[level-membership-for-critical-care-medicine-category]
15 Hematologic and Oncologic Emergencies Requiring Critical Care
 Be sure to check out the supplementary content available at
Be sure to check out the supplementary content available at Pearls
• Severe anemia associated with a hemoglobin (Hgb) concentration less than 5 g/dL and a hematocrit (Hct) less than 15% is likely to produce congestive heart failure (CHF). Transfuse packed red blood cells (PRBCs) slowly (3 mL/kg per hour).
• Complete bone marrow failure produces a fall in Hgb of approximately 0.7-1 g/dL per day. A more precipitous fall in Hgb is likely to be caused by bleeding or increased red blood cell (RBC) destruction.
• The risk of spontaneous bleeding is increased if the platelet count is less than 20,000/mm3 and intracranial hemorrhage is more likely once the platelet count is less than 5000/mm3.
• Rule out intracranial hemorrhage in any child with severe thrombocytopenia and unilateral headache. If hemorrhage is suspected, you will often need to begin to stabilize first; obtain scans and radiographs later.
• Acute tumor lysis syndrome produces hypocalcemia, hyperkalemia, hyperuricemia, and hyperphosphatemia. Disseminated intravascular coagulation (DIC) is also possible.
• An absolute neutrophil count less than 500/mm3 is associated with a significant risk of infection. Monitor these patients closely for evidence of infection.
• Whenever blood products are administered, label the crossmatch specimen carefully at the bedside and double-check patient and blood product identification (an extra time).
Essential anatomy and physiology
Blood Components
Blood is the fluid that sustains life. This highly specialized body fluid has many functions, including transporting oxygen and nutrition, eliminating waste, acid-base buffering, and maintaining homeostasis. The hematopoietic system consists of the bone marrow, liver, spleen, lymph nodes, and thymus gland.27 Each of these components plays a specific role in the regulation of blood.
Blood is composed of a liquid phase called plasma and a formed or cellular phase. Approximately 90% of plasma consists of water, and the remaining portions are solutes including factors that form clots (e.g., clotting factors, fibrinogen, globulins) and serum that contains electrolytes, hormones, antibodies, nutrients, and other factors. The cellular portion of the blood consists of the formed elements: RBCs, white blood cells (WBCs), and platelets (for an illustration, refer to Evolve Fig. 15-1 in the Chapter 15 Supplement on the Evolve Website).
The body’s first hematopoietic stem cell is produced in the yolk sac of the embryo. The fetal liver becomes the site for hematopoiesis at approximately the second month of fetal life. The liver is the main organ producing blood cells from the second to the fifth month of fetal life.27 At approximately the fourth month, the bone marrow begins producing blood cells and remains the production site throughout life. Bone marrow, one of the largest organs in the body, is located in the cavities of all bones.
Hematopoiesis consists of the production, differentiation, and development of blood cells; it normally occurs in the bone marrow.27 RBCs, WBCs, and platelets are all thought to arise from multipotential stem cells that inhabit the bone marrow. These stem cells have the ability to differentiate into any of the three cell lines, based on the needs of the body. The process of cell differentiation by the pluripotent stem cell is normally (i.e., in the absence of disease) self regulatory. In children, all bones produce blood cells. When bone growth ceases, often by 18 years old, only the ribs, sternum, vertebrae, and pelvis continue to produce blood cells.27
WBCs, also called leukocytes, defend the body against infectious agents. Their main function is to fight infection by migrating to the site of inflammation to assist in defending the body against foreign antigens. There are several different types of WBCs, each with a specific purpose. The differential of a complete blood cell count (CBC) reveals the percentages of neutrophils, lymphocytes, monocytes, and eosinophils. The numeric value of the WBCs is not as important as an evaluation of the absolute neutrophil count (ANC), a more accurate measure of the body’s ability to fight infection (see Neutrophils below and formula for ANC calculation in Box 15-1).
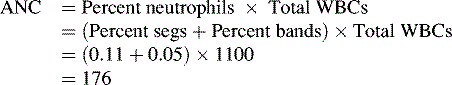
Neutrophils provide the primary defense against bacterial infections, because they engulf and destroy invading organisms. The ANC is determined from a WBC differential by multiplying the percent of neutrophils plus bands by the total WBC count (Box 15-1 shows an example of ANC calculation). In general, children with an ANC of less than 500/mm3 have a high risk of developing life-threatening bacterial infections. The risk of infection is particularly high in patients with neutropenia as the result of failure of neutrophil production, rather than those with immune-mediated neutropenia.
Clotting Cascade
Normal hemostasis is maintained by a complex balance of procoagulant and anticoagulant factors. Together, these factors provide rapid and localized control of bleeding at sites of injury while preventing the clotting process in unaffected tissues. A simplified scheme of the coagulation process is shown in Evolve Fig. 15-2 in the Chapter 15 Supplement on the Evolve Website.
Common clinical conditions
Acute Anemia
Etiology
Anemia is present when the number of circulating RBCs is reduced, resulting in decreased oxygen-carrying capacity of the blood. Anemia is the most common hematologic condition of infancy and childhood. Most patients with anemia have an underlying disease, so anemia is a symptom rather than a disease. Causes of anemia include acute or chronic blood loss, decreased production of RBCs, splenic sequestration, and hemolysis (Box 15-2).
Box 15-2 Etiology of Severe Anemia
Decreased Production
• Bone marrow replacement (leukemia, other malignancies)
• Bone marrow failure (aplastic anemia)
• Bone marrow suppression (chemotherapy, radiation)
• Transient erythroblastopenia of childhood
• Congenital red cell aplasia (Diamond-Blackfan syndrome)
• Aplastic crisis (sickle cell anemia, hemoglobin sickle cell disease, hereditary spherocytosis)
Pathophysiology
Blood Loss
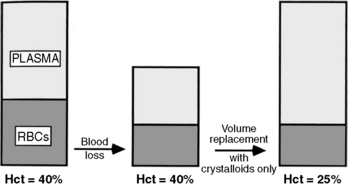
When a patient experiences acute blood loss, the Hgb and Hct are not initially affected because both plasma and RBCs have been lost. If only crystalloid and colloids are used (without blood) for fluid resuscitation, then the Hct will fall because whole blood loss has been replaced with fluid that does not contain RBCs (Fig. 15-1). The patient may remain anemic until blood is administered.
Decreased RBC Production
When complete bone marrow failure is present, the Hgb will fall approximately 0.7 to 1 g/dL per day7; this usually produces a chronic rather than an acute anemia. If the Hgb or Hct falls precipitously (greater than 1 g/dL per day) in the patient with bone marrow suppression, then hemorrhage or increased RBC destruction is probably present.
Thrombocytopenia
Etiology and Pathophysiology
Patients with thrombocytopenia have a decrease in circulating platelets. This condition can occur as a result of decreased production of platelets, increased destruction of platelets, or sequestration of platelets in the spleen or liver (Box 15-3). The cause of the thrombocytopenia affects the treatment.
Box 15-3 Most Common Causes of Thrombocytopenia
Decreased Production
• Bone marrow failure (aplastic anemia)
• Bone marrow replacement (leukemia, other malignancies)
• Bone marrow suppression (chemicals, insecticides, chemotherapy, radiation therapy, chloramphenicol, alcohol)
• Hereditary thrombocytopenia (Alport’s syndrome, Fanconi syndrome, Wiskott-Aldrich syndrome)
Increased Destruction
From Turgeon ML: Clinical hematology: theory and procedures, ed 4. Philadelphia, 2005, Lippincott Williams and Wilkins.
Clinical Signs and Symptoms
Reticulated platelets (RPs) are newly synthesized platelets with increased ribonucleic acid content. These new platelets can be identified and quantified, and the RP value can aid in determining the cause of the thrombocytopenia. A decrease in RP value in the patient with thrombocytopenia suggests bone marrow suppression (i.e., platelets are not being made despite a fall in platelet count), whereas a high RP value in a patient with thrombocytopenia suggests immune-mediated thrombocytopenia (i.e., platelets are being made but are not correcting the thrombocytopenia).21
Management
Prophylactic platelet transfusion is provided if the platelet count is less than 10,000/mm3, to reduce the risk of spontaneous intracranial hemorrhage. Although published data indicate no difference in outcomes for patients with leukemia when platelet transfusion thresholds were 10,000/mm3 or 20,000/mm3 in the absence of active bleeding, institutional practices vary widely based on local practice guidelines.28 In general, lower thresholds are used for platelet transfusion for children with conditions such as fever, central nervous system tumors, bleeding, or coagulopathies or those requiring procedures.2 Patients can safely have “major invasive procedures” such as bone marrow aspirate and biopsies with platelet counts of 50,000/mm3; the thresholds can vary slightly among institutions.23,24,28
Patients with immune-mediated platelet destruction can be treated with high-dose steroids (prednisone, 4 to 8 mg/kg per day), intravenous immune gamma globulin (0.5 to 1 g/kg of immunoglobulin G), or both. Children with exceptionally low platelet counts and acute bleeding require inpatient hospitalization with close observation. Bone marrow aspirate is recommended before initiating high-dose methylprednisolone (30 to 50 mg/kg per day) to treat severe bleeding. These children may also receive a 2- or 3-day course of intravenous immune gamma globulin.13 Frequent monitoring of laboratory tests may be necessary until the platelet count improves. Monitor the patient closely for signs of bleeding.
