7.7 Hepatic failure
Introduction
Acute liver failure (ALF) is a rare but devastating presentation in children. The major functions of the liver include synthetic and metabolic functions. Synthetic functions include production of coagulation factors and albumin; while metabolic functions include: glucose metabolism, and waste product processing (e.g. bilirubin, nitrogenous compounds, drug elimination). ALF in children may be due to many causes (Table 7.7.1). The manifestations of coagulopathy, hypoglycaemia, jaundice, encephalopathy and hypoalbuminaemia, reflect common disturbances of liver function.1 ALF may be an immediate life-threatening process or a subacute process, with a spectrum of severity between those extremes. Medical management is multifaceted and focuses on supporting vital functions while hepatic recovery occurs or liver transplantation can be performed.
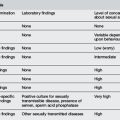
ALF has been defined in adults by clinical and laboratory criteria:


The Pediatric Acute Liver Failure Study Group2 has defined ALF in children as:




ALF classification, using the time interval between the onset of jaundice and encephalopathy, has aetiological and prognostic importance (Table 7.7.2), despite the difficulties in identifying encephalopathy mentioned above. O’Grady et al3,4 and Poddar et al5 found that, in comparison with patients suffering acute or subacute liver failure, those with hyperacute liver failure had a better prognosis.
| Interval between onset of jaundice and encephalopathy | Classification |
|---|---|
| 7 days or less | Hyperacute |
| 8 to 28 days | Acute |
| 5 to 12 weeks | Subacute |
Aetiology
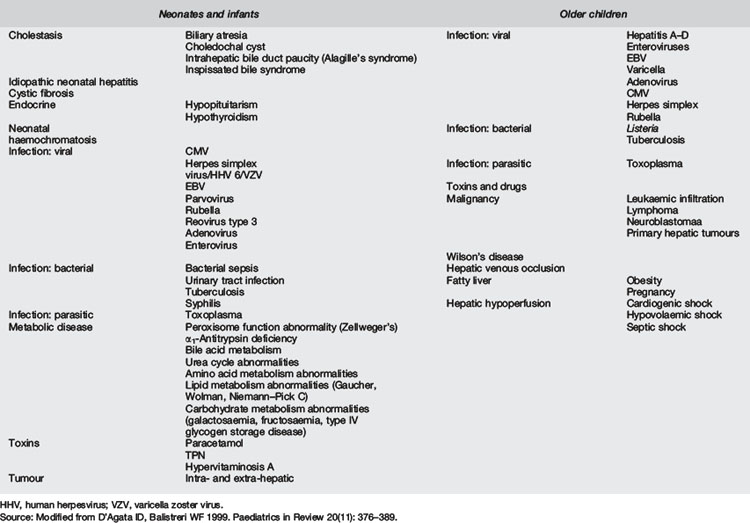
Table 7.7.1 demonstrates the variety of diagnoses that may cause ALF in children. The aetiology can be grouped according to onset prior to or after the first year of life. In broad terms, infection, immune dysregulation, toxicity (including medication), infiltration, and inborn errors of metabolism are the causative pathways that may lead to ALF. Cases where the cause is not determined predominate in children under 3 years.
Neonates and infants
In the neonatal population, the estimated incidence of liver disease is approximately 1:2500. Biliary atresia and neonatal idiopathic hepatitis contribute 60% of all cases of cholestasis. In 80 infants under 12 months with ALF, inherited metabolic conditions were responsible for 42.5% of cases: neonatal haemochromatosis 16%, acute viral hepatitis 15%, and miscellaneous causes (toxins, autoimmune, malignancy) 10%. 16% of neonatal cases were undetermined.6 Metabolic causes of liver failure include: disorders of the mitochondrial electron transport chain; disorders of protein, carbohydrate and lipid metabolism; and inherited causes of cholestasis.
