[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 50 Helicobacter pylori
Helicobacter pylori are unique bacteria ideally suited to live in the acidic environment of the human stomach. Their spiral shape and multiple unipolar flagella allow them to move freely through the gastric mucous layer, where they remain protected from low gastric pH.1 Organisms produce large amounts of urease, an enzyme that hydrolyzes urea to alkaline ammonia and CO2. This permits the bacteria to further control the pH of their microenvironment. Urease is also the basis of clinical diagnostic tests (urea breath test and rapid urea biopsy tests) for infection. H. pylori remain difficult and tedious to culture because they grow slowly and require specialized culture media and a controlled microaerophilic environment.
When they gain access to a human host, H. pylori recognize and attach to various gastric epithelial surface receptors, thereby chronically colonizing the mucosa, disrupting cell function, inciting an intense local inflammatory and systemic immune response, and altering acid secretory physiology.2,3 The ultimate clinical manifestations of H. pylori infection include gastric and duodenal ulcer, gastric mucosa–associated lymphoid tissue (MALT) lymphoma, and adenocarcinoma; yet most infected individuals remain asymptomatic for life despite developing chronic histologic gastritis.1,2,4,5 What factors determine why some develop disease in response to infection and others do not remains a mystery, but host genetics, bacterial characteristics, and environmental features undoubtedly can influence clinical outcome.5,6 Research focusing on epidemiology, pathogenesis, management, and prevention of H. pylori infection and associated clinical conditions continues to be fueled by the tremendous worldwide prevalence of infection (especially in less-developed countries), the huge health and economic burden imposed by ulcer disease and gastric cancer, and heightened prominence and awareness of the bacteria accompanying Drs. Robin Warren and Barry Marshall’s receipt of the 2005 Nobel Prize in Physiology or Medicine for seminal contributions to the field.7
EPIDEMIOLOGY
Helicobacter pylori infection remains one of the most common chronic bacterial infections in humans. Estimates suggest that more than 50% of the world’s population is infected with the bacterium and genetic sequence analysis proposes that humans have been infected for more than 58,000 years at a time when they first migrated from Africa.8
While H. pylori have been demonstrated worldwide in individuals of all ages, infection is more common and acquired at an earlier age in developing countries compared with industrialized nations.9,10 In developing nations, the majority of children become infected before the age of 10, and during early childhood spontaneous elimination of bacteria and subsequent reinfection is quite common. Infection persists in older children and adults so that in the developing areas of the world H. pylori prevalence can reach more than 80% by age 50. In developed countries, such as the United States, young children can also acquire H. pylori, but usually before age 5.11 Spontaneous clearance often occurs and there is less chance of reinfection; thus, persistent childhood infection is much less frequently seen than in less-developed countries.9 In fact, serologic evidence of H. pylori is uncommon in children before age 10, but rises to 10% in adults between 18 and 30 years of age and further increases to 50% in those 60 or older.9 This increased prevalence of infection with age was initially thought to represent continuing acquisition throughout adult life. However, new adult infection and reinfection are quite uncommon, especially in developed countries. Epidemiologic evidence supports childhood-acquired infection even in developed nations, so the frequency of H. pylori infection for any age group in any locality reflects that particular birth cohort’s rate of bacterial acquisition early in life.9 In the United States, within any age group, infection appears to be more common in blacks than whites.12 Also Hispanic immigrants and their first-generation children are more likely to harbor H. pylori than their second-generation relatives.13 These differences probably relate to factors early in life that are linked to acquiring infection.
The risk of acquiring H. pylori is associated with living conditions and the family’s socioeconomic status during one’s childhood.9 Housing density, crowded conditions in the home, number of siblings, sharing a bed, and lack of hot or running water have been linked to higher rates of infection. In Japan, the rapidly declining prevalence of H. pylori appears to parallel the nation’s postwar economic progress and improvement in hygiene and sanitation. Of the Japanese born before 1950, more than 70% are infected compared with 45% born between 1950 and 1960 and 25% born between 1960 and 1970.14 Presently, childhood infection in Japan is rare. Predictions based on a similar declining prevalence in the United States suggest that the organism could eventually become extinct here and in other areas of the world, which would affect infection-related illness.15
Twin studies support genetic susceptibility to H. pylori infection because monozygotic twins who were raised in different households have a greater concordance of infection than dizygotic twins also raised separately.16 However, twins growing up together have a higher concordance of H. pylori status than twins growing up separately, suggesting childhood environmental factors are also important for acquisition.
Humans appear to be the major reservoir of H. pylori. However, domestic cats and captive primates and sheep can harbor these organisms,10 although it is possible that these animals actually acquired H. pylori from a human source. In the case of cats, isolation of viable bacteria from saliva and gastric juice suggests at least the possibility of transmission to humans.10
Especially in developing countries, contaminated water might serve as an environmental source of bacteria because the organism can remain viable for several days in water.17 Bacterial deoxyribonucleic acid (DNA) can be found in samples of municipal water from endemic areas of infection but whether viable H. pylori are present remains to be proven.5 In countries where infection is common, children who drink untreated stream water, eat uncooked vegetables, or swim in rivers and streams are more likely to harbor the bacteria, providing further indirect evidence of an environmental source of organisms.
Person-to-person transmission of bacteria from fecal-oral, oral-oral, or gastro-oral exposure seems the most probable explanation for infection.5,10 Within-family clustering of infection (often with genetically identical strains of H. pylori) supports person-to-person transmission.9 Also infected individuals more often have infected spouses or children than uninfected individuals. Support for sibling-to-sibling transmission comes from studies reporting that likelihood of infection correlated with number of children in the household and that younger children were more apt to be infected if older siblings were also infected.9 Mother-to-child transmission is also quite likely.11,18
Fecal-oral transmission of bacteria is a possible mechanism by which H. pylori gain access to the human host. The bacterium can be cultured from diarrheal stools and vomitus, suggesting the potential for transmission.19 Exposure to an infected family member during an acute gastrointestinal illness, especially with vomiting, appears to be a risk factor for subsequent infection.20
How frequently bacteria are transmitted through oral-oral contact is not known. Although organisms can be identified in dental plaque and saliva, the prevalence is low and it is questionable if the mouth serves as a source or reservoir for H. pylori.21 Also dentists and oral hygienists who continually have occupational exposure to dental plaque and oral secretions do not have a higher prevalence of H. pylori.22 In developed countries, spousal transmission of infection also appears to be uncommon.
Infected gastric secretions can serve as a source of bacterial transmission. Iatrogenic infection has occurred during the use of a variety of inadequately disinfected gastric devices, endoscopes, and endoscopic accessories.10 Also gastroenterologists and nurses appear to be at greater risk for acquiring H. pylori, presumably due to occupational contact with infected gastric secretions.23 Mandated universal precautions, standardized equipment disinfection, and use of video-endoscopes that reposition the instrument channel away from the mouth should reduce such iatrogenic and occupational transmission. Natural transmission could occur through contact with infected vomitus during an acute illness20 or with regurgitated material from an infected child. Such contact could explain the higher concordance of maternal/child H. pylori infection and the presumed child-to-child transmission that occurs in an infant daycare setting.24
COLONIZATION AND VIRULENCE FACTORS
One of the interesting aspects of this pathogen is how it confers disease when the organism resides, for the most part, in the lumen. Studies describing the genome of two distinct strains of H. pylori have helped to advance our understanding of the ecology of the organism and the potential gene expression patterns that can affect the pathogenesis of disease.25,26 Importantly, bacterial genes expressed in gastric mucosa differ from the pattern observed in vitro,27 whereas exposure of the bacterium to low pH increases its expression of genes encoding proteins involved in the motility apparatus and of genes encoding urease.28 The urease helps the organism adapt to the gastric milieu as it retains optimal function at two different pH values: usually pH 7.2 and pH 3.29 H. pylori is further adapted to the gastric pH by producing the molecular machinery required to migrate rapidly to a more favorable environment below the mucus layer. Motility is one of the few H. pylori characteristics shown to be necessary for successful colonization of the host.
