CHAPTER 74 Hemochromatosis
Trousseau was the first to describe a case of hemochromatosis in the French pathology literature in 1865.1 Almost 25 years later, in 1889, von Recklinghausen, thinking that the disease was a blood disorder that caused increased skin pigmentation, introduced the term hemochromatosis.1 In 1935, Sheldon published a description of all 311 cases of the disease that had been reported in the world’s literature to that point, including several from his own records. He recognized that hemochromatosis was an inborn error of iron metabolism and that all the pathologic manifestations of the disease were caused by increased iron deposition in the affected organs.1 In 1976, Simon and coworkers2 demonstrated that the gene for hereditary hemochromatosis (HH) was linked to the HLA region on the short arm of chromosome 6. The benefit of early diagnosis on survival was shown in a classic paper by Niederau and colleagues,3 who demonstrated that if HH was identified and treated before the development of cirrhosis or diabetes mellitus, survival of affected patients was equivalent to that of an age- and gender-matched population.
In 1996, the HFE gene was identified on chromosome 6, thereby permitting genetic testing for the two major mutations (C282Y, H63D) that are responsible for HFE-related HH.4 Several prospective population studies have shown that the frequency of the C282Y homozygous state is approximately 1 in 250 in white populations of northern European descent.5 It is now recognized that C282Y homozygosity has incomplete clinical penetrance, with a strong male predominance for symptomatic disease.6 HH is characterized by increased intestinal iron absorption that results from low expression of the iron-regulatory protein hepcidin.5–7 In addition to the discovery of HFE and hepcidin, several additional genes and proteins involved in the regulation of iron homeostasis have been identified, contributing to a better understanding of cellular iron uptake and release. Also, numerous clinical and pathophysiologic studies have been performed and have led to improved diagnosis, better family screening, and new insights into normal and abnormal iron homeostasis. HFE-related HH is a common autosomal recessive disorder of iron metabolism; if it is diagnosed early and treated appropriately, every patient with the disorder can have a normal lifespan.
CAUSES OF IRON OVERLOAD
Hereditary hemochromatosis comprises several inherited disorders of iron homeostasis characterized by increased intestinal iron absorption that results in tissue iron deposition (Table 74-1). The older terms primary hemochromatosis and idiopathic hemochromatosis should no longer be used. The liver is always the principal recipient of most of the absorbed iron and is always involved in symptomatic HH. The most common form of HH by far is HFE-related HH.5–9 It is an autosomal recessive disorder usually identified in adults of northern European ancestry. Most patients who present with HH are homozygous for the C282Y mutation of HFE, although some persons who are compound heterozygotes (C282Y/H63D) also have iron overload.
| Hereditary hemochromatosis | HFE-related hereditary hemochromatosis (type 1) |
| C282Y homozygosity | |
| C282Y/H63D compound heterozygosity | |
| Other HFE mutations | |
| Non–HFE-related hereditary hemochromatosis: | |
| Hemojuvelin (HJV) mutations (type 2A) | |
| Hepcidin (HAMP) mutations (type 2B) | |
| Transferrin receptor 2 (TFR2) mutations (type 3) | |
| Ferroportin (SLC40A1) mutations (type 4) | |
| Loss-of-function mutations | |
| Gain-of-function mutations | |
| African iron overload | |
| Secondary iron overload | Iron-loading anemias |
| Aplastic anemia | |
| Chronic hemolytic anemia | |
| Pyridoxine-responsive anemia | |
| Pyruvate kinase deficiency | |
| Sideroblastic anemia | |
| Thalassemia major | |
| Parenteral iron overload | |
| Iron-dextran injections | |
| Long-term hemodialysis | |
| Red blood cell transfusions | |
| Chronic liver disease | |
| Alcoholic liver disease | |
| Hepatitis B | |
| Hepatitis C | |
| Nonalcoholic steatohepatitis | |
| Porphyria cutanea tarda | |
| Portacaval shunt | |
| Insulin resistance syndrome with iron overload | |
| Dietary iron overload | |
| Miscellaneous | Aceruloplasminemia |
| Congenital alloimmune hepatitis (neonatal hemochromatosis) | |
| Congenital atransferrinemia |
Other inherited forms of iron overload, classified as non-HFE-related HH, are juvenile hemochromatosis and iron overload resulting from mutations in the genes for hepcidin,10 transferrin receptor 2 (TFR2),11 or ferroportin.12 Juvenile HH is characterized by rapid iron accumulation. Mutations in two different genes have been shown to cause forms of juvenile HH. The more common mutation occurs in the HJV gene on chromosome 1q; this gene encodes a protein called hemojuvelin.13 Mutations in the hepcidin gene (HAMP) also produce a form of juvenile HH10; hepcidin is a hepatic peptide that acts to down-regulate iron absorption (see later). Mutations of the gene TFR2 produce an autosomal recessive form of HH that is clinically similar to HFE-related HH.11 How these TFR2 mutations result in iron overload is not yet known; they possibly cause abnormal iron sensing by hepatocytes, the predominant site of TFR2 expression.14 A rare autosomal dominant form of HH results from two categories of mutations in the gene for the iron transporter ferroportin.15 “Loss-of-function” mutations decrease the cell surface localization of ferroportin, thereby reducing its ability to export iron. The result is iron deposition primarily in macrophages, and this disorder is sometimes termed ferroportin disease. The second category includes “gain-of-function” ferroportin mutations that abolish hepcidin-induced ferroportin internalization and degradation; the distribution of excess iron is similar to that in HFE-related HH, primarily parenchymal.