Disseminated Intravascular Coagulation
Etiology
Disseminated intravascular coagulation is characterized by the intravascular consumption of platelets and plasma clotting factors.13 DIC is not a primary disease but a complication of other disease processes.27 DIC can complicate sepsis, hypoxemia, major trauma with severe tissue injury, malignancy, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome (HUS), extensive burns, and severe viral infections (Box 15-4).
Pathophysiology and Clinical Signs and Symptoms
DIC is characterized by an abnormal coagulation process—the entire clotting mechanism is triggered inappropriately. Unrestrained clotting causes systemic or local formation of fibrin clots. This development of fibrin clots leads to microthrombi and excessive bleeding secondary to the consumption of the platelets and clotting factors. The fibrin clot development triggers the clotting cascade process to break down the clots and fibrin that have been formed. Fibrinolysis results in the development of fibrin degradation products, which in turn will act as anticoagulants and promote more bleeding. Organ injury may be the end result of intravascular clots, which cause microvascular occlusions and anoxia.20 In addition, when the RBCs flow through obstructed vessels the end result is additional hemolysis that can lead to hemolytic anemia (Fig. 15-2).
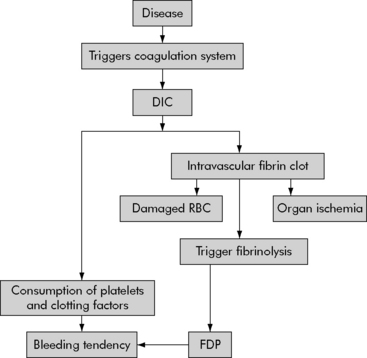
Fig. 15-2 Pathophysiology of disseminated intravascular coagulation.
DIC, Disseminated intravascular coagulation; FDP, fibrin degradation products.
(From Pagana K, Pagana T: Mosby’s manual of diagnostic and laboratory tests, St Louis, 2006, Mosby-Elsevier.)
No single test can be used to diagnose DIC. Laboratory abnormalities consistent with the diagnosis of DIC include thrombocytopenia, prolonged PT or activated partial thromboplastin time (aPTT), decreased fibrinogen, elevated fibrin degradation products (fibrin split products), and anemia (Table 15-1). The presence of fibrin monomer indicates that fibrinogen is being broken down by thrombin and that clot is being formed. A rise in fibrin monomer is an early indication of DIC.
Table 15-1 Disseminated Intravascular Coagulation Blood Tests
| Test | Result |
| Bleeding time | Prolonged |
| Platelet count | Decreased |
| Prothrombin time | Prolonged |
| Activated partial thromboplastin time | Prolonged |
| Coagulation factors | I, II, V, VIII, X, and XIII decreased |
| Fibrin degradation products | Increased |
| Red blood smear | Damaged red blood cells |
| Euglobulin lysis time | Normal or prolonged |
| D-dimer | Increased |
| Thrombin time | Prolonged |
| Fibrinopeptide A | Increased |
| Prothrombin fragment | Increased |
Hyperleukocytosis
Clinical Signs and Symptoms
Patients with hyperleukocytosis often have clinical complications from clumping of the leukemia cells in the lungs and brain,27 with resultant pulmonary sequestration or cerebral thromboembolic events. Signs and symptoms of pulmonary sequestration include hypoxia, restlessness, agitation, shortness of breath, tachypnea, cyanosis, decreased lung sounds, and increased work of breathing. Respiratory distress will persist despite oxygen administration.
Management
Perform frequent pulmonary and neurologic assessment to detect early respiratory or neurologic compromise. Evaluate oxygenation and respiratory effort, and evaluate the patient’s ability to follow commands, pupil size, and response to light. Next, immediately investigate any change in level of consciousness or headache (see Increased Intracranial Pressure in Chapter 11). In addition, monitor systemic perfusion and fluid and electrolyte balance.
Acute Tumor Lysis Syndrome
Etiology
Acute tumor lysis syndrome (ATLS) is a metabolic condition that can occur as a result of rapidly proliferating malignancies, such as Burkitt’s lymphoma, or following induction chemotherapy for malignancies that have a large tumor burden, leukemia with high WBC count, and tumors that respond rapidly to chemotherapy. ATLS can occur immediately after therapy, but most often will be observed 2 to 3 days after initiating treatment. The syndrome typically lasts approximately 7 days.4
Pathophysiology
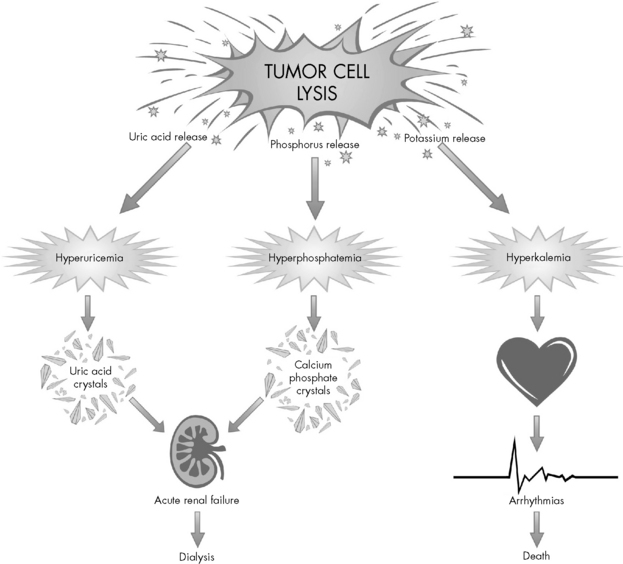
Destroyed cells release a large amount of potassium, producing hyperkalemia that can rapidly worsen with the development of renal failure (Fig. 15-3). Hyperkalemia is one of the most dangerous electrolyte imbalances, and fatal arrhythmias may occur with potassium levels greater than 7.0 mEq/L. Hyperphosphatemia can cause the formation of calcium phosphate crystals. Similar to uric acid crystals, calcium phosphate crystals can also obstruct the renal tubules compromising renal function. The precipitation of calcium phosphate crystals also may cause a secondary hypocalcemia.
Management
For asymptomatic hyperkalemia, treatment includes diuretics such as furosemide (Lasix) or mannitol or the administration of sodium polystyrene sulfonate (Kayexalate) orally or rectally. The rectal route (i.e., enema) will reduce the serum potassium faster than oral administration, but the oral route will result in a greater reduction over several hours.12 Symptomatic hyperkalemia requires more aggressive interventions such as calcium, sodium bicarbonate, and insulin and glucose infusion, which shift potassium back into cells. Other interventions for symptomatic hyperkalemia can include peritoneal dialysis, hemodialysis, and continuous venovenous hemofiltration. For additional information about hyperkalemia, see Chapter 12; for additional information about renal failure, see Chapter 13.
Hyperphosphatemia does not produce symptoms, but it will produce hypocalcemia and related symptoms. The phosphate combines with calcium and forms a precipitate in tissues. The serum ionized calcium is considered a more sensitive and reliable indicator of effective calcium concentration than the total calcium concentration.20 Signs of hypocalcemia include: a positive Chvostek’s sign (spasm of facial muscle following a tap over facial nerve, seen in tetany), a positive Trousseau sign (carpal-pedal spasms resulting from compression of an extremity artery, indicative of latent tetany), and prolongation of the Q-T interval on the electrocardiogram. Correction of the hypocalcemia is usually contraindicated unless the patient is symptomatic with the neuromuscular irritability (e.g., seizures, positive Chvostek or Trousseau signs).
Hypercalcemia
Etiology
Severe hypercalcemia is defined as a total calcium greater than 15 mg/dL. Total calcium concentration above this level may be life threatening.14 Hypercalcemia is associated with malignancies such as acute lymphoblastic leukemia, lymphomas, some soft tissue sarcomas, renal cell carcinoma, and lung and breast cancer. It is less common in pediatric than in adult oncology patients.
Pathophysiology
Mild hypercalcemia (total calcium less than 15 mg/dL) is generally not life threatening; however, significant elevations in calcium levels (greater than 15 mg/dL) can produce cardiovascular and renal complications. Hypercalcemia develops when a parathormone-like substance is released from malignant cells. This substance stimulates bone reabsorption and the release of calcium. Hypercalcemia can also result from malignancies causing bone destruction and from decreased renal calcium excretion.16,20,26
Management
With life-threatening hypercalcemia, more aggressive therapy is needed to reduce the serum calcium. Biphosphonate inhibits bone reabsorption, calcitonin inhibits calcium reabsorption from bone, mithracin reduces osteoclast activity, and gallium nitrate interferes with osteoclast function (Table 15-2).14 These drugs typically reduce the serum calcium concentration over several days.
| Drug | Description and Action | Dose* |
| Pamidronate | Second-generation biphosphate inhibits bone reabsorption; effective in malignancy-induced hypercalcemia | 1 mg/kg, may be repeated once in 24 h |
| Calcitonin | Hormone that inhibits calcium reabsorption from bone | 4 IU/kg body weight, given subcutaneously or intramuscularly |
| Plicamycin (Mithramycin) | Osteoclast RNA synthesis inhibitor that reduces osteoclast activity | Short infusion of 25 mcg/kg, given over 30 min or longer; dose may be repeated after 48 h |
| Gallium nitrate | Hydrated gallium salt that interferes with osteoclast function | Continuous infusion of 200 mg/m2 body surface area per day for a total of 5 days |
* Pamidronate dose from Kerdudo C and others: Hypercalcemia and childhood cancer: a 7-year experience. J Pediatr Hematol Oncol 27:23-27, 2005. Remaining doses from National Cancer Institute: Hypercalcemia (PDQ). Available at: http://www.cancer.gov/cancerinfo/pdq/supportivecare/hypercalcemia. Accessed January 4, 2012.