H. pylori show a strict tropism for the gastric mucosa or intestinal sites in which there is gastric metaplasia. The corollary is also true, as H. pylori do not colonize epithelium in the stomach that has undergone intestinal metaplastic change, possibly due to the production of antimicrobial factors that select against colonization. This possibility is supported by the fact that H. pylori rarely colonize the deeper portions of the gastric glandular mucosa, where O-glycans that impair H. pylori growth are found.30 This concept is also supported by another study that shows H. pylori decreases the expression of the antibacterial molecule, secretory leukocyte protease inhibitor,31 thereby removing an element of the host response that would be detrimental to the persistent infection.
Another important factor that controls colonization is the expression of receptors on host cells that allow H. pylori to bind. Lewis (Le) antigens expressed by host cells serve as a receptor for bacterial binding.32 Specific bacterial gene products, most notably BabA, act as the bacterial ligand for the Leb receptor.33 The Leb receptor may be located in the gastric mucus because strains bearing BabA also bind to the mucin MUC5AC.34 Some studies suggest that the babA2 genotype is more frequently associated with inflammation, duodenal ulcer and gastric cancer.2 However, observations that binding of H. pylori to epithelial cells freshly isolated from human gastric biopsy specimens is unaffected by the expression of Le antigen, that infection is not increased in subjects with the Leb phenotype, and that individuals who do not express Leb can clearly be infected with H. pylori cast doubt on the true role of Leb and BabA.2 Additionally, in one report, the majority of strains infecting the individuals studied did not induce ulcers or cancer, despite expressing the babA2 gene.35
H. pylori also bind to the molecular complex of invariant chain and class II human leukocyte antigens (HLAs) expressed on the surface of gastric epithelial cells.36 Class II major histocompatibility complex (MHC) molecules, with their expression increased by infection, were the first epithelial cell receptor for H. pylori demonstrated to directly affect signaling in host cells. Binding of urease to epithelial cells via class II MHC was sufficient to induce apoptosis.37 More recently, the gastric trefoil protein TFF1 was shown to serve as a receptor for H. pylori.38 This molecule is predominantly expressed in the gastric mucosa and found in association with gastric mucus. In genetically engineered mice with TFF1 deficiency, a spontaneous, antral adenoma develops, suggesting that this molecule provides an element of control over gastric epithelial cell growth.39,40
The pathogen-associated molecular receptors (PAMPS) have also been examined for their role in binding of H. pylori to the host epithelial cells. The Toll-like receptors (TLRs) are a family of PAMPS, with an apparent different specificity for various bacterial molecules.41 For example, TLR4 is able to recognize the LPS of many bacteria, with cytokines and H. pylori particles increasing the expression of TLR4.42 H. pylori LPS stimulates monocytes and gastric epithelial cell responses via TLR4.2 Other PAMPs, including TLR2, are also activated by highly purified H. pylori LPS and have even been described as being more important than TLR4.43,44 These receptors may bind bacterial products, and thereby enhance bacterial binding and cell signaling.42 TLR5 binds bacterial flagellins and, similar to TLR2, induces a signaling response that can trigger acute inflammation. H. pylori produce flagellin that binds TLR5 and activates a response in vitro.43 There is disagreement on which TLR is stimulated by H. pylori LPS or whether H. pylori flagellin is ever able to bind TLR5,45 and this may reflect the different conditions used for bacterial culture and LPS or flagellin purification. Further studies are needed to establish the significance of H. pylori binding to TLRs in the pathogenesis of infection.
After H. pylori migrate to the gastric epithelium, the organism attaches to host cells and may damage them in order to obtain nutrients from the subsequent inflammatory exudate or transudate. A key interaction between the bacteria and gastric epithelium involves a segment of bacterial DNA referred to as the cag pathogenicity island (cag PAI). Genes within the cag PAI encode proteins that provide a type IV secretion apparatus (i.e., cagE) that allows bacterial macromolecules to translocate into the host cell (i.e., cagA).26,46 cag PAI plays an important role in the pathogenesis of gastritis in humans26,47 because H. pylori bearing the cag PAI are associated with increased interleukin-8 (IL-8) expression and inflammation in gastric mucosal biopsy specimens and increased IL-8 expression and apoptosis in vitro.48 Infecting gerbils with mutated strains lacking cagE reduces the severity of gastritis and the development of gastric ulcers, intestinal metaplasia, and gastric cancer compared with gerbils infected with the wild-type strain.48,49 Human studies in which duodenal ulceration occurred more frequently in children carrying strains expressing cagE associated with higher levels of gastric IL-850 corroborate animal and in vitro studies.
All strains of H. pylori possess the vacA gene, with more than half expressing the vacuolating cytotoxin (VacA), which attaches to epithelial cells via an interaction with protein-tyrosine phosphatases.51 Although the majority of the VacA is secreted, some may remain on the surface of the bacteria and serve as a ligand for bacterial attachment via this protein-tyrosine phosphatase receptor. Several studies have examined the structure and function of VacA and its association with disease.26,52,53 For example, mice deficient in protein-tyrosine phosphatase beta do not develop ulceration when exposed to VacA.54 Different vacA alleles have been classified in the 5′ signal region (s-region) and the middle region (m-region) of the vacA gene.52 The s-region is present as s1 (which can be further distinguished as s1a, s1b, s1c) or s2, whereas the m-region is present as m1 or m2. Production of VacA is designated by the allelic combination s1/m1 and s1/m2. Specific vacA alleles (s1 and m1) are associated with disease55 and the induction of epithelial cell apoptosis.56 The interaction between VacA and its receptor(s) appears to be important in the pathogenesis of gastroduodenal disease, whether it serves as a ligand for bacterial attachment or as a secreted virulence factor.
HOST RESPONSE TO INFECTION
Increasing evidence suggests that the host response to H. pylori infection is an intrinsic component of the pathogenesis of gastrointestinal disease (Chapters 29, 52, and 54). The possibility that the host response may play a direct role in gastric cancer is supported by the observation that heterogeneity in the regions of the genome that control the magnitude of inflammation is associated with cancer linked to H. pylori infection.57 Polymorphisms in the regions controlling IL-1β58 were shown to be associated with an increased incidence of hypochlorhydria and gastric cancer. This seminal observation has been replicated in other studies in which IL-1β polymorphisms were associated not only with gastric cancer but also a decrease in recurrence of duodenal ulcer.59 An increase in IL-1 may not only drive inflammation but also lead to a physiologic state known to precede gastric cancer development because IL-1 potently inhibits gastric acid secretion. Increased gastric IL-1, more severe gastritis, gastric atrophy, and greater colonization with H. pylori strains have been associated with gastric cancer.2 Other genes that regulate the magnitude of the inflammatory response, including IL-10, tumor necrosis factor-α (TNF-α), and IL-8, have also been associated with the sequence of events leading to cancer.60,61
It is apparent that epithelial cells play an integral part in the host response to H. pylori infection as well as being the target of infection. The epithelial response to infection is complex, as it is driven by several variables: bacterial virulence factors; the signaling linked to specific receptors that recognize the bacterial components; and the local milieu of hormones, neurotransmitters, immune or inflammatory cytokines and mediators, as well as stromal factors. The epithelial cell responses include changes in epithelial cell morphology (the hummingbird phenotype),62 disruption of the tight junctional complexes,63 the production of cytokines,46 increased epithelial cell proliferation, increased rates of epithelial cell death via apoptosis, and the induction of numerous genes associated with the stress encountered in response to infection.2 The best overview of the epithelial cell response can be ascertained from the broad changes in the expression of hundreds of genes demonstrated with high-throughput gene expression systems.2 Detailed analyses of the effects of infection, different cell lines and various inflammatory mediators have yet to be carried out over the wide range of time points that would be relevant to the conditions epithelial cells face in vivo.
