Chapter 29 Hematologic Malignancy
The Lymphomas
Epidemiology
Incidence
In the United States, NHL annually accounts for 5% of new cancers in men and 4% of new cancers in women. It is estimated that there were 66,360 new cases of NHL and 19,320 deaths in 2011.1 HL accounts for approximately 15% of all lymphomas. It is estimated that there will be 8830 new cases in 2011 and 1300 deaths.1 Men are affected somewhat more than women: for HL, the male-to-female ratio is 1.4:1; for NHL, it is 1.1:1.
The incidence of HL has been stable for years, but that of NHL has risen by approximately 60% in the United States since 1960. This is evident for all age groups, but particularly for older persons. It occurs more often in men and in white ethnic groups, but has been observed in all geographic areas of the United States.2 Geographically, the incidence of NHL varies 8- to 10-fold, being much more common in the West. The overall mortality for NHL has also risen over the last few decades, especially in older patients, despite the fact that survival rates for each subtype of NHL have improved, reflecting advances in treatment.
The reasons for the increasing incidence are only partly understood. Some may be artifactual, in that new lymphoma classifications have led to a diagnosis of NHL in patients who would previously have had other diagnoses,3 including HL in up to 15% of cases. Part of the increase from the late 1980s resulted from the increased incidence of lymphomas associated with immune deficiency, notably secondary to human immunodeficiency virus (HIV) infection. However, the occurrence of some HIV-associated NHLs has started to fall since the introduction of highly active antiretroviral therapies.4
Key Points Incidence
• Incidence of NHL has increased by 60% since the 1960s in the United States and the United Kingdom.
• HL has a peak incidence between the ages of 20 and 40 years and also in those older than 50 years.
• NHLs are seen in children and in those older than 50 years.
• There is a link between Epstein-Barr virus (EBV) and lymphoma, especially Burkitt lymphoma (BL) and HL.
Etiology
There is an association between EBV and HL, but the exact etiologic role of the virus is uncertain. Patients with HL have a higher antibody titer to EBV viral capsular antigen, and the risk of HL among patients who have had infectious mononucleosis is trebled. EBV can also be found in the malignant cells of HL. Other infective agents such as human herpesvirus-6 (HHV-6) and HIV-1, both associated with the mixed cellularity subtype of classic HL, may also have roles in development of this disease. Infective agents are also associated with the development of certain subtypes of NHL. EBV is found in virtually 100% of cases of endemic African BL; in the sporadic form, the incidence is 15% to 30%. The rare primary effusion lymphomas are associated with HHV-8. Helicobacter pylori infection is necessary for the development of gastric lymphoma of mucosa-associated lymphoid tissue (MALT) type. The human T-cell lymphotrophic virus (HTLV-1) retrovirus has a causal relationship with adult T-cell leukemia/lymphoma, seen in South Japan and the Caribbean. The disease is believed to represent clonal expansion of HTLV-1–infected T lymphocytes.
Pathology
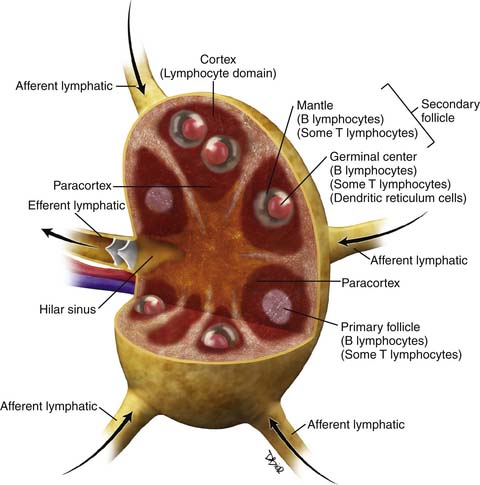
There have been several pathologic classifications of lymphoma in the past; however, the functional anatomy of the lymph node is key to understanding the pathologic classification of lymphomas (Figure 29-1).
Non-Hodgkin Lymphoma
Over 90% of NHLs in the Western world are of B-cell origin. In general, those arising at stages of development within the germinal center of the node have a follicular or nodular architecture, whereas those arising outside the germinal center have a diffuse pattern. New insights into the pathogenesis of NHL, in terms of cellular and immunologic origins, have resulted in the introduction of the Revised European American Classification of Lymphoid Neoplasms (REAL) classification in 1994. This, in turn, led to the adoption of the definitive World Health Organization (WHO) Classification of Tumors of Hematopoietic and Lymphoid Tissues in 1995,5 which is an updated version of the REAL classification (Table 29-1).
Table 29-1 Summary of the World Health Organization Classification of Tumors of Lymphoid Tissue
| Precursor B-Cell Neoplasms | Precursor T-Cell Neoplasms |
| B lymphoblastic leukemia/lymphomas | T lymphoblastic leukemia/lymphoma |
| Mature B-Cell Neoplasms | Mature T-Cell and Natural Killer Neoplasms |
|
CLL/small lymphocytic lymphoma B-cell prolymphocytic leukemia Splenic B-cell marginal zone lymphoma Extranodal marginal zone B-cell lymphoma of Primary cutaneous follicle center lymphoma DLBCL associated with chronic inflammation Primary mediastinal (thymic) LBCL LBCL arising in HHV-8–associated multicentric Castleman’s disease B-cell lymphoma, unclassifiable, intermediate between DLBCL and BL B-cell lymphoma, unclassifiable, intermediate between DLBCL and classical HL |
T-cell prolymphocytic leukemia
T-cell large granular lymphocytic leukemia
Systemic EBV-positive T-cell lymphoproliferative disease of childhood
Adult T-cell leukemia/lymphoma
Extranodal NK/T-cell lymphoma, nasal type
Enteropathy-type T-cell lymphoma
Subcutaneous panniculitis-like T-cell lymphoma
Primary cutaneous CD30-positive T-cell lymphoproliferative disorders
Primary cutaneous gamma-delta T-cell lymphoma
Peripheral T-cell lymphoma, NOS
Langerhans cell histiocytosis
Langerhans cell sarcoma
Interdigitating dendritic cell sarcoma
Follicular dendritic cell sarcoma
Fibroblastic reticular cell tumor
Indeterminate dendritic cell tumor
Classical HL
Nodular sclerosis classical HL
Lymphocyte-rich classical HL
Mixed cellularity classical HL
Lymphocyte-depleted classical HL
ALK, anaplastic lymphoma kinase; BL, Burkitt lymphoma; CNS, central nervous system; CLL, chronic lymphocytic leukemia; DLBCL, diffuse large B-cell lymphoma; EBV, Ebstein-Barr virus; HHV-8, human herpesvirus-8; HL, Hodgkin lymphoma; LBCL, large B-cell lymphoma; MALT, mucosa-associated lymphoid tissue; NK, natural killer; NOS, not otherwise specified; PTLD, posttransplant lymphoproliferative disorder.
The WHO classification is a list of distinct disease entities defined by a combination of morphology, immunophenotype, and genetic and clinical features. It stratifies neoplasms according to myeloid, lymphoid, and histiocytic/dendritic cell lineages and recognizes three major categories of lymphoid neoplasms: B cell, T cell, and natural killer (NK) cell; and separately, HL. Precursor neoplasms, corresponding to the early stages of differentiation and including lymphoblastic leukemias and lymphomas, are separated from the more mature or peripheral neoplasms. Conversely, chronic lymphocytic leukemia (CLL) is the circulating form of small lymphocytic lymphoma, a mature B-cell neoplasm, and is classified as such. Histologic grade may influence therapeutic decision-making. For example, follicular lymphoma (FL) is graded according to the number of centroblasts per high-power field.
This comprehensive approach has significantly improved the consistency of classification of lymphoma, such that, if given sufficient material, expert hematopathologists agree on classification of entities in over 95% of cases.6 Further refinements may facilitate appropriate patient management.7 For example, gene expression profiling for diffuse large B-cell lymphoma (DLBCL) enables recognition of discrete subsets (germinal center B-cell type and activated B-cell type) that have independent prognostic significance.8,9 Other recent additions to the classification include pediatric FL, primary DLBCL of the central nervous system (PCNSL), and two so-called gray zone lymphomas: B-cell lymphoma with features intermediate between DLBCL and classic HL, and B-cell lymphoma with features intermediate between DLBCL and BL.
Hodgkin Lymphoma
Investigators have demonstrated that HL is a true lymphoma; hence, the term Hodgkin lymphoma is preferred to Hodgkin disease. Indeed, the distinction between HL and NHL is not always straightforward, and composite cases occur. Diagnosis depends upon the demonstration of malignant Reed-Sternberg and Hodgkin cells against a background of non-neoplastic inflammatory cells. The WHO classification recognizes two distinct entities that differ in clinical features, behavior, morphology, and immunophenotype: classical Hodgkin lymphoma (CHL, 95%) and nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL, 5%). CHL is, in turn, divided into four subgroups, based on the proportion of lymphocytes in relation to the number of malignant cells and on the connective tissue background (Table 29-2).10 All share the same immunophenotype, with expression of CD30 by the malignant cells.
Table 29-2 Rye Classification of Classical Hodgkin Lymphoma and Approximate Frequency
| HISTOLOGY | FREQUENCY (%) |
|---|---|
| Lymphocyte rich | 5 |
| Nodular sclerosis | 65 |
| Mixed cellularity | 25 |
| Lymphocyte depletion | 5 |
From Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting—Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835-3849.
Key Points Classification of lymphomas
• HL comprises two distinct entities: NLPHL (5%) and CHL (95%).
• CHL consists of four subtypes: nodular sclerosing (70%), mixed cellularity (20-25%), lymphocyte-rich (5%), and lymphocyte-depleted (<5%).