Neutropenia
Etiology and Pathophysiology
Neutropenia is defined as an absolute neutrophil count of less than 1000/mm3 in infants younger than 1 year and 1500/mm3 in children older than 1 year.13 The ANC is the total number of WBCs multiplied by the percentage of neutrophils (segmented neutrophils or segs plus bands). (For calculation, see Box 15-1.) The National Cancer Institute neutropenia grading system developed a classification scale for neutropenia (Box 15-5). The risk of serious bacterial infection increases dramatically when the ANC is less than 500/mm3.
Box 15-5 National Cancer Institute Neutropenia Grading System
In addition to the classification of severity, neutropenia can be further classified as acquired or congenital; acquired neutropenia is more common than the congenital form. Acute acquired neutropenia can be caused by an acute transient neutropenia or infections. Viral infections that may cause acquired neutropenia include respiratory syncytial virus, hepatitis A and B, Epstein-Barr virus, measles, rubella, and varicella. Bacterial infections that can cause acquired neutropenia include typhoid, paratyphoid, tuberculosis, and rickettsia infection.13 Acquired chronic neutropenia can be caused by bone marrow aplasia, bone marrow infiltration, or treatment such as chemotherapy, radiation therapy, or immunosuppressive medications. Bone marrow infiltration leading to neutropenia can result from diseases such as neuroblastoma, lymphoma, or rhabdomyosarcoma.
Management
Treatment of neutropenia is primarily supportive with a focus on evaluating risk for infection and on detecting and treating infection. Patients may be treated with colony-stimulating factors such as granulocyte colony-stimulating factor (GCSF; Neupogen [Amgen]) that stimulate the bone marrow to produce more neutrophils.13
Fever, defined as an oral temperature greater than 38° C, in the patient with neutropenia should be treated as a potential medical emergency because the patient may develop septic shock.9 After cultures are obtained, administer broad-spectrum antibiotics while waiting for culture results; administer antibiotics within 1 hour of the child’s first medical contact. Dosing is often adjusted based on reported culture and sensitivity results. Monitor the child’s vital signs and appearance to detect clinical signs of sepsis or septic shock, such as hyperthermia, hypothermia, tachycardia, hypotension, and changes in perfusion. Note that children with gram-negative bacteremia may deteriorate after antibiotic administration, because lysis of the gram-negative bacteria results in endotoxemia that can perpetuate the septic cascade.
Treatment of septic shock requires immediate antibiotics, aggressive fluid resuscitation, and hemodynamic support with vasoactive agents. Survival from septic shock has increased dramatically in recent years following appreciation of the need for repeated fluid bolus therapy (often totaling 80 mL/kg or more in the first hour of therapy and 240 mL/kg or more in the first 8 h of therapy) and early vasoactive support. For further information, see Chapter 6, and Fig. 6-8.
Spinal Cord Compression
Clinical Signs and Symptoms
A preliminary spine radiograph is the often the first and most convenient diagnostic test, but it is not definitive. To image the soft tissues involved, a CT scan is definitive, and an MRI study can further delineate the size and location of the tumor. If the patient with suspected spinal cord compression has possible increased intracranial pressure, such as caused by brain tumors with spinal (drop) metastases, then a CT scan should be obtained to rule out increased intracranial pressure before a lumbar puncture is performed to evaluate the cerebral spinal fluid. CSF protein will be elevated in patients with complete spinal cord obstruction.22
Obstructive Mediastinal Mass
Etiology
A mediastinal mass is a tumor that may be growing rapidly and thus causing respiratory compromise and airway obstruction. Some mediastinal masses also produce superior vena cava (SVC) obstruction. Childhood cancers most often causing mediastinal mass are non-Hodgkin’s lymphoma, Burkitt’s lymphoma, T cell lymphoblastic lymphoma or leukemia, neuroblastoma, and sarcomas.22 The most common cause of mediastinal mass in pediatric oncology is non-Hodgkin’s lymphoma.
Clinical Signs and Symptoms
A chest radiograph will usually reveal a large mass in the anterior mediastinum and possible tracheal deviation. Be extremely careful during positioning of the child for diagnostic procedures, because the supine position will often exacerbate tumor compression of the airways and increase respiratory distress. A CT or MRI scan may be useful in determining the extent of tracheal compression, but the patient’s condition may be too unstable to tolerate the transport to the radiology department or the time or positioning required for the studies (see Fig. 10-24).
Management
Patients with a mediastinal mass require immediate treatment and may deteriorate rapidly as treatment is provided. The goal of treatment is to prevent further respiratory deterioration; the basic principle of management is to keep the patient breathing spontaneously if possible. Emergent tracheal intubation may be required, but will not maintain the airway if the mass compresses the trachea distal to the end of the tube. If such distal compression is present, the airway can be maintained by rigid bronchoscopy. For severe distal airway compression, rapid initiation of extracorporeal membrane oxygenation (ECMO) support may be required (see Chapter 7).
Syndrome of Inappropriate Antidiuretic Hormone
Patients with SIADH usually exhibit hyponatremia, a decreased urine output, increased urine specific gravity with high urine sodium concentration, adequate intravascular volume, and possible increased weight without noticeable edema. Worrisome signs and symptoms include deterioration in neurologic function and seizures. Management includes fluid restriction, careful monitoring of fluid intake and output and correction of serum electrolytes, and treatment of the underlying cause. See Chapter 12 for more information.
Anaphylaxis
Anaphylaxis is an allergic hypersensitivity reaction to a foreign protein or drug that causes a systemic response. This reaction is presented in more detail in the Chapter 15 Supplement on the Evolve Website and in Chapters 6 and 16.
For patients with a known allergy or hypersensitivity reaction, premedications can be prescribed before the exposure to the potential allergen. Medications commonly used for pretreatment are corticosteroids, antihistamines, and antipyretics.6 Patients with known hypersensitivity responses should wear a medical alert jewelry and should have an epinephrine pen readily available.
Hematopoietic Stem Cell Transplantation
Preparation and Procedure
The preparative regimen consists of near-lethal doses of chemotherapy, radiation, or both. This regimen is designed to ablate the defective bone marrow and create space for new healthy cells to populate. In addition, it is an immunosuppressant to reduce the risk of graft rejection and to decrease the risk of graft-versus-host disease.17 The combination of the patient’s illness and conditioning regimen can produce severe complications that require critical care. These patients often experience severe myelosuppression and multisystem failure.
The third type of HSCT is an umbilical cord blood stem cell transplant obtained from a cord blood bank. A cord blood HSCT is also an allogeneic transplant. These stems cells are obtained in the delivery room after childbirth. Stem cells have only recently become available as a viable type of bone marrow transplantation.3
Management
The phases of the bone marrow transplant process are classified as early, immediate, and late. Complications can occur during any phase of the transplant process; see Table 15-3 for complications of the bone transplant process. These patients require numerous admissions to the critical care setting for management of their disease.
Table 15-3 Complications of Hematopoietic Stem Cell Transplant
| Early Complications (Pretransplant to Engraftment) | |
| Bone marrow suppression | All cell lines may be depressed; patient may be neutropenic, thrombocytopenic, and anemic; patient will require blood transfusions for support; lab values should be closely monitored |
| Gastrointestinal toxicities (nausea, vomiting, diarrhea, mucositis) | Gastrointestinal imbalances may result in critical electrolyte abnormalities; mucositis may compromise the airway and is extremely painful |
| Infections | Related to neutropenia and immunosuppressive agents; must be aggressively treated with appropriate therapy (i.e., antibacterial, antifungal, antiviral, antiprotozoan); can progress to life-threatening sepsis |
| Skin erythema | Radiation and chemotherapy may alter the skin integrity, resulting in vulnerability of the body’s protection |
| Capillary leak syndrome | Radiation and chemotherapy may cause tissue damage that results in cytokine release, which increases the permeability of cells; clinical symptoms may include systemic and pulmonary edema, fluid retention, and ascites |
| Acute renal insufficiency | Radiation, chemotherapy, and nephrotoxicity medications may result in decreased renal function; dialysis may be required if toxicity is severe |
| Hemorrhagic cystitis | The metabolite acrolein from cyclophosphamide-ifosfamide is a known irritant to the bladder and can cause acute bleeding; provide hydration and administer 2-mercaptoethane sufonate sodium (mesna) per protocol or physician orders |
| Venoocclusive disease | Toxicity to the liver caused by radiation, chemotherapy, and preexisting liver conditions can result in narrowing or fibrosis of the vessels |
| Seizures | Certain chemotherapy medications (e.g., busulfan) and electrolyte imbalances can predispose the patient to changes in neurologic status |
| Intermediate Complications (Engraftment to 100 Days) | |
| Infections | Related to immunosuppressive agents and compromised function of the new bone marrow; must be aggressively treated with appropriate therapy; can progress to life-threatening sepsis |
| Acute GVHD | An immune-mediated response of the donor T cells that attack the host antigens; the body systems affected are the skin, liver, and gut; initial presentation is a usually a maculopapular rash on the palms and soles that progresses to a generalized rash; diarrhea, abdominal pain, and abnormal liver function tests are other characteristics; degree of severity is based on organ involvement; skin or liver biopsy may be required to confirm diagnosis; treatment consists of immunosuppressive therapy and symptom management |
| Graft failure | Can result if the complete ablation is not achieved, stem dose is too low, infection occurs, or disease reoccurs; treatment can include an additional infusion of donor cells if available |
| Interstitial pneumonitis | Can result from infection or damage to the lung tissue from the preparative regime; respiratory status may be compromised; may require ventilatory support |
| Late Complications (>100 Days) | |
| Immunosuppression and infections | Immunosuppression is directly related to the extent of infections and the presence of GVHD; patients with GVHD will require additional immunosuppressive agents, which increases their risk for this complication |
| Chronic GVHD | This autoimmune syndrome is similar to a patient with collagen vascular disease; organs affected may include the skin, mouth, gastrointestinal tract, liver, lungs, eyes, and vaginal mucosa; treatment consists of immunosuppressive therapy; numerous experimental treatments are now available |
| Endocrine dysfunction | As a result of the preparative regimen, patients may experience thyroid dysfunction, growth and development delays, and gonadal dysfunction |
| Disease recurrence | Patients with aggressive disease before transplant may experience an increased rate of disease reoccurrence; poor prognosis if this occurs within 1-2 years following transplant |
| Secondary malignancies | Patients treated with specific cytotoxic drugs, radiation therapy, and immunosuppressants are at increased risk for developing secondary malignancies throughout their lifetime |
GVHD, Graft-versus-host disease.