The expression of genes in epithelial cells stimulated with H. pylori is regulated by transcription factors that are controlled by a series of signaling mechanisms. While many transcription factors are likely activated by infection, the most studied are nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1) which regulate the expression of a wide variety of proinflammatory cytokines and cellular adhesion molecules in response to infection or the local cytokine milieu. H pylori activates NF-κB in gastric epithelial cells, both in vitro and in vivo in patients with H. pylori gastritis; gastric epithelial cell NF-κB activity is markedly enhanced, correlating with the intensity of neutrophil infiltration and IL-8 protein levels.46 This pathway is of particular interest given the recent report that polymorphisms in the IL-8 gene lead to increased mucosal IL-8 expression, inflammation, and other premalignant changes associated with gastric cancer. H. pylori infection appears to activate NF-κB and AP-1 in gastric epithelial cell lines through various signaling mechanisms including mitogen-activated protein (MAP) kinases.47,64 The MAP kinase cascades regulate a wide range of cell functions, including proliferation, inflammatory responses, and cell survival. For example, cag PAI-positive H. pylori activate the ERK, JNK, and p38 MAP kinase pathways, and ERK and p38 regulate IL-8 production in gastric epithelial cells.65,66 An inhibitor of p38α MAP kinase, FR167653, reduced both neutrophil infiltration and gastric mucosal injury in H. pylori–infected Mongolian gerbils.67
One of the novel concepts emerging is the role of oxidative stress in regulating gene expression during H. pylori infection. Apurinic-apyrimidinic endonuclease-1 (also referred to as redox factor-1) plays a key role in the regulation of redox-sensitive signaling and is expressed in gastric epithelial cells during infection with H. pylori,68 contributing to activation of AP-1 and NF-κB required for the host response to infection, including IL-8 production.69 However, the role it and other redox-regulatory molecules play in the pathogenesis of diseases associated with H. pylori has yet to be clearly defined. Oxidation of DNA by reactive oxygen species such as hydroxyl radicals are thought to play a causal role in malignant transformation through the induction of DNA damage. Oxidative DNA damage is increased in gastric epithelial cells by H. pylori infection. There is growing interest in the role of antioxidants in disease prevention or treatment because infection is associated with decreased levels of a tissue antioxidant scavenger, vitamin C. Moreover, there is evidence that diets high in antioxidants70 or “nutraceuticals” of the isothiocyanate group, such as sulforaphane,71 can antagonize oxidative stress and protect the host from gastric cancer, perhaps by decreasing inflammation and attenuating bacterial load.
CagA protein translocates into the host cell cytoplasm where it is tyrosine phosphorylated by host Src kinases, and through other interactions, regulates epithelial cell morphology.72–76 CagA in strains from distinct geographic populations appears to be phosphorylated in a different manner resulting in different effects on intracellular signaling.77,78 It is intriguing to speculate that heterogeneity in the CagA protein may lead to distinct effects on the host response that account for some of the geographic differences in disease. Although phosphorylation of the CagA protein may be important, it is not the only mechanism whereby this molecule regulates the host response. A phosphorylation-independent effect on gene transcription, which is also attributed to the CagA protein, also has been reported.79 Moreover, greater than 30% of host gene expression is altered independently of the phosphorylation of CagA, at least in intestinal epithelial cell lines.80
Outer inflammatory protein A (OipA) is another bacterial product that induces epithelial cell IL-8 production81 and strains that express OipA are associated with increased bacterial density, mucosal IL-8 levels, and neutrophil infiltration, as well as more severe clinical consequences.82 Peptidoglycan has been reported to translocate into gastric epithelial cells via the type IV secretion system encoded by the cag PAI. Once inside the cell these bacterial compounds are recognized by nucleotide-binding oligomerization domain-1 (NOD1), thereby providing a novel mechanism of bacterial sensing.83 Binding to NOD1 can lead to activation of NF-κB and the subsequent expression of various host genes encoding proinflammatory molecules.
As discussed in Chapter 49, gastric acid secretion is a major function of the gastric mucosa that is regulated by a variety of neural, endocrine, and immune factors.3 Elevated fasting and meal- or hormone-stimulated levels of gastrin are well documented in H. pylori infection, and there is evidence that gastrin expression is regulated by bacterial factors and cytokines. Expression of somatostatin, an acid-inhibitory peptide, is diminished in infected individuals as is duodenal bicarbonate secretion. The net effect of H. pylori infection on acid secretion is complex and varies depending on the duration and distribution of infection and presence of mucosal atrophy. Secretion of mucus is also affected by H. pylori infection with decreased amounts of mucus and gastric mucosal hydrophobicity; these abnormalities reverse after eradication of infection. Epithelial barrier function is altered during H. pylori infection as a consequence of both direct effects of H. pylori and the accompanying inflammatory response that collectively increase epithelial cell proliferation and programmed cell death.2
Infection with H. pylori results in a unique inflammatory response in which infection persists despite the recruitment and activation of T and B lymphocytes, phagocytic cells, and other immune cell populations. Whereas several epithelial cell responses to H. pylori have been described earlier, they do not appear to fully account for the magnitude of the inflammatory response to an organism that resides predominantly in the lumen. Some bacteria may infect epithelial cells, and significant amounts of bacterial material may “leak” around epithelial cells and reach the lamina propria, where it can activate underlying phagocytes, including neutrophils and macrophages. One bacterial factor is the H. pylori neutrophil-activating protein (HP-NAP). This 150-kd decamer protein promotes neutrophil adhesion to endothelial cells and stimulates chemotaxis of monocytes and neutrophils, NADPH oxidase complex assembly at the plasma membrane, and the subsequent production of reactive oxygen intermediates (ROIs).84 In the inflammatory environment present during H. pylori gastritis, TNF-α and interferon-γ (IFN-γ) can prime neutrophils and potentate the effects of HP-NAP.
When cells undergo apoptosis and die, they are removed by phagocytes. This response occurs in the digestive tract as epithelial cells migrate toward the lumen and undergo apoptosis, providing another means whereby the host can sample the antigenic challenges facing epithelial cells. Viral antigens are presented to T cells when infected apoptotic epithelial cells overlie the Peyer’s patch. Engulfment of H. pylori infected epithelial cells by phagocytes may also be an important mechanism by which H. pylori can activate the host response, and it has been shown that macrophages bind and then engulf gastric epithelial cells that undergo apoptosis due to infection.2
Recruitment and activation of macrophages and neutrophils cause the release of other inflammatory mediators. Increased expression of inducible nitric oxide synthase (iNOS) is observed in the gastric mucosa during infection with H. pylori.2 Nitric oxide (NO) and superoxide (O2−), which may be produced by infiltrating neutrophils, react to form peroxynitrite (ONOO−), a potent oxidant and reducing agent. Although these products have potent antimicrobial effects, uncontrolled or inappropriate production could play a role in the gastric mucosal damage observed during H. pylori infection. The catabolism of urea by urease provides CO2, which rapidly neutralizes the bactericidal activity of the peroxynitrate by reacting with it to form ONO-OCO2. Urease may favor bacterial colonization by neutralizing some host responses but this also enhances the nitration potential of ONOO− and may favor mutagenesis of host cell DNA.
