34 Hematologic Symptoms
who maintains the healthy attitude,
But don’t discard feeling from a patient’s history.
symptoms and contraindications
Evoked responses, unsuppressed,
Anemia
Primary anemia
Primary anemias result from production of red blood cells that have a structural defect, leading to increased red cell destruction such as in sickle cell anemia, beta-thalassemia, and others (Box 34-1). Primary anemias are no longer strictly childhood illnesses. The ability to replace defective red blood cells with transfusion or partial exchange transfusion has revolutionized the care of these children, extending life expectancy well into the sixth decade or beyond. As these diseases have been converted into chronic illnesses, patients suffering from these diseases are rarely included in the dialogue of palliative care.
Frequent transfusions have brought new challenges to the care of children and young adults affected by primary anemias. During the 1980s and 1990s, blood-borne infection with Hepatitis B and C, and HIV infected many people who received transfusions due to primary anemia. Although the risk of transfusion-associated infection is now decreased to approximately 1 in 2 million transfusions,1,2 relieving symptoms of primary anemia continues to carry a significant cost in the form of iron overload. The deposits of iron in tissues, especially the liver and heart, ultimately compromises end-organ function leading to heart failure and death. Development and clinical use of first intravenous and more recently oral iron chelators are once again revolutionizing delivery of care to patients with primary anemia. Nonetheless, significant challenges and opportunities for improving palliative care to this often overlooked population remain. David Nathan has written a poignant description of one patient’s journey navigating the waves of innovation in care for patients with thalassemia. He discusses the burdens of subcutaneous desferoxamine infusion and how much his patient hated it, to the extent of refusing to take the desferoxamine. That decision resulted in repeated episodes of heart failure. Dr. Nathan talks about the development of the oral chelator, desferisirox and the positive impact on quality of life for his patient to be free of the iron-chelator infusion pump.3
Secondary anemia
The most common cause of secondary anemia is exposure to marrow-suppressive chemotherapy and radiation therapy in the course of treatment for childhood cancer. Overall survival for childhood cancer has improved significantly during the past 30 years and is 80%.4 Cure rates of some childhood cancers, such as low-risk acute lymphoblastic leukemia (ALL), Hodgkin Lymphoma, and low stage Wilms tumor are 95 percent or better.4 However, many chemotherapeutic agents used in the treatment of childhood cancers have bone marrow suppression or transient bone marrow aplasia as a side effect. Children receiving intensive chemotherapy are frequently at risk for development of anemia, bleeding, and infection. For many modern chemotherapy regimens, transfusions of packed red blood cells (pRBC) and apheresed platelets are an integral and anticipated component of supportive care.
symptoms associated with anemia
Fatigue during treatment for malignancy, in part related to anemia, is the most frequently reported symptom for adults with cancer. A survey of major pediatric hematology- oncology centers in Europe documented more than 80 percent of children with cancer as being anemic.5 For children, symptoms related to anemia can extend far beyond a complaint of fatigue (Box 34-2). Children manifest different symptoms of anemia in different age groups. Many young school-age and toddler-age children appear very active and apparently feel well with moderate anemia where hemoglobin is in the 8 g/dL range. For infants and toddlers, symptomatic anemia may be expressed by a decreased ability to nurse, take a bottle, or eat. Many a mother has made the simple statement that their infant or toddler “probably needs to be transfused” because the child just isn’t eating as well as usual. Administration of a transfusion can be the difference between a child who is able to eat consistently with his or her personal baseline and one who is not eating well. For other children, anemia may be recognized by longer nap times, change in temperament or limitations in ability to engage in activities or play. All of these are important issues of quality of life for families of children with life- threatening illness, whether they are pursuing curative therapy or during terminal care.
Diagnostic evaluation
Hemoglobinopathies are frequently identified because of presence of family history or newborn screening. However, some defects of red cell structure, such as hereditary spherocytosis, may cause sufficiently mild anemia to go undetected into adulthood in otherwise healthy parents. Diagnostic evaluation includes a thorough patient and family history, physical exam, examination of the blood smear, hemoglobin electrophoresis, evaluation of nutritional status, and osmotic fragility studies to name the most frequently used tests. A detailed diagnostic algorithm is beyond the scope of this text. Readers are referred to other resources such as Practical Algorithms in Pediatric Hematology and Oncology for more information.6
Guidelines for Transfusion
Primary anemia
The most recent recommendations for medical management of sickle cell disease are available from the National Heart, Lung, and Blood Institute of the National Institutes of Health (NIH).7 General indications for transfusion of children with sickle cell anemia include hemoglobin less than 5 to 6 g/dL, development of acute chest syndrome, aplastic crisis, preoperative prophylaxis, or to resolve protracted pain crises. Similarly, transfusion goals for patients with β-thalassemia major is to maintain a hemoglobin in the range of 9 to 9.5 g/dL.8,9
Secondary anemia
Several transfusion guidelines exist.8,9 While many centers use a guideline of hemoglobin less than 7 to 8 g/dL as a parameter for transfusion, the ultimate indication for transfusion is a symptomatic patient who is unlikely to correct the anemia in a timely manner without medical intervention. Symptoms of anemia include: tachycardia, tiredness, orthostatic hypotension, increased fatigue, and sleeping more hours per day (see Box 34-2).
Specifications of the Product
Leukoreduced ABO and D blood group appropriate and cross-matched pRBC are used in primary as well as secondary anemia to decrease alloimmunization and febrile transfusion reactions. Contaminating white blood cells, especially lymphocytes, are responsible for the majority of allergic transfusion reactions. Leukocyte reduction can be achieved either during processing soon after collection of the product or before administration to a patient. The American Association of Blood Bank Standards requires leukoreduced units to have less than 5 million leukocytes per unit.10 Packed RBC for immunocompromised individuals should be leukoreduced. In some geographic regions, blood banks provide exclusively leukoreduced red cells or platelets.