African iron overload occurs primarily in sub-Saharan Africa and is now considered to be the result of a non-HFE–related genetic trait that can be exacerbated by dietary iron loading.16 Some persons who manifest African iron overload consume an iron-rich fermented maize beverage, but iron overload also can occur in people who do not drink this beverage. In most cases, iron-loaded Kupffer cells are prominent in African iron overload; by contrast, Kupffer cells are relatively spared in HFE-related HH. A similar form of iron overload has been suggested to occur in African Americans,17 and further investigations are needed to determine the genetic basis, prevalence, and clinical consequences of this condition.
Persons who absorb excessive amounts of iron as a result of an underlying cause other than any of the previously mentioned inherited defects have secondary iron overload18 (see Table 74-1). Examples are persons with disorders of ineffective erythropoiesis, liver disease (in some cases), increased oral intake of iron, or the rare condition congenital atransferrinemia. Both HH and secondary iron overload should be distinguished from parenteral iron overload, which is always iatrogenic and which leads to iron deposition that is found initially in the reticuloendothelial system. In patients with ineffective erythropoiesis who require red blood cell transfusions, parenchymal and reticuloendothelial iron overload coexists because these people have a stimulus to increased iron absorption and receive iron in the form of red blood cell transfusions. Congenital alloimmune hepatitis is responsible for most cases of neonatal hemochromatosis.19 In these cases, immune-mediated liver injury in the fetus is associated with the development of iron overload. Treatment with intravenous immunoglobulin during pregnancy markedly slows or prevents the development of this condition.
PATHOPHYSIOLOGY
INTESTINAL IRON ABSORPTION
An increase in intestinal iron absorption is a pathogenic characteristic of HFE-related HH.5–9 Understanding the pathogenesis of HH, therefore, requires a review of the determinants of duodenal iron absorption. Because there are no important physiologic mechanisms to regulate iron loss, iron homeostasis depends on a tight linkage between body iron requirements and intestinal iron absorption. Nearly all absorption of dietary iron occurs in the duodenum, where iron may be taken up as either ionic iron or heme.14,20 The absorption of both forms of iron is increased in patients with HFE-related HH.
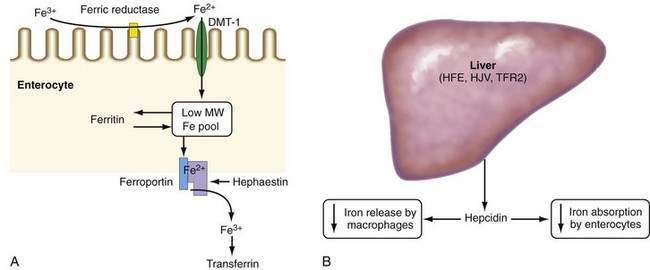
Absorption of ionic iron across the enterocyte occurs in two stages: uptake across the apical membrane and transfer across the basolateral membrane (Fig. 74-1A). Before uptake, ionic iron must be reduced from the ferric to the ferrous state; this step is accomplished by ferric reductases that are expressed on the luminal surface of duodenal enterocytes. The ferrous iron crosses the apical membrane via divalent metal transporter 1 (DMT-1). Iron taken up by the enterocyte may be stored as ferritin (and excreted in the feces when the senescent enterocyte is sloughed) or transferred across the basolateral membrane to the plasma. This latter process occurs via the transporter, ferroportin. The basolateral transfer of iron requires oxidation of iron to the ferric state by the ferroxidase hephaestin. Uptake of heme occurs by a transporter whose identity remains uncertain. Once internalized, the heme is degraded and the liberated iron is handled by the enterocyte in the same manner as absorbed ionic iron. Patients with HFE-related HH demonstrate increased basolateral transfer of iron from the enterocytes to the plasma. This increased transfer may be the driving force behind the increased intestinal iron absorption that is characteristic of HH. Some studies of patients with HFE-related HH have demonstrated higher duodenal expression of ferroportin and DMT-1.14 The major regulator of intestinal iron absorption is the peptide hormone hepcidin.
HEPCIDIN
Hepcidin is an iron-regulatory hormone that plays a central role in iron homeostasis by coordinating iron absorption, mobilization, and storage to meet the iron requirements of erythropoiesis and other iron-dependent processes20–23 (see Fig. 74-1B). Hepcidin is expressed predominantly in hepatocytes and is secreted into the circulation. It binds to ferroportin, which is highly expressed on macrophages and the basolateral surface of enterocytes, thereby causing ferroportin to be internalized and degraded, thus inhibiting iron export. Hepcidin expression is regulated by total body iron, erythropoiesis, hypoxia, and inflammation. Excess iron and inflammation induce hepcidin expression, which, in turn, results in decreased intestinal iron absorption and diminished iron release from macrophages. By contrast, hepcidin expression is decreased by iron deficiency, erythropoiesis, and hypoxia, with resulting increases in iron absorption from the intestine and release of iron from macrophages.
In all types of HH, iron overload results from impairment in the hepcidin regulatory pathway. In humans and mice, mutations or knockout of the genes for HFE, hemojuvelin, hepcidin, or TFR2 decrease hepcidin expression, with a resulting increase in intestinal iron absorption via upregulation of ferroportin levels.20–25
Studies have revealed that iron-induced regulation of hepcidin expression involves a bone morphogenetic protein (BMP)-dependent signaling pathway.20,23 BMPs bind to specific receptors on hepatocytes, thereby triggering SMAD protein-dependent activation of hepcidin expression. Selective inhibition of BMP signaling abrogates iron-induced upregulation of hepcidin. Hemojuvelin is a BMP co-receptor and facilitates the binding of BMP to its receptor; knockout of the hemojuvelin gene markedly decreases BMP signaling and hepcidin expression and causes iron overload.