Infectious hepatitis
Worldwide, infectious hepatitis is the greatest cause of ALF. Five RNA viruses (hepatitis A, C, D, E and G) and one DNA virus (hepatitis B) can infect the liver. Transmission of A and E is via the faecal-oral route. The remainder are transmitted via body fluids. Acute viral hepatitis is a clinical syndrome with systemic symptoms occurring after a virus-dependent incubation period. Jaundice ensues after hepatocyte necrosis reduces the liver’s capacity to metabolise bilirubin. Fulminant hepatitis occurs in less than 1% of children with hepatitis A and in 1–2% of cases of hepatitis B. Fulminant disease occurs with hepatitis D in approximately 10% of cases and is more likely with superinfection. Hepatitis C can cause acute and chronic infection and rarely fulminant hepatitis. Severe acute hepatitis E infection is a leading cause of ALF in the tropics. Epstein–Barr virus (EBV), cytomegalovirus (CMV), herpes simplex virus, varicella zoster virus, human herpesvirus 6 and parvovirus B-19 are non-hepatotropic viruses that can rarely cause ALF. Consideration of, and investigation for, these viruses is important because specific therapy with antiviral medication is available for some of these pathogens.7
Toxins and medication
Paracetamol
Paracetamol toxicity is the most common cause of ALF in the developed world. Paracetamol is an analgesic and antipyretic freely available in many countries. It is metabolised by the hepatocyte and toxicity exhausts hepatic glutathione stores. Generation of toxic metabolites leads to centrilobular necrosis. Toxicity is unlikely with single doses under 150 mg kg–1. Children are often given multiple doses of paracetamol and this can lead to toxicity if the cumulative daily dose is greater than 60 mg kg–1 day–1. Factors predictive of hepatotoxicity include: age (lower incidence in children under 5), genetics (cytochrome isoenzyme polymorphisms are inherited), alcohol and tobacco use (relevant in adolescents), other medications and nutritional status.8 Treatment of toxicity is discussed elsewhere in the text.
Anticonvulsants
Genetic predisposition has been purported for anticonvulsant induced hepatotoxicity.2 Sodium valproate causes intracellular fat accumulation within the hepatocyte, and may be related to a primary defect of respiratory chain enzyme function.2 Impaired metabolic functions within the cell may lead to necrosis. Children under 2 years and those on multiple medications are at highest risk. Carbamazepine may cause hepatitis and/or cholestasis during the first months of therapy. Clinically significant hepatotoxicity is rare.
Aspirin and Reye’s syndrome
Mitochondrial dysfunction leading to acute encephalopathy, selective hepatic dysfunction and visceral fatty infiltration has been called Reye’s syndrome.9 Metabolic disorders have been later identified in some children initially diagnosed with Reye’s syndrome. Mitochondrial oxidative phosphorylation and fatty acid β-oxidation are the metabolic pathways affected in Reye’s syndrome. Preceding viral infection (classically varicella), immune mediators and aspirin (or its metabolites) all can limit normal functioning of these pathways. The association of aspirin with this disorder remains unclear despite a study by Forsyth et al,10 which identified a dose–response relationship, and population studies that demonstrate that the decline in Reye’s syndrome mirrors a decline in aspirin usage.9,11
Metabolic diseases associated with liver failure
Zellweger’s syndrome (cerebrohepatorenal syndrome)
Autosomal recessive inheritance of this peroxisomal abnormality leads to abnormal bile acid synthesis, and abnormal fatty-acid oxidation. Multiorgan involvement (cardiac, pulmonary, neurological, renal) with failure to thrive and hypotonia are major early features. Jaundice occurs in 50% of cases. Therapy has not been shown to prolong life although histological improvement may occur on liver biopsy. Biopsy demonstrates abnormal mitochondria and absent peroxisomes. More information can be found at http://www.ncbi.nlm.nih.gov/omim/214100.
Tyrosinaemia
Linked to a gene on chromosome 15, tyrosinaemia is an autosomal recessive condition due to deficiency of fumarylacetoacetase which is the last enzyme in the processing of phenylalanine (pathway available at http://www.ncbi.nlm.nih.gov/omim/Images/tyrosine.html). This autosomal recessive disorder is characterised by progressive liver parenchymal damage and renal tubulopathy with generalised aminoaciduria. It is particularly common in parts of Quebec. Rapid progression may occur in infancy or an indolent course leading to hepatic cell carcinoma may occur in up to 37% of those over 2 years.