Secondary anemia
Even with leukoreduction, small T lymphocytes, which have a diameter similar to erythrocytes, can pass through the filter and ultimately into the recipient. These lymphocytes have the potential in immunocompromised individuals to cause GVHD.11 To prevent transfusion-associated GVHD, pRBC are irradiated at 25 to 50 Gy.10 In large urban areas, hospital blood banks have their own blood product irradiators. Smaller urban areas may rely on a single unit centrally located at the local Red Cross Center. More rural areas may have to special order irradiated units from distant Red Cross Centers, creating a delay in availabity of the product of hours to days.
Transfusion Side Effects
Acute
Minor allergic reactions vary from appearance of few to abundant hives. Minor allergic reactions usually respond quickly to 0.5 to 1 mg/kg diphenhydramine administered intravenously or orally. Alternatively, hydroxyzine 0.5 to 1 mg/kg intravenously or 2 mg/kg orally can be used. Allergic reactions leading to shortness of breath and bronchospasm are more common with transfusion of platelet products, due to the greater volume of donor plasma, which contains antibodies. Anaphylaxis may require administration of hydrocortisone 1 to 5 mg/kg/day intravenously or, in severe cases, epinephrine according to resuscitation protocols. Patients with a history of allergic reaction may do better with prophylactic diphenhydramine or hydroxyzine before transfusion.7 Washing pRBC may lessen the amount of plasma in the product, decreasing allergic and hemolytic reactions, at the cost of also decreasing the number of RBC in the unit. The most severe form of allergic reaction is transfusion-related acute lung injury (TRALI), which occurs during or immediately after transfusion and is characterized by difficulty breathing and pulmonary infiltrates on chest x-ray. Patients experiencing TRALI may require intubation and ventilator support.
Profoundly neutropenic patients who develop febrile reactions during a transfusion are generally committed to a minimum of 24 to 48 hours of intravenous antibiotic therapy, until it is clear the fever is not due to bacterial contamination of the product or other bacterial infection in the patient. Culturing the transfusion product bag is ideal. In practice though, febrile reactions often occur after completion of the transfusion, at which point the product bag has been discarded. Many clinicians have reasoned that administration of prophylactic acetaminophen should decrease the febrile inflammatory response to the blood product. If we could decrease the incidence of febrile transfusion reaction, we may be able to spare immunocompromised patients from hospital admission and empiric antibiotics. However, several studies have failed to demonstrate benefit.12,13 Therefore, routinely administering acetominophen before transfusion is not recommended unless the child has a personal history of transfusion reaction.
Hemolytic transfusion reactions can also start with fever accompanied by abdominal or flank pain. Patients may experience a general sense of feeling unwell or agitation. Tea-colored or cola-colored urine is a supportive finding of intravascular hemolysis. The product infusion should be stopped and returned to the blood bank with new patient samples so evaluation for a hemolytic reaction can be pursued. Routine blood counts demonstrate a decreased hemoglobin. Other supporting laboratory studies include increased haptoglobin levels. Urinalysis may demonstrate urine hemoglobin in the absence of red blood cells. When these symptoms occur in a patient with sickle cell disease, it can be challenging to sort out the presence of a hemolytic transfusion reaction from underlying pathophysiology of hemolysis due to vaso-occlusive crisis. The hallmark of a transfusion reaction is that the patient becomes Coombs positive. Additionally, patients with sickle cell disease can experience delayed transfusion reactions with a fall in hemoglobin below their personal baseline days to weeks after a transfusion. Adding to the challenge of managing these patients is that both acute and chronic transfusion reactions can precipitate an acute vaso-occlusive pain crisis or acute chest syndrome.7
Chronic
Other adverse events associated with transfusions include transmission of infectious agents, particularly viruses. Risks of known viral infectious agents such as hepatitis B, hepatitis C, and HIV are approximately 1 in 2 million units.1,2
Those who are regularly transfused, especially for primary anemia, battle with chronic complications of transfusion. For adults transfused pre-1990, transfusion-acquired viral infection may add additional complexity to their healthcare. Teenagers and young adults with extensive transfusion histories may develop extensive alloimmunization that makes finding an appropriately matched unit challenging. The incidence of alloimmunization in patients with sickle cell disease is 25%, higher than the general population.7 The higher incidence of alloimmunization is due in part to the difference in surface expression of red cell antigens between sickle cell patients who are predominantly African American and blood donors, who are predominantly Caucasian.14 Additionally, alloimmunization makes allergic, acute, and chronic hemolytic transfusion reactions much more frequent in this population than in children receiving transfusions for chemotherapy-induced anemia.
For children and young adults with any primary anemia who receive frequent pRBC transfusions, the major complication is from iron overload. Iron deposition leads to end organ dysfunction, particularly in the heart and liver. Patients receiving regular transfusions should be monitored closely for elevated ferritin levels, our closest noninvasive surrogate measure for assessing iron deposition in tissue. Other noninvasive measures, such as cardiac and liver MRI to measure organ iron loads, are under investigation. Introduction of desferoxamine, an iron chelator, revolutionized care of patients with thalassemia and sickle cell anemia.15 However, desferoxamine must be administered intravenously or through subcutaneous injections or subcutaneous continuous infusion. These methods are cumbersome and problematic for many patients. Administration of desferoxamine was identified as a source of discomfort and decreased quality of life in several studies.16–18 Nearly 50% of patients identified iron chelator injections as the most disliked component of therapy. More than 40% identified missed work or school as a quality of life issue whether receiving transfusions or not.16 In one study, quality of life measures were higher for Malaysian patients with β-thalassemia who were receiving optimal desferoxamine regimens compared with patients receiving suboptimal desferoxamine.18 Healthcare providers would identify frequent transfusions with a greater medical burden as compared with transfusion independence. One study used the Dartmouth primary care cooperative information chart system (COOP) questionnaire found reported complaints of moderate pain in both transfused and nontransfused patients. Interestingly, 27% of transfused patients reported moderately impaired overall health versus 42% of transfusion independent patients.17 Additionally, physical fitness and better performance of daily activities were reported by patients receiving regular transfusions. Despite the complication of iron overload, regular transfusions appear to improve the quality of life in at least some populations of patients with primary anemia.