The inflammatory cytokine interleukin-6 upregulates hepcidin via STAT3 (signal transducer and activator of transcription-3) signaling, causing iron retention in macrophages and decreased intestinal iron absorption. The resulting hypoferremia plays a major causal role in the anemia of chronic disease.20–23 Reactive oxygen species inhibit hepcidin expression via a C/EBPα (CCAAT/enhancer binding protein alpha)-mediated mechanism, which may account for the hepatic iron loading associated with alcoholic liver disease and chronic hepatitis C.26
HFE PROTEIN
Studies of HFE protein structure and function were a direct consequence of the cloning of the HFE gene. The HFE gene encodes a 343-amino acid protein consisting of a 22-amino acid signal peptide, large extracellular domain, single transmembrane domain, and short cytoplasmic tail (Fig. 74-2).4 The extracellular domain of HFE protein consists of three loops (α1, α2, and α3), with intramolecular disulfide bonds within the second and third loops. The structure of the HFE protein is similar to that of other major histocompatibility complex (MHC) class I proteins, but evidence indicates that HFE protein does not participate in antigen presentation. Like MHC class I molecules, however, HFE protein is physically associated with β2-microglobulin (see Fig. 74-2). The major mutation responsible for HH results in the substitution of tyrosine for cysteine at amino acid 282 in the α3 loop (C282Y) and abolishes the disulfide bond in this domain.4 Loss of this disulfide bond interferes with the interaction of HFE protein with β2-microglobulin, and the C282Y mutant protein demonstrates decreased presentation at the cell surface, increased retention in the endoplasmic reticulum, and accelerated degradation.27 A second mutation associated with HH results in the change of a histidine to an aspartate at position 63 in the α1 chain (H63D), but this mutation has less biological impact than the C282Y mutation. Like HH patients, HFE-knockout mice manifest higher hepatic iron levels, elevated transferrin saturation (TS), increased intestinal iron absorption, and relative sparing of iron loading in reticuloendothelial cells.14
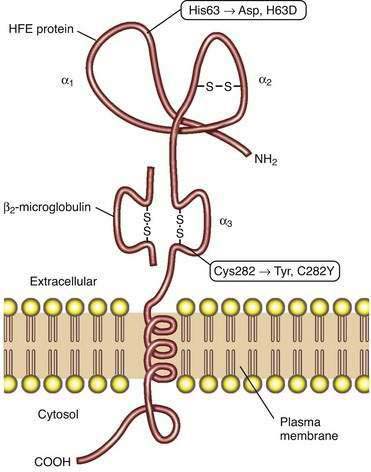
The molecular mechanisms by which HFE influences iron-dependent regulation of hepcidin remain unclear. HFE can bind to both TFR2 and the classic transferrin receptor, TFR1.20 In addition, both HFE and TFR2 may interact with hemojuvelin, suggesting that a complex of HFE and TFR2 may play a regulatory role in BMP signaling. One proposed model suggests that the complex of TFR1 and HFE acts as an iron sensor at the cell membrane of the hepatocyte; as the TS increases, diferric transferrin displaces HFE from TFR1, thereby making HFE available to bind to TFR2. The complex of HFE and TFR2 is then postulated to influence hepcidin expression.20,23
IRON-INDUCED TISSUE INJURY AND FIBROSIS
Another major pathophysiologic mechanism in HH relates to the liver damage that results from iron overload. In patients with advanced HH, hepatic fibrosis and cirrhosis are the principal pathologic findings. A number of studies of experimental hepatic iron overload have identified iron-dependent lipid peroxidation and associated impairment of membrane-dependent functions of mitochondria, microsomes, and lysosomes.28 A relationship between iron-induced lipid peroxidation and fibrosis has been shown in several studies.29,30 One hypothesis is that iron-induced lipid peroxidation occurs in hepatocytes and causes hepatocellular injury or death. Kupffer cells may become activated by products released from injured iron-loaded hepatocytes and produce profibrogenic cytokines, which can, in turn, stimulate hepatic stellate cells to synthesize larger amounts of collagen, thereby leading to pathologic fibrosis.29,30
Studies of iron-induced tissue damage in organs other than the liver, such as the heart, pancreas, and endocrine glands, have been limited. Studies in myocardial cells have shown functional abnormalities resulting from iron-induced peroxidation.31
CLINICAL FEATURES
Many patients with HFE-related HH come to medical attention without any symptoms or physical findings. They are identified as homozygous relatives of probands during family screening studies or by the results of serum iron studies in routine screening blood chemistry panels (Table 74-2).32,33 Nonetheless, the clinician should know the typical clinical manifestations in patients who do present with symptomatic disease. Most patients with symptomatic HFE-related HH are 40 to 50 years of age at the time of detection. Although C282Y homozygosity is distributed equally between men and women, the clinical penetrance is much lower in women, as a result of iron loss from normal menses and childbirth and possible gender-related disease modifier genes (Table 74-3).34–39
Table 74-2 Clinical Features of Hereditary Hemochromatosis in Three Studies from Different Time Periods
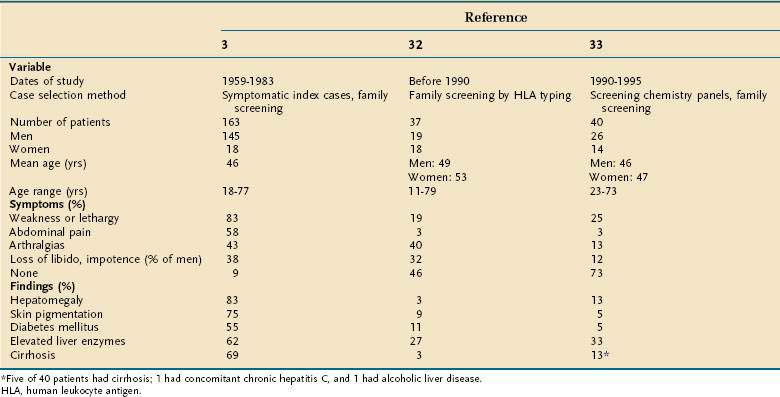
Table 74-3 Clinical Penetrance of C282Y Homozygosity in Women and Men
| FINDING | WOMEN (%) | MEN (%) |
|---|---|---|
| Iron-overload–related disease*39 | 1 | 28 |
| Liver fibrosis34,37–39 | 0-5 | 11-18 |
| Cirrhosis34,35,37–39 | 0-2 | 1-12 |
| Abnormal metacarpophalangeal joints34,37,39 | 2-12 | 4-26 |
Superscript numbers are references.