Cytokines secreted by epithelial cells complement those released in the lamina propria. For example, neutrophils are not only activated by IL-8 but also by chemokines such as ENA-7885 and Gro-α,86 which can derive from the epithelium, the adjacent myofibroblasts or the macrophages within the lamina propria. Cytokine induction in macrophages includes induction of TNF-α and IL-6 by urease,87 whereas heat shock protein 60 induces IL-6.88 Intact bacteria can induce the production of chemokines that recruit T cells,89 as well as IL-1290,91 and IL-18,92 two cytokines that favor the selection of Th1 cells. Thus, intact bacteria or bacterial factors trigger a broad cytokine response within the lamina propria.
As adaptive responses develop, different T lymphocyte helper (Th) cell subsets emerge, with characteristic patterns of cytokine secretion. Th1 cells promote cell-mediated immune responses through the production of IFN-γ and TNF-α, whereas Th2 cells produce IL-4, IL-5, IL-10, and transforming growth factor-β (TGF-β). Th2 cells can promote mucosal IgA or IgE responses, as well as diminish the inflammation caused by Th1 cytokines. Previous studies suggest that the infected gastric mucosa is preconditioned to favor Th1 cell development.90 One possible hypothesis to account for this tendency is that infection selectively blocks Th2 development. H. pylori can interfere with STAT6 activation by IL-4,93 which could impair Th2 development, and IL-12 and IL-18 induced in response to infection may positively select for the predominant Th1 response. Other cytokines that may enhance Th1 responses, such as IL-23 and IL-27, have yet to be studied in human tissue.
T cell activation by H. pylori infection may contribute to more severe inflammation and gastroduodenal diseases. Increased levels of biologically active IL-17, a cytokine produced by activated CD4+ T lymphocytes, are found in the mucosa of H. pylori–infected patients.94,95 IL-17, in turn, induces IL-8 expression by gastric epithelial cells, thereby enhancing neutrophil recruitment. Activation of transcription factors by IL-17 may also contribute to the increased levels of numerous other proinflammatory cytokines and enzymes observed during H. pylori infection, such as IL-1β, TNF-α, and cyclooxygenase-2 (COX-2). IFN-γ and TNF-α produced by Th1 cells can increase the expression of many genes in the epithelium, including IL-8. These cytokines also enhance bacterial binding36 and may contribute to enhanced bacterial load.96 In animal models Th1 cells increase epithelial cell apoptosis36 as well as inflammation, atrophy, and dysplasia.97 TNF-α, IFN-γ, and IL-1β up-regulate gastric mucosal Fas antigen expression.98 Since Th1 cells express higher levels of Fas ligand (FasL) than Th2 cells, the relative increase in Th1 cells during H. pylori infection may induce epithelial cell death through Fas-FasL interactions.98,99 This notion is substantiated by the observation that proton pump H+,K+-ATPase–specific Th1 cells in the gastric mucosa kill target cells via Fas-FasL interactions and may act as effector cells in autoimmune gastritis.100
Because Th1 cells cannot clear H. pylori, some other T cell subset may have to be stimulated in order to confer immunity. Studies in animal models indicate that protective immunity was induced by vaccines for Helicobacter spp. via Th cells other than Th1 cells, possibly including Th2 cells. The anti-inflammatory cytokines associated with Th2 cells or other regulatory subsets of Th cells can attenuate the pathogenic effects of Th1 cells.101 More direct evidence suggests that IL-4 can decrease gastritis, an effect that may be mediated by the release of somatostatin.97 As gastric responses can be modified by Th2 cells, the role of other T cell subsets, such as regulatory T cells (Treg), in the pathogenesis of disease associated with H. pylori infection is being addressed. Depletion of Treg in neonatal mice leads to autoimmune gastritis,102 and infection with H. pylori alleviates autoimmune gastritis induced in neonatal mice.103 This suggests that infection may stimulate a subset of anti-inflammatory T cells that impair excessive inflammation, which could otherwise lead to the spontaneous clearance of the organism, an effect that appears to occur in the human mucosa in response to H. pylori infection.104
Antibodies in the gastrointestinal tract are normally of the immunoglobulin A (IgA) isotype, which are highly adapted for mucosal protection, conferring protective immunity without activating complement and stimulating deleterious amounts of inflammation. During infection with H. pylori, the number of IgA producing cells increases. IgG and IgM are also detected, along with activated complement. It has been suggested that the level of autoantibodies in humans correlates with the severity of gastritis.2 Local immune complexes contribute to gastroduodenal inflammation and tissue damage during infection and may contribute to autoimmune gastritis. Monoclonal antibodies that recognize H. pylori cross-react with human and murine gastric epithelial cells.105,106 Adoptive transfer of these antibodies to recipient mice induces gastritis,105 as does the transfer of B cells that recognize heat shock proteins from individuals with maltoma.107 Anti-Le antibodies have been described in humans and occur independently of the Le phenotype of the host but they do not appear to be autoreactive. Autoantibodies induced in mice may recognize different targets within the gastric mucosa and even though they may cross-react with human gastric tissue, autoantibodies induced in humans may have a completely different specificity.
With few exceptions, infection with H. pylori persists for the life of the host unless there is some intervention with antibiotics. This observation has led to investigations as to whether immunity is impaired by immunologic avoidance or tolerance. Several bacterial factors, including catalase and urease, antagonize innate host responses. Production of the enzyme arginase by H. pylori inhibits NO production and may favor bacterial survival,108 whereas virulent strains of H. pylori have also been shown to alter mucus production109 and phagocytosis.110 The VacA toxin can impair antigen presentation by macrophages by inhibiting the Ii-dependent pathway, which is mediated by newly synthesized class II MHC molecules.111 Moreover, H. pylori express antigenic molecules that mimic host molecules, such as Lewis antigens that theoretically could stimulate T cells to release cytokines that inhibit autoimmune reactions. However, the cytokine profile associated with H. pylori infection is not one that would be expected to occur in a tolerant environment. For example, IL-4, IL-10, and TGF-β (which could mediate an anti-inflammatory effect) are not expressed to the same levels as proinflammatory cytokines such as IFN-γ and TNF-α.2 Because the infected gastric mucosa is characterized by chronic active inflammation, tolerance, if it has occurred, may favor persistent infection even though it cannot prevent the chronic inflammatory response.