Interdisciplinary team considerations
Each institution has its own policy regarding infusion of blood components, though there are common themes. Some issues relate to safety of administration, including confirmation of appropriate blood type of unit to be transfused, confirmation of identity of recipient, appropriateness of intravenous access and frequent monitoring for signs of transfusion reaction. For details on infusion protocols, the reader is referred to their institutional transfusion policy and Essentials of Pediatric Oncology Nursing.19 Nurses contribute to team assessment of the patient’s and family’s religious or cultural beliefs, which may affect transfusion administration. Nurses also have an important ongoing role in educating patients and families with regard to symptoms of anemia, what to expect from a transfusion, and signs of transfusion reactions. Some individuals have very strong visceral reactions to the sight of blood, whether their own or someone else’s. In the context of blood transfusion, both nursing and child life specialists have helped address these concerns by finding creative ways to disguise transfusions, such as decorating a pillowcase to cover the pRBC bag.
Alternatives to Transfusion
Erythropoietin is produced by the kidney in response to anemia. Hematopoietic stem cells differentiate along the erythroid lineage in response to erythropoietin. Erythropoietin was first licensed in 1989 for treatment of anemia associated with chronic renal failure. There are two erythropoietin formulations, epoetin alpha is marketed by Amgen as Epogen and by Ortho Biotech as Procrit. Epoetin alpha is administered 2 to 3 times a week. The second formulation, Darbepoetin, is longer acting and is marketed by Amgen as Aranesp. These agents are frequently used in adults receiving chemotherapy. In fact they have become the first and second ranked expenditures for individual drugs by Medicare Part B. However, recent studies have led to FDA warnings about increased thromboembolic events and increased risk for cardiovascular events.20 Poorer survival in some studies where epoetin was used has again raised questions about whether epoetin may be a growth factor for some types of cancer. The American Society of Hematology/American Society of Clinical Oncology clinical practice update cautions against the use of epoetins in patients with malignancy who are not receiving either chemotherapy or radiation therapy.21
There are fewer randomized studies using erythropoietin in pediatric cancer patients. Epoetin alpha has been shown to be well tolerated by pediatric oncology patients and results in increased hemoglobin levels.22,23 However, results differed with respect to affecting the number of transfusions administered or quality of life parameters. In one study of patients with solid tumors receiving platinum-based chemotherapy regimens, epoetin alpha decreased transfusion requirements.22 Another study reported 224 patients receiving chemotherapy for nonmyeloid malignancy who were randomized to receive either epoetin alpha or placebo. The group receiving epoetin alpha had greater improvement in hemoglobin and a higher percentage of the patients were independent of transfusions at 4 weeks. Pediatric Quality of Life Inventory Generic Core Scales (PedsQL-GCS) did not differ between treatment groups.23 However, further analysis demonstrated correlation between PedsQL-GCS and improved hemoglobin.24 Use of epoetin alpha in combination with granulocyte colony stimulating factor (G-CSF) for children with high-risk neuroblastoma resulted in an increased number of transfusions compared with patients in the control group receiving G-CSF without epoetin alpha.25 After reviewing these studies and others, the French National Cancer Institute’s evidenced-based practice guideline does not recommend systematic administration of erythropoietin for prevention of chemotherapy associated anemia in children with cancer.26
Although FDA warnings and mixed results in pediatric oncology studies raise concerns, erythropoietin may be useful for patients who object to blood transfusions on ethical or religious grounds, as do many of the Jehovah’s Witness faith.5 Indeed, patients of the Jehovah’s Witness faith have taught us that much more severe anemia can be tolerated than was initially supposed.27–31 Additionally, they have helped drive the interest in development of blood conservation programs and blood alternatives. Blood alternatives such as human and bovine hemoglobin based oxygen carriers (HBOC), which are acellular cross-linked hemoglobin molecules, have been described as bridging the gap between life-threatening anemia and recovery of normal red cell mass after trauma.32 These products are in clinical trials in Africa and other countries. As yet, none are available for clinical use in the United States, but may have a future role in palliation of anemia.
Caring for patients of the Jehovah’s Witness faith who refuse transfusion may cause ethical conflicts for medical personnel who feel strongly that transfusion is medically indicated.33 The moral distress caused by discordance between the values and goals of the medical staff and the values and goals of the patient can be destructive to delivery of patient-centered care as well as to the medical team striving to provide care. It can take considerable emotional and ethical work for the team to honor a patient’s autonomy and freedom to refuse specific treatments without destroying staff-patient or staff-staff relationships. At times it may be necessary to use the experience and expertise of resources such as patient advocates, the hospital ethics committee, and human resources for the staff.