• WHO classification stratifies lymphoma according to myeloid, lymphoid, and histiocytic/dendritic lineage and provides distinction between disease entities.
Clinical Features
Both HL and NHL are predominantly diseases of lymph nodes, which may present as a localized process involving a single nodal group or organ or as widely disseminated disease. However, there are recognizable differences in the clinical presentation of the two diseases (Table 29-3).
Table 29-3 Key Differences between the Clinical Features of Hodgkin Lymphoma and Non-Hodgkin Lymphoma
Hodgkin Lymphoma
Most patients with HL present with painless asymmetrical lymph node enlargement, which may be accompanied by sweats, fever, weight loss (“B” symptoms), and pruritus in approximately 40% of patients. B symptoms are more common in advanced-stage disease and thus in the mixed cellularity and lymphocyte-depleted subtypes of CHL. Alcohol-induced pain is rare.
Staging Systems and Prognostication
Because lymphomas are primarily neoplasms of lymphoid tissues (whether nodal or extranodal), the tumor-node-metastasis (TNM) staging system is not appropriate. The Ann Arbor staging system for HL was introduced in 1970 and took into account the extent of nodal disease and the presence of extranodal extension. Increasing recognition of the influence of tumor bulk as an independent prognostic indicator within each stage and the routine application of computed tomography (CT) in the 1980s led to a modification of the classification in 1989, the Cotswolds classification.11 This system is similar to the original Ann Arbor classification, but stage III is subdivided and an additional qualifier “X” denotes bulky disease (Table 29-4). The prognosis of HL depends upon a number of factors, including
• Age. Older patients have a worse prognosis (for early-stage disease, 5-year survival is 45% in patients older than age 65, as opposed to over 90% in younger patients).
• Tumor subtype. Mixed cellularity and lymphocyte-depleted HL have a poorer prognosis.
• Raised erythrocyte sedimentation rate (ESR).
• Multiple sites of involvement (more than three or four involved regions).
| STAGE | AREA OF INVOLVEMENT | |
|---|---|---|
| I | One lymph node region or extralymphatic site | |
| II | Two or more lymph node regions on the same side of the diaphragm | |
| III | ||
From Lister TA, Crowther D, Sutcliffe SB, et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol. 1989;7:1630-1636.
The modified Ann Arbor classification is also currently applied to NHL, but is less useful, because the prognosis in NHL is more dependent on the histologic subtype, tumor bulk, and specific organ involvement, as well as stage. In NHL, the critical questions are whether or not disease is limited, the effect of the disease on end organs such as bone marrow, and symptomatology. Because NHL spreads more randomly than HL, the Ann Arbor staging system is less helpful in defining prognostic subgroups. Therefore, considerable effort has been applied to the development of robust prognostic indices that are accurate, simple, and discriminant. Such indices not only aid in the management of the individual patient but also facilitate meaningful comparison of results from clinical trials. The International Prognostic Index (IPI) was developed by an international collaborative group.12 For DLBCL, five factors have prognostic significance: age older than 60 years, elevated serum lactate dehydrogenase (LDH), Eastern Cooperative Oncology Group (ECOG) performance status greater than 1 (i.e., nonambulatory), advanced stage (III or IV) disease, and more than one extranodal site of disease.
Four risk groups are recognized depending on the number of adverse prognostic features that are present. Such a stratification enables choice of more intensive therapies for those at higher risk. Prior to the monoclonal antibody era, patients in the low risk group (no or one prognostic factor present) had a 5-year survival greater than 70%, whereas patients in the high risk group (four or five factors present) had only a 25% 5-year survival. More recently, the prognostic importance of gene expression profiling within individual subtypes of NHL has become evident, as described previously for DLBCL,8 where the better prognosis of the germinal center–like subtype compared with the activated B-cell subtype is independent of the IPI. A similar prognostic index has been developed for FL,13 where the important factors are age older than 60, elevated serum LDH, hemoglobin less than 12 g/dL, stage 3 or 4 disease, and more than four nodal sites of disease.
The clinical spectrum of childhood lymphoma differs somewhat from adult lymphoma with more frequent extranodal involvement, especially of the gastrointestinal tract, solid viscera including the kidneys and pancreas, and extranodal sites in the head and neck.14 In these situations, the St. Jude or Murphy’s staging classification is applied, because it takes into account the increased frequency of extranodal disease (Table 29-5).
Table 29-5 Murphy’s Staging System for Childhood Non-Hodgkin Lymphoma
| STAGE | CRITERIA FOR EXTENT OF DISEASE |
|---|---|
| I | A single tumor (extranodal) or single nodal region except in the mediastinum or abdomen |
| II | A single tumor (extranodal) with regional nodal involvement Two or more nodal areas on the same side of the diaphragm Two single (extranodal) tumors with or without regional node involvement on the same side of the diaphragm A primary GI tract tumor, usually ileocecal, with or without involvement of associated mesenteric nodes only, grossly completely resected |
| III | Two single tumors (extranodal) on opposite sides of the diaphragm Two or more nodal areas above and below the diaphragm ALL primary intrathoracic tumors (mediastinal, pleural, thymic) ALL extensive primary intra-abdominal disease, unresected ALL paraspinal or epidural tumors, regardless of other tumor site(s) |
| IV | Any of the above with initial CNS and/or bone marrow involvement |
CNS, central nervous system; GI, gastrointestinal.
Patterns of Tumor Spread
NHL is generally a disseminated disease involving lymph node groups through hematogenous spread, and multiple organs may be involved as well as the bone marrow. Thus, nodal enlargement might be seen at CT in the neck and pelvis, with no abnormality in the chest or abdomen, whereas this distribution would be most unusual for HL. This unpredictability makes a whole body staging technique imperative. However, individual subtypes of NHL are associated with certain patterns of disease. Marked splenomegaly is a feature of splenic marginal zone lymphoma and MCL, the latter often in association with bowel involvement. The constellation of a large anterior mediastinal mass with central venous obstruction and disease in the liver, kidneys, or adrenal glands but little or no nodal disease makes primary mediastinal large B-cell lymphoma (PMBL) the most likely diagnosis, whereas widespread peritoneal disease at presentation, with involvement of the viscera and female genital tract, suggests BL. Certain types of NHL are strongly associated with central nervous system (CNS) or meningeal disease, especially testicular and head and neck lymphoma, and this association may warrant screening of the craniospinal axis or prophylactic intrathecal therapy. With disease progression, nodal lymphoma may spread to involve adjacent structures. In the retroperitoneum, this can affect the paravertebral and paraspinal structures with resultant neural compression. In the mesentery, spread into adjacent bowel loops is common, causing displacement, encasement, or compression. As disease advances, peritoneal involvement can occur, radiologically indistinguishable from peritoneal carcinomatosis.
Often, the pattern of disease at CT may suggest the diagnosis. Thus, involvement of cervical lymph nodes and the tissues of Waldeyer’s ring suggests NHL rather than HL. Nodal enlargement in the anterior and middle mediastinum suggests HL, whereas disease in the mesentery, with or without concomitant bowel involvement, strongly favors NHL (see Table 29-3).
Imaging Techniques
Nodal Disease
Cross-sectional Imaging
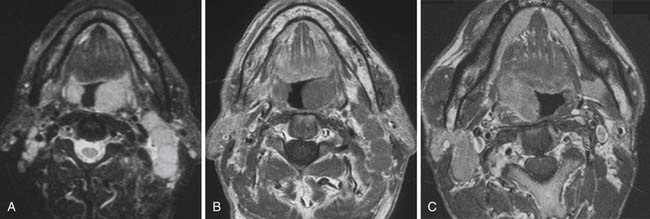
For some time, CT has been the modality of choice for the staging and follow-up of lymphoma. It enables localization of the most appropriate lesion for consideration of percutaneous image-guided biopsy. Ultrasound has limited value in staging. Involved lymph nodes have nonspecific appearances, although the pattern of nodal vascular perfusion on power Doppler sonography may suggest the diagnosis. The main value of ultrasound is in providing image guidance for biopsy. Although magnetic resonance imaging (MRI) is as accurate as CT in detecting lymph node enlargement, its role is essentially adjunctive. As with CT and ultrasound, involved lymph nodes cannot be diagnosed other than by size criteria (Figure 29-2). Advances in scanner technology including high–field strength magnets permit whole body MRI for staging lymphoma. Whole body diffusion-weighted imaging of lymphoma may have a role in the differentiation of lymphoma from other causes of malignant nodal enlargement.15
Functional Imaging with Positron-Emission Tomography (PET) and PET/CT
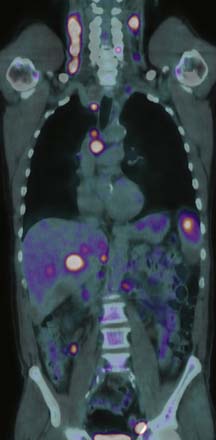
Detection of disease in normal-sized nodes is not possible with cross-sectional imaging, nor is it possible to differentiate between nodal enlargement secondary to lymphoma or reactive hyperplasia. Functional radioisotope studies permit this distinction. Gallium-67 (67Ga) citrate has largely been replaced by positron-emission tomography (PET) using 2-[18F]fluoro-2-deoxy-D-glucose (FDG-PET). In most lymphomas, increased glucose metabolism results in increased cellular uptake of FDG with an accuracy comparable with or better than CT for the detection of nodal and extranodal disease.16,17 In NHL, staging FDG-PET gives important information by indicating tumor burden as well as the presence of extranodal disease (Figure 29-3). Most NHLs show increased uptake, the exceptions being some MALT, cutaneous, and small lymphocytic lymphomas. PET/CT allows accurate co-localization of morphologic abnormalities and their associated functional changes. Debate continues as to whether it is always necessary to carry out a full diagnostic CT scan as part of the PET/CT study or whether a low-dose CT for the purposes of attenuation correction and anatomic correlation is sufficient.18
Key Points Nodal imaging: general
• Recognition of nodal disease with CT and MRI depends on size criteria alone.