An alternative possibility is that rather than the T cell responses being inappropriate, they lack a level of coordination necessary to achieve immunity. Under certain circumstances, bacteria can inhibit the growth of T cells and actually induce their apoptosis.112–114 This process has been demonstrated using cell lines, and attributed to very specific patterns of gene expression, including the induction of FasL and Fas on T cells, which enables apoptosis.98,114 The studies by Wang and associates suggest this can occur directly,114 but peptides from H. pylori may activate monocytes to produce oxygen radicals which impair the expression of CD3 zeta chain of the T cell receptor complex and the induction of T cell apoptosis.113 The loss of T cells by apoptosis as an explanation is further supported by the presence of apoptotic T cells in the gastric mucosa,112 as well as the expression of Fas and FasL on biopsy specimens.98
More recently, VacA has been shown to impair the growth of a T cell line, independently of any effect on apoptosis, by inhibiting nuclear translocation of a transcription factor, NFAT, that is required for IL-2 production.115,116 However, these events are complex, with VacA toxin impairing IL-2–dependent expansion in T cells freshly isolated from human peripheral blood, but not their survival or ability to produce IL-2.117 H. pylori can also impair T cell function directly due to arginase118 as well as indirectly, as a consequence of inducing hydrogen peroxide in adjacent macrophages that leads to T cell apoptosis.113 These observations support a model in which H. pylori interferes with normal T cell activation in several ways. Despite the evidence that H. pylori can impair T cell responses, some antigen-specific T cell responses are found in the gastric mucosa.100 Nonetheless, it is still possible that a combination of the effects of H. pylori and the cytokine milieu disrupt the coordination required for the development of an effective, antigen-specific T cell response.
CONDITIONS ARISING FROM INFECTION
Infection of the human stomach by H. pylori leads to gastritis, which initially affects the superficial layers of the mucosa (see Chapter 51). In some instances the infection is short lived, but typically the infection results in a unique pattern of gastritis, so-called chronic active gastritis, which is essentially a lifelong condition. As discussed, chemokines induced by infection lead to a persistent acute inflammatory infiltrate with neutrophils and other cells (active inflammation) coexisting with cells characteristic of chronic inflammation (lymphocytes, macrophages). In spite of this robust immune response bacteria persist in most infected humans, and the factors that result in clearance of some cases of acute infection remain largely unknown.2 Such insights might have augmented the efforts of the past two decades to develop vaccines to prevent chronic H. pylori infection and enhance eradication that have been unfortunately, largely unsuccessful. Most chronically infected individuals are asymptomatic with somewhere between 10% and 15% going on to develop peptic ulcer disease, gastric adenocarcinoma, and lymphoma of the gastric mucosa–associated lymphoid tissue. As discussed in the previous section, factors that contribute to whether an individual develops these complications of chronic infection include bacterial factors, although these are not yet well defined; host factors including genes that regulate the host immune response (cytokines and their receptors); and environmental factors.
As mentioned, H. pylori organisms colonize only gastric-type epithelium within the human host and may colonize tissues outside the stomach when there is gastric metaplasia of the esophagus or duodenum, or in a Meckel’s diverticulum. The pattern of colonization within the stomach appears to be an important determinant of H. pylori disease manifestations. It is unclear exactly what leads to duodenal ulcers associated with H. pylori infection, but it is thought that hyperacidity associated with antral colonization leads to gastric metaplasia of the duodenum, which can then become colonized, leading to duodenal ulcer in some instances. Distal gastric infection may also present with erosions and small ulcers in the gastric antrum, similar in appearance to lesions associated with anti-inflammatory drug use. Gastric ulcers and gastric adenocarcinoma occur more often when there is proximal colonization of the stomach (pan-gastritis), which results in injury to the gastric glands, leading to atrophic gastritis and associated hypo- or achlorhydria (see Chapter 54). Precursor lesions of gastric cancer, including atrophic gastritis, intestinal metaplasia, and dysplasia,119 result from infection although most individuals with intestinal metaplasia in North American populations do not have evidence of infection.120 As such, there are no general recommendations to screen individuals with intestinal metaplasia in the United States.
The patterns of infection-associated gastritis and resultant effects on acid secretion are thought to account for the reported differences of the impact of infection and eradication on pre-existing and new symptoms of gastroesophageal acid reflux.121 Those with antral-predominant infection have hyperacidity, which may promote acid reflux that improves after eradication therapy. In contrast, those with pan-gastritis do not apparently suffer from heartburn but after clearance of infection there is a slow return of secretory function, which may be associated with the development of heartburn. Perhaps even more contentious is the inverse association of H. pylori infection with Barrett’s esophagus and esophageal adenocarcinoma (see Chapters 43, 44, and 46). This may be a related to acid exposure but studies indicate that Cag A–positive strains are more strongly associated with a reduced frequency of Barrett’s esophagus and esophageal adenocarcinoma than Cag A–negative strains. This negative association seems to be greatest in Eastern countries.122 Of note, gastric cancer remains the second major cause of cancer death in the world. The burden of risk of gastric cancer is considered largely attributable to H. pylori infection, with cag PAI–bearing strains having a higher association with gastric cancer than cag PAI–negative strains.123 Given the burden of gastric cancer worldwide, the risk of infection seemingly outweighs the benefits in terms of the development of proximal gastrointestinal tract cancer.
Over the past two decades a large number of associations with nongastric diseases and H. pylori infection have been reported including Raynaud’s, scleroderma, idiopathic urticaria, acne rosacea, migraines, thyroiditis, and Guillain-Barré syndrome, but the data supporting an association for this group of conditions are weak or nonexistent.124 Associations that have somewhat better levels of evidence for an association include coronary artery disease, immune thrombocytopenic purpura,125,126 and iron deficiency anemia127 and for the latter two conditions, eradication of infection may be considered when other treatments have failed. The proposed mechanisms leading to these various conditions range from systemic immune reactions, cross-reactivity of bacterial and host proteins, and events secondary to gastric mucosal injury.
DIAGNOSIS
The National Institutes of Health (NIH) proposed consensus recommendations for H. pylori testing applicable to the United States in 1994.128 H. pylori management in other parts of the world is generally concordant with the United States, but there are some exceptions related to regional differences in clinical practice and specific infection-related disease prevalence (gastric cancer and premalignant gastric histology).129,130
The American College of Gastroenterology published updated U.S. guidelines in 2007 that recommend testing for H. pylori only if a clinician is prepared to treat a patient with a positive test result.131 Specific indications for testing include patients with active or documented history of uncomplicated or complicated peptic ulcer, early gastric cancer, or gastric MALT lymphoma. Testing for H. pylori is often recommended in younger patients with uninvestigated dyspepsia and no “alarm features” (i.e., early satiety, unexplained weight loss, dysphagia, recurrent vomiting, family history of gastric cancer)132 and in patients with functional dyspepsia (symptoms and negative endoscopy).133 However, the clinical and cost benefits of H. pylori in the setting of dyspepsia remain controversial (see Chapter 13), especially in regions where prevalence of infection is relatively low and gastroesophageal reflux disease (GERD) as a cause of symptoms is high.134 Testing for infection prior to starting nonsteroidal anti-inflammatory drugs (NSAIDs) may reduce subsequent ulcers, but this not generally recommended or often done in the United States, where prevalence of H. pylori is low.131 Also there is no general recommendation to test asymptomatic persons, with the possible exception of those with a family history of gastric cancer,135 particularly individuals of Asian, Eastern European, or Mesoamerican descent, for whom the risk of gastric malignancy is highest. Occasionally immune thrombocytopenic purpura126,136 and refractory iron deficiency anemia127 respond to eradication of infection, so decisions to test for H. pylori in these conditions are made on a case-by-case and regional basis. Finally a potpourri of conditions is linked to H. pylori infection, but causal evidence and biologic plausibility are sufficiently weak or lacking in these clinical situations to recommend testing.124 Indications for testing and treatment for H. pylori are summarized in Table 50-1.
Table 50-1 Indications for Testing and Treatment of Helicobacter pylori Infection
GERD, gastroesophageal reflux disease; MALT, mucosa-associated lymphoid tissue; NSAIDs, nonsteroidal anti-inflammatory drugs.
Adapted from references 129 and 131.