Biliary atresia
Complete absence of all extrahepatic biliary structures is the usual malformation, leading to a clinical picture of jaundice, pale (acholic) stools and dark urine due to cholestasis. A choledochal cyst may present in identical fashion and may co-exist with biliary atresia. Presentation is usually with prolonged neonatal jaundice or delayed onset of jaundice (age 2–3 weeks). This occurs in approximately 1:10 000 to 1:20 000 live births, with equal gender incidence. Following diagnostic testing (ultrasonography, nuclear medicine scan, occasionally liver biopsy), management is by surgical hepatoportoenterostomy (Kasai procedure) prior to 8 weeks of age, if possible, as earlier surgery improves outcomes.12
Mushrooms
Edible and inedible mushrooms can be difficult to distinguish. Amanita phalloides produces amatoxin, which is hepatotoxic. The toxin is a heat stable octapeptide. After a period of 6–48 hours the affected patient will start vomiting, complain of abdominal pain, develop diarrhoea, before neurological symptoms (coma, seizures) and hepatic failure commence, with a mortality of up to 30%. Cholinergic symptoms via muscarinic receptors may also occur and respond to atropine. Charcoal should be given to reduce absorption. Silibinin and high-dose penicillin G may assist in limiting hepatic damage.13 Identification of the mushroom is important and may require referral to local botanists or mycologists.
Pathophysiology
Exposure to hepatotoxic agents such as: drugs, products of metabolism or infectious particles, in addition to immune responses, initiates hepatocyte injury that may progress to necrosis. Biopsy, when performed, reveals multilobular or bridging necrosis with reticulin framework collapse. Patterns related to aetiology can be seen, such as centrilobular necrosis in paracetamol toxicity or with circulatory shock. When normal regenerative processes do not occur, liver failure follows.14 Astrocyte oedema may be seen and may be due to altered cell wall permeability, glutamate, ammonia, and neurotransmitter balance.15
Hepatic encephalopathy occurs through the interplay of three factors:



Contributions from ammonia, inflammatory cytokines, benzodiazepine-like compounds and manganese16 lead to neuronal dysfunction and altered interaction of astrocytes with neurons. This leads to the clinical manifestations of hepatic encephalopathy. The balance of inhibitory (e.g. GABA) versus excitatory (e.g. glutamate) neurotransmission is altered in hepatic encephalopathy.9 Ammonia appears to augment inhibitory neurotransmission. In addition, the role of sepsis, either via systemic immune response17 or via lipopolysaccharides directly,18 hypoglycaemia and raised intracranial pressure is important in the development of encephalopathy.
Presentation
Examination
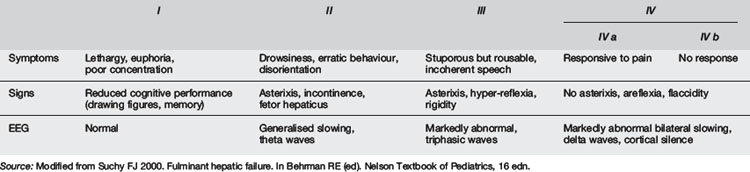
As discussed above, specific diseases associated with liver failure will have other features. The phenotypes of galactosaemia, Alagille’s syndrome, α1-antitrypsin deficiency and Wilson’s disease may be identified by careful clinical examination. Neurological evaluation is essential and the presence of asterixis important in a child with liver failure in order to assess for, and classify a stage of, hepatic encephalopathy, if present (Table 7.7.3). In young children encephalopathy, which occurs late, is difficult to detect. Cutaneous features such as bruising, petechiae or bleeding may indicate an associated coagulopathy. In children with chronic liver disease there may be signs such as spider naevi, caput medusae or finger clubbing.
Investigations
Investigation of the child with hepatic failure focuses on identifying the extent of liver dysfunction and on identifying the cause. History and examination findings must guide investigation. ED screen should include a full blood examination, liver function tests including conjugated and unconjugated bilirubin, blood glucose, coagulation screen, renal function and arterial blood gases. Further potentially relevant investigations such as viral serology, copper and caeruloplasmin levels, Pi typing, lactate level, drug screens, urine metabolic screen, or imaging should be determined after discussion with a paediatric hepatologist. Note that the international normalised ratio for coagulation and the serum bilirubin concentration are not predictive of post-transplant survival, while renal dysfunction requiring dialysis is associated with a higher mortality.14 Escudie et al19 found a prothrombin index below 10% (INR approx 6) 4 days after Amanita phalloides ingestion was predictive of fatal outcome.