Blood and blood component therapy
Pediatric critical care nurses must be familiar with indications for use of blood products, methods of administration, safety precautions, and appropriate volumes to administer. In addition nurses must be familiar with monitoring needed during transfusion (Table 15-4), and they must be able to detect and manage potential complications and adverse reactions.
| Blood Product, Dose | Clinical Indications | Nursing Interventions |
| Red blood cells (10-15 mL/kg) Acute massive blood loss (10-20 mL/kg) |
Hgb <7-8 g/dL in a stable patient with a chronic anemia Children requiring increased oxygen carrying capacity (i.e., complex congenital heart, intracardiac shunting, severe pulmonary disease-ARDS): • Shock states (decreased BP, increased peripheral vasoconstriction, pallor, cyanosis, diaphoresis, clamminess, mottled skin, increased oxygen requirement, decreased urine output) • Respiratory failure requiring significant ventilatory support Postoperative anemia |
Verify blood unit and patient
Use PPE
Monitor vital signs per hospital policy and procedure
Monitor Hgb and Hct
During infusion, observe for signs of adverse reactions
• Mild: fever or allergic reaction
• Severe: hemolysis (fever, chills, hemoglobinuria, DIC, renal failure)
Blood can only be stored in a designated blood refrigerator
For general surgical procedures the recommended Hgb is 10 g/dL or greater; for orthopedic surgery the recommended Hgb is 11.5 g/dL or greater.
Patient identification and administration process is the same as all other blood products
Use PPE
Hypovolemia from acute blood loss nonresponsive to crystalloids
• Hypovolemia from acute massive blood loss (i.e., major trauma)
• History of blood loss at delivery or large amount of blood drawn for lab studies (10% blood volume)
• Cardiac patients: Hct <40% (structural heart disease, cyanosis, or CHF)
Drop in Hgb to <10 g/dL intraoperatively
Exchange transfusion
Same nursing actions as for red blood cell infusions: verify unit and patient
For major trauma, may transfuse with O-negative blood (the universal donor) until crossmatch is complete
Use PPE
Use blood warmer and rapid infuser if available
During infusion, observe for signs of adverse reactions
Platelet pheresis (single donor unit, may be split for small infants and neonates)
Random donor (1-8 units per transfusion)
Active bleeding with symptoms of DIC or other significant coagulopathies
Platelet count <100,000/mm3 with planned invasive procedure (i.e., surgical procedure, central line insertion), not including drawing blood, intramuscular injection, or intravenous catheter insertion.
Prevention or treatment of bleeding associated with thrombocytopenia (secondary to chemotherapy, radiation, or bone marrow failure)
Treatment of patients with severe thrombocytopenia secondary to increased platelet destruction or immune thrombocytopenia associated with complication of severe trauma
Massive transfusion with platelet dilution
Verify platelet unit and patient
Use PPE
Monitor for allergic reaction
During infusion, observe for signs of adverse reactions
• Mild: fever or allergic reaction
• Severe: hemolysis (fever, chills, hemoglobinuria, DIC, renal failure)
Hemolysis may develop if sufficient incompatible plasma is present in transfusion
Bleeding, invasive procedure, or surgery with documented plasma clotting protein deficiency (i.e., liver failure, DIC, septic shock)
Prolonged PT and/or aPTT without bleeding
Significant intraoperative bleeding (>10% blood volume/hour) in excess of normally anticipated blood loss with high risk of clotting-factor deficiency
Massive transfusion
Therapeutic plasma exchanges
Warfarin anticoagulant overdose
Verify unit and patient
Use PPE
Monitor vital signs per hospital policy and procedure
Monitor coagulation studies
Observe for adverse reactions
Bleeding or prophylaxis in von Willebrand disease or in factor VIII deficiency (hemophilia A) unresponsive to or unsuitable for DDAVP or factor VII concentrates
Replacement therapy, bleeding, or invasive procedure in patients with factor XIII deficiency
Patients with active intraoperative hemorrhage in excess of normally anticipated blood loss who are at risk of clotting factor deficiency
Use appropriate PPE
Monitor vital signs per hospital policy and procedure
Monitor coagulation studies
Observe for adverse reactions during cryoprecipitate infusions
Infection (proven or strongly suspected) unresponsive to antimicrobial therapy
Verify unit and patient
Premedications may be ordered, such as antihistamines or acetaminophen
(90 units/kg)
Treatment of factor VIII inhibitors
Treatment of factor IX inhibitors
Idiopathic uncontrolled bleeding
Assess for signs and symptoms of bleeding
Use appropriate PPE, even with recombinant product
Monitor coagulation studies
If undiluted, dilute vial with indicated amount of sterile water and administer intravenously per manufacturer’s guidelines
Patient with factor VIII inhibitors
Patients with von Willebrand disease
Check product to determine whether refrigeration is necessary
Record expiration date and lot number of product
Assess for signs and symptoms of bleeding
Use appropriate PPE
Monitor coagulation studies
Hemophilia A with factor VIII inhibitors
Patients with congenital deficiency of prothrombin, factor VII, and factor X
Record expiration date and lot number of product
Assess for signs and symptoms of bleeding
Use appropriate PPE
Monitor coagulation studies
Immunologic disorders such as idiopathic thrombocytopenia, Kawasaki disease
Posttransplant patients (used prophylactically)
Newborns with severe bacterial infections
Record expiration date and lot number of product
Use appropriate PPE
Monitor vital signs per hospital policy and procedure
Start infusion slowly and increase rate; titrate per physician orders
During IVIG infusion, observe for adverse reactions such as fever, chills, and headache
ARDS, Acute respiratory distress syndrome; BP, blood pressure; CHF, congestive heart failure; DDAVP, l-deamino-8-arginine vasopressin; DIC, disseminated intravascular coagulation; FFP, fresh frozen plasma; Hct, hematocrit; Hgb, hemoglobin; IVIG, intravenous immunoglobulin; PPE, personal protective equipment; PRBC, packed red blood cell; WBC, white blood cell.
From Norville R, Bryant R: In Baggott CR, et al, editors: Blood component deficiencies in nursing care of the child with cancer, ed 3. Philadelphia, 2002, WB Saunders, Association of Pediatric Oncology Nurses.
Red Blood Cell Transfusion
A general threshold suggesting a need for RBCs transfusion in stable critically ill children is an Hgb concentration of 7 g/dL or less, typically associated with an Hct of 20% to 21% or less. This threshold was evaluated in stable children in critical care units11; different thresholds may be appropriate for children with severe hypoxemia (including those with cyanotic heart disease), hemodynamic instability, or active blood loss. Children with respiratory disease and cyanotic heart disease may require higher Hgb concentration; these children may require an Hgb concentration greater than 12 g/dL and Hct greater than 35% to maintain oxygen carrying capacity, although such thresholds are determined individually. Other thresholds may be appropriate for children with sickle cell anemia or noncyanotic heart defects.
Obtaining a Type and Crossmatch
Before a transfusion is needed, send a patient blood sample for type and crossmatch. The typing identifies the patient’s general blood type (A, B, O, or AB) and whether the patient’s blood type is Rh-negative or Rh-positive (Table 15-5). When blood is sent for typing and crossmatch, the patient’s blood is crossmatched with donor blood to determine compatibility. To reduce error, hospitals require labeling of the patient blood specimen that is sent for typing at the bedside, and placement of a blood bracelet containing the crossmatch number at the time the blood sample is obtained (Box 15-6). In addition, most facilities require a double check with the patient identification and two signatures to prevent fatal errors associated with incorrect blood typing.
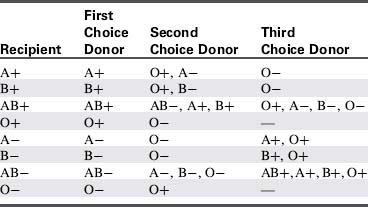
In general, to reduce Rh-sensitization in females, Rh-negative blood and blood products are ideally used for female patients of child-bearing age who are Rh-negative. Rh-positive blood and blood products are used for male patients and for female patients who are beyond child-bearing age. This attention to Rh sensitization can reduce the risk of Rh incompatibility during pregnancy.24 Patients who receive multiple transfusions can develop antibodies, and additional blood samples may be required to complete the necessary crossmatching.