* Defined as iron overload accompanied by at least one of the following conditions: hepatocellular carcinoma, liver fibrosis or cirrhosis, characteristic arthropathy, raised serum aminotransferase levels, or diagnosis due to symptoms of hereditary hemochromatosis.
When patients present with symptoms, the most common are weakness and lethargy, arthralgias, abdominal pain, and loss of libido or potency in men.3,40 Patients with HFE-related HH may have nonspecific right upper quadrant abdominal pain that is most likely caused by hepatic capsular distention. Hepatomegaly is found on physical examination in a majority of patients; splenomegaly and other complications of chronic liver disease, including ascites, edema, and jaundice, may be present. Diabetes mellitus has decreased in frequency with earlier diagnosis of hemochromatosis and is typically not seen in the absence of cirrhosis (see later). Detection of the often subtle bronzed or slate gray skin pigmentation of HFE-related HH requires astuteness on the part of the clinician (see later). Organ damage and symptoms are usually related to the extent of iron loading. When patients are identified prospectively by either family or population screening, the frequency of patients who are asymptomatic increases dramatically.34–39
All patients with HFE-related HH who have an elevated serum ferritin value should also have increased hepatic iron stores, but the extent of hepatic iron loading is often not high enough to cause liver damage. In the late 1960s, cirrhosis was found in more than 50% of the patients identified with HH3; in studies from the 1970s through the 1990s, cirrhosis was found in only 5% to 10% of patients.32,33 Later population screening studies have reported an even lower frequency of cirrhosis in C282Y homozygotes.35,38,39 Serum aminotransferase elevations are usually mild. With regular phlebotomy and depletion of excess iron stores, elevated liver enzyme values typically revert to normal. When HFE-related HH is diagnosed and treated before the development of hepatic fibrosis or cirrhosis, long-term hepatic abnormalities do not develop. When HFE-related HH is detected after cirrhosis has developed, however, hepatocellular carcinoma can occur even after successful phlebotomy,41 thus emphasizing the importance of early diagnosis and treatment.
Other clinical manifestations that can occur relate to the level of iron loading in nonhepatic organs. In older series, diabetes mellitus was a common complication of pancreatic iron loading,3 but in later series in which the diagnosis of HFE-related HH was made earlier in its course, diabetes mellitus has rarely been present.32,33 Other endocrinologic abnormalities are loss of libido and impotence in men, owing to both primary testicular failure and gonadotropin insufficiency resulting from the effects of iron on pituitary function,42 and occasionally hypothyroidism; adrenal function is typically normal. Other endocrinologic effects can occur as a result of complications of cirrhosis (see Chapter 92).
Cardiac manifestations occur rarely because patients are now diagnosed earlier in the course of HFE-related HH than in the past. Cardiomyopathy, atrial and ventricular dysrhythmias, and congestive heart failure can occur.43 Characteristic of the arthropathy of HFE-related HH are changes in the second and third metacarpophalangeal joints. Joint space narrowing, chondrocalcinosis, subchondral cyst formation, osteopenia, and swelling of the joints may be seen.8,9,40 Unfortunately, the arthritic symptoms of HFE-related HH typically do not improve with phlebotomy. The skin pigmentation of HFE-related HH, which can be subtle, is characterized by either a bronze discoloration due to predominant melanin pigmentation or a gray pigmentation resulting from iron deposition in the basal layers of the epidermis.40 The frequency of certain infections, including those caused by Vibrio vulnificus, Listeria monocytogenes, Yersinia enterocolitica, and Yersinia pseudotuberculosis, is more common in iron-loaded patients, although still rare.
DIAGNOSIS
The requirements for diagnosis of HH have changed since the availability of HFE mutation analysis.8,9 As in the past, the disorder must be considered in any patient with typical symptoms or abnormal screening iron test results. If the physical examination or family history raises suspicions, then the appropriate serum iron tests along with HFE mutation analysis should be obtained. With the advent of genetic testing, the need for liver biopsy has diminished. In symptomatic patients, as discussed earlier, the most common symptoms are fatigue, malaise, right upper quadrant abdominal pain, and arthralgias. Less commonly, symptoms of chronic liver disease, diabetes mellitus, and congestive heart failure are identified. Because many of these symptoms are nonspecific or are related to other common diseases, HH is often overlooked by clinicians.