• CT and MRI have equal sensitivity below the diaphragm.
• Ultrasound has a role in problem-solving and guidance for biopsy.
• FDG-PET/CT may enable identification of disease in normal-sized lymph nodes.
• Nodal disease in the lymphomas may involve any anatomic lymph node site.
Neck
Cervical lymph node enlargement is seen in 60% to 80% of patients with HL at presentation. It typically involves the internal jugular chain of nodes initially, with further spread to the spinal accessory and transverse cervical chains. In NHL, the pattern of involvement is more haphazard than in HL (see Figure 29-2). Lymph nodes greater than 1 cm in short-axis diameter (SAD) are generally considered enlarged. Enhancement after administration of contrast medium is usually mild to moderate, and central necrosis within a lymph node is rare. MRI may be particularly useful for defining the extent of lymphomatous masses in the lower neck and supraclavicular fossa.
Thorax
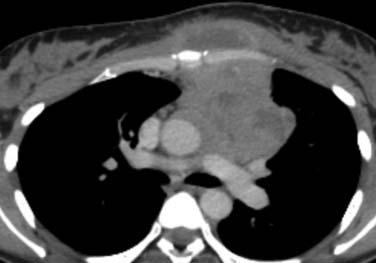
Intrathoracic nodes are involved at presentation in 60% to 85% of patients with HL and 25% to 40% of patients with NHL. Almost all patients with nodular sclerosing HL have disease in the anterior mediastinum. Nodal involvement in mediastinal presentations includes prevascular and paratracheal (84%; Figure 29-4A), hilar (28%), subcarinal (22%), and other sites (~5%), including aortoplumonary, anterior diaphragmatic, and internal mammary (see Figure 29-4B and C).19 In NHL, the incidence varies, but may include superior mediastinal (34%), hilar (9%), subcarinal (13%), and other sites (≤10%).20
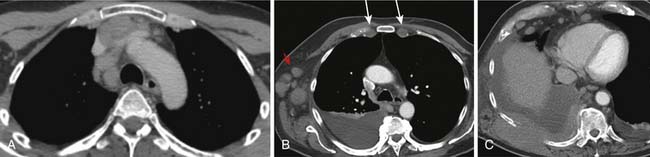
The majority of cases of HL show enlargement of two or more nodal groups, whereas only one nodal group is involved in up to half of the cases of NHL. Hilar nodal enlargement is rare without associated mediastinal involvement, particularly in HL. Although nodes in the internal mammary chains and paracardiac regions are rarely involved at presentation, they become important as sites of recurrence because they may not be included in conventional radiation fields (see Figure 29-4C). It is important to review these sites because minimally enlarged nodes are easily overlooked.
Nodal calcification before therapy is extremely rare. Cystic degeneration may be seen in both HL and NHL, especially with large anterior mediastinal masses, and is a particular feature of PMBL21 (Figure 29-5). Thymic infiltration occurs in both HL and NHL and is often indistinguishable from anterior mediastinal nodal disease. A large anterior mediastinal mass is an adverse prognostic factor in HL. Axillary nodes are also frequently detected on CT in HL and NHL (see Figure 29-4B). Preservation of the normal fatty hilum may be helpful in distinguishing benign reactive hyperplasia from lymphomatous involvement.
Key Points Supradiaphragmatic nodal disease
• Sixty percent to 80% of patients with HL present with enlarged neck nodes. This is also a common presentation in NHL.
• Sixty percent to 80% of patients with HL and 25% to 40% of patients with NHL have prevascular and paratracheal lymphadenopathy at diagnosis.
• A large anterior mediastinal mass may represent lymphomatous infiltration of the thymus.
Abdomen and Pelvis
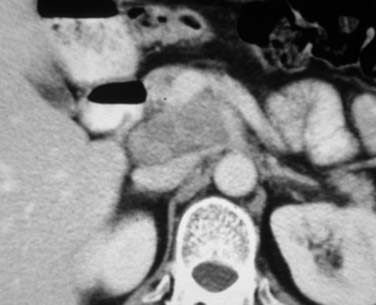
At presentation, retroperitoneal nodes are involved in 25% to 35% of patients with HL and in 45% to 55% of patients with NHL.22 Mesenteric lymph nodes are involved in more than half of patients with NHL and fewer than 5% of patients with HL. Other intra-abdominal sites are also less frequently involved in HL than in NHL. Given the mode of spread of HL from one nodal group to the next in anatomic contiguity, involvement of retrocrural nodes should prompt close scrutiny of the celiac axis nodes. The celiac axis, splenic hilar, and porta hepatis nodes are involved in approximately 33% of patients, and splenic hilar nodal involvement is almost always associated with diffuse splenic infiltration. In the porta hepatis, the node of the foramen of Winslow (portocaval node) should be carefully evaluated. It lies between the portal vein and the inferior vena cava (Figure 29-6) and has a triangular shape, with a normal transverse diameter up to 3 cm. Enlargement of this node is easily overlooked.
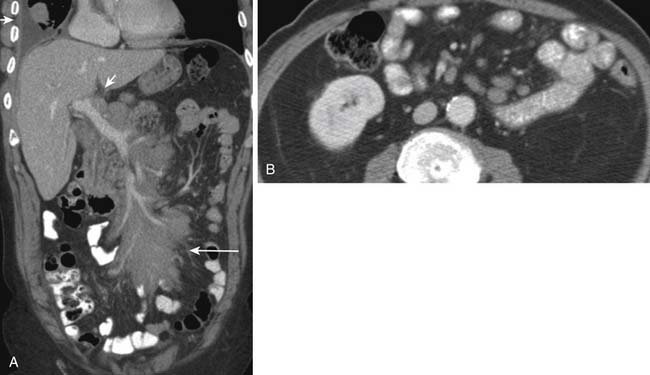
In NHL, nodal involvement is frequently noncontiguous, bulky, and often associated with extranodal disease (Figure 29-7A). Discrete mesenteric nodal enlargement or masses may be seen with or without retroperitoneal nodal enlargement. Regional nodal involvement is frequently observed in primary extranodal lymphoma involving an abdominal viscus. In a patient with lymphoma, the presence of multiple normal-sized mesenteric nodes is suspicious for involvement, as is nodularity and streakiness within the mesentery, lymphoma being one cause of the misty mesentery (see Figure 29-7B). Any pelvic nodal group may be involved in HL and NHL. Presentation with enlarged inguinal/femoral lymphadenopathy is seen in fewer than 20% of cases of HL.
Extranodal Disease
Approximately 30% of cases of NHL arise primarily in extranodal sites other than the spleen or bone marrow. The most common pathologic subtypes with extranodal involvement are DLBCL, MALT, and FL, and the most common locations affected in decreasing order of frequency are Waldeyer’s ring, the stomach and small intestine, the soft tissues, and the orbit. Secondary extranodal lymphoma occurs owing to spread of lymph node disease into adjacent structures and organs or as part of widespread disease. Although the latter is an adverse prognostic factor in the IPI,12 stage IE or IIE DLBCL does not have as poor a prognosis as stage IV disease unless it arises in the testis. There has been a marked increase in incidence of extranodal NHL, especially in the gastrointestinal (GI) tract and CNS. Visceral lymphoma can mimic many other disease entities, and it is important to recognize its protean radiologic appearances.
FDG-PET is generally more sensitive than CT in the staging of extranodal disease, largely because of its ability to demonstrate bone marrow involvement.17 FDG-PET and PET/CT are also more sensitive than CT in identifying organ involvement, reaching sensitivities of up to 85%, with only 37% sensitivity for CT (see Figure 29-3). Use of PET or PET/CT can result in upstaging in up to 40% of cases. However, in some instances of low-grade NHLs, CT is more sensitive.
Thorax
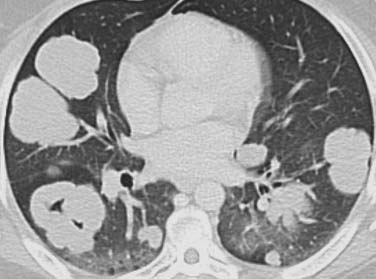
Secondary involvement of the pulmonary parenchyma in HL is most commonly by direct invasion from involved hilar or mediastinal nodes. On chest radiography, lung parenchymal involvement is seen three times more frequently in HL (12%) than in NHL. Parenchymal involvement in HL is almost invariably accompanied by intrathoracic adenopathy unless there has been prior mediastinal irradiation, whereas in NHL, pulmonary or pleural lesions occur in isolation in up to 50% of cases. The radiographic changes in both HL and NHL are extremely varied. The most common pattern is one or more discrete nodules resembling primary or metastatic carcinoma, which may cavitate (Figure 29-8). Primary (or isolated) pulmonary lymphoma is uncommon (1% of all extranodal presentations) and is usually due to NHL, particularly low-grade B-cell lymphomas, the DLBCL accounting for the remaining 15% to 20%. The diverse appearances of pulmonary lymphoma provide a particular diagnostic challenge because many of these patients have other reasons for developing lung disease.