Blood Product Administration
Blood should be administered only after verifying that the correct blood product is available and that the patient’s clinical condition is appropriate to receive a blood transfusion. The nurse must also complete the preassessment process for blood administration and should record baseline patient vital signs, including body temperature. Several clinical conditions may delay the administration of a blood product, including body temperature greater than 101° F (Box 15-7).
Box 15-7 Checklist for Blood Administration
Before the Transfusion
• Verify physician orders and consent for blood products.
• Obtain a complete set of vital signs including temperature.
• Verify whether there are any orders for pre-medications.
• Obtain blood product (i.e., blood bank, pharmacy, unit blood refrigerator). Check for special instructions such as autologous, leukocyte reduced, or irradiated.
• The transfusion must be started within 30 min after the blood has left the refrigerator.



• Follow hospital policy for obtaining, verifying, and transporting blood products obtained from sources outside the critical care unit.
• Ensure accurate patient identification—most commonly name and date of birth. Two appropriate healthcare team members must check all blood products at the patient’s bedside per institutional policy.
• Confirm R number (for red and white blood cells) with patient’s bracelet and blood product.
• Always use personal protective equipment, including eye shields and gloves.
During the Transfusion
• Never infuse any solutions (other than normal saline) through the same catheter with blood.1
• Never add any medications to blood.
• Monitor vital signs per the institution’s policy and procedures.
• Leave all identification information attached to the blood product until the transfusion is complete.
• Monitor for signs and symptoms of adverse reactions (see the discussion under Transfusion Reactions).
After the Transfusion
• Save the transfusion bag for at least 1 h after transfusion has ended, in the event of an adverse reaction.
• Complete the blood slip as indicated per institution’s required information.
• According to the 2006 American Association of Blood Banks guidelines,1 information to be included in the patient’s medical record must include the transfusion order, the type of blood product, the donor unit number, date and time of transfusion, vital signs before and after transfusion, the volume infused, required signatures, and any transfusion adverse events.
• Required documentation should be placed in the medical record and sent to the blood bank for record keeping per American Association of Blood Banks guidelines.
Platelet Transfusions
Indications for the administration of platelets to the patient with thrombocytopenia include bleeding or a platelet count of less than 10,000 to 20,000/mm3. 24 Prophylactic platelet transfusion is provided if the platelet count is less than 5000/mm3 to reduce the risk of spontaneous intracranial hemorrhage. Although published data indicate no difference in outcomes for patients with leukemia whose platelet transfusion thresholds were 10,000/mm3 or 20,000/mm3 in the absence of active bleeding, institutional practices vary widely based on local practice guidelines.28 In general, lower thresholds are used for platelet transfusion for children with conditions such as fever, central nervous system tumors, bleeding, or coagulopathies or those requiring procedures.2 According to the American Society of Clinical Oncology, patients can safely have “major invasive procedures” (such as bone marrow aspirate and biopsies) with platelet counts of 50,000/mm3,24 although the threshold varies among institutions.23,28 Platelet preparations are listed in Box 15-8.
Transfusion Reactions
Critical care nurses must be able to immediately recognize and respond to transfusion reactions. Most reactions occur during the first minutes of a transfusion, although they can occur at any time, including after completion of the transfusion. Reactions can vary from mild (e.g., febrile or allergic) to life-threatening with severe hemolysis, anaphylaxis, and death. Patients who have had multiple transfusions are at higher risk for developing a febrile transfusion reaction.10 Although general recommendations are provided in Box 15-9, nurses should follow institutional guidelines for management and documentation of transfusion reactions.
Box 15-9 Treatment of Transfusion Reaction
• Stop transfusion, keep vein open (administer intravenous fluids per order or protocol).
• Support airway, oxygenation, ventilation, and circulation if compromised.
• Notify physician; activate emergency response team, if indicated.
• Treat symptoms per physician order; prepare to administer diphenhydramine, hydrocortisone, and epinephrine if applicable.
• Recheck identification labels.
• Obtain blood and urine sample if indicated.
• Complete transfusion reaction documentation.
• Follow institutional policies for blood transfusion reactions.
Febrile Nonhemolytic Reaction
Febrile reactions can be caused by bacterial contamination of a unit of blood, because blood products provide an excellent medium for bacterial growth. Bacterial contamination is more common with platelets than other blood products because platelets are stored at room temperature, which allows more bacterial growth than refrigeration.24 Contamination of products can occur at any phase of the transfusion process, from collection to administration. Guidelines from the American Association of Blood Banks1 require strict adherence to the completion of all transfusions within 4 hours or less to reduce the risk of contamination and cell lysis. In addition, blood collection centers should function within strict guidelines for screening of potential donors and for collection and storage of blood products.
Anaphylaxis
In rare cases, an allergic transfusion reaction can progress to life-threatening anaphylaxis, a medical emergency. Patients with immunoglobulin A deficiency may be more susceptible to these reactions. A classic anaphylactic reaction has a sudden onset after administration of only a few milliliters of the blood product. The patient develops bronchospasm, shortness of breath, respiratory distress, and hypotension. The designated staff member at the bedside during the first 15 minutes of the transfusion should be prepared to rapidly identify and respond to these severe adverse reactions. In severe anaphylaxis, stop the transfusion and administer intravenous fluids per physician order or protocol); support airway, oxygenation, ventilation, and circulation as needed; evaluate vital signs; notify a physician; and activate the emergency response system, as appropriate. For further information, see Anaphylaxis earlier in this chapter and in Chapters 6 and 16.
Transfusion-Related Acute Lung Injury
Transfusion-related acute lung injury (TRALI) is a serious blood transfusion reaction characterized by acute onset of pulmonary edema, a similar clinical picture to acute respiratory distress syndrome. TRALI and clerical error causing mismatched blood transfusion reaction are the most common causes of transfusion-related deaths.5 TRALI occurs when there is an atypical antigen-antibody reaction caused by human leukocyte antibodies in the donated blood. These donor antibodies are transfused to the patient during the transfusion; they attach to the patient’s WBCs and form microaggregates. When the microaggregates circulate to the lungs, they trigger an inflammatory response that causes increased vascular permeability, pulmonary edema, and life-threatening respiratory failure.10
Patients experiencing TRALI can develop sudden signs of respiratory distress such as shortness of breath, hypoxia, hypotension, fever, and abnormal breath sounds. This reaction typically occurs within 1 to 2 hours after the transfusion has started, and full blown acute respiratory distress syndrome can develop within 6 hours.10 For mild cases of TRALI, the patient may respond to oxygen administration (perhaps with bilevel positive airway pressure) and diuretics. In severe cases, patients will also require intubation and mechanical ventilation with positive end-expiratory pressure. If TRALI is suspected, this transfusion reaction is reportable to the FDA.
Transfusions and Jehovah Witnesses
Jehovah Witnesses believe that blood transfusions—even in life-threatening situations—are forbidden by scripture. They also believe that if blood is removed from the body, it cannot be re-infused, such as with the cell-saver technology used in operating rooms. In addition, they believe that self-donation is also prohibited. Further information is available in the Chapter 15 Supplement on the Evolve Website.
If the child requires a life-saving transfusion, physicians can obtain a court order that will enable blood product administration. With the court order, decisions related to the administration of blood and blood products are made by a court-appointed guardian.15
Apheresis
The word apheresis is derived from Greek, meaning “to separate or remove.” The apheresis procedure involves removing blood from a donor or patient and separating its components. One or more of the components are selectively retained, and the remaining components are recombined and returned to the donor.18,19 Apheresis is often used in the critical care setting for patients with hematologic, oncologic, and other emergencies.
Intermittent flow centrifugation19 is a form of apheresis that uses a single intravenous access site. Intermittent flow centrifugation is performed in cycles (pull in and pull out): blood is removed with the assistance of a pump and placed in a centrifuge separator for component removal; the needed components are collected for storage, and the remaining product is returned to the patient through the intravenous access. Another method of apheresis is continuous flow centrifugation. In this method blood is withdrawn through one catheter, processed, and returned through a second venous catheter.
Erythrocytapheresis (Red Blood Cell Exchange)
The patient’s RBCs are removed from the patient’s blood and replaced by donor RBCs that are infused into the patient with the patient’s plasma, WBCs, and platelets. This treatment can be used for sickle cell anemia with acute crises, such as acute chest syndrome, cerebral vascular accident, or severe priapism. Other diseases that can benefit from an RBC exchange include erythrocytosis polycythemia vera and severe malaria.19
Adverse Effects of Apheresis
During the apheresis process, there is potential for adverse effects. Table 15-6 summarizes potential complications of apheresis.
| Complication | Nursing Intervention |
| Air embolism | Closely monitor connection sites and tubing Prime line with normal saline or compatible fluid |
| Citrate toxicity | Monitor for signs and symptoms (numbness and tingling around mouth) Decrease re-infusion rate to diminish symptoms |
| Hypocalcemia | Verify ionized calcium before pheresis, correct abnormalities Monitor ionized calcium concentration hourly during pheresis (or per physician orders) Consider calcium infusion if clinical condition indicates |
| Hypotension | Have fluids available in case of rapid onset of hypotension May need to increase rate of vasopressors |
| Hypothermia | Monitor body temperature frequently to prevent or promptly treat hypothermia Use blood warmer on pheresis machine Keep patient warm with blankets, external warmers, or both |
| Infection | Strict hand washing Maintain sterile technique with all invasive lines. |
| Risk for bleeding | Monitor coagulation profile before prior to pheresis therapy Platelet or other blood products such as clotting factors may be required during the procedure |
| Transfusion reaction | Use leukodepleted, irradiated blood products if indicated Monitor for transfusion reactions from the replacement products; follow the transfusion reaction protocol if this occurs Consider administration of an antihistamine for patients receiving multiple treatments |
| Thrombus | Obtain platelet count before catheter placement and be aware when possible transfusion of platelets is necessary Flush vigorously with normal saline per hospital policy |
Specific diseases
Sickle Cell Disease
Etiology
Sickle cell anemia is one of the most common genetic hematologic conditions in children. The disease is transmitted by an autosomal recessive pattern of inheritance; both parents are carriers of the sickle cell gene (For more information, see Evolve Fig. 15-4 in the Chapter 15 Supplement on the Evolve Website).13,27
Patients with sickle cell disease are homozygous for the sickle cell gene; this means that both genes are abnormal.27 Patients with sickle cell trait are heterozygous for the sickle cell gene; these patients are generally asymptomatic because they possess one sickle and one normal gene.