There are endoscopic and nonendoscopic means to diagnose infection, and techniques can directly (histologic demonstration of organisms, presence of bacterial antigen in the stool, culture) or indirectly (using urease or an antibody response as a marker of bacteria) detect H. pylori.137,138 The appropriate method to choose depends on the clinical situation, population prevalence, and pretest probability of infection as well as test availability and cost. In addition, recent use of antibiotics or proton pump inhibitors can influence results of certain tests.131 The advantages and disadvantages of commonly used diagnostic tests for H. pylori are summarized in Table 50-2.
| NONENDOSCOPIC TESTS | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| Serology (qualitative or quantitative immunoglobulin G [IgG]) | Widely available, inexpensive, good NPV | Poor PPV if HP prevalence is low, not useful after treatment |
| Urea breath test (13C or 14C) | Identifies active infection, accuracy (PPV, NPV) not affected by H. pylori prevalence, useful both before and after treatment | Availability and reimbursement inconsistent, accuracy affected by PPI and antibiotic use, small radiation dose with 14C test |
| Stool antigen test | Identifies active infection; accuracy (PPV, NPV) not affected by H. pylori prevalence; useful both before and after treatment (monoclonal test) | Fewer data available for polyclonal test, accuracy affected by PPI and antibiotic use |
| ENDOSCOPIC TESTS | ADVANTAGES | DISADVANTAGES |
|---|---|---|
| Histology | Excellent sensitivity and specificity, especially with special and immune stains; provides additional information about gastric mucosa | Expensive (endoscopy and histopathology costs), interobserver variability, accuracy affected by PPI and antibiotic use |
| Rapid urease test | Rapid results, accurate in patients not using PPIs or antibiotics, no added histopathology cost | Requires endoscopy, less accurate after treatment or in patients using PPIs |
| Culture | Specificity 100%, allows antibiotic sensitivity testing | Difficult and tedious to perform; not widely available; expensive |
| Polymerase chain reaction (PCR) assay | Excellent sensitivity and specificity, permits detection of antibiotic resistance | Not widely available; technique not standardized; expensive |
NPV, negative predictive value; PPI, proton pump inhibitor (see Table 50-3); PPV, positive predictive value.
Adapted from Chey WD, Wong BC. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol 2007; 102:1808-25.
Performing endoscopy solely to diagnosis H. pylori infection is not appropriate; there are three methods—biopsy urease test, histology, and (less often) culture—to identify the organism during an otherwise indicated endoscopic procedure. The choice of method depends on the clinical situation, cost, and test accuracy.131 Guidelines propose initially using a biopsy urease test because the method is quick, easy to perform, relatively inexpensive, and generally accurate. Gastric biopsy material is tested for urease activity by placing several pieces of tissue in a medium containing urea and a pH reagent. Bacterial urease hydrolyzes urea-liberating ammonia, producing an alkaline pH and a resultant color change of the test medium.137 Test results are often positive within minutes to hours. Several urease test kits are commercially available based on the methodology described here, differing only with regard to medium (agar gel or membrane pad) and testing reagents.137 These test kits are generally inexpensive but there are added costs associated with obtaining gastric tissue samples, for example, up-coding diagnostic esophagogastroduodenoscopy (EGD), to EGD with biopsy. Nevertheless, biopsy urease testing is less expensive than histology so one proposed cost-saving measure is to obtain specimens for histology but delay sending them to the laboratory pending urease test results. Specificity of the urease tests is 95% to 100% with false-positive tests uncommon.137,139 Although reported sensitivity of urease tests is 90% to 95%, accuracy can be negatively affected by blood in the stomach,140 and current or recent use of medications such as antibiotics, bismuth-containing compounds, or acid inhibitors, especially PPIs.141 Therefore, a negative urease test does not necessarily exclude H. pylori infection in an individual taking antisecretory medication, a common scenario in patients referred for endoscopy. Testing samples from multiple regions of the stomach or stopping offending medication and delaying endoscopy for several weeks may improve test sensitivity in such patients.
Evaluation of gastric mucosal histology is generally not necessary to diagnose H. pylori, but it can provide information regarding the activity and severity of mucosal inflammation (see Fig. 51-3C). Histology can also detect metaplasia, dysplasia, and neoplasia.137 In addition to biopsying “clinically suspicious” areas, taking multiple biopsies and sampling lesser and greater curvatures of gastric antrum and body are important, especially when looking for evidence of multifocal atrophic gastritis and/or intestinal metaplasia (see Chapter 51). Histologic examination had been considered the gold standard for identifying infection, with reported sensitivity and specificity as high as 95% and 98%, respectively.142 However, the distribution and density of organisms can vary within the stomach resulting in sampling error, particularly in patients taking antisecretory medications.131,137 Detecting organisms can be difficult when standard hematoxylin and eosin staining is used alone, but is less of an issue when processing tissue with special stains such as Giemsa, silver, or Genta (Fig. 50-1) or specific immune stains.138,142,143
H. pylori are difficult to culture because the organism is fastidious, slow growing, and requires specialized media and growth environment.137,138 In fact the initial isolation of H. pylori occurred by happenstance when plated cultures incubated over a long holiday weekend. When culturing for H. pylori, tissue should be obtained before biopsy forceps become contaminated with formalin and placed in a container with only a few drops of saline to preserve the specimen during transport to a local or offsite microbiology facility.138 Although culture is not generally recommended, in those with refractory disease culture with antibiotic sensitivity testing can guide subsequent treatment, although in vitro sensitivity testing does not always predict clinical treatment outcome.138,144
Most often nonendoscopic tests are used to diagnose H. pylori infection, and serology remains the most popular method used, although use of other noninvasive methods that can detect active infection has increased. Infection incites a systemic immune response, and enzyme-linked immunosorbent assay (ELISA) technology can detect IgG antibodies to a variety of bacterial antigens in serum samples.137,138 Tests for IgA and IgM class antibodies are less reliable and not recommended.137 Office-based kits that test whole blood can provide results within 30 minutes and permit “point of service” testing. Although serology is inexpensive, noninvasive and ideally suited to a primary care setting, the prevalence of H. pylori in the population being tested influences its accuracy.131 The sensitivity of serology is generally quite high (90% to 100%) but specificity is variable (76% to 96%), especially if prevalence of H. pylori is low. Therefore, in places where infection is less common (most areas of the United States), the negative predictive value of serology is high. On the other hand, the corresponding positive predictive value is poor, suggesting most often positive results are actually falsely positive.131 So it is best to confirm positive serology results with another method such as a stool antigen or urea breath test before starting treatment or to use a test that detects active infection in the first place. Conversion of a positive serology to negative after treatment suggests bacterial cure, but in most instances serology remains positive for months to years even after successful treatment of infection.145 This “serologic scar” effectively precludes use of serology to confirm bacterial eradication after treatment, a practice that is unfortunately still quite common in the primary care setting even though better tests to confirm eradication are more widely available.
The urea breath test (UBT) detects active H. pylori infection and so it is useful for making the primary diagnosis, confirming the accuracy of serology, and documenting successful treatment.131 UBT relies on bacterial hydrolysis of orally administered urea tagged with a carbon isotope, either 13C or 14C (Fig. 50-2). Hydrolysis generates ammonia and tagged CO2, which can be detected in breath samples.137,138 The 13C test is best for children and pregnant women because it uses a nonradioactive isotope, whereas the radiation dose with the 14C test is 1 microCi138 equivalent to one day of background radiation exposure. The specificity of UBT is more than 95%131,137; therefore, false-positive results are uncommon. The sensitivity of the test is 88% to 95% with false-negative results reported in patients taking antisecretory therapy such as PPIs,131,141 bismuth, or antibiotics. To improve accuracy, antibiotics should be stopped at least four weeks and PPIs at least one week before breath testing.131 UBT is not accurate in patients who have had gastric resective surgery.