In sickle cell anemia, administration of hydroxyurea switches on production of fetal hemoglobin, decreasing percentage of hemoglobin S. Some patients experience a significant decrease in acute vaso-occlusive and acute chest syndrome episodes, therefore decreasing the need for transfusion.7
Donor directed transfusions
Families of children diagnosed with cancer also often inquire about direct donation. Here, too, the frequency of required transfusions is generally too often for any one parent or family member to safely donate. For patients with either malignant disorders or with primary hematologic failure disorders who are candidates for hematopoietic stem cell transplantation (HCST), transfusions from family members are strongly discouraged. Siblings and, in some cases, parents are potential HSCT donors. Family-member blood transfusions before HSCT sensitize the patient to minor histocompatibility antigens, which increases the risk of graft failure. Similarly, because of shared histocompatibility antigen, related donor transfusions carry a risk of inducing transfusion associated graft versus host disease even in immunocompetent individuals. Therefore, all transfusions from a donor related to the recipient must be irradiated, adding cost to the transfusion. Finally, due to the emotional incentives for family and friends to donate for a specific patient, there may be a higher risk of transfusion-related infections than from a blood bank unit. A safer product is likely to come from a donor who has donated multiple times and therefore been frequently screened.34
Palliative Care Considerations in Secondary Anemia
Despite the incredible improvement in disease-free survival achieved with modern chemotherapy and multi-modality treatment, nearly 20% of all children diagnosed with childhood cancer will die of their malignancy. In fact, among all children with life-limiting illness, cancer remains the leading cause of disease-related death.35,36 Early in the trajectory of treatment of malignancy, transfusion may be largely a matter of supportive care, allowing administration of more myelosuppressive or more frequent chemotherapy. When cure is not possible, transfusion may be used to maintain the quality of life rather than prolong life. During the days, weeks, or months after the realization that cure can no longer be the endpoint, many short- and long-term goals are reassessed. Transfusion frequency varies considerably during this phase of illness. Transfusions may not be required or may be infrequent for a patient with a solid tumor that is not invading the bone marrow space. Conversely, for a child with a bone marrow failure syndrome or malignancy that involves the marrow space, transfusions may become increasingly frequent. Reassessing the goals with regard to transfusion is an important discussion to have with the patient and family.
Continuing transfusion support may complicate other decisions as a family prepares for end-of-life care such as enrollment in hospice care. Although attitudes and practices are changing, some hospice programs still do not support such services as transfusion, parenteral nutrition, or continuing chemotherapy.37 In recent years, the attitude toward providing transfusions for patients enrolled in hospice has begun to shift. In some hospice organizations, transfusion is now allowable if the motivation for administration of the transfusion is solely for relief of symptoms.
Several programs have demonstrated successful administration of transfusions at home.38,39 However, in many areas, providing transfusion with blood or platelets at home remains technically challenging, if not impossible. In some regions, even if a child is enrolled in hospice with Do Not Resuscitate (DNR) or Allow Natural Death (AND) directives in place, there are no agencies willing to provide transfusions in the patient’s home. This means families must go to an outpatient infusion center or be admitted to the hospital for transfusion. When admission is required to provide a transfusion, some third-party payers require discharge from hospice during the time the child is admitted to the hospital. Upon discharge from the hospital, nursing resources are then questionably used to repeat paperwork for readmission to hospice. For families in more rural areas, many hours may be spent traveling to and from the site for transfusion, receiving a transfusion, and repeating paperwork to resume hospice care. Further efforts are needed for development and dissemination of procedures and protocols for delivery of transfusions at home. Administrative support will be necessary for a personnel intensive investment for home transfusion programs. Payer mechanisms will also have to see the benefit and provide reimbursement commensurate with skill level.
Palliative Care Considerations for Primary Anemia
Children with primary anemias are not typically included in the discussion of palliative care. Now that children with hemoglobinopathies can expect to reach adulthood and have the potential to live into their fifth decade and beyond, we are met with new challenges for providing high-quality palliative care for this population. The principles of palliative care offer new opportunities to address the evolving needs of these patients.40,41
The hemoglobinopathies are characterized by largely unpredictable episodic acute pain from vaso-occlusion. Adequately addressing these painful episodes remains a challenge to the medical system. Many physicians are uncomfortable managing both acute and chronic episodes of pain.41 The problem is compounded by an often adversarial patient-physician relationship.41 Parents of children with sickle cell disease are more frequently dissatisfied with their child’s care in the hospital.42 The care of this population is complicated by decreased access to care, resulting in increased emergency room use, and therefore inconsistent care.41,43 New models of care such as the “Day Hospital” design used by the Bronx Sickle Cell Center are needed to provide rapid and consistent access to pain medications.41 Additionally, when children with sickle cell disease are not followed by a hematologist, they miss out on the benefit of preventative care.44
Contrast as well the social experience of the child with cancer who is in school. Often the school and even the broader community rallies around this child, celebrating each day he or she is in school, working hard to arrange alternate education plans for the days, weeks, or months when therapy or illness prevents school attendance. The child with sickle cell disease doesn’t receive that community and school support. Missed school days are difficult to make up. Where patients with cancer may miss school in large blocks of time, patients with primary hematologic disease miss smaller blocks of time but multiple times throughout the year and throughout their school career. There are relatively few studies examining the impact of sequelae of sickle cell disease on school and school performance. Several small studies suggest that adolescents with sickle cell disease miss an average of 12% to 21% of school days.45,46 In a recent study, 35% of the children followed missed a month of school from one school year.46 Children with sickle cell disease miss more days of school than their siblings.47 Falling behind in schoolwork missed because of pain, hospitalization, and clinic follow-up may develop into a pattern of school avoidance for some. Many missed days can leave the child who has primary anemias socially alienated and educationally delayed. There are only limited studies evaluating healthcare quality of life in children with sickle cell disease. However, in two studies using the health related quality of life questionnaire (HRQOL) parent report form, parents identified their children as being more limited in physical, psychological, and social well-being and having more limitations on schoolwork and interaction with peers than healthy children.48,49
Cancer affects 14 to 16 in 100,000 children. Sickle cell disease affects 500 in 100,000 African American children, and African American children are 18% of the population. Hemophilia affects 1 child in every 5000 male births, which translates to 10 in 100,000 children. There are 141 federally funded hemophilia treatment centers for children and adults; the listing is available at www.cdc.gov/ncbddd/hbd/htc_list.htm. Funding for these centers is specifically focused on provision of medical care. Comprehensive hemophilia treatment centers were first established in 1973. There were 10 funded Comprehensive Sickle Cell Centers for treatment of children and adults with sickle cell disease. Only a portion of affected children received care in centers specialized in providing treatment for those affected by sickle cell anemia. Recently, these programs have been eliminated and are now converted to funded Basic Translation Research Programs for sickle cell centers primarily engaged in clinical research. After reorganization, funding for provision of care to patients with sickle cell disease is through HRSA and the CDC on a limited and competitive basis. Children not treated by specialists are often missing the opportunity for the preventive care so essential for preventing life-altering complications from end organ damage, such as stroke.44 The most effective management occurs in the context of a consistent albeit unique interdisciplinary approach. Within the medical field we need to continue to build and strengthen medical homes for not only children but also adults with chronic anemias.