Pathophysiology
Sickled RBCs have reduced flexibility and tend to be “sticky” so that they will adhere more readily to the blood vessel walls and to each other, causing occlusion of small vessels and tissue ischemia. Sickled RBCs are more fragile (i.e., they readily hemolyze) so turnover of RBCs is more rapid, causing chronic anemia.19 These changes in RBC morphology compromise microvascular blood flow and tissue oxygenation that can lead to ischemia and infarcts, in a cycle of hypoxia and RBC sickling called a vasoocclusive crisis.
The definitive diagnostic test for sickle cell disease or trait is Hgb electrophoresis.19 This test measures the various types and percentages of Hgb present.
Clinical Signs and Symptoms
Acute Chest Syndrome
Acute chest syndrome can cause the sudden onset of respiratory distress, characterized by dyspnea, tachypnea, cough, fever, arterial oxygen desaturation (oxy-hemoglobin saturation less than 90%), hypoxemia, and confusion. The chest radiograph may show increased infiltrates and pleural effusion. Clinical signs may be difficult to distinguish from pneumonia. With acute chest syndrome, onset and deterioration may be more sudden, and the radiograph often reveals multilobar infiltrates. Mortality is high.27
Management
RBC exchange is used for severe crises, to reduce the concentration of Hgb S. Generally the goal is to reduce the concentration of Hgb S to less than 30%, although this goal is tailored to the patient and the patient’s clinical condition. Pain control is essential, ideally with a combination of both pharmacologic and nonpharmacologic therapies (see Chapter 5); intravenous narcotic administration may be needed. It is helpful to determine the pain control measures that have worked previously.
Pathophysiology
This group of bleeding disorders is characterized by a deficiency of either factor VIII or IX that results in the inability to form a clot at a site of injury. Hemophilia A, also known as classic hemophilia, is characterized by a deficiency of factor VIII. Hemophilia B, also known as Christmas disease, is a deficiency of factor IX.25 Children with hemophilia have prolonged bleeding.
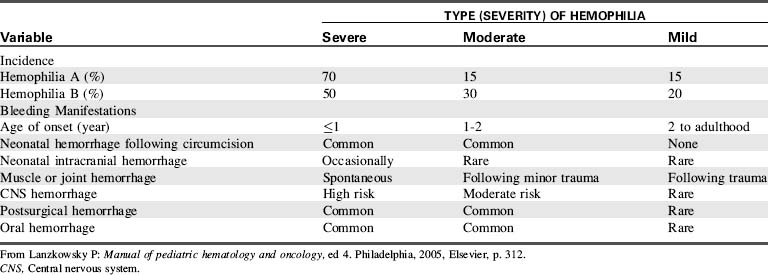
Hemophilia is classified as mild, moderate, or severe; the severity of the bleeding disorder is directly related to the degree of factor deficiency (Table 15-7). The child with a significant factor deficiency will experience more bleeding episodes than the child with mild deficiency. Bleeding is not faster in these patients; bleeding is prolonged and causes the clinical manifestations and risks of complications such as intracranial bleeding, hemorrhage, and hemarthrosis.
Table 15-7 Relationship of Factor Levels to Severity of Clinical Manifestations of Hemophilia A and B
| Type | Percentage of Factor VIII/IX (%) | Type of Hemorrhage |
| Severe | <1 | Spontaneous: hemarthroses and deep soft-tissue hemorrhages |
| Moderate | 1-5 | Gross bleeding following mild to moderate trauma; some hemarthroses; rare spontaneous hemorrhage |
| Mild | 5-25 | Severe hemorrhage only after moderate to severe trauma or surgery |
| High-risk carrier females | Variable | Gynecologic and obstetric hemorrhage common; other symptoms depend on plasma factor level |
From Lanzkowsky P: Manual of pediatric hematology and oncology, ed 4. Philadelphia, 2005, Elsevier, p. 311.
Clinical Signs and Symptoms
The most obvious sign of a bleeding disorder is persistent oozing, associated with small lacerations or minor procedures. Infant males may be diagnosed after prolonged bleeding after circumcision; the older child may exhibit bleeding after a dental procedure or tooth loss (Table 15-8). Other potential clinical signs and symptoms of factor deficiency include pain, swelling, bleeding into a joint after injury, bleeding from the mouth or nose, and menorrhagia. Life-threatening bleeding events for hemophiliacs include head injury, neck and throat bleeding, an acute abdomen, and bleeding into the iliopsoas muscle near the femoral artery.
Patients with bleeding disorders who develop any head, neck, or torso injury must be evaluated immediately, because bleeding in these areas can be life threatening. When treating life-threatening bleeding, the first and most important step is to stop the bleeding; the diagnostic analysis is completed after replacement factors are initiated.25
Common laboratory tests include aPTT and PT; both will be elevated (prolonged). The most important test is the direct assay of plasma factor activity level for hemophilia A and B.8
Management
Some patients with hemophilia may benefit from factor prophylaxis to prevent complications and bleeding episodes. Patient treatment plans are individualized, but typically call for infusions three times per week for patients with hemophilia A and infusions twice per week for patients with hemophilia B.25
Rarely, patients with hemophilia can develop inhibitors to coagulation proteins after receiving numerous (more than 20 to 30) doses of replacement factor products.8 You should suspect development of inhibitors in patients who have continued prolongation of the aPTT despite receiving 100% of corrective dose of factor products. If the patient develops inhibitors, changes in or additions to the treatment regimen may be necessary to provide adequate replacement factors.
Hemolytic Uremic Syndrome
Hemolytic uremic syndrome is an uncommon renal disease that is seen most often in children younger than 5 years. The causative agent is usually Escherichia coli O157:H7,27 and the dominant clinical feature of this disease is acute renal failure. These patients generally have a prodromal illness, such as acute gastroenteritis or an upper respiratory illness. Most cases of HUS present with bloody diarrhea and gastric symptoms. These patients have a history of consumption of undercooked meat, specifically ground beef. Other possible sources of E. coli infection include unpasteurized milk or juices, alfalfa sprouts, lettuce, and drinking or swimming in water contaminated with sewage.
The clinical signs of HUS itself can include the acute onset of purpura, irritability, lethargy, marked pallor, oliguria, hemolysis, and eventual renal failure. Diagnosis is usually confirmed by the clinical features of anemia, thrombocytopenia, and renal failure. The renal failure is characterized by an elevated blood urea nitrogen and creatinine, proteinuria, hematuria, and casts in urine. A CBC often reveals a decrease in Hgb and Hct and an increase in reticulocyte count, indicative of HUS. Management of HUS in young children may differ slightly than for older adolescents and adults. Generally, young children will recover with only hemodialysis support and minimal complications. The older adolescent and adult with HUS should be managed with apheresis (plasma exchange) and hemodialysis. Most patients with HUS recover without long-term sequelae, although a potential long term complication may be the development of chronic renal failure (for further information, see Chapter 13).
Common diagnostic tests
Lumbar Puncture
Procedure
The patient is positioned in the side with the head flexed (chin to chest) and the knees drawn; this position opens the intravertebral spaces. Alternatively, patients can be positioned sitting upright, with the buttocks placed on the edge of the procedure table with the head flexed (chin to chest). Nurses will need to assist the child in maintaining proper position (Fig. 15-4).
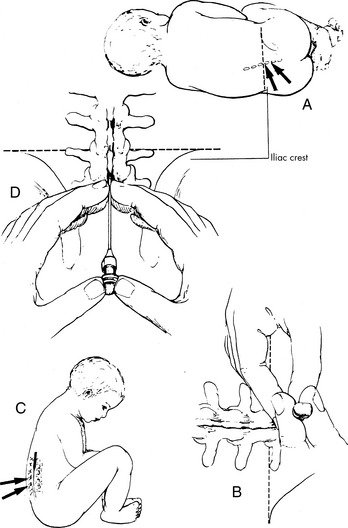
Often, pediatric patients will be sedated for this procedure (see Chapter 5). If the patient is not sedated, a topical anesthetic is applied to the puncture site and additional local anesthetic may be used.
Following sterile preparation and administration of local anesthetic, an appropriately sized spinal needle with stylet is inserted through the skin and into the intervertebral space at the midline, between the third and fourth or the fourth and fifth lumbar vertebrae. The spinal needle is then inserted through the dura and into the subarachnoid space. The stylet is removed and, if indicated, the opening CSF pressure is measured with a manometer. A pressure measurement of 20 cm of water or greater is considered abnormal and is suggestive of increased intracranial pressure.20 The appropriate collection tubes are placed under the needle to collect the cerebral spinal fluid. Providers should describe the color and clarity (or opacity) of the fluid in the chart. For cancer patients who require intrathecal therapy, the appropriate volume of CSF is removed and the chemotherapy is administered.