Pleural effusions are nearly always accompanied by mediastinal lymph node enlargement and, by CT, are present in over 50% of cases with mediastinal nodal disease (see Figure 29-4B). Most effusions are unilateral exudates from venous or lymphatic obstruction rather than direct neoplastic involvement.23 Chest wall involvement in HL is usually as a result of direct extension from an anterior mediastinal mass. Less commonly, NHL may arise in the thoracic wall (see Figure 29-4C). Bony destruction is relatively uncommon. Pericardial effusions are seen on CT in 6% of patients with HL at presentation and are associated with coexistent bulky mediastinal adenopathy abutting the pericardium. Intracardiac masses can occur with T-cell lymphomas and aggressive lymphomas, especially in the setting of AIDS-related lymphomas (ARL).
Thymic involvement occurs in 30% of patients with newly diagnosed HL24 and is a particular feature of PMBL (see Figure 29-5). Cysts and calcification may be seen within the thymus at presentation or during follow-up on both CT and MRI.25 Benign thymic rebound hyperplasia after completion of chemotherapy can be difficult to differentiate from recurrent disease.
Key Points
• Secondary involvement of the lung parenchyma is seen three times more frequently in HL than in NHL.
• Lung involvement in HL is almost invariably associated with mediastinal lymphadenopathy.
• Most primary low-grade lymphomas of the lung are MALT or bronchus-associated lymphoid tissue (BALT) tumors.
Abdomen
Spleen and Liver
The spleen is involved in approximately 30% of patients with HL, usually with nodal disease above and below the diaphragm. The spleen is regarded as a lymph node in the Ann Arbor classification and is denoted by the suffix “S,” but an enlarged spleen is not a reliable indicator of disease in HL. Cross-sectional imaging techniques are unreliable in the detection of splenic involvement because infiltration is usually microscopic. Occasionally, focal splenic nodules larger than 1 cm are seen on cross-sectional imaging (Figure 29-9). However, the failure of imaging to detect splenic infiltration in HL is now of less clinical importance, because most patients with early-stage disease and occult splenic infiltration will receive multiagent chemotherapy.
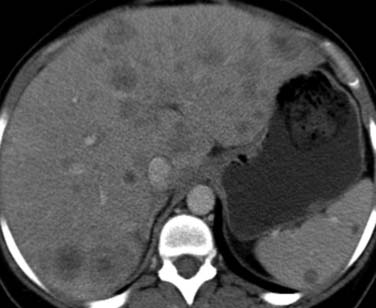
Figure 29-9 Axial contrast-enhanced CT in a patient with HL demonstrates focal hepatic and splenic nodules.
Up to 40% of patients with NHL have splenic involvement at some stage. It is a particular feature of MCL and splenic marginal zone lymphoma, where massive splenomegaly can occur. Primary splenic lymphoma usually presents as a mass or masses rather than splenomegaly alone, and miliary nodules can also be seen. The presence of splenomegaly generally indicates diffuse infiltration, and infarction is a frequent complication. FDG-PET detects splenic infiltration more accurately than CT or 67Ga scintigraphy.17,26
As with splenic disease, hepatic lymphoma usually takes the form of microscopic infiltration around the portal triads and detection by cross-sectional imaging is usually difficult, although periportal low attenuation may be seen at CT.14 The presence of hepatomegaly suggests infiltration. Large focal areas of involvement are seen in only 5% to 10% of patients and resemble metastatic disease from other sources (see Figure 29-9).
Key Points Spleen and liver involvement
• In 10% of HL, the spleen is the only site of subdiaphragmatic disease.
• An enlarged spleen is not a reliable indicator of disease in HL.
• Liver involvement is almost invariably associated with splenic infiltration.
• Only 5% to 10% of patients with liver disease have focal lesions detectable on cross-sectional imaging, resembling metastases.
• Enlargement of the liver is a strong indicator of lymphomatous infiltration.
Primary lymphoma of the stomach accounts for approximately 2% to 5% of gastric tumors, commonly appearing radiologically as multiple nodules, some with central ulceration; a large fungating lesion; diffuse infiltration with marked mural thickening, sometimes with extension into the duodenum; and localized polypoid forms.27 A rare pattern is gastric rugal hypertrophy, similar to the pattern seen in hypertrophic gastritis. CT often shows extensive gastric wall thickening as well as associated nodal disease (Figure 29-10A). Spread into adjacent organs is variable. Gastric MALT lymphomas are usually localized at diagnosis and often result in minimal gastric mural thickening, which may not be recognizable even with dedicated CT studies,28 but which can be recognized with endoscopic ultrasound.29 Multiorgan involvement occurs in up to 25% of patients, so extensive imaging for staging may be necessary.30
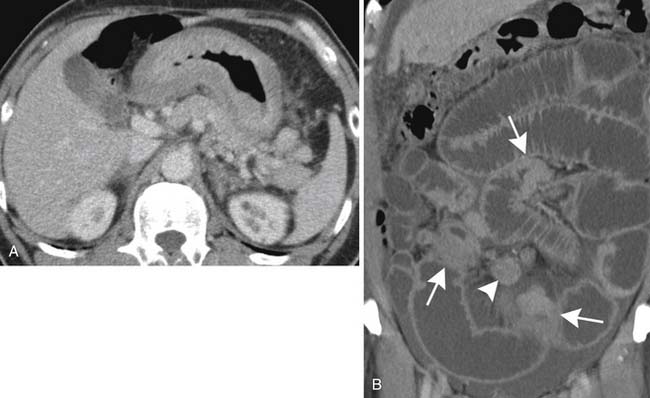
Lymphoma accounts for up to 50% of all primary tumors of the small bowel, occurring most frequently in the terminal ileum and becoming less frequent proximally. Mural thickening results in constriction of segments of bowel with obstructive symptoms, which are common at presentation (see Figure 29-10B). Alternating areas of dilatation and constriction are the most common manifestations.31 Occasionally, multiple submucosal nodules or polyps of varying size are seen, predominantly in the terminal ileum. This form is prone to intussusception (usually ileocecal or ileoileal).
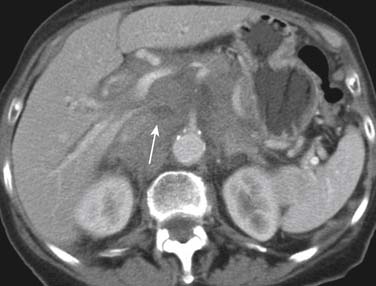
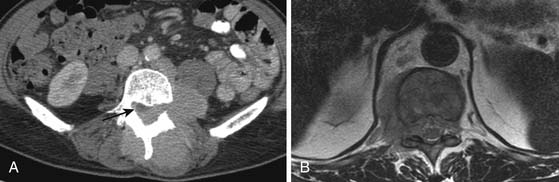
Primary pancreatic lymphoma usually results in a solitary mass lesion or diffuse infiltration and is extremely rare, accounting for only 1.3% of all cases of pancreatic malignancy and 2% of patients with NHL. Secondary pancreatic involvement is most commonly due to direct infiltration from adjacent nodal masses and may be focal or massive (Figure 29-11).
Genitourinary Tract
Although the genitourinary tract is infrequently involved at the time of presentation, in end-stage disease more than 50% of cases will have involvement of some part of it. The testicle is the most commonly involved organ, followed by the kidney and perirenal space. Involvement of the bladder, prostate, uterus, vagina, and ovaries is extremely rare.32
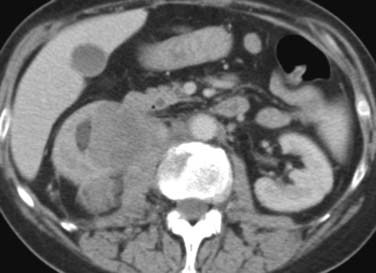
Close to 90% of renal cases are due to high-grade NHL (DLBCL, BL). In more than 40% of patients, the disease occurs at the time of recurrence only. The most frequent pattern of disease is multiple masses (60%)33 followed by solitary masses (10–20%) and direct infiltration from the retroperitoneum into the renal hilum and sinus (25%; Figure 29-12). In over 50% of cases, there is no accompanying retroperitoneal lymph node enlargement on CT.33 Frequently, a soft tissue mass is seen in the perirenal space, occasionally encasing the kidney without evidence on CT of invasion of the parenchyma. Diffuse infiltration of the kidneys with global renal enlargement is a rare manifestation. The appearance after intravenous contrast medium injection is variable, but usually homogeneous nonenhancing tissue replaces the normal enhancing parenchyma. A particularly rare form of disease is isolated periureteric lymphoma, described in both NHL and HL.
Primary lymphoma of the bladder is extremely rare, although secondary lymphoma of the bladder is found in 10% to 15% of patients with lymphoma at autopsy.32 The appearances on CT and MRI are nonspecific with either diffuse widespread thickening of the bladder wall or a large nodular mass, both patterns indistinguishable from transitional cell carcinoma.
In advanced, widespread lymphomatous disease, the female genital organs are frequently involved secondarily. Isolated lymphomatous involvement is rare. Tumors originate predominantly in the uterine cervix, and CT and MRI may demonstrate a large mass (Figure 29-13). Involvement of the uterine body usually produces diffuse enlargement, often with a lobular contour similar to that of a fibroid, with relatively homogeneous signal intensity at MRI on all sequences despite the large tumor size (see Figure 29-13).34 Similarly, primary lymphoma of the cervix and/or vagina is characterized by a large, exophytic mass. Ovarian lymphoma is less common and carries a poorer prognosis, because tumors are usually more advanced at the time of discovery. As with primary epithelial ovarian carcinoma, involvement is often bilateral and the appearances on cross-sectional imaging are identical.