What accounts for the difference between the approach to treatment for hemophilia and treatment for sickle cell disease? In part, the existence of the National Hemophilia Foundation (NHF) established 1948. NHF is a very strong advocate for patients with hemophilia, and lobbies for healthcare legislation that improves care for those who suffer from hemophilia. Goals of NHF are: to increase lifetime insurance caps, increase funding for hemophilia treatment centers, and eliminate any travel ban for people with HIV (www.hemophilia.org/nhfweb). NHF is an organization of consumers, that is people with hemophilia, with an advisory board of providers. There are numerous sickle cell foundations that are more recently formed, smaller, and generally local or regional organizations. These foundations primarily focus on community education and testing for sickle cell trait and disease. As yet, they do not have as extensive private-sector funding or federal lobbying influence. Such efforts on behalf of patients with sickle cell disease would result in more resources focused on addressing the palliative care needs of these patients.
Public education, treatment and palliative efforts in sickle cell disease could benefit from a well-known celebrity or sports figure that championed the cause of children and adults affected with sickle cell disease. Recently, Michael J. Fox has raised public awareness, participated in raising money for research and lobbying the federal government to increase the investment in Parkinson disease research. Similarly, Lance Armstrong’s “LiveStrong” foundation and the Susan G. Komen Foundation raise public awareness and billions of dollars in cancer research support. Examples of more long-term celebrity champions include Jerry Lewis, whose 2009 telethon raised $60.5 million dollars for the Muscular Dystrophy Association, which supports research, provision of care and services, and education (www.mda.org).
Thrombocytopenia and Functional Platelet Disorders
Platelet failure may be due to abnormal numbers or abnormal function (Box 34-3). Both of these may be primary, due to an intrinsic platelet disorder or secondary to underlying disease, such as malignancy. As with secondary anemia, poor bone marrow reserve and hematopoietic toxicities from chemotherapeutic agents can lead to thrombocytopenia. Frequent platelet transfusion can lead to development of alloimmunization against platelet antigens, which in turn can worsen thrombocytopenia due to increased platelet destruction.
Primary thrombocytopenia
Amegakaryocytic thrombocytopenia is a rare inherited failure of platelet production due to mutations in c-mpl, the thrombopoietin receptor, in about 50% of cases.50 The disorder has a tendency for malignant transformation. Platelet transfusions are essential in the support of these patients as they await definitive therapy with HSCT.
Primary Platelet Dysfunction
Primary platelet dysfunction in general causes mild bleeding problems. Only two disorders, Glanzmann thrombasthenia and Bernard-Soulier Syndrome, could be classified as life threatening.51 Both are genetic defects affecting the critical platelet receptors for fibrin and von Willebrand factor, respectively. Infants with severe Glanzmann thrombasthenia present early in life including the neonatal period. Severe hemorrhages are responsive to platelet transfusion, but sensitization to platelet antigens makes the long term use of platelet transfusion therapy difficult. Judicious use of platelet transfusion allows these children to have a reasonably normal life. Because of the risk of platelet sensitization, definitive therapy with hematopoietic stem cell transplantation is indicated for children with the severe form of Glanzmann thrombasthenia. Bernard-Soulier syndrome is less severe and patients have a reasonable quality of life when supported with the judicious use of platelet support.52
Hemorrhage secondary to platelet dysfunction is best exemplified by renal disease. As renal function decreases and blood urea nitrogen levels increases to above 50 mg/dL, platelet dysfunction becomes clinically significant.53 Menorrhagia and nosebleeds are the most common clinical manifestation and the most disturbing to the quality of day to day living. Several strategies can improve these patients’ quality of life. The hematocrit has an inverse relation to the bleeding time in renal disease. Thus, improving hemoglobin levels with erythropoietin not only improves quality of life from disease fatigue but also decreases the risk of hemorrhage. Clinical hemorrhage can also be treated by increasing the levels of von Willebrand factor with desmopressin acetate (DDAVP) therapy or by transfusions of cryoprecipitate. Finally, anti-fibrinolytic therapy with ∊-amino caproic acid or tranexemic acid can decrease both nasal hemorrhage as well as menorrhagia.
Symptoms of thrombocytopenia
Thrombocytopenia first manifests with skin and mucosal membrane petechiae, which can coalesce into purpuric lesions. Gum and nosebleeds can result from thrombocytopenia. Other bleeding manifestations can include hematuria, menorrhagia, scleral hemorrhage, and retinal hemorrhage. Intracranial hemorrhage is the most significant and life-threatening bleeding (Box 34-4).
Diagnostic evaluation
Diagnostic evaluation for primary thrombocytopenia includes past medical history, family history, physical exam, complete blood counts, evaluation of the blood smear, and may include a bone marrow aspirate as well as genetic testing. Specifics of the differential diagnostic evaluation of primary thrombocytopenia is beyond the scope of this chapter, and for further diagnostic algorithms the reader is referred to other sources.6
Guidelines for transfusion
Interdisciplinary Considerations
As with pRBC, every medical facility has its own policy regarding infusion of blood components. The reader is referred to their institutional transfusion policy and Essentials of Pediatric Oncology Nursing.19 Nurses educate patients with severe thrombocytopenia to consider not flossing teeth and using soft toothbrushes to minimize gum bleeding. When a nosebleed happens, the patient is advised to sit up and lean forward so blood loss can be more accurately monitored. Also, this decreases the amount of swallowed blood. Rectal bleeding can occur with the passage of hard stool, so stool softeners are encouraged to decrease bleeding with constipation. Encourage patients to avoid use of rectal thermometers in thrombocytopenic and neutropenic infants. Counsel teens to avoid high-impact sports, and activities associated with high G-forces or high impact such as rollercoasters, mountain biking, and paintball.