In the early 1990s, patients with HH commonly presented after the discovery of abnormal results of screening blood chemistry tests obtained as part of routine health maintenance or for another reason.33 Many commercial laboratories added iron and total iron-binding capacity (TIBC), with TS calculated as iron ÷ TIBC × 100%, to their panel of screening serum chemistry tests, and in many patients, a TS was obtained inadvertently even though the test had not been specifically ordered. In one series, 62% of patients newly identified between 1990 and 1995 came to medical attention in this way.33 Another 14% of cases were identified through screening of family members of a known proband. Therefore, as many as 75% of patients came to medical attention by way of screening laboratory tests. The majority of these patients were asymptomatic and had no physical findings of HH, and the frequency of end-stage complications of HFE-related HH, such as cirrhosis and diabetes mellitus, was much lower than that reported in earlier series of patients who presented with symptoms of the disease.33 In 1998, the Health Care Finance Administration (now the Center for Medicare and Medicaid Services) stopped providing reimbursement for screening tests of any kind, and since then, fewer American patients with HH have been identified through routine screening.
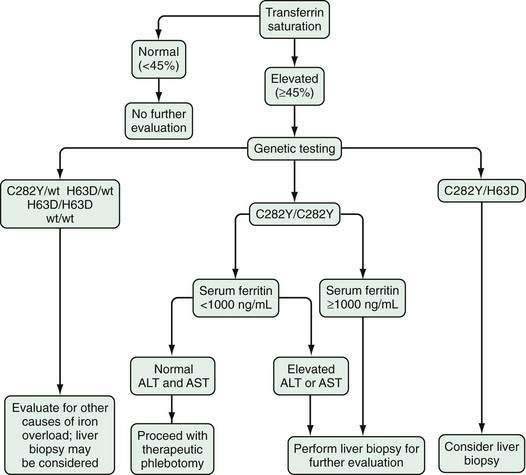
When HFE-related HH has been considered, the diagnosis is relatively straightforward. Measurements of serum iron and TIBC or transferrin, with calculation of TS, and serum ferritin should be obtained (Table 74-4). Studies have shown that it is not necessary for blood samples to be drawn in the fasting state. A TS value greater than 45% is the earliest phenotypic manifestation of HFE-related HH. As a result, TS is a more sensitive and specific test for HFE-related HH than serum ferritin, which can be normal in young persons with HFE-related HH or elevated in unaffected persons for a variety of reasons, including various types of necro-inflammatory liver disease (e.g., chronic viral hepatitis, alcoholic liver disease, nonalcoholic steatohepatitis), certain malignancies, and other inflammatory conditions. An elevated serum ferritin level with a normal TS value in a person who has an inflammatory disorder generally suggests that the person does not have HFE-related HH. On the other hand, an elevated TS value with a normal ferritin value in a young person does not exclude HFE-related HH. A large North American population screening study demonstrated that 1 in 227 white persons was homozygous for the C282Y mutation, but only 57% and 88% of the female and male homozygotes, respectively, had an elevated ferritin value.36 This finding indicates that a higher proportion of C282Y homozygotes do not express iron overload than had previously been thought. The proportion of the non-expressing cohort that will subsequently show evidence of iron loading is uncertain. In a longitudinal follow-up study of patients identified by genetic screening, progressive iron loading, as indicated by rising serum ferritin levels, developed in 40% of C282Y homozygotes.44
Table 74-4 Representative Iron Measurements in Serum and Liver and HFE Mutation Analysis Findings in Patients with Phenotypic HFE-Related Hereditary Hemochromatosis
| TEST | NORMAL VALUE/RESULT | VALUE/RESULT IN HFE-RELATED HEMOCHROMATOSIS |
|---|---|---|
| Serum | ||
| Iron | ||
| (µg/dL) | 60-180 | 180-300 |
| (µmol/L) | 11-32 | 32-54 |
| Transferrin saturation (%) | 20-45* | 45-100 |
| Ferritin | ||
| Men (ng/mL; µg/L) | 20-200 | 300-3000 |
| Women (ng/mL; µg/L) | 15-150 | 250-3000 |
| Liver | ||
| Iron staining | 0, 1+ | 3+, 4+ |
| Iron concentration | ||
| (µg/g dry weight) | 300-1500 | 3000-30,000 |
| (µmol/g dry weight) | 5-27 | 53-536 |
| Hepatic iron index ([µmol/g dry weight] ÷ age in years) | <1.1 | >1.9 |
| HFE Mutation Analysis | ||
| wt/wt | C282Y/C282Y | |
| C282Y/wt | C282Y/H63D | |
| H63D/wt |
wt, wild type.
* A value of 45% is considered elevated.
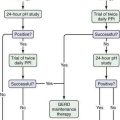
As soon as serum iron parameters have been determined to be abnormal, HFE mutation analysis should be performed. If the patient is a C282Y homozygote or a compound heterozygote (C282Y/H63D) and has a serum ferritin level lower than 1000 ng/mL and normal liver enzyme values, a liver biopsy is not needed.45–47 If, however, the serum ferritin value is higher than 1000 ng/mL or liver enzymes are elevated, liver biopsy is indicated. If liver biopsy is determined to be appropriate, sufficient tissue for histopathologic evaluation and biochemical measurement of the hepatic iron concentration (HIC) should be obtained. With the advent of genetic testing, liver biopsy is performed solely to assess the damage (if any) to the liver. A proposed algorithm for evaluating people for possible HFE-related HH is shown in Figure 74-3.
When a liver biopsy specimen is obtained, Perls’ Prussian blue stain is used for the determination and localization of storage iron. Iron stores in HFE-related HH are typically found in periportal hepatocytes, with little or no iron found in Kupffer cells (Fig. 74-4).48 In patients with a higher HIC, iron distribution becomes panlobular, and storage iron can be seen in Kupffer cells and bile duct cells. Grade 1 or 2 Perls’ Prussian blue staining can be seen in specimens from normal livers or specimens from patients with very early HH confirmed by HFE mutation analysis. Grade 3 stainable iron occasionally can be seen in specimens from patients with alcoholic cirrhosis or end-stage liver disease, in which iron staining correlates poorly with HIC. In the absence of other disorders, grade 3 to 4 stainable iron in an HFE pattern is consistent with HFE-related HH.