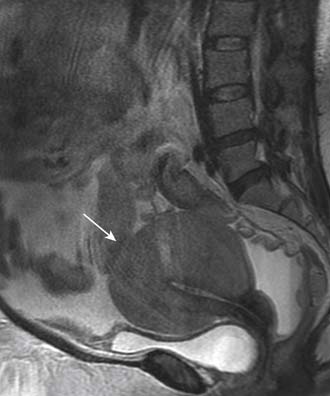
Key Points Genitourinary tract
• Testis is the most frequent site of involvement of the genitourinary tract with lymphoma, although it accounts for only 5% of all testicular tumors.
• Lymphomatous involvement of the kidneys is not usually associated with renal impairment.
• Primary lymphoma of the prostate carries a poor prognosis, whereas primary lymphoma of the bladder has a good prognosis.
• Primary lymphoma of the female genital tract is rare, best demonstrated on MRI.
Primary adrenal lymphoma is rare, usually occurring in men older than age 60. Secondary involvement of the adrenal glands is usually demonstrated on routine abdominal CT for staging (where it is seen in ≤ 6% of cases of NHL), although adrenal insufficiency is extremely rare.32 Involvement is usually bilateral and indistinguishable from metastases, although it is readily distinguishable from adenomas.
Central Nervous System
Primary central nervous system lymphoma (PCNSL) is initially localized to the CNS. It occurs almost exclusively within the brain rather than the spinal cord35 and presents most frequently between the fourth and the sixth decades, with a separate peak in the first decade of life. It now accounts for over 3% of brain tumors and up to 30% of cases of NHL in some series.
On CT or MRI, more than 50% of lesions are seen within the cerebral white matter, close to or within the corpus callosum (Figure 29-14). Most occur about the ependymal surface of the ventricles. A butterfly distribution with spread across the corpus callosum is characteristic.36 In approximately 15% of cases, the deep cortical gray matter of the basal ganglia and thalamus are involved. In 10%, lymphoma develops in the posterior fossa and is multifocal in 20%. In AIDS-related PCNSL, multifocality is much more common, being seen in up to 50% of cases. On CT, the typical tumor mass demonstrates increased density on unenhanced CT with homogeneous enhancement after intravenous injection of contrast medium; only approximately 10% of lesions do not enhance.35 Calcification is very rare and surrounding vasogenic edema relatively mild.36 On MRI, the typical appearance is of a tumor mass, hyper- or isointense relative to the surrounding normal tissue on T2-weighted sequences. There is usually uniform enhancement after injection of gadolinium–diethylenetriamine-penta-acetic acid (DTPA), but ring enhancement can be a feature of AIDS-related PCNSL.
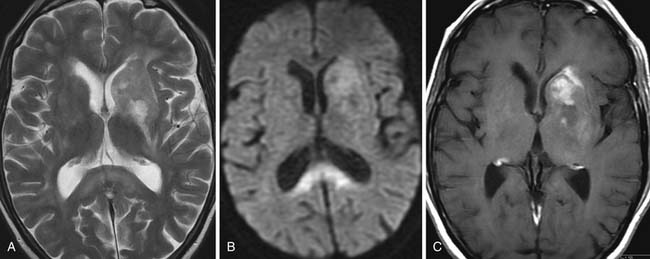
Secondary cerebral involvement occurs in 10% to 15% of patients with NHL at some time during the course of their disease. Secondary cerebral involvement is so rare in HL that a space-occupying lesion in the brain of a patient with known HL should prompt a second diagnosis. Secondary lymphoma is distinguishable from the primary form to some extent by its propensity to involve the extracerebral spaces (epidural, subdural, and subarachnoid; Figure 29-15A) and the spinal epidural and subarachnoid spaces.35 MRI typically shows extracerebral plaquelike tumor deposits in the subdural or epidural spaces, made more obvious on gadolinium-DTPA–enhanced T1-weighted images. CT is less sensitive, not only in the detection of these extracerebral lesions but also in demonstrating leptomeningeal deposits around the cranial nerves,37 particularly when resulting in cranial nerve palsies.
Gadolinium-enhanced MRI is relatively sensitive in demonstrating spinal leptomeningeal cord and nerve root involvement. Epidural extension of tumor into the spinal canal from a paravertebral nodal mass may also be elegantly demonstrated on MRI (see Figure 29-15B). Compression of the spinal cord or cauda equina due to lymphoma is a late manifestation of HL, but it is often an earlier manifestation of NHL. In both conditions, the usual cause is extension of nodal disease through the intervertebral foramina. Tumor compresses the dura, but the latter usually acts as an effective barrier to the intrathecal spread of tumor.
NHL is the most common primary orbital malignancy in adults, accounting for 10% to 15% of orbital masses35 and approximately 5% of all primary extranodal NHLs. Primary orbital lymphomas occur most commonly in patients aged 40 and 70 years. They can arise from the conjunctiva, eyelids, lacrimal glands, or retrobulbar tissues. Up to 50% will be found to have an extra-CNS primary site of origin. Secondary orbital involvement occurs in approximately 3% to 5% of NHL. Involvement of the lacrimal glands is bilateral in over 20% of cases. Involvement of the eyelids and subconjunctival spaces is readily assessed on clinical examination, whereas MRI best depicts the presence and extent of any intracranial extension.
True extranodal involvement of sites in the head and neck in HL is very rare. In NHL, 10% of patients present with extranodal head and neck involvement and NHL accounts for approximately 5% of head and neck malignancies. Waldeyer’s ring is the most common site of disease, and there is a pronounced link with involvement of the gut, which may be synchronous or metachronous. A diagnosis of NHL is suggested by circumferential involvement or multifocality (see Figure 29-2). Middle-aged women are most often affected.
NHL accounts for 2% to 5% of malignant tumors of the thyroid.38 There is an association with Hashimoto’s disease, so the disease tends to occur in women in their 60s with MALT types. However, DLBCL also occurs, and these patients present with a rapidly growing mass involving adjacent structures causing obstructive symptoms. On CT, these masses usually have a lower attenuation than the normal gland and peripheral enhancement can be seen after administration of intravenous contrast.
Primary lymphoma of bone is almost exclusively due to NHL. The criteria for the diagnosis of primary lymphoma require that only a single bone is involved, there is unequivocal histologic evidence of lymphoma, other disease is limited to regional areas at the time of diagnosis, and the primary tumor precedes metastasis by at least 6 months. The mean age at diagnosis is 50 years; most patients present in their 50s and 60s. Males are affected more often than females. Secondary involvement of bones is present in 5% to 6% of patients with NHL, but is more common in children with NHL.14 Systemic (secondary) NHL involves the axial skeleton more frequently than the appendicular skeleton.
Primary NHL of bone is radiologically indistinguishable from secondary lymphoma or other bone tumors. However, whereas bone lesions in NHL (primary or secondary) are usually permeative osteolytic (77%) or mixed lytic/sclerotic (16%), bony involvement in HL typically gives a sclerotic or mixed picture (86%) and is infrequently lytic. In HL, soft tissue disease typically may involve adjacent bones. A classic finding is the sclerotic ivory vertebra. MRI is the imaging modality of choice for staging and follow-up39 (Figure 29-16). Bone scintigraphy is of limited usefulness compared with FDG-PET or PET/CT, and there is also some evidence that FDG-PET can demonstrate response to treatment earlier and more accurately than conventional modalities including MRI.40
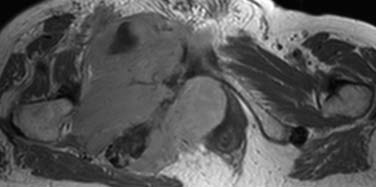
Involvement of the bone marrow indicates stage IV disease. It is rare at presentation in HL but is found in 20% to 40% of patients with NHL41 and is associated with a worse prognosis than involvement of the liver, lung, or osseous bone. During the course of HL, marrow involvement occurs in 5% to 15% of patients. Bone marrow biopsies may not be indicated as part of the initial staging of early-stage HL, but the high incidence in NHL justifies its use as a staging procedure.42 Marrow involvement in FL is typically paratrabecular, whereas in other subtypes, it is more likely to be diffuse.
MRI is extremely sensitive in the demonstration of bone marrow involvement (Figure 29-17). Tumor infiltration is of low signal on T1-weighted sequences and high signal intensity on short tau inversion recovery (STIR). T1-weighted spin echo sequences are the most sensitive.43 MRI can upstage as many as 33% of patients with negative iliac crest biopsies (see Figure 29-17).
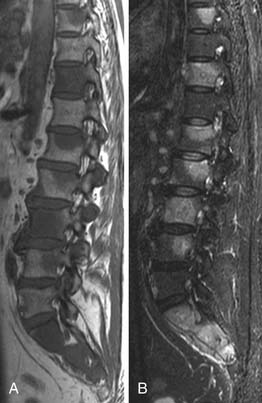
FDG-PET to date has shown only moderate accuracy for bone marrow disease, as demonstrated by a recent meta-analysis.44 False-negative PET findings occur with indolent lymphoma, often PET-negative elsewhere, or with microscopic infiltration, whereas reactive marrow hyperplasia can lower specificity.45,46 Despite the superior performance of these techniques compared with CT, their precise role in the staging of patients with lymphoma is unclear, given the obvious need to examine the cytology of bone marrow in these patients.
Key Points Bone and bone marrow
• Primary lymphoma of bone accounts for only 1% of all cases of NHL; secondary involvement is seen in 5% to 6% of cases.
• Secondary bone involvement is seen in 20% of patients with HL. Primary HL of bone is extremely rare.
• In HL, soft tissue disease may invade adjacent bone, but this is rare in NHL.
• Bone marrow involvement indicates stage IV disease and is present in 20% to 40% of patients with NHL at presentation.