Bleeding in terminal care
Bleeding, particularly visible bleeding, is a very distressing event for families particularly for those involved in terminal care. A United Kingdom Hospice delivered platelet transfusions at home for children with cancer in terminal care, who had known thrombocytopenia and bleeding lasting more than 1 hour.39 There is greater potential for transfusion-related reactions with platelets than with pRBC because of the greater plasma volume associated with a unit of apheresed platelets. However, the authors were encouraged by a low frequency of transfusion reactions, most of which were mild hives or mild lip swelling. Of all their pediatric oncology patients that died at home, less than 20% received platelet transfusions. The transfusion was effective in stopping the bleeding most times.
Bleeding Disorder
Primary
Primary plasma coagulation disorders are best represented by hemophilia. A discussion of the care of patients with hemophilia and of subjects suffering from rare factor deficiencies is beyond the scope of this chapter and readers are referred to other sources.54–56 However, the principles of therapy of hemophilia as life-saving and improving quality of life are worth discussing.
The advent of recombinant factors and viral inactivation strategies for clotting factor concentrates has improved safety during the past 20 years. Once again, prophylaxis with factor replacement became possible and has improved the quality of life of people with hemophilia. This generation of children with hemophilia can expect freedom from infusion-acquired viral infection and good joint health, in addition to a life expectancy nearly equivalent to the general population.57 Arthropathy, pain, arthritis, school performance, employment, and psychological adjustment to a chronic illness are cultural aspects of hemophilia care that are becoming the focus of hemophilia centers. The hemophilia growth and development study outlined the school problems of children with hemophilia.58 Early intervention and educational consultations are critical in the well being of children with hemophilia. Dutch children with hemophilia are more likely to participate in sports than their peers.59 How do we prepare these children and families to cope with sports and the tendency for risk-taking behavior in children with hemophilia? Arthropathy is still a problem with pain. Hemophilia programs have developed techniques to improve pain, and use arthroscopy to improve pain and function and cryo-cuffs to provide pain relief, decrease swelling, and inflammation.
Guidelines for Transfusion
Primary
Primary plasma coagulation disorders are now treated by transfusion of specific factors to replace the deficient factor. A discussion of the particular products and dosing of these factors is beyond the scope of this chapter and the interested reader is referred to several excellent recent reviews.54,55,60 However, there are two principles of the use of these factors that are important when we discuss palliative care in the broader sense. The contamination of these factors with HIV created not only a devastating medical crisis but also left the families of patients with primary plasma coagulation disorders with fear and often distrustful of their care providers. For these patients it is important that their providers are familiar with the products and transparent regarding side effects, particularly transfusion-transmitted infections. The safety of the product becomes very important. The second concept is the purity of the factor. Although often both go together, they are not necessarily the same.
Mentoring
Clinical Vignette
A 4-year-old girl who had recently been internationally adopted was brought to the clinic. Limited translations of available records indicated she had received blood transfusions approximately monthly. The average hemoglobin pretransfusion was 3 to 5 gm/dL. On initial presentation, she was pale and jaundiced with frontal bossing and mid-face bony overgrowth characteristic of thalassemia major. She was tachycardic with a palpable thrill and heart rate of 110 and greater with 6-cm hepatomegaly and 8-cm splenomegaly (Fig. 34-1). She was a quiet, polite little girl. Although she cried with IV starts, everyone was impressed with the fact that she was so well behaved and really didn’t offer much resistance. She sat in clinic for hours entertaining herself with coloring and puzzles. She showed little interest in television. She did not walk about to explore the clinic. We assumed that was due to recent changes of environment, unfamiliar people, and even recently new caretakers, her newly adoptive parents. In her new home, she also preferred to sit and play with crayons and small toys. Sometimes she fell asleep on the floor while playing. She refused to walk more than 10 feet before she would sit down and cry. She went to the second floor of the house only when carried. She did not participate in running or playing games with the other children. In fact, there wasn’t much interaction with the other six children in the household. When they were out of the house, she rode in a stroller. Mom had been told prior to the adoption that the girl’s activity level was low. Because she had an international change of surroundings and caretakers, the parents assumed the quiet behavior was due partly to the adjustment to new surroundings and partly to her unique personality. They had a picture of her in the orphanage at age 16 months.
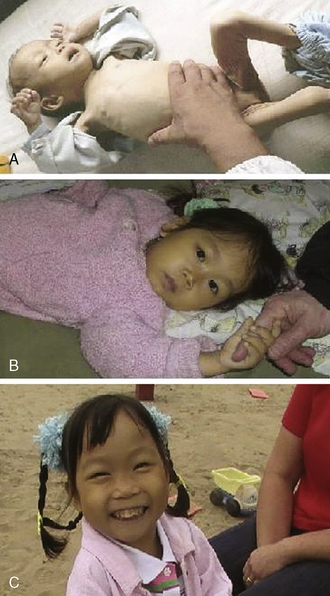
After several weeks, her hemoglobin before transfusion was 8. Her heart rate was under 100 for the first time since we had begun her care. Her spleen had returned to 8 cm. Her murmur was less prominent and she no longer had a palpable thrill. We proceeded with the transfusion as scheduled. At the end of the transfusion, her heart rate had finally improved to an age-appropriate rate in the 80s. A follow up hemoglobin a few days later was 10 (Fig. 34-1). On the next clinic visit, her mother excitedly described how she was a “totally different” little girl. She had walked much more that week. In fact, she had run for the first time since arriving in the United States. She had engaged in pillow fights with the siblings. She had engaged in a game of her own making called “monster” with the other children. For the next visit to the clinic, she walked through the door. The nurses were struck with how much more physical resistance she put up when they started her IV. Over the next several clinic visits her parents continued to comment on being aware of how much more active she was now that her hemoglobin was higher. They also repeatedly commented on how she now appeared tan consistent with her ethnic heritage rather than pale and yellow. The child also no longer complained of being cold.
