recipients. Chronic viremia does not occur. | 28,29 |
| Bile | HAV has been detected in the bile of chimpanzees infected with HAV. | 30 |
| Urine | HAV is detected in low titer during the viremic phase. A urine sample was reported to be infectious after oral inoculation. Urine contaminated with blood was also infectious. | 31,32 |
| Nasopharyngeal secretions | Unknown in humans. HAV has been identified in the oropharynx of experimentally infected chimpanzees. | 33 |
| Semen, vaginal fluid | Uncertain. HAV may be detectable during the viremic phase. | — |
From 11% to 22% of patients with acute hepatitis A require hospitalization, with an average length of stay of 4.6 days, costing on average $7926 per patient in 2004. In one outbreak involving 43 persons, the total cost was approximately $800,000. On average, 27 workdays are lost per adult case of hepatitis A. In adolescents and adults, the combined direct and indirect costs associated with HAV infection in the United States totaled approximately $488.8 million in 1997, compared with $93 million in 2006.25,34,35 The decline in costs is a direct result of the dramatic reduction in the number of infections seen since the introduction of the HAV vaccine combined with changes in vaccination policies in the United States (see later).18
PATHOGENESIS
After HAV is ingested and survives gastric acid, it traverses the small intestinal mucosa and reaches the liver via the portal vein. The precise mechanism of hepatic uptake in humans is unknown (see earlier). In an experimental model using African green monkey kidney cells,11 the putative cellular receptor for HAV has been identified as a surface glycoprotein. Once the virus enters the hepatocyte, it starts replicating in the cytoplasm, where it is seen on electron microscopy as a fine granular pattern, but it is not present in the nucleus. HAV is distributed throughout the liver. Although HAV antigen has been detected in other organs (lymph nodes, spleen, kidney), the virus appears to replicate exclusively in hepatocytes. When the virus is mature, it reaches the systemic circulation via the hepatic sinusoids and is released into the biliary tree through bile canaliculi, passed into the small intestine, and eventually excreted in the feces.
CLINICAL FEATURES
Infection with HAV does not result in chronic infection, only in an acute self-limited episode of hepatitis. Rarely, acute hepatitis A can have a prolonged or a relapsing course, and occasionally profound cholestasis can occur.36 The incubation period is commonly 2 to 4 weeks, rarely up to 6 weeks. The mortality rate is low in previously healthy persons. Morbidity can be significant in adults and older children.
The clinical characteristics of cases of hepatitis A reported in 2002 were similar to those in previous years, with a preponderance of cases in men in all age groups. Overall, 72% of patients manifested jaundice, 25% required hospitalization, and 0.5 % died.37 The need for hospitalization rose with age, from 5% among children younger than 5 years to 34% among persons 60 years or older.37
Patients with HAV infection usually present with one of the following five clinical patterns: (1) asymptomatic without jaundice, (2) symptomatic with jaundice and self-limited after approximately 8 weeks, (3) cholestatic, with jaundice lasting 10 weeks or more,36 (4) relapsing, with two or more bouts of acute HAV infection occurring over a 6- to 10-week period, and (5) FHF.
[/not-level-membership-for-gastroenterology-and-hepatology-category]