• Bone marrow biopsy in NHL increases the stage of disease in up to 30% of cases (usually from stage III to IV).
• In HL, bone marrow involvement occurs in 5% to 15% of patients during the course of disease.
• Although FDG-PET is more sensitive than CT or bone scintigraphy, false-negative examinations occur with microscopic infiltration and indolent lymphomas.
Lymphoma in the Immunocompromised
The WHO classification recognizes four broad groupings associated with an increased incidence of lymphoma and lymphoproliferative disorders: lymphoproliferative diseases associated with primary immune disorders, lymphomas associated with HIV infection, posttransplant lymphoproliferative disorders (PTLDs), and other iatrogenic immunodeficiency-associated lymphoproliferative disorders. The development of lymphoma in these settings is multifactorial, but mostly related to defective immune surveillance, with or without chronic antigenic stimulation.
AIDS-related Lymphomas (ARL)
Most ARL are aggressive, presenting with advanced stage, bulky disease, and a marked propensity to involve extranodal sites, especially the GI tract, CNS (less frequent with the advent of HAART), liver, and bone marrow. Multiple sites of extranodal involvement are seen in over 75% of cases. Peripheral lymph node enlargement is seen in only 30%. In the chest, NHL is usually extranodal; pleural effusion and lung disease are common, often with nodules, and acinar and interstitial opacities. There is a wide differential diagnosis, especially mycobacterial infection.47 Within the abdomen, the GIT, lower genitourinary tract, and major viscera are commonly involved. Regarding PCNSL, features such as rim enhancement and multifocality are seen more often than in the immunocompetent population. The differential diagnosis includes cerebral toxoplasmosis, although the location of PCNSL in the deep white matter is suggestive. Quantitative FDG-PET uptake may help in the differentiation of PCNSL, toxoplasmosis, and progressive multifocal leucoencephalopathy (PML), being higher in the former.48
Posttransplant Lymphoproliferative Disorders
PTLDs occur in 2% to 4% of solid organ transplant recipients depending on the type of transplant, the lowest frequency being seen in renal transplant recipients (1%). Marrow allograft recipients have a low risk (<1%). Most appear to represent EBV-induced monoclonal or, more rarely, polyclonal B-cell or T-cell proliferation in a setting of reduced immune surveillance. The clinical features are variable, correlating with the type of allograft and type of immunosuppression. In all cases, extranodal disease is common. In patients receiving azathioprine, the allograft itself and the CNS are often involved, whereas in patients receiving cyclosporin A, the GIT is affected more often. The bone marrow, liver, and lung are frequently involved. In the lung, pulmonary nodules or airspace opacity and pleural effusions are common, with or without mediastinal adenopathy (see Figure 29-8). Abdominal PTLD is characterized by frequent extranodal disease. Multiple segments of bowel may be affected along with the allograft itself.49
Key Points Lymphoma in the immunocompromised
• Lymphomas associated with HIV have a propensity to involve extranodal sites (especially the GIT, CNS, liver, and bone marrow).
• Most tumors are aggressive with advanced-stage bulky disease.
• PTLD occurs in 2% to 4% of solid organ and 1% of marrow allograft recipients and typically causes solitary or multiple lung nodules. In the abdomen, this disorder is characterized by involvement of the GIT and liver.
Treatment
Hodgkin Lymphoma
There has been a dramatic improvement in survival from HL in the past 40 years, with mortality rates falling by over 50% since 1975. Current mortality rates are approximately 0.5/100,000 for men and 0.3/100,000 for women. Early-stage disease has a 5-year survival rate of over 90%. The histologic subtype does not usually affect treatment decisions except for NLPHL.
Patients with HL have traditionally been divided into two or three prognostic groups, each associated with a standard treatment strategy; but the latter has changed markedly in recent years with the development of more efficacious chemotherapy and with recognition of the late effects of radiotherapy. HL is highly radiosensitive and, for many years, mantle radiotherapy for cervical disease was the treatment of choice. It included irradiation to the cervical, supraclavicular, axillary, and mediastinal nodes, down to the lower border of the T10 vertebral body. However, with the prolonged survival of most patients with HL, the long-term consequences of this treatment have become apparent, including a huge excess of breast cancer in women and thyroid cancer in men and women treated with mantle radiotherapy.50 In patients with early-stage disease treated when they were younger than 50, the absolute excess mortality actually increases with time because of the increased incidence of cardiac disease and second tumors consequent upon treatment.51 New chemotherapeutic regimens have, therefore, been developed with the aim of reducing toxicity while maintaining efficacy. Only in NLPHL, which commonly presents with stage I disease, would involved field radiotherapy or surgical excision alone be considered now. Both yield excellent results with long-term progression-free survival (PFS) in over 80%.
The most common chemotherapy regimen is ABVD (doxorubicin, bleomycin, vinblastine, and dacarbazine). For patients with more advanced disease, 50% to 80% will achieve complete remission, but of these, 30% to 50% will relapse within 5 years. Therefore, investigators have intensified chemotherapy regimens for patients at poor risk in whom increased toxicity of treatment can be justified, including the BEACOPP (bleomycin, etoposide, adriamycin, cyclophosphamide, oncovin, procarbazine, and prednisolone) regimen. The Stanford V protocol utilizes polypharmacy with low-dose small-field irradiation of all sites originally larger than 5 cm or to the spleen if it is clinically involved. Another strategy is to treat all such patients with ABVD but use an interim FDG-PET scan to identify nonresponders.52 In patients who achieve a complete response (CR) with chemotherapy alone, radiotherapy may not be necessary, but if there is only a partial response (PR), radiotherapy as consolidation improves survival. Patients failing initial chemotherapy for advanced HL have a poor prognosis, but salvage chemotherapy with high-dose treatment and hemopoietic stem cell rescue is being increasingly employed for those with chemosensitive disease, although its long-term benefits have not been established in a randomized trial.
Non-Hodgkin Lymphoma
As indicated previously, the prognosis and, therefore, treatment of NHL varies according to histologic subtype. FL may be regarded as the paradigm for other subtypes of indolent lymphoma. The median survival for patients with FL was 10 years at the end of the 20th century, but survival is improving with the introduction of new therapies. With time, FL may undergo histologic transformation, with increasing numbers of blast cells in the affected lymph node. Such transformation has a grave prognosis and may be a terminal event in up to 70% of cases.53
Monoclonal antibodies have also been conjugated to radioactive isotopes to form radioimmunoconjugates, the antibody delivering the isotope to the tumor cells. The anti-CD20 antibody tositumomab combined with iodine-131 (Bexxar), has been used successfully to treat FL. It is a gamma emitter and can be used for imaging as well as treatment. Ibritumomab, combined with yttrium-90 (ibritumomab tiuxetan [Zevalin]) has also been used for FL. This is a beta emitter, which has higher energies and is better for larger tumors as well as being safe to administer on an outpatient basis. Both drugs induce high response rates and improve progression-free survival. However, recurrence remains a problem, and increasingly, rituximab is being used to maintain remission, although development of rituximab-refractory disease is well recognized.
Experimental Therapies
Key Points Treatment
• In HL, disease stage is the most important prognostic factor and determines the intensity and nature of treatment.
• HL is curable in the vast majority of cases.
• NHL, the histologic subtype, is the major determinant of treatment.
• Paradoxically, cure is most often achieved in the more aggressive large cell lymphomas.
Surveillance
Radiographic Predictors of Outcome
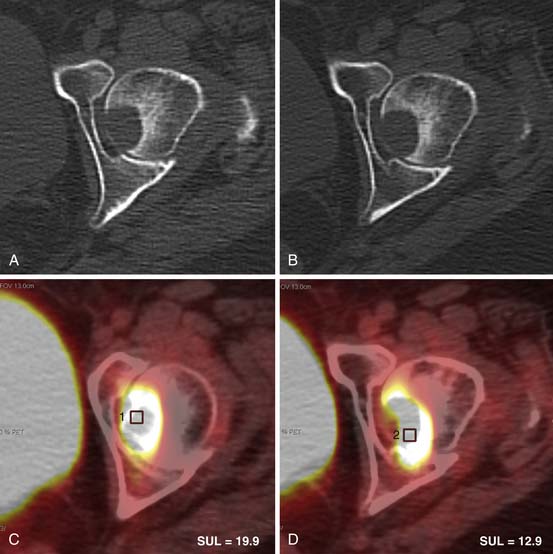
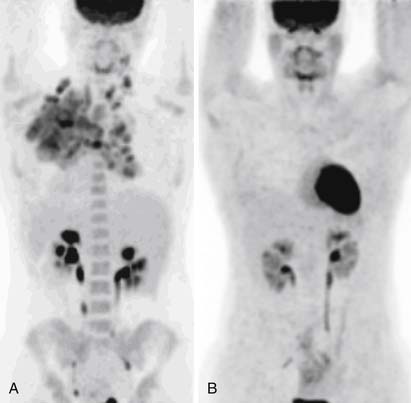
Many centers favor an interim CT scan after a few cycles of chemotherapy to confirm some response to treatment, but recently, numerous studies have shown that an interim FDG-PET scan yields much more prognostic information. Early FDG-PET in aggressive NHL is a more accurate predictor of PFS and overall survival than other prognostic indicators.54,55 A similar association has been shown in patients with HL,56,57 even after just one cycle of treatment58 (Figure 29-18). However, the existing data are heterogeneous and, therefore, interim scans should be done only in large clinical trials, especially because there is evidence that, since the widespread use of rituximab, the positive predictive value (PPV) of interim scans for NHL has diminished.