1 Kleinman S., Chan P., Robillard P. Risks associated with transfusion of cellular blood components in Canada. Transfus Med Rev. 2003;17:120-162.
2 Busch M.P., Glynn S.A., Stramer S.L., et al. A new strategy for estimating risks of transfusion-transmitted viral infections based on rates of detection of recently infected donors. Transfusion. 2005;45:254-264.
3 Nathan D.G. Lessons from an unexpected life: a doctor, a patient, and a formerly fatal disease. Harvard Magazine. 2009:36-41.
4 Horner M.J.R.L., Krapcho M., Neyman N., Aminou R., Howlader N., Altekruse S.F., Feuer E.J., Huang L., Mariotto A., Miller B.A., Lewis D.R., Eisner M.P., Stinchcomb D.G., Edwards B.K., editors. SEER Cancer Statistics Review, 1975–2006. Bethesda, Md: National Cancer, Institute, 2009.
5 Michon J. Incidence of anemia in pediatric cancer patients in Europe: results of a large, international survey. Med Pediatr Oncol. 2002;39:448-450.
6 Sills R.H., editor. Practical algorithms in pediatric hematology and oncology. New York: Karger, 2003.
7 The Management of Sickle Cell Disease. 4th edition. June 2002. National Institutes of Health: National Heart, Lung, and Blood Institute.
8 Miller Y., Bachowski G., Benjamin R., et al. Practice guidelines for blood transfusion: a compilation from recent peer-reviewed literature. American National Red Cross, 2007.
9 Liumbruno G., Bennardello F., Lattanzio A., Piccoli P., Rossetti G. Recommendations for the transfusion of red blood cells. Blood Transfus. 2009;7:49-64.
10 Roseff S. Pediatric transfusion, a physicians handbook. D. Triulzi. 2003. American Association of Blood Banks: Bethesda.
11 Ruhl H., Bein G., Sachs U.J. Transfusion-associated graft-versus-host disease. Transfus Med Rev. 2009;23:62-71.
12 Sanders R.P., Maddirala S.D., Geiger T.L., et al. Premedication with ace-taminophen or diphenhydramine for transfusion with leucoreduced blood products in children. Br J Haematol. 2005;130:781-787.
13 Geiger T.L., Howard S.C. Acetaminophen and diphenhydramine premedication for allergic and febrile nonhemolytic transfusion reactions: good prophylaxis or bad practice? Transfus Med Rev. 2007;21:1-12.
14 Vichinsky E.P., Haberkern C.M., Neumayr L., et al. A comparison of conservative and aggressive transfusion regimens in the perioperative management of sickle cell disease. The Preoperative Transfusion in Sickle Cell Disease Study Group. N Engl J Med. 1995;333:206-213.
15 Brittenham G.M., Griffith P.M., Nienhuis A.W., et al. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N Engl J Med. 1994;331:567-573.
16 Telfer P., Constantinidou G., Andreou P., Christou S., Modell B., Angastiniotis M. Quality of life in thalassemia. Ann N Y Acad Sci. 2005;1054:273-282.
17 Pakbaz Z., Treadwell M., Yamashita R., et al. Quality of life in patients with thalassemia intermedia compared to thalassemia major. Ann N Y Acad Sci. 2005;1054:457-461.
18 Dahlui M., Hishamshah M.I., Rahman A.J., Aljunid S.M. Quality of life in transfusion-dependent thalassaemia patients on desferrioxamine treatment. Singapore Med J. 2009;50:794-799.
19 Kline N.E., O’Neill J.E.B., Hooke M.C., Norville R., Wilson K., editors. Essentials of Pediatric Oncology Nursing, ed 2, Glenview, Ill: Association of Pediatric Oncology Nurses, 2004.
20 Steinbrook R. Erythropoietin, the FDA, and oncology. N Engl J Med. 2007;356:2448-2451.
21 Rizzo J.D., Somerfield M.R., Hagerty K.L., et al. Use of epoetin and darbepoetin in patients with cancer: 2007 American Society of Hematology/American Society of Clinical Oncology clinical practice guideline update. Blood. 2008;111:25-41.
22 Buyukpamukcu M., Varan A., Kutluk T., Akyuz C. Is epoetin alfa a treatment option for chemotherapy-related anemia in children? Med Pediatr Oncol. 2002;39:455-458.
23 Razzouk B.I., Hord J.D., Hockenberry M., et al. Double-blind, placebo-controlled study of quality of life, hematologic end points, and safety of weekly epoetin alfa in children with cancer receiving myelosuppressive chemotherapy. J Clin Oncol. 2006;24:3583-3589.
24 Hinds P.S., Hockenberry M., Feusner J., Hord J.D., Rackoff W., Rozzouk B.I. Hemoglobin response and improvements in quality of life in anemic children with cancer receiving myelosuppressive chemotherapy. J Support Oncol. 2005;3:10-11.
25 Wagner L.M., Billups C.A., Furman W.L., Rao B.N., Santana V.M. Combined use of erythropoietin and granulocyte colony-stimulating factor does not decrease blood transfusion requirements during induction therapy for high-risk neuroblastoma: a randomized controlled trial. J Clin Oncol. 2004;22:1886-1893.
26 Marec-Berard P., Chastagner P., Kassab-Chahmi D., et al. Standards, options, and recommendations: use of erythropoiesis-stimulating agents (ESA: epoetin alfa, epoetin beta, and darbepoetin) for the management of anemia in children with cancer,. Pediatr Blood Cancer. 2009;53:7-12. 2007
27 Howell P.J., Bamber P.A. Severe acute anaemia in a Jehovah’s Witness. Survival without blood transfusion. Anaesthesia. 1987;42:44-48.
28 Penson R.T., Amrein P.C. Faith and freedom: leukemia in Jehovah Witness minors. Onkologie. 2004;27:126-128.
29 Knuti K.A., Amrein P.C., Chabner B.A., Lynch T.J.Jr, Penson R.T. Faith, identity, and leukemia: when blood products are not an option. Oncologist. 2002;7:371-380.