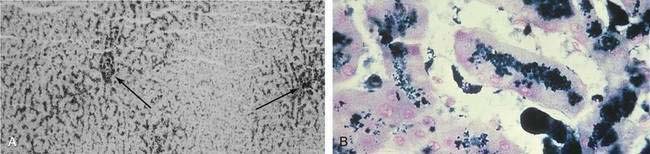
In addition to histochemical staining, biochemical iron measurement in the liver is important (see Table 74-4). Typically, patients with HFE-related HH who present with symptoms have a HIC greater than 10,000 µg/g (dry weight) (normal < 1500 µg/g); HIC values may be more than 30,000 µg/g. Fibrosis and cirrhosis are usually not seen until the HIC exceeds 20,000 µg/g.49 In patients with both HFE-related HH and other forms of chronic liver disease, such as alcoholic liver disease or chronic viral hepatitis, increased fibrosis or cirrhosis can occur at a much lower HIC and at a younger age.8,9,50,51 In asymptomatic or younger patients with early HFE-related HH, HIC is increased to a lesser degree and often is much less than 10,000 µg/g.
A common diagnostic dilemma occurs when it is not clear whether a patient has liver disease with abnormal iron parameters or HFE-related HH with elevations in liver enzymes. In this setting, HFE mutation analysis is extremely useful. If the patient is a C282Y homozygote or a compound heterozygote (C282Y/H63D), the iron loading is most likely caused predominantly by the genetic abnormality. On the other hand, if the patient has underlying liver disease, is a C282Y heterozygote, or is an H63D heterozygote or H63D homozygote or has neither mutation, it is likely that the iron loading is caused by the underlying liver disease, perhaps with a minor contribution from the HFE genotype. In the past, the hepatic iron index (HIC in µmol/g ÷ the patient’s age in years) was used to distinguish HH (>1.9) from secondary iron overload (⩽1.9).49 With HFE mutation analysis, the value of the hepatic iron index has diminished.
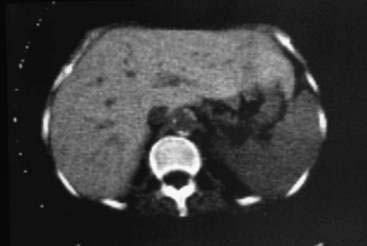
Computed tomography (CT), magnetic resonance imaging (MRI), and magnetic susceptibility testing have all been proposed as techniques to quantify the HIC without the need for a liver biopsy (Fig. 74-5). Magnetic susceptibility testing is available in only a few centers in the United States and Europe as a research tool. In early studies, CT and MRI were generally not reliable for detecting mild iron overload, but newer MRI techniques have shown improved sensitivity.52,53
TREATMENT AND PROGNOSIS
The treatment of HFE-related HH is relatively straightforward; most patients can be treated with routine therapeutic phlebotomy8,9 (Table 74-5). Ideally, diagnosis and initiation of treatment should begin before the development of hepatic fibrosis or cirrhosis; if they are, patients will have a normal lifespan. Each unit of whole blood (500 mL) contains approximately 200 to 250 mg of iron, depending on the hemoglobin concentration; therefore, C282Y homozygotes who have 10 to 20 g of excess storage iron require extended phlebotomy regimens (40 to 80 units of blood removed). Most patients can tolerate weekly phlebotomy of 1 unit of whole blood, and occasional younger patients can tolerate the removal of 2 or 3 units per week. Some older patients and occasional patients with a coexisting underlying hematologic disorder resulting in anemia can tolerate phlebotomy of only 0.5 unit per week or every other week. The iron-chelating drug deferoxamine is used in patients with HFE-related HH and cardiac manifestations or in patients who cannot tolerate phlebotomy. Deferoxamine, 20 to 50 mg/kg/day, is administered 5 days per week as a continuous subcutaneous infusion (over a 12-hour period each day) via a portable pump.
Table 74-5 Treatment of HFE-Related Hereditary Hemochromatosis
The prognosis for patients with HFE-related HH is improved significantly by therapeutic phlebotomy.3,54,55 Life expectancy is reduced in patients who present with cirrhosis or diabetes mellitus, and the risk of death from hepatocellular carcinoma is higher in patients with HFE-related HH. Hepatocellular cancer is usually seen only in patients who already have cirrhosis. Established cirrhosis typically does not reverse with phlebotomy, but many patients will have a decrease in hepatic fibrosis with aggressive treatment.37 Unfortunately, neither arthritis nor hypogonadism improves; however, management of diabetes mellitus may become easier after iron removal.
The value of screening for hepatocellular carcinoma in cirrhotic patients with HH is controversial because the cost-effectiveness of the screening approaches has not been validated. Most authorities suggest ultrasonography or CT scanning and a serum alpha fetoprotein measurement every six months in cirrhotic patients with HFE-related HH. With improved methods of detection and treatment of small, early hepatocellular carcinomas (e.g., radiofrequency ablation, chemoembolization, resection, and liver transplantation), screening seems to be reasonable (see Chapter 94).
When diagnosis and treatment are delayed and complications of end-stage liver disease develop, liver transplantation (LT) may be undertaken (see also Chapter 95). In the era before HFE genotyping was available, studies addressing the outcome of iron-loaded patients after LT reported that the survival rate was substantially lower than that for other patients. Significant hepatic iron loading is now known to occur in 35% to 78% of patients with end-stage liver disease, regardless of the cause of cirrhosis.56 Only about 10% of patients with iron overload and end-stage liver disease are C282Y homozygotes.57 Two studies have analyzed post-LT outcome in patients with confirmed HFE-related HH and found five-year survival rates of 34% and 55%, which were lower than the overall post-LT survival rate of 72% to 75%.58,59 The most common causes of death were infections, cardiovascular complications, and recurrence of hepatocellular carcinoma. The five-year survival rate for patients with non-HH iron overload was higher (63%) than that for patients with HFE-related HH (34%) but still somewhat reduced compared with the overall survival rate.58 If iron overload of any cause is diagnosed early, it should be treated to reduce the chance of death following LT.