Management
The management of acute liver failure involves supportive care, complication management and specific treatment modalities where they exist. Therapy needs to be initiated in the ED prior to transfer to a paediatric intensive-care environment. The initial management issues are listed in Table 7.7.4. Intubation and ventilation may be required due to coma or respiratory failure. Respiratory failure itself is multifactorial: altered cardiac output; capillary leak; possible oliguria; and significant fluid requirements contribute. Fluid management may be complex in the face of renal failure and electrolyte imbalances. Vitamin K, fresh frozen plasma or cryoprecipitate may be required to correct symptomatic coagulopathy. Intravenous fluids containing 10% dextrose or more are usually required to correct hypoglycaemia and then maintain normoglycaemia. Lactulose is given in hepatic encephalopathy to reduce absorption of nitrogenous wastes. Neomycin and/or metronidazole, given enterally, reduce the enteric bacterial load and the production of nitrogenous wastes. Proton pump inhibitors, H2-receptor blockers and/or sucralfate are given to limit the risk of gastric ulceration in the context of coagulopathy. Sepsis is a common and serious complication, and will exacerbate liver failure and requires aggressive antimicrobial management. Raised intracranial pressure and, more specifically, reduced cerebral perfusion pressure are important complications of ALF. Both these effects may be improved by therapeutic cooling. Therapeutic cooling (32–33°C) reduces brain energy metabolism, normalises cerebral blood flow, reduces ammonia delivery, reduces oxidative pressure on astrocytes and reduces brain glutamate.14
Pulse oximetry
Level of consciousness
Urine output (may need indwelling catheter)
Blood glucose
Acid/base status (blood gases)
Coagulation
Liver function tests
Serum electrolytes
Nasogastric tube (for gastric drainage and administration of neomycin and lactulose)
Potassium
Albumin
Coagulation factors (vitamin K, frozen plasma, cryoprecipitate, platelets)
Oxygen (may require endotracheal intubation)
Intravascular volume if required
Evolving encephalopathy
Raised intracranial pressure
A bridge to liver transplantation may be created via the use of liver support devices. These have not undergone extensive testing as yet. Artificial systems using filtration, dialysis or ion exchange or bioartificial systems (human or non-human hepatocytes within an artificial framework) are available. Anecdotal reports of these systems include reduction in serum copper in Wilson’s disease-induced ALF that allowed stabilisation prior to transplantation.20
The development of liver transplantation has allowed children with irreversible liver failure to survive. The paucity of available donors and contraindications to transplantation limit the number of children able to receive liver transplants in the acute setting. Contraindications to transplantation include: uncontrolled systemic infection; extrahepatic metastasis in liver tumours, irreversible neurological injury; and multiorgan failure. Living donor transplantation has increased the number of liver transplants in children. Auxiliary partial orthotopic transplantation allows the transplanted liver segment to function while a diseased native liver recovers and regenerates. This technique also may allow for the discontinuity of antirejection medication once the native liver recovers.21 Disease-specific treatment is available for paracetamol toxicity. Intravenous N-acetylcysteine (NAC) is given in addition to supportive measures. Herpesvirus-induced fulminant hepatitis may respond to aciclovir. Children with Wilson’s disease may respond to chelation therapy pending transplantation. Certain metabolic diseases can be managed through dietary manipulation or metabolic pathway manipulation.
Prognosis
The mortality of ALF is approximately 60%. Of children with Stage 4 hepatic encephalopathy, 80% will die. Sepsis is the cause of death in approximately 10% of children with ALF. The King’s College criteria (Table 7.7.5), based on multivariate analysis of 588 patients with fulminant hepatic failure, provide clinical and laboratory parameters predictive of mortality. A single adverse factor was associated with 80% mortality; three adverse factors were associated with 95% mortality. Paracetamol toxicity was associated with a better prognosis. If paracetamol toxicity was the reason for ALF then the presence of a single adverse risk factor places mortality risk at 55%. It is important to compare these figures with the 20–30% mortality rate of all children undergoing liver transplantation.
Source: Data from O’Grady et al 1989. Gastroenterology 97: 439–445.
The United Network for Organ Sharing (UNOS) has developed the Model for End-Stage Liver Disease (MELD) for patients over 12 years. The Pediatric End-Stage Liver Disease score is for patients under 12. Both assess risks of death while waiting for transplantation in order to prioritise available organs. The PELD score uses: albumin, bilirubin, INR, growth failure and age at listing (for transplantation) while the MELD score uses creatinine, bilirubin and INR in predictive modelling (www.unos.org).