Criteria for Response
In 1999, a report set out standardized international criteria for assessment of response in NHL, similar to those already in use for HL.59 The International Workshop Radiological Criteria (IWC) defined specific criteria for response:
• Complete remission, unconfirmed (CRu)
Functional Imaging
Persistent gallium uptake within a residual mass usually indicates persistent disease (reflected in a PPV for relapse double that of CT), but the low accuracy of gallium-67 and the significant proportion of non–gallium-avid tumors limits its use in the evaluation of the residual mass. By contrast, FDG-PET has a high predictive value in the differentiation between active tumor and fibrosis (Figure 29-19). Indeed, it has a high PPV for relapse, with or without a residual mass at CT.60,61 However, small volumes of disease can produce false-negative examinations whereas reactive and inflammatory changes can give false-positive results. Recent evidence suggests that the PPV of FDG-PET may be lower in patients treated with rituximab.62 Therefore, clinical correlation is essential when interpreting FDG-PET results.63
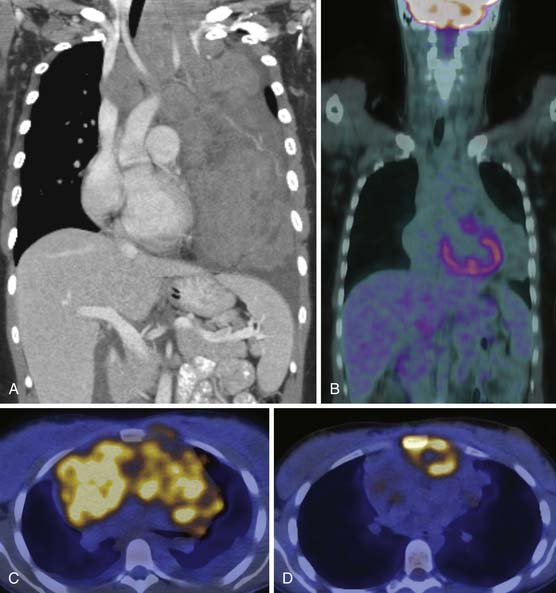
FDG-PET in Response Assessment
Generally, a positive FDG-PET scan at the end of therapy strongly predicts for early disease relapse,60 with a PPV at least double that of CT. In one follow-up study, all PET-positive/CT-negative patients relapsed, whereas only 5% of PET-negative/CT-positive patients developed relapse.64
Incorporation of the end of treatment FDG-PET scan result into the IWC criteria has a significant impact on response categorization. In one study, patients with a PR by standard IWC who were PET-negative did as well as those who were in CR by IWC and IWC + PET, indicating superior discriminative ability.65 As a result of such studies, an International Harmonization Project (IHP) was convened to revise the IWC criteria.66 The IHP guidelines support the use of FDG-PET for end of therapy response assessment in DLBCL and HL, but not for other NHLs, unless the CR rate is a primary endpoint of a clinical trial. In the revised criteria, patients with residual masses are assigned to the CR category, provided that it is PET-negative at the end of treatment and was, or can reasonably be expected to have been, PET-positive before treatment. The CRu category is eliminated. A PR exists when there is residual FDG-PET positivity in at least one previously involved site. Importantly, the group has also issued guidance on timing, performance, and interpretation of FDG-PET scans.67 Specifically, simple visual assessment of tracer uptake is deemed sufficient and it is not necessary to measure the standardized uptake value (SUV).
FDG-PET can also be used to predict outcome prior to high-dose treatment.68 Patients commencing high-dose therapy with a positive pretreatment PET scan have a much poorer prognosis.
Detection of Recurrence
Relapse following satisfactory response to initial treatment occurs in 10% to 40% of patients with HL and in approximately 50% of patients with NHL. In HL, relapse usually occurs within the first 2 years after treatment and patients are followed up closely during this period. For patients with HL or aggressive NHL in whom CR is achieved, there is very little evidence for routine surveillance with imaging.69,70 Routine follow-up tests, including CT, only rarely detect relapse before patients become symptomatic.70–72
It might be anticipated that functional imaging could identify asymptomatic relapse before anatomic cross-sectional imaging (especially in the presence of a residual mass), but there is only limited evidence for this.73 In one series of a cohort of patients treated for HL, relapses were identified by FDG-PET before there was any other evidence of relapse74 but the false-positive rate was high. In another study, it was concluded that there was no benefit from surveillance studies for HL or aggressive NHL beyond 18 months.75 In cases of clinically suspected relapse, the development of a positive PET scan is highly suggestive, but at the moment, the IHP does not recommend the use of PET/CT for posttreatment surveillance.
Complications of Therapy
Key Points Complications of therapy
Short Term
• Drug-induced pulmonary toxicity (diffuse alveolar damage, organizing pneumonia, pulmonary hemorrhage, nonspecific interstitial pneumonia)
• Opportunistic pulmonary infection (Pneumocystis spp., viral pneumonia, invasive fungal infection, tuberculosis)
• Enterocolitis, pseudomembranous colitis, neutropenic typhlitis
1. American Cancer Society. Cancer Facts and Figures 2011. Atlanta: American Cancer Society; 2011:4.
2. Ries L.A., Kosary C.L., Hankey B.P., et al. SEER Cancer Statistics Review, 1973-1996. Bethesda, MD: National Cancer Institute, 1999.
3. Rabkin C.S., Devesa S.S., Zahm S.H., Gail M.H. Increasing incidence of non-Hodgkin’s lymphoma. Semin Hematol. 1993;30:286-296.
4. International collaboration on HIV and cancer. Highly active antiretroviral therapy and the incidence of cancer in HIV-infected adults. J Natl Cancer Inst. 2000;92:1823-1830.
5. Harris N.L., Jaffe E.S., Diebold J., et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting—Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835-3849.
6. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin’s Lymphoma Classification Project. Blood. 1997;89:3909-3918.
7. Jaffe E.S. The 2008 WHO Classification of lymphomas: implications for clinical practice and translational research. Hematology Am Soc Hematol Educ Program. 2009:523-531.
8. Alizadeh A.A., Eisen M.B., Davis R.E., et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503-511.
9. Swerdlow S.H., Campo E., Harris N.L., et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed., Lyon, France: IARC; 2008:233-237.
10. Lukes R.J., Butler J.J. The pathology and nomenclature of Hodgkin’s disease. Cancer Res. 1966;26:1063-1083.
11. Lister T.A., Crowther D., Sutcliffe S.B., et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol. 1989;7:1630-1636.
12. Shipp M.A., Harrington D.P., Anderson J.R., et al. A predictive model for aggressive non-Hodgkin’s lymphoma. The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. N Engl J Med. 1993;329:987-994.
13. Solal-Celigny P. Follicular Lymphoma International Prognostic Index. Curr Treat Option Oncol. 2006;7:270-275.
14. Ng Y.Y., Healy J.C., Vincent J.M., et al. The radiology of non-Hodgkin lymphoma in childhood: a review of 80 cases. Clin Radiol. 1994;49:594-600.
15. King A.D., Ahuja A.T., Yeung D.K., et al. Malignant cervical lymphadenopathy: diagnostic accuracy of diffusion-weighted MR imaging. Radiology. 2007;245:806-813.
16. Moog F., Bangerter M., Diederichs C.G., et al. Lymphoma: role of whole-body 2-deoxy-2-[F-18]fluoro-D-glucose (FDG) PET in nodal staging. Radiology. 1997;203:795-800.
17. Moog F., Bangerter M., Diederichs C.G., et al. Extranodal malignant lymphoma: detection with FDG PET versus CT. Radiology. 1998;206:475-481.
18. Rodríguez-Vigil B., Gómez-León N., Pinílla I., et al. PET/CT in lymphoma: prospective study of enhanced full-dose PET/CT versus unenhanced low-dose PET/CT. J Nucl Med. 2006;47:1643-1648.
19. Castellino R.A., Blank N., Hoppe R.T., Cho C. Hodgkin disease: contributions of chest CT in the initial staging evaluation. Radiology. 1986;160:603-605.
20. Castellino R.A., Hilton S., O’Brien J.P., Portlock C.S. Non-Hodgkin’s lymphoma: contribution of chest CT in the initial staging evaluation. Radiology. 1996;199:129-132.
21. Shaffer K., Smith D., Kirn D., et al. Primary mediastinal large-B-cell lymphoma: radiologic findings at presentation. AJR Am J Roentgenol. 1996;167:425-430.
22. Harell G.S., Breiman R.S., Glatstein E.J., et al. Computed tomography of the abdomen in the malignant lymphomas. Radiol Clin North Am. 1977;15:391-400.
23. Lewis E.R., Caskey C.I., Fishman E.K. Lymphoma of the lung: CT findings in 31 patients. AJR Am J Roentgenol. 1991;156:711-714.
24. Wernecke K., Vassallo P., Rutsch F., et al. Thymic involvement in Hodgkin disease: CT and sonographic findings. Radiology. 1991;181:375-383.
25. Spiers A.S., Husband J.E., MacVicar A.D. Treated thymic lymphoma: comparison of MR imaging with CT. Radiology. 1997;203:369-376.
26. Rini J.N., Manalili E.Y., Hoffman M.A., et al. F-18 FDG versus Ga-67 for detecting splenic involvement in Hodgkin’s disease. Clin Nucl Med. 2002;27:572-577.
27. Fishman E.K., Urban B.A., Hruban R.H. CT of the stomach: spectrum of disease. Radiographics. 1996;16:1035-1054.
28. Kessar P., Norton A., Rohatiner A.Z., et al. CT appearances of mucosa-associated lymphoid tissue (MALT) lymphoma. Eur Radiol. 1999;9:693-696.