FAMILY SCREENING
When a proband with HFE-related HH has been identified and therapy has been initiated, the clinician still has a responsibility to the patient’s family.8,9,60 For asymptomatic C282Y homozygotes and compound heterozygotes (C282Y/H63D) identified by HFE mutation analysis within a sibship, a liver biopsy is unnecessary. In family members in whom serum ferritin levels are increased, proceeding to therapeutic phlebotomy is reasonable. Liver biopsy should be reserved for patients in whom another underlying liver disease is possible. Persons who are C282Y heterozygotes are not at risk for progressive iron overload. If the spouse of a C282Y homozygote is a C282Y heterozygote, their offspring have a 50% chance of being homozygous for C282Y. HFE mutation analysis in the children can eliminate the need for subsequent serum iron testing if a genotype of C282Y/C282Y or C282Y/H63D is not found in a proband’s spouse, although the issues of genetic discrimination and stigmatization must be acknowledged and considered. In children who are C282Y homozygotes or compound heterozygotes, serum ferritin measurements should be obtained yearly and phlebotomy instituted when ferritin values become elevated.
Adams PC, Barton JC. Haemochromatosis. Lancet. 2007;370:1855-60. (Ref 5.)
Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769-78. (Ref 36.)
Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221-30. (Ref 39.)
Andrews NC. Forging a field: The golden age of iron biology. Blood. 2008;112:219-30. (Ref 20.)
Bacon BR. Hemochromatosis: Diagnosis and management. Gastroenterology. 2001;120:718-25. (Ref 8.)
Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of 845G→A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211-18. (Ref 35.)
Feder JN, Gnirke A, Thomas W, et al. A novel MHC class 1-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399-408. (Ref 4.)
Ganz T. Iron homeostasis: Fitting the puzzle pieces together. Cell Metab. 2008;7:288-90. (Ref 23.)
Gordeuk VR. African iron overload. Semin Hematol. 2002;39:263-9. (Ref 16.)
Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323-42. (Ref 21.)
Olynyk JK, Trinder D, Ramm GA, et al. Hereditary hemochromatosis in the post-HFE era. Hepatology. 2008;48:991-1001. (Ref 6.)
Philippe MA, Ruddell RG, Ramm GA. Role of iron in hepatic fibrosis: One piece in the puzzle. World J Gastroenterol. 2007;13:4746-54. (Ref 30.)
Pietrangelo A. Hereditary hemochromatosis: A new look at an old disease. N Engl J Med. 2004;350:2383-97. (Ref 7.)
Pietrangelo A. Non-HFE hemochromatosis. Hepatology. 2004;39:21-9. (Ref 12.)
Powell LW. Hereditary hemochromatosis and iron overload diseases. J Gastroenterol Hepatol. 2002;17(Suppl):S191-5. (Ref 9.)
1. Bacon BR, Joseph H. Sheldon and hereditary hemochromatosis: Historical highlights. J Lab Clin Med. 1989;113:761-2.
2. Simon M, Bourel M, Fauchet R, Genetet B. Association of HLA-A3 and HLA-B14 antigens with idiopathic hemochromatosis. Gut. 1976;17:332-4.
3. Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic and noncirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313:1256-62.
4. Feder JN, Gnirke A, Thomas W, et al. A novel MHC class 1-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399-408.
5. Adams PC, Barton JC. Haemochromatosis. Lancet. 2007;370:1855-60.
6. Olynyk JK, Trinder D, Ramm GA, et al. Hereditary hemochromatosis in the post-HFE era. Hepatology. 2008;48:991-1001.
7. Pietrangelo A. Hereditary hemochromatosis: A new look at an old disease. N Engl J Med. 2004;350:2383-97.
8. Bacon BR. Hemochromatosis: Diagnosis and management. Gastroenterology. 2001;120:718-25.
9. Powell LW. Hereditary hemochromatosis and iron overload diseases. J Gastroenterol Hepatol. 2002;17(Suppl):S191-5.
10. Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21-2.
11. Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25:14-15.
12. Pietrangelo A. Non-HFE hemochromatosis. Hepatology. 2004;39:21-9.
13. Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77-82.
14. Fleming RE, Britton RS, Waheed A, et al. Pathogenesis of hereditary hemochromatosis. Clin Liver Dis. 2004;8:755-73.
15. De Domenico I, Ward DM, Nemeth E, et al. The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci U S A. 2005;102:8955-60.
16. Gordeuk VR. African iron overload. Semin Hematol. 2002;39:263-9.
17. Barton JC, Acton RT, Rivers CA, et al. Genotypic and phenotypic heterogeneity of African Americans with primary iron overload. Blood Cells Mol Dis. 2003;31:310-19.
18. Bottomley SS. Secondary iron overload disorders. Semin Hematol. 1998;35:77-86.
19. Whitington PF. Neonatal hemochromatosis: A congenital alloimmune hepatitis. Semin Liver Dis. 2007;27:243-50.
20. Andrews NC. Forging a field: The golden age of iron biology. Blood. 2008;112:219-30.
21. Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323-42.
22. Pietrangelo A. Hemochromatosis: An endocrine liver disease. Hepatology. 2007;46:1291-301.
23. Ganz T. Iron homeostasis: Fitting the puzzle pieces together. Cell Metab. 2008;7:288-90.
24. Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669-73.