The CSF specimen is sent to the laboratory to determine WBC count, protein and glucose values (see Table 11-11) and, if appropriate, for cytologic examination for cancer cells and cultures for possible infections. CSF is examined for color and presence of blood. Normal CSF is clear and colorless.
Bone Marrow Aspirate and Biopsy
Procedure
Both of these procedures are performed under sterile conditions, and both use large bore needles that contain stylets. After the site is prepared and appropriate local or systemic analgesia is provided, the needle is inserted through the soft tissue of the skin, into the outer layer of the pelvic bone, and into the marrow cavity. The most common site used is the posterior superior iliac crest. Alternative collection sites include the anterior iliac crest, proximal tibia, and sternum. Once the needle is inside the marrow, the stylet is removed and a syringe is attached to obtain specimens. A portion of these specimens is often placed on slides for microscopic examination.20 When the procedure is complete, the bone marrow needle is removed and a dressing is applied. If the patient has a low platelet count, less than 50,000/mm3, then a pressure dressing is recommended.
1 American Association of Blood Banks Standards for Blood Banks and Transfusion Services. ed 24, Bethesda, MD; American Association of Blood Banks Press:2006
2 Beutler E. Platelet transfusions: the 20,000/µL trigger. Blood. 1993;81:1411-1413.
3 Bollard C.M., Krance R.A., Heslop H.E. Hematopoietic stem cell transplantation in pediatric oncology. In Pizzo PA, Poplack D.G., editors: Principles and practice of pediatric oncology, ed 5, Philadelphia: Lippincott Williams and Wilkins, 2006.
4 Cairo M.S., Bishop M. Tumor lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
5 Department of Health and Human Services: Transfusion related acute lung injury (TRALI). 2001 FDA Patient Safety Newsletter http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/BloodSafety/ucm095556.htm October
6 Gobel B. Chemotherapy induced hypersensitivity reactions. Oncol Nurs Forum. 2005;32:1027-1055.
7 Hastings C., Lubin B., Feusner J.: Hematologic supportive care for children with cancer, Chapter 40, Pizzo P.A., Poplack D.G., editors. Principles and practice of pediatric oncology, ed 5, Philadelphia: Lippincott Williams and Wilkins, 2006.
8 Hillman R.S., Ault K.A., Leporrier M., Rinder H.M. Hematology in clinical practice, ed 4, New York: McGraw Hill, 2005.
9 Khilnani P. Practical approach to pediatric intensive care. New York: Oxford University Press; 2005.
10 Knippen M. Transfusion-related acute lung injury. Am J Nurs. 2006;6:61-64.
11 Lacroix J., et al. Transfusion strategies for patients in pediatric intensive care units. N Engl J Med. 2007;356:1609-1619.
12 Lacy C.F., et al, editors. Lexi-Comp’s drug information handbook: a comprehensive resource for clinicians and all healthcare professionals, ed 18, Hudson, OH: Lexi-Comp, 2009.
13 Lanzkowsky P. Manual of pediatric hematology and oncology, ed 4. Philadelphia: Elsevier; 2005.
14 Leyland-Jones B. Treating cancer-related Hypercalcemia with gallium nitrate. J Support Oncol. 2004;2:509-520.
15 Linnard-Palmar L., Kools S. Parents’ refusal of medical treatment based on religious and/or cultural beliefs: the law, ethical principles and clinical implications. J Pediatr Nurs. 2004;19:351-356.
16 National Cancer Institute: Hypercalcemia (PDQ). 2009 Available at http://www.cancer.gov/cancerinfo/pdq/supportivecare/hypercalcemia Accessed January 21
17 Norville R. Hematopoietic stem cell transplantation in essentials of pediatric hematology/oncology nursing: a core curriculum, ed 3. Glenview, IL: APON Press; 2008.
18 Norville R., Bryant R. Blood component deficiencies. In Baggott C.R., et al, editors: Nursing care of the child with cancer, ed 3, Philadelphia: Association of Pediatric Oncology Nurses; WB Saunders, 2002.
19 Orkin S., et al. Nathan and Oski’s hematology of infancy and childhood, ed 7. Philadelphia: Saunders-Elsevier; 2009.
20 Pagana K., Pagana T. Mosby’s manual of diagnostic and laboratory tests. St Louis: Mosby-Elsevier; 2006.
21 Peterec S.M., et al. Reticulated platelet values in normal and thrombocytopenic neonates. J Pediatr. 1996;129:269-274.
22 Rheingold S., Lange B. Oncologic emergencies. In Pizzo P.A., Poplack D.G., editors: Principles and practice of pediatric oncology, ed 5, Philadelphia: Lippincott Williams and Wilkins, 2006.
23 Rogers C. Supportive care in essentials of pediatric hematology/oncology nursing: a core curriculum, ed 3. Glenview, IL: APON Press; 2008.
24 Schiffer C., et al. Platelet transfusion for patients with cancer: clinical practice guidelines of the American Society of Clinical Oncology. J Clin Oncol. 2001;19:1519-1538.
25 Sevier N. Inherited coagulation factor abnormalities: a pediatric review. J Pediatr Oncol Nurs. 2005;22:137-144.
26 Smith M. Bisphosphonates. In: Chabner B., Longo D., editors. Cancer chemotherapy and biotherapy: principles and practice. Philadelphia: Lippincott Williams and Wilkins, 2006.
27 Turgeon M.L. Clinical hematology: theory and procedures, ed 4. Philadelphia: Lippincott Williams and Wilkins; 2005.
28 Wong E.C.C., et al. Transfusion management strategies: a survey of practicing pediatric hematology/oncology specialists. Pediatr Blood Cancer. 2005;44:118-127.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
15 Hematologic and Oncologic Emergencies Requiring Critical Care
Pearls
• Severe anemia associated with a hemoglobin (Hgb) concentration less than 5 g/dL and a hematocrit (Hct) less than 15% is likely to produce congestive heart failure (CHF). Transfuse packed red blood cells (PRBCs) slowly (3 mL/kg per hour).
• Complete bone marrow failure produces a fall in Hgb of approximately 0.7-1 g/dL per day. A more precipitous fall in Hgb is likely to be caused by bleeding or increased red blood cell (RBC) destruction.
• The risk of spontaneous bleeding is increased if the platelet count is less than 20,000/mm3 and intracranial hemorrhage is more likely once the platelet count is less than 5000/mm3.
• Rule out intracranial hemorrhage in any child with severe thrombocytopenia and unilateral headache. If hemorrhage is suspected, you will often need to begin to stabilize first; obtain scans and radiographs later.
• Acute tumor lysis syndrome produces hypocalcemia, hyperkalemia, hyperuricemia, and hyperphosphatemia. Disseminated intravascular coagulation (DIC) is also possible.
• An absolute neutrophil count less than 500/mm3 is associated with a significant risk of infection. Monitor these patients closely for evidence of infection.
• Whenever blood products are administered, label the crossmatch specimen carefully at the bedside and double-check patient and blood product identification (an extra time).
Essential anatomy and physiology
Blood Components
Blood is the fluid that sustains life. This highly specialized body fluid has many functions, including transporting oxygen and nutrition, eliminating waste, acid-base buffering, and maintaining homeostasis. The hematopoietic system consists of the bone marrow, liver, spleen, lymph nodes, and thymus gland.27 Each of these components plays a specific role in the regulation of blood.
Blood is composed of a liquid phase called plasma and a formed or cellular phase. Approximately 90% of plasma consists of water, and the remaining portions are solutes including factors that form clots (e.g., clotting factors, fibrinogen, globulins) and serum that contains electrolytes, hormones, antibodies, nutrients, and other factors. The cellular portion of the blood consists of the formed elements: RBCs, white blood cells (WBCs), and platelets (for an illustration, refer to Evolve Fig. 15-1 in the Chapter 15 Supplement on the Evolve Website).
The body’s first hematopoietic stem cell is produced in the yolk sac of the embryo. The fetal liver becomes the site for hematopoiesis at approximately the second month of fetal life. The liver is the main organ producing blood cells from the second to the fifth month of fetal life.27 At approximately the fourth month, the bone marrow begins producing blood cells and remains the production site throughout life. Bone marrow, one of the largest organs in the body, is located in the cavities of all bones.
Hematopoiesis consists of the production, differentiation, and development of blood cells; it normally occurs in the bone marrow.27 RBCs, WBCs, and platelets are all thought to arise from multipotential stem cells that inhabit the bone marrow. These stem cells have the ability to differentiate into any of the three cell lines, based on the needs of the body. The process of cell differentiation by the pluripotent stem cell is normally (i.e., in the absence of disease) self regulatory. In children, all bones produce blood cells. When bone growth ceases, often by 18 years old, only the ribs, sternum, vertebrae, and pelvis continue to produce blood cells.27
WBCs, also called leukocytes, defend the body against infectious agents. Their main function is to fight infection by migrating to the site of inflammation to assist in defending the body against foreign antigens. There are several different types of WBCs, each with a specific purpose. The differential of a complete blood cell count (CBC) reveals the percentages of neutrophils, lymphocytes, monocytes, and eosinophils. The numeric value of the WBCs is not as important as an evaluation of the absolute neutrophil count (ANC), a more accurate measure of the body’s ability to fight infection (see Neutrophils below and formula for ANC calculation in Box 15-1).