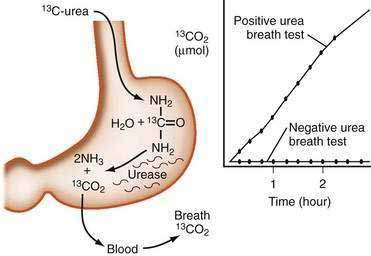
Figure 50-2. The urea breath test.
(From Walsh JH, Peterson WL. Drug therapy: The treatment of Helicobacter pylori infection in the management of peptic ulcer disease. N Engl J Med 1995; 333:984.)
An immunoassay that detects the presence of bacterial antigens in stool of infected patients is an alternative nonendoscopic method to diagnose active H. pylori infection as well as confirm eradication following treatment. Overall sensitivity and specificity of the stool test are comparable to the UBT (94% and 97%, respectively).131,138,140 A rapid H. pylori stool antigen test is available that permits testing during a clinic visit but it is slightly less accurate than a traditional laboratory based stool test.146 The sensitivity of stool testing is negatively affected by PPIs, bismuth, and antibiotics, which can decrease bacterial load, so similar precautions as described earlier for UBT are appropriate when using stool tests.131,138,141
Polymerase chain reaction is a sensitive method to detect H. pylori in gastric mucosal biopsies, but it is not practical for routine clinical diagnosis. It is, however, used for research purposes to identify bacteria when ordinary culture is difficult, as when testing stool or drinking water in a community setting, to type organisms during epidemiologic or transmission studies or for “real time” antibiotic resistance testing of tissue.138,147
TREATMENT
Because there is currently no “pylori specific” or single antibiotic available to cure infection, treatment requires combining several medications. Recommended regimens usually include two antibiotics dosed several times daily for 7 to 14 days along with acid-suppressive medication.131,148 Attempts to simplify regimens or shorten treatment duration generally reduce effectiveness. Compliance can be a problem because taking multiple medications is difficult, and minor medication-related side effects are frequent. Treatment success can vary among countries and even within regions of countries, possibly related to antibiotic-resistant organisms that are more common than previously appreciated.148,149 Despite these concerns, treatment regimens are available that cure H. pylori infection in more 75% of individuals.131,148,150,151
After cure, annual adult reinfection especially in developed countries is uncommon, probably less than 1%. Higher rates of reinfection are reported, but these often include cases that actually represent recrudescence of the original infection that failed to clear during antibiotic treatment.152 Reinfection tends to be higher in children especially after spontaneous clearance of a primary infection, and it is reported to be higher in adults living in areas of the world with high H. pylori prevalence.9,11,153
Commonly used treatment regimens are summarized in Table 50-3.129,131,148,150,151 Triple therapy, composed of two antibiotics, clarithromycin (500 mg twice daily) and amoxicillin (1 g twice daily) along with a PPI (e.g., omeprazole 20 mg twice daily, lansoprazole 30 mg twice daily, pantoprazole 40 mg twice daily, rabeprazole 20 mg twice daily, or esomeprazole 40 mg every day) for 7 to 14 days, is currently the most popular initial treatment for H. pylori. PPI triple therapy consistently cures more than 80% of infections, especially if organisms are sensitive to clarithromycin and longer treatment duration (14 or 10 days vs. 7 days) is used. Metronidazole (500 mg twice daily) can be substituted for either amoxicillin or clarithromycin, but this is appropriate only for penicillin-allergic or macrolide-intolerant individuals because metronidazole resistance is common and can reduce treatment success.129,131,148,150
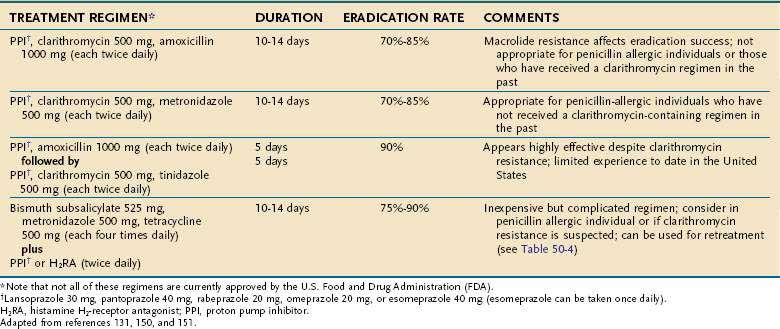
A 10-day sequential regimen (a PPI and amoxicillin 1 g, each given twice daily for the first 5 days, followed by the PPI, clarithromycin 500 mg, and tinidazole 500 mg, each given twice daily for the remaining 5 days) improved overall eradication rates compared with standard PPI triple therapy (89% vs. 77 %), but was particularly better for clarithromycin-resistant bacteria (89% vs. 29%).154 A pooled analysis of studies evaluating sequential therapy confirmed its superior efficacy especially with macrolide-resistant bacterial strains.155 Such results are encouraging, although most experience with this treatment is geographically limited to Mediterranean countries. However, there is no reason to expect different efficacy in other regions.131,154 Although used more than a decade ago, dual regimens consisting of a single antibiotic (amoxicillin or clarithromycin) and a PPI are no longer recommended because eradication is significantly less than with three drug regimens.150
Bismuth-based therapy, which combines a bismuth salt, metronidazole 500 mg and tetracycline 500 mg each given four times a day, and daily acid suppression (usually a PPI every day) for two weeks was actually one of the first therapies used to treat H. pylori. Although it remains effective (more than 80% eradication), the number of daily pills and associated frequent minor side effects negatively affect tolerability and compliance. For that reason, in the United States it is usually reserved as a second-line or retreatment regimen.129,131,148,150
A combination capsule that contains bismuth subcitrate 140 mg, metronidazole 125 mg, and tetracycline 125 mg is available in the United States and Canada, simplifying bismuth-based treatment. In a comparative study, patients treated with three combination capsules four times daily and PPI twice daily for 10 days had comparable H. pylori eradication with those treated with traditional PPI triple therapy (88% vs. 83%).156 Short course (1 to 7 days) bismuth-based treatment157 has been evaluated, but consistent long-term cure of infection has not been confirmed, so abbreviated treatment cannot be recommended.150
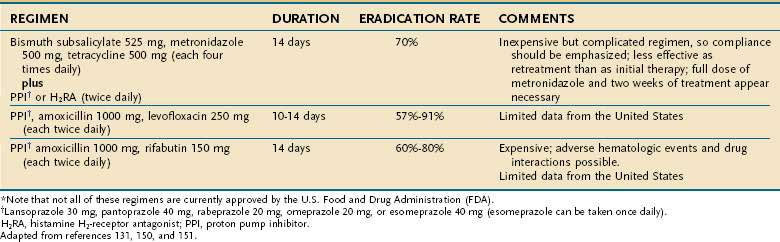
Initial treatment of H. pylori infection fails in up to 25% of patients as a result of an infection with antibiotic-resistant organisms, poor compliance with medication, and patient demographics such as younger age, smoking, prior antibiotic use, and underlying condition (functional dyspepsia vs. peptic ulcer).149,158,159 A review of various retreatment regimens (Table 50-4) reported eradication rates of 46%, 70%, 80%, and 76 % percent for PPI dual therapies, PPI triple therapies, ranitidine bismuth citrate–based triple therapy and bismuth-based therapy, respectively.160 Ranitidine bismuth citrate is no longer available in the United States. When two new antibiotics are used during retreatment, cure of infection appears to be superior compared with when only one new antimicrobial is used. One more recently recommended “rescue therapy” includes a PPI, levofloxacin 250 mg, and amoxicillin 1 g, all given twice daily for 10 days. This combination cures infection in up to 80% of patients who have failed one or more prior treatment attempts. Less well studied, but reported to be 85% effective when used as retreatment, is a combination of PPI and amoxicillin 1 g, each twice daily, along with rifabutin 300 mg every day for 10 days. A lower dose of rifabutin (150 mg) appears to be less effective. Successful retreatment with regimens substituting furazolidone for metronidazole has also been reported, but furazolidone is no longer commercially available in the United States.