30 Varela J.E., Gomez-Marin O., Fleming L.E., Cohn S.M. The risk of death for Jehovah’s Witnesses after major trauma. J Trauma. 2003;54:967-972.
31 Collins S.L., Timberlake G.A. Severe anemia in the Jehovah’s Witness: case report and discussion. Am J Crit Care. 1993;2:256-259.
32 Gannon C.J., Napolitano L.M. Severe anemia after gastrointestinal hemorrhage in a Jehovah’s Witness: new treatment strategies. Crit Care Med. 2002;30:1893-1895.
33 Bodnaruk Z.M., Wong C.J., Thomas M.J. Meeting the clinical challenge of care for Jehovah’s Witnesses. Transfus Med Rev. 2004;18:105-116.
34 Wong E.C., Baxter C.A., Frey C., Criss V.R., Luban N.L. Transfusion transmitted viral disease and deferral rates in parental, directed and community donors for pediatric patients. Transfusion. 2001;41:114S.
35 Friebert S. NHPCO Facts and Figures: Pediatric Palliative and Hospice Care in America. Alexandria, Va: National Hospice and Palliative Care Organization, 2009.
36 Field M.J., Behrman R.E.E. When children die: improving palliative and end-of-life care for children and their families. Washington, D.C.: Institute of Medicine of the National Academies; National Academies Press, 2003.
37 Fowler K., Poehling K., Billheimer D., et al. Hospice referral practices for children with cancer: a survey of pediatric oncologists. J Clin Oncol. 2006;24:1099-1104.
38 Benson K. Home is where the heart is: do blood transfusions belong there too? Transfus Med Rev. 2006;20:218-229.
39 Brook L., Vickers J., Pizer B. Home platelet transfusion in pediatric oncology terminal care. Med Pediatr Oncol. 2003;40:249-251.
40 McClain B.C, Kain Z.N. Pediatric palliative care: a novel approach to children with sickle cell disease. Pediatrics. 2007;119:612-614.
41 Benjamin L. Pain management in sickle cell disease: palliative care begins at birth? Hematology Am Soc Hematol Educ Program. 2008:466-474.
42 Brousseau D.C., Mukonje T., Brandow A.M., Nimmer M., Panepinto J.A. Dissatisfaction with hospital care for children with sickle cell disease not due only to race and chronic disease. Pediatr Blood Cancer. 2009;53:174-178.
43 Raphael J.L., Dietrich C.L., Whitmire D., Mahoney D.H., Mueller B.U., Giardino A.P. Healthcare utilization and expenditures for low income children with sickle cell disease. Pediatr Blood Cancer. 2009;52:263-267.
44 Hankins J., Wang W. The painful face of poverty. Pediatr Blood Cancer. 2009;52:157-158.
45 Shapiro B.S., Dinges D.F., Orne E.C., et al. Home management of sickle cell-related pain in children and adolescents: natural history and impact on school attendance. Pain. 1995;61:139-144.
46 Schwartz L.A., Radcliffe J., Barakat L.P. Associates of school absenteeism in adolescents with sickle cell disease. Pediatr Blood Cancer. 2009;52:92-96.
47 Ogunfowora O.B., Olanrewaju D.M., Akenzua G.I. A comparative study of academic achievement of children with sickle cell anemia and their healthy siblings. J Natl Med Assoc. 2005;97:405-408.
48 Palermo T.M., Schwartz L., Drotar D., McGowan K. Parental report of health-related quality of life in children with sickle cell disease. J Behav Med. 2002;25:269-283.
49 Panepinto J.A., O’Mahar K.M., DeBaun M.R., Rennie K.M., Scott J.P. Validity of the child health questionnaire for use In children with sickle cell disease. J Pediatr Hematol Oncol. 2004;26:574-578.
50 Geddis A.E. Congenital amegakaryocytic thrombocytopenia and thrombocytopenia with absent radii. Hematol Oncol Clin North Am. 2009;23:321-331.
51 Nurden A.T. Glanzmann thrombasthenia. Orphanet J Rare Dis. 2006;1:10.
52 Simon D., Kunicki T., Nugent D. Platelet function defects. Haemophilia. 2008;14:1240-1249.
53 Borawski J., Rydzewski A., Mazerska M., Kalinowski M., Pawlak K., Mysliwiec M. Inverse relationships between haemoglobin and ristocetin-induced platelet aggregation in haemodialysis patients under erythropoietin therapy. Nephrol Dial Transplant. 1996;11:2444-2448.
54 Tarantino M.D., Aledort L.M. Advances in clotting factor treatment for congenital hemorrhagic disorders. Clin Adv Hematol Oncol. 2004;2:363-368.
55 Srivastava A. Guideline for the Management of Hemophilia: World Federation of Hemophilia. Montréal, Québec: World Federation of Hemophilia, 2005.
56 Acharya S.S, Coughlin A., Dimichele D.M. Rare bleeding disorder registry: deficiencies of factors II, V, VII, X, XIII, fibrinogen and dysfibrinogenemias. J Thromb Haemost. 2004;2:248-256.
57 Tarantino M., Ma A., Aledort L. Safety of human plasma-derived clotting factor products and their role in haemostasis in patients with haemophilia: meeting report. Haemophilia. 2007;13:663-669.
58 Su Y., Wong W.Y, Lail A., Donfield S.M., Konzal S., Gomperts E. Long-term major joint outcomes in young adults with haemophilia: interim data from the HGDS. Haemophilia. 2007;13:387-390.
59 Heijnen L., Mauser-Bunschoten E.P., Roosendaal G. Participation in sports by Dutch persons with haemophilia. Haemophilia. 2000;6:537-546.
60 Manco-Johnson M. Hemophilia management: optimizing treatment based on patient needs. Curr Opin Pediatr. 2005;17:3-6.