29. Fujishima H., Chijiiwa Y. Endoscopic ultrasonographic staging of primary gastric lymphoma. Abdom Imaging. 1996;21:192-194.
30. Raderer M., Vorbeck F., Formanek M., et al. Importance of extensive staging in patients with mucosa-associated lymphoid tissue (MALT)-type lymphoma. Br J Cancer. 2000;83:454-457.
31. Dodd G.D. Lymphoma of the hollow abdominal viscera. Radiol Clin North Am. 1990;28:771-783.
32. Charnsangavej C. Lymphoma of the genitourinary tract. Radiol Clin North Am. 1990;28:865-877.
33. Reznek R.H., Mootoosamy I., Webb J.A., Richards M.A. CT in renal and perirenal lymphoma: a further look. Clin Radiol. 1990;42:233-238.
34. Kim Y.S., Koh B.H., Cho O.K., Rhim H.C. MR imaging of primary uterine lymphoma. Abdom Imaging. 1997;22:441-444.
35. Zimmerman R.A. Central nervous system lymphoma. Radiol Clin North Am. 1990;28:697-721.
36. Jenkins C.N., Colquhoun I.R. Characterization of primary intracranial lymphoma by computed tomography: an analysis of 36 cases and a review of the literature with particular reference to calcification haemorrhage and cyst formation. Clin Radiol. 1998;53:428-434.
37. Chamberlain M.C., Sandy A.D., Press G.A. Leptomeningeal metastasis: a comparison of gadolinium-enhanced MR and contrast-enhanced CT of the brain. Neurology. 1990;40:435-438.
38. Takashima S., Ikezoe J., Morimoto S., et al. Primary thyroid lymphoma: evaluation with CT. Radiology. 1988;168:765-768.
39. Mengiardi B., Honegger H., Hodler J., et al. Primary lymphoma of bone: MRI and CT characteristics before and after successful treatment. AJR Am J Roentgenol. 2005;184:185-192.
40. Park Y.H., Kim S., Choi S.J., et al. Clinical impact of whole-body FDG-PET for evaluation of response and therapeutic decision-making of primary lymphoma of bone. Ann Oncol. 2005;16:1401-1402.
41. Kaplan H.S. Essentials of staging and management of the malignant lymphomas. Semin Roentgenol. 1980;15:219-226.
42. Pond G.D., Castellino R.A., Horning S., Hoppe R.T. Non-Hodgkin’s lymphoma: influence of lymphography, CT, and bone marrow biopsy on staging and management. Radiology. 1989;170:159-164.
43. Yasumoto M., Nonomura Y., Yoshimura R., et al. MR detection of iliac bone marrow involvement by malignant lymphoma with various MR sequences including diffusion-weighted echo-planar imaging. Skeletal Radiol. 2002;31:263-269.
44. Pakos E.E., Fotopoulos A.D., Ioannidis J.P. 18F-FDG PET for evaluation of bone marrow infiltration in staging of lymphoma: a meta-analysis. J Nucl Med. 2005;46:958-996.
45. Elstrom R., Guan L., Baker G., et al. Utility of FDG-PET scanning in lymphoma by WHO classification. Blood. 2003;101:3875-3876.
46. Moog F., Bangerter M., Kotzerke J., et al. 18-F-fluorodeoxyglucose-positron emission tomography as a new approach to detect lymphomatous bone marrow. J Clin Oncol. 1998;16:603-609.
47. Jasmer R.M., Gotway M.B., Creasman J.M., et al. Clinical and radiographic predictors of the etiology of computed tomography-diagnosed intrathoracic lymphadenopathy in HIV-infected patients. J Acquir Immune Defic Syndr. 2002;31:291-298.
48. O’Doherty M.J., Barrington S.F., Campbell M., et al. PET scanning and the human immunodeficiency virus-positive patient. J Nucl Med. 1997;38:1575-1583.
49. Pickhardt P.J., Siegel M.J. Abdominal manifestations of posttransplantation lymphoproliferative disorder. AJR Am J Roentgenol. 1998;171:1007-1013.
50. Henry-Amar M. Second cancer after the treatment for Hodgkin’s disease: a report from the International Database on Hodgkin’s Disease. Ann Oncol. 1992;3(Suppl 4):117-128.
51. Ng A.K., Bernardo M.P., Weller E., et al. Long term survival and competing causes of death in patients with early stage Hodgkin’s disease treated at age 50 or younger. J Clin Oncol. 2002;20:2101-2108.
52. The RATHL Trial. UK Clinical Research Network: Portfolio database. RATHL. Available at http://public.ukcrn.org.uk/Search/studyDetail.aspx?studyID=4488
53. Bastion Y., Sebban C., Berger F., et al. Incidence, predictive factors and outcome of lymphoma transformation in follicular lymphoma patients. J Clin Oncol. 1997;15:1587-1594.
54. Spaepen K., Stroobants S., Dupont P., et al. Early restaging positron emission tomography with (18)F-fluorodeoxyglucose predicts outcome in patients with aggressive non-Hodgkin’s lymphoma. Ann Oncol. 2002;13:1356-1363.
55. Haioun C., Itti E., Rahmouni A., et al. [18F]fluoro-2-deoxy-d-glucose positron emission tomography (FDG-PET) in aggressive lymphoma: an early prognostic tool for predicting patient outcome. Blood. 2005;106:1376-1381.
56. Hutchings M., Loft A., Hansen M., et al. FDG-PET after two cycles of chemotherapy predicts treatment failure and progression-free survival in Hodgkin lymphoma. Blood. 2006;107:52-59.
57. Gallamini A., Hutchings M., Rigacci L., et al. Early interim 2-[18F]fluoro-2-deoxy-d-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol. 2007;25:3746-3752.
58. Kostakoglu L., Goldsmith S.J., Leonard J.P., et al. FDG-PET after 1 cycle of therapy predicts outcome in diffuse large cell lymphoma and classic Hodgkin disease. Cancer. 2006;107:2678-2687.
59. Cheson B.D., Horning S.J., Coiffier B., et al. Report of an international workshop to standardize response criteria for Non-Hodgkin’s lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17:1244.
60. Naumann R., Vaic A., Beuthien-Baumann B., et al. Prognostic value of positron emission tomography in the evaluation of post-treatment residual mass in patients with Hodgkin’s disease and non-Hodgkin’s lymphoma. Br J Haematol. 2001;115:793-800.
61. Weihrauch M.R., Re D., Scheidhauer K., et al. Thoracic positron emission tomography using 18F-fluorodeoxyglucose for the evaluation of residual mediastinal Hodgkin disease. Blood. 2001;98:2930-2934.
62. Han H.S., Escalon M.P., Hsiao B., et al. High incidence of false positive PET scans in patients with aggressive non-Hodgkin’s lymphoma treated with rituximab-containing regimens. Ann Oncol. 2009;20:309-318.
63. Bakheet S.M., Powe J. Benign causes of 18-FDG uptake on whole body imaging. Semin Nucl Med. 1998;28:352-358.
64. Zinzani P.L., Chierichetti F., Zompatori M., et al. Advantages of positron emission tomography (PET) with respect to computed tomography in the follow-up of lymphoma patients with abdominal presentation. Leuk Lymphoma. 2002;43:1239-1243.
65. Juweid M., Wiseman G.A., Vose J., et al. Response assessment of aggressive non-Hodgkin’s lymphoma by Integrated International Workshop criteria and fluorine-18-fluordeoxyglucose positron emission tomography. J Clin Oncol. 2005;23:4652-4661.
66. Cheson B.D., Pfistner B., Juweid M.E., et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579-586.
67. Juweid M.E., Stroobants S., Hoekstra O.S., et al. Use of positron emission tomography for response assessment of lymphoma: consensus of the Imaging Subcommittee of International Harmonization Project in Lymphoma. J Clin Oncol. 2007;25:571-578.
68. Becherer A., Mitterbauer M., Jaeger U., et al. Positron emission tomography with [18F]2-fluoro-D-2-deoxyglucose (FDG-PET) predicts relapse of malignant lymphoma after high-dose therapy with stem cell transplantation. Leukemia. 2002;16:260-267.
69. Weeks J.C., Yeap B.Y., Canellos G.P., Shipp M.A. Value of follow-up procedures in patients with large-cell lymphoma who achieve a complete remission. J Clin Oncol. 1991;9:1196-1203.
70. Guadagnolo B.A., Punglia R.S., Kuntz K.M., et al. Cost-effectiveness analysis of computerized tomography in the routine follow-up of patients after primary treatment for Hodgkin’s disease. J Clin Oncol. 2006;24:4116-4122.
71. Radford J.A., Eardley A., Woodman C., Crowther D. Follow up policy after treatment for Hodgkin’s disease: too many clinic visits and routine tests? A review of hospital records. Br Med J. 1997;314:343-346.
72. Elis A., Blickstein D., Klein O., et al. Detection of relapse in non-Hodgkin’s lymphoma: role of routine follow-up studies. Am J Hematol. 2002;69:41-44.
73. Setoain F.J., Pons F., Herranz R., et al. 67Ga scintigraphy for the evaluation of recurrences and residual masses in patients with lymphoma. Nucl Med Commun. 1997;18:405-411.
74. Jerusalem G., Beguin Y., Fassotte M.F., et al. Early detection of relapse by whole-body positron emission tomography in the follow-up of patients with Hodgkin’s disease. Ann Oncol. 2003;14:123-130.
75. Zinzani P.L., Stefoni V., Ambrosini V., et al. FDG-PET in the serial assessment of patients with lymphoma in complete remission. Blood. 2007;110:71. (abstract 216)