Neutrophils provide the primary defense against bacterial infections, because they engulf and destroy invading organisms. The ANC is determined from a WBC differential by multiplying the percent of neutrophils plus bands by the total WBC count (Box 15-1 shows an example of ANC calculation). In general, children with an ANC of less than 500/mm3 have a high risk of developing life-threatening bacterial infections. The risk of infection is particularly high in patients with neutropenia as the result of failure of neutrophil production, rather than those with immune-mediated neutropenia.
Clotting Cascade
Normal hemostasis is maintained by a complex balance of procoagulant and anticoagulant factors. Together, these factors provide rapid and localized control of bleeding at sites of injury while preventing the clotting process in unaffected tissues. A simplified scheme of the coagulation process is shown in Evolve Fig. 15-2 in the Chapter 15 Supplement on the Evolve Website.
Common clinical conditions
Acute Anemia
Etiology
Anemia is present when the number of circulating RBCs is reduced, resulting in decreased oxygen-carrying capacity of the blood. Anemia is the most common hematologic condition of infancy and childhood. Most patients with anemia have an underlying disease, so anemia is a symptom rather than a disease. Causes of anemia include acute or chronic blood loss, decreased production of RBCs, splenic sequestration, and hemolysis (Box 15-2).
Box 15-2 Etiology of Severe Anemia
Decreased Production
• Bone marrow replacement (leukemia, other malignancies)
• Bone marrow failure (aplastic anemia)
• Bone marrow suppression (chemotherapy, radiation)
• Transient erythroblastopenia of childhood
• Congenital red cell aplasia (Diamond-Blackfan syndrome)
• Aplastic crisis (sickle cell anemia, hemoglobin sickle cell disease, hereditary spherocytosis)
Pathophysiology
Blood Loss
When a patient experiences acute blood loss, the Hgb and Hct are not initially affected because both plasma and RBCs have been lost. If only crystalloid and colloids are used (without blood) for fluid resuscitation, then the Hct will fall because whole blood loss has been replaced with fluid that does not contain RBCs (Fig. 15-1). The patient may remain anemic until blood is administered.
Decreased RBC Production
When complete bone marrow failure is present, the Hgb will fall approximately 0.7 to 1 g/dL per day7; this usually produces a chronic rather than an acute anemia. If the Hgb or Hct falls precipitously (greater than 1 g/dL per day) in the patient with bone marrow suppression, then hemorrhage or increased RBC destruction is probably present.
Thrombocytopenia
Etiology and Pathophysiology
Patients with thrombocytopenia have a decrease in circulating platelets. This condition can occur as a result of decreased production of platelets, increased destruction of platelets, or sequestration of platelets in the spleen or liver (Box 15-3). The cause of the thrombocytopenia affects the treatment.
Box 15-3 Most Common Causes of Thrombocytopenia
Decreased Production
• Bone marrow failure (aplastic anemia)
• Bone marrow replacement (leukemia, other malignancies)
• Bone marrow suppression (chemicals, insecticides, chemotherapy, radiation therapy, chloramphenicol, alcohol)
• Hereditary thrombocytopenia (Alport’s syndrome, Fanconi syndrome, Wiskott-Aldrich syndrome)
Increased Destruction
From Turgeon ML: Clinical hematology: theory and procedures, ed 4. Philadelphia, 2005, Lippincott Williams and Wilkins.
Clinical Signs and Symptoms
Reticulated platelets (RPs) are newly synthesized platelets with increased ribonucleic acid content. These new platelets can be identified and quantified, and the RP value can aid in determining the cause of the thrombocytopenia. A decrease in RP value in the patient with thrombocytopenia suggests bone marrow suppression (i.e., platelets are not being made despite a fall in platelet count), whereas a high RP value in a patient with thrombocytopenia suggests immune-mediated thrombocytopenia (i.e., platelets are being made but are not correcting the thrombocytopenia).21
Management
Prophylactic platelet transfusion is provided if the platelet count is less than 10,000/mm3, to reduce the risk of spontaneous intracranial hemorrhage. Although published data indicate no difference in outcomes for patients with leukemia when platelet transfusion thresholds were 10,000/mm3 or 20,000/mm3 in the absence of active bleeding, institutional practices vary widely based on local practice guidelines.28 In general, lower thresholds are used for platelet transfusion for children with conditions such as fever, central nervous system tumors, bleeding, or coagulopathies or those requiring procedures.2 Patients can safely have “major invasive procedures” such as bone marrow aspirate and biopsies with platelet counts of 50,000/mm3; the thresholds can vary slightly among institutions.23,24,28
Patients with immune-mediated platelet destruction can be treated with high-dose steroids (prednisone, 4 to 8 mg/kg per day), intravenous immune gamma globulin (0.5 to 1 g/kg of immunoglobulin G), or both. Children with exceptionally low platelet counts and acute bleeding require inpatient hospitalization with close observation. Bone marrow aspirate is recommended before initiating high-dose methylprednisolone (30 to 50 mg/kg per day) to treat severe bleeding. These children may also receive a 2- or 3-day course of intravenous immune gamma globulin.13 Frequent monitoring of laboratory tests may be necessary until the platelet count improves. Monitor the patient closely for signs of bleeding.
Disseminated Intravascular Coagulation
Etiology
Disseminated intravascular coagulation is characterized by the intravascular consumption of platelets and plasma clotting factors.13 DIC is not a primary disease but a complication of other disease processes.27 DIC can complicate sepsis, hypoxemia, major trauma with severe tissue injury, malignancy, thrombotic thrombocytopenic purpura, hemolytic uremic syndrome (HUS), extensive burns, and severe viral infections (Box 15-4).
Pathophysiology and Clinical Signs and Symptoms
DIC is characterized by an abnormal coagulation process—the entire clotting mechanism is triggered inappropriately. Unrestrained clotting causes systemic or local formation of fibrin clots. This development of fibrin clots leads to microthrombi and excessive bleeding secondary to the consumption of the platelets and clotting factors. The fibrin clot development triggers the clotting cascade process to break down the clots and fibrin that have been formed. Fibrinolysis results in the development of fibrin degradation products, which in turn will act as anticoagulants and promote more bleeding. Organ injury may be the end result of intravascular clots, which cause microvascular occlusions and anoxia.20 In addition, when the RBCs flow through obstructed vessels the end result is additional hemolysis that can lead to hemolytic anemia (Fig. 15-2).
Fig. 15-2 Pathophysiology of disseminated intravascular coagulation.
DIC, Disseminated intravascular coagulation; FDP, fibrin degradation products.
(From Pagana K, Pagana T: Mosby’s manual of diagnostic and laboratory tests, St Louis, 2006, Mosby-Elsevier.)
No single test can be used to diagnose DIC. Laboratory abnormalities consistent with the diagnosis of DIC include thrombocytopenia, prolonged PT or activated partial thromboplastin time (aPTT), decreased fibrinogen, elevated fibrin degradation products (fibrin split products), and anemia (Table 15-1). The presence of fibrin monomer indicates that fibrinogen is being broken down by thrombin and that clot is being formed. A rise in fibrin monomer is an early indication of DIC.
Table 15-1 Disseminated Intravascular Coagulation Blood Tests
| Test | Result |
| Bleeding time | Prolonged |
| Platelet count | Decreased |
| Prothrombin time | Prolonged |
| Activated partial thromboplastin time | Prolonged |
| Coagulation factors | I, II, V, VIII, X, and XIII decreased |
| Fibrin degradation products | Increased |
| Red blood smear | Damaged red blood cells |
| Euglobulin lysis time | Normal or prolonged |
| D-dimer | Increased |
| Thrombin time | Prolonged |
| Fibrinopeptide A | Increased |
| Prothrombin fragment | Increased |
Hyperleukocytosis
Clinical Signs and Symptoms
Patients with hyperleukocytosis often have clinical complications from clumping of the leukemia cells in the lungs and brain,27 with resultant pulmonary sequestration or cerebral thromboembolic events. Signs and symptoms of pulmonary sequestration include hypoxia, restlessness, agitation, shortness of breath, tachypnea, cyanosis, decreased lung sounds, and increased work of breathing. Respiratory distress will persist despite oxygen administration.
Management
Perform frequent pulmonary and neurologic assessment to detect early respiratory or neurologic compromise. Evaluate oxygenation and respiratory effort, and evaluate the patient’s ability to follow commands, pupil size, and response to light. Next, immediately investigate any change in level of consciousness or headache (see Increased Intracranial Pressure in Chapter 11). In addition, monitor systemic perfusion and fluid and electrolyte balance.
Acute Tumor Lysis Syndrome
Etiology
Acute tumor lysis syndrome (ATLS) is a metabolic condition that can occur as a result of rapidly proliferating malignancies, such as Burkitt’s lymphoma, or following induction chemotherapy for malignancies that have a large tumor burden, leukemia with high WBC count, and tumors that respond rapidly to chemotherapy. ATLS can occur immediately after therapy, but most often will be observed 2 to 3 days after initiating treatment. The syndrome typically lasts approximately 7 days.4
Pathophysiology
Destroyed cells release a large amount of potassium, producing hyperkalemia that can rapidly worsen with the development of renal failure (Fig. 15-3). Hyperkalemia is one of the most dangerous electrolyte imbalances, and fatal arrhythmias may occur with potassium levels greater than 7.0 mEq/L. Hyperphosphatemia can cause the formation of calcium phosphate crystals. Similar to uric acid crystals, calcium phosphate crystals can also obstruct the renal tubules compromising renal function. The precipitation of calcium phosphate crystals also may cause a secondary hypocalcemia.