Prevention
Controversies and future directions
 Transplantation remains the gold standard therapy and case reports of auxiliary partial orthotopic liver transplantation demonstrated that a transplanted liver lobe functioned satisfactorily until native liver recovery occurred in a case of mushroom toxicity.22
Transplantation remains the gold standard therapy and case reports of auxiliary partial orthotopic liver transplantation demonstrated that a transplanted liver lobe functioned satisfactorily until native liver recovery occurred in a case of mushroom toxicity.22
 Prognostic scores have been developed according to aetiology of ALF and need more prospective research: in paracetamol hepatotoxicity, arterial pH, serum lactate and prothrombin time and creatinine; in mushroom toxicity, prothrombin time and creatinine appear more predictive. A patient with Wilson’s disease requires transplantation if encephalopathy is present, although serum bilirubin, INR, aspartate aminotransferase and white cell count are important predictors of the need for transplantation. In general, predictors of poor outcome include: poor renal function, a PELD score >25, and age under 24 months and onset of encephalopathy within 7 days of onset of jaundice.4
Prognostic scores have been developed according to aetiology of ALF and need more prospective research: in paracetamol hepatotoxicity, arterial pH, serum lactate and prothrombin time and creatinine; in mushroom toxicity, prothrombin time and creatinine appear more predictive. A patient with Wilson’s disease requires transplantation if encephalopathy is present, although serum bilirubin, INR, aspartate aminotransferase and white cell count are important predictors of the need for transplantation. In general, predictors of poor outcome include: poor renal function, a PELD score >25, and age under 24 months and onset of encephalopathy within 7 days of onset of jaundice.41 D’Agata I., Balistreri W. Evaluation of liver disease in the pediatric patient. Pediatr Rev. 1999;20(11):376-389.
2 Bucuvalas J., Yazigi N., Squires R.H. Acute liver failure in children. Clin Liver Dis. 2006;10(1):149-168.
3 O’Grady J., Schalm S., Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273-275.
4 O’Grady J. Modern management of acute liver failure. Clin Liver Dis. 2007;11(2):291-303.
5 Poddar U., Thapa B., Prasad A., et al. Natural history and risk factors in fulminant hepatic failure. Arch Dis Child. 2002;87(1):54-56.
6 Durand P., Debray D., Mandel R., et al. Acute liver failure in infancy: a 14 year experience of a pediatric liver transplantation center. J Pediatr. 2001;139(6):871-876.
7 Fontana R. Acute liver failure including acetaminophen overdose. Med Clin North Am. 2008;92(4):761-794.
8 Larsen A.M. Acetaminophen hepatotoxicity. Clin Liver Dis. 2007;11(3):525-548.
9 Glasgow J., Middleton B. Reye syndrome – insights on causation and prognosis. Arch Dis Child. 2001;85:351-353.
10 Forsyth B.W., Horwitz R.I., Acampora D., et al. New epidemiologic evidence confirming that bias does not explain the aspirin/Reye’s syndrome association. JAMA. 1989;261(17):2517-2524.
11 Belay E., Bresee J., Holman R., et al. Reye’s syndrome in the United States from 1981 through 1997. N Engl J Med. 1999;340(18):1377-1382.
12 Serinet M., Wildhaber B., Broue P., et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics. 2009;123(5):1280-1286.
13 Schilsky M.L., Honiden S., Arnott L., Emre S. ICU management of acute liver failure. Clin Chest Med. 2009;30(1):71-87.
14 Suchy FJ. Fulminant hepatic failure. In Kliegman R., Rudolf M., editors: Nelson Textbook of Pediatrics, 18th ed., Saunders, 2007.
15 Sundaram V., Shaikh O. Hepatic encephalopathy: pathophysiology and emerging therapies. Med Clin North Am. 2009;93:819-836.
16 Munoz S.J. Hepatic encephalopathy. Med Clin North Am. 2008;92(8):795-812.
17 Shawcross D.L., Davies N.A., Williams R. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis. J Hepatol. 2004;40:247-254.
18 Pedersen H.R., Ring-Larsen H., Olsen N.V. Hyperammonemia acts synergistically with lipopolysaccharide in inducing changes in cerebral hemodynamics in rats anaesthetised with pentobarbital. J Hepatol. 2007;47:245-252.
19 Escudie L., Francoz C., Vinel J.-P., et al. Amanita phalloides poisoning: Reasessment of prognostic factors and indications for emergency liver transplantation. J Hepatol. 2007;46(3):466-473.
20 Sen S., Felldin M., Steiner C., et al. Albumin dialysis and Molecular Adsorbents Recirculating System (MARS) for acute Wilson’s disease. Liver Transpl. 2002;8(10):962-967.
21 Kerkar N., Emre S. Issues unique to pediatric liver transplantation. Clin Liver Dis. 2007;11(2):323-335.
22 Rosenthal P., Roberts J., Ascher N., Emond J. Auxiliary liver transplant in fulminant failure. Paediatrics. 1997;100(2):E10.