25. Ahmad KA, Ahmann JR, Migas MC, et al. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol Dis. 2002;29:361-6.
26. Harrison-Findik DD. Role of alcohol in the regulation of iron metabolism. World J Gastroenterol. 2007;13:4925-30.
27. Waheed A, Parkkila S, Zhou XY, et al. Hereditary hemochromatosis: Effects of C282Y and H63D mutations on association with β2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells. Proc Natl Acad Sci USA. 1997;94:12384-9.
28. Bacon BR, Britton RS. The pathology of hepatic iron overload: A free radical-mediated process? Hepatology. 1990;11:127-37.
29. Britton RS, Bacon BR. Role of free radicals in liver diseases and hepatic fibrosis. Hepatogastroenterology. 1994;41:343-8.
30. Philippe MA, Ruddell RG, Ramm GA. Role of iron in hepatic fibrosis: One piece in the puzzle. World J Gastroenterol. 2007;13:4746-54.
31. Hershko C, Link G, Cabantchik I. Pathophysiology of iron overload. Ann N Y Acad Sci. 1998;850:191-201.
32. Adams PC, Kertesz AE, Valberg LS. Clinical presentation of hemochromatosis: A changing scene. Am J Med. 1991;90:445-9.
33. Bacon BR, Sadiq SA. Hereditary hemochromatosis: Presentation and diagnosis in the 1990s. Am J Gastroenterol. 1997;92:784-9.
34. Bulaj ZJ, Ajioka RS, Phillips JD, et al. Disease-related conditions in relatives of patients with hemochromatosis. N Engl J Med. 2000;343:1529-35.
35. Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of 845G→A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211-18.
36. Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769-78.
37. Powell LW, Dixon JL, Ramm GA, et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med. 2006;166:294-301.
38. Asberg A, Hveem K, Kannelonning K, Irgens WO. Penetrance of the C28Y/C282Y genotype of the HFE gene. Scand J Gastroenterol. 2007;42:1073-7.
39. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221-30.
40. Edwards CQ, Cartwright GE, Skolnick MH, Amos DB. Homozygosity for hemochromatosis: Clinical manifestations. Ann Intern Med. 1980;93:519-25.
41. Deugnier Y. Iron and liver cancer. Alcohol. 2003;30:145-50.
42. Lufkin EG, Baldus WP, Bergstralh EJ, Kao PC. Influence of phlebotomy treatment on abnormal hypothalamic-pituitary function in genetic hemochromatosis. Mayo Clin Proc. 1987;62:473-9.
43. Olson LJ, Edwards WD, Holmes DRJr, et al. Endomyocardial biopsy in hemochromatosis: Clinicopathologic correlates in six cases. J Am Coll Cardiol. 1989;13:116-20.
44. Olynyk JK, Hagan SE, Cullen DJ, et al. Evolution of untreated hereditary hemochromatosis in the Busselton population: A 17-year study. Mayo Clin Proc. 2004;79:309-13.
45. Guyader D, Jacquelinet C, Moirand R, et al. Noninvasive prediction of fibrosis in C282Y homozygous hemochromatosis. Gastroenterology. 1998;115:929-36.
46. Bacon BR, Olynyk JK, Brunt EM, et al. HFE genotype in patients with hemochromatosis and other liver diseases. Ann Intern Med. 1999;130:953-62.
47. Morrison ED, Brandhagen DJ, Phatak PD, et al. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med. 2003;138:627-33.
48. Brunt EM. Pathology of hepatic iron overload. Semin Liver Dis. 2005;25:392-401.
49. Bassett ML, Halliday JW, Powell LW. Value of hepatic iron measurements in early hemochromatosis and determination of the critical iron level associated with fibrosis. Hepatology. 1986;6:24-9.
50. Diwarkaran HH, Befeler AS, Britton RS, et al. Accelerated hepatic fibrosis in patients with combined hereditary hemochromatosis and chronic hepatitis C infection. J Hepatol. 2002;36:687-91.
51. Fletcher LM, Dixon JL, Purdie DM, et al. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology. 2002;122:281-9.
52. St Pierre TG, Clark PR, Chua-anusorn W, et al. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood. 2005;105:855-61.
53. Positano V, Salani B, Pepe A, et al. Improved T2* assessment in liver iron overload by magnetic resonance imaging. Magn Reson Imaging. 2008 Jul 28. [Epub ahead of print]
54. Adams PC, Speechley M, Kertesz AE. Long-term survival analysis in hereditary hemochromatosis. Gastroenterology. 1991;101:368-72.
55. Niederau C, Fischer R, Pürschel A, et al. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology. 1996;110:1107-19.
56. Deugnier Y, Turlin B. Pathology of hepatic iron overload. World J Gastroentrol. 2007;13:4755-60.
57. Brandhagen DJ, Alvarez W, Therneau TM, et al. Iron overload in cirrhosis: HFE genotypes and outcome after liver transplantation. Hepatology. 2000;31:456-60.
58. Kowdley KV, Brandhagen DJ, Gish RG, et al. Survival after liver transplantation in patients with hepatic iron overload: The National Hemochromatosis Transplant Registry. Gastroenterology. 2005;129:494-503.
59. Crawford DHG, Fletcher LM, Hubscher SG, et al. Patient and graft survival after liver transplantation for hereditary hemochromatosis: Implications for pathogenesis. Hepatology. 2004;39:1655-62.
60. Galhenage SP, Viiala CH, Olynyk JK. Screening for haemochromatosis: Patients with liver disease, families, and populations. Curr Gastroenterol Rep. 2004;6:44-51.