Primary resistance to antibiotics commonly used to treat H. pylori varies widely throughout the world.138,144,148 In the United States resistance to metronidazole can be detected in up to 40% of stains, whereas clarithromycin resistance is approximately 11%. Resistance to tetracycline and amoxicillin is unusual, generally less than 1%.148,161,162 In the United States, clarithromycin and metronidazole resistance increase with age and are more common in women than in men. Clarithromycin resistance is more common in the mid-Atlantic and northeast regions of the country. Metronidazole resistance is more common in Hispanics and Asians.
Antibiotic resistance significantly affects the success of PPI triple regimens but is less important with bismuth-based regimens.138,149 A bacterial point mutation(s) that prevents reduction of metronidazole to its active metabolite is responsible for drug resistance.138,144 Resistance to metronidazole appears to be a relative condition that can be overcome in most instances by using a higher dose (500 mg) or combining the drug with a bismuth preparation. On the other hand, clarithromycin resistance appears to be an absolute situation that cannot be easily overcome by increasing the macrolide dose. One of three bacterial point mutations within its conserved loop of 23S strand of ribosomal RNA (A2143G, A2142G, and A2142C) can interfere with ribosomal macrolide binding and lead to clarithromycin resistance.138,144 The A2143G mutation appears to have the greatest negative effect on treatment and is likely the major reason for PPI triple therapy failure. Testing for specific mutations is not clinically available, so if clarithromycin resistance is suspected or confirmed by culture, non-macrolide regimens or sequential therapy are appropriate treatment options. Table 50-4 summarizes rescue treatments for persistent infection.
Failed attempts at eradication generally result in secondary antibiotic resistance.148 Therefore, one can assume when treatment with clarithromycin or metronidazole-containing regimens is unsuccessful, specific drug resistance has emerged which should influence any subsequent choice of therapy.138,144
Treatment-related side effects can occur in as many as 50% of patients taking one of the treatment regimens described previously, but generally these are mild and do not require discontinuation of therapy. Some of the more common side effects include taste alteration and gastrointestinal (GI) upset with metronidazole and clarithromycin and allergic reactions and diarrhea with amoxicillin. In addition, tetracycline should not be prescribed to children or pregnant women. Side effects of H. pylori treatment have recently been extensively reviewed elsewhere.150,151
Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306-23. (Ref 5.)
Blaser MJ, Atherton JC. Helicobacter pylori persistence: Biology and disease. J Clin Invest. 2004;113:321-33. (Ref 26.)
Chey WD, Wong BC. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol. 2007;102:1808-25. (Ref 131.)
Correa P, Houghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology. 2007;133:659-72. (Ref 119.)
Ernst PB, Peura DA, Crowe SE. The translation of Helicobacter pylori basic research to patient care. Gastroenterology. 2006;130:188-206. (Ref 2.)
Gillen D, McColl KE. Gastroduodenal disease, Helicobacter pylori, and genetic polymorphisms. Clin Gastroenterol Hepatol. 2005;3:1180-6. (Ref 6.)
Jafri NS, Hornung CA, Howden CW. Meta-analysis: Sequential therapy appears superior to standard therapy for Helicobacter pylori infection in patients naive to treatment. Ann Intern Med. 2008;148:923-31. (Ref 155.)
Jodlowski TZ, Lam S, Ashby CR. Emerging therapies for the treatment of Helicobacter pylori infections. Ann Pharmacother. 2008;42:1621-39. (Ref 151.)
Malfertheiner PF, Megraud C, O’Morain F, et al. Current concepts in the management of Helicobacter pylori infection: The Maastricht III Consensus Report. Gut. 2007;56:772-81. (Ref 129.)
Megraud F, Lehours P. Helicobacter pylori detection and antimicrobial susceptibility testing. Clin Microbiol Rev. 2007;20:280-322. (Ref 138.)
Naumann M, Crabtree JE. Helicobacter pylori–induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 2004;12:29-36. (Ref 46.)
Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175-86. (Ref 4.)
Talley NJ, Vakil N. Guidelines for the management of dyspepsia. Am J Gastroenterol. 2005;100:2324-37. (Ref 132.)
Vakil N, Megraud F. Eradication therapy for Helicobacter pylori. Gastroenterology. 2007;133:985-1001. (Ref 148.)
Versalovic J. Helicobacter pylori. Pathology and diagnostic strategies. Am J Clin Pathol. 2003;119:403-12. (Ref 137.)
1. Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449-90.
2. Ernst PB, Peura DA, Crowe SE. The translation of Helicobacter pylori basic research to patient care. Gastroenterology. 2006;130:188-206.
3. Schubert ML, Peura DA. Control of gastric acid secretion in health and disease. Gastroenterology. 2008;134:1842-60.
4. Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175-86.
5. Amieva MR, El-Omar EM. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology. 2008;134:306-23.
6. Gillen D, McColl KE. Gastroduodenal disease, Helicobacter pylori, and genetic polymorphisms. Clin Gastroenterol Hepatol. 2005;3:1180-6.
7. Megraud F. A humble bacterium sweeps this year’s Nobel Prize. Cell. 2005;123:975-6.
8. Linz B, Balloux F, Moodley Y, et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature. 2007;445:915-18.
9. Kivi M, Tindberg Y. Helicobacter pylori occurrence and transmission: A family affair? Scand. J Infect Dis. 2006;38:407-17.
10. Brown LM. Helicobacter pylori: Epidemiology and routes of transmission. Epidemiol Rev. 2000;22:283-97.
11. Rowland M, Daly L, Vaughan M, et al. Age-specific incidence of Helicobacter pylori. Gastroenterology. 2006;130:65-72.
12. Everhart JE, Kruszon-Moran D, Perez-Perez GI, et al. Seroprevalence and ethnic differences in Helicobacter pylori infection among adults in the United States. J Infect Dis. 2000;181:1359-63.
13. Tsai CJ, Chang MH. Seroepidemiologic study of Helicobacter pylori infection in children in Taipei city. Acta Paediatrica Sinica. 1995;36:254-6.
14. Asaka M, Kimura T, Kudo M, et al. Relationship of Helicobacter pylori to serum pepsinogens in an asymptomatic Japanese population. Gastroenterology. 1992;102:760-6.
15. Rupnow MF, Shachter RD, Owens DK, Parsonnet J. A dynamic transmission model for predicting trends in Helicobacter pylori and associated diseases in the United States. Emerg Infect Dis. 2000;6:228-37.
16. Malaty HM, Graham DY, Isaksson I, et al. Co-twin study of the effect of environment and dietary elements on acquisition of Helicobacter pylori infection. Am J Epidemiol. 1998;148:793-7.
17. Bellack NR, Koehoorn MW, MacNab YC, Morshed MG. A conceptual model of water’s role as a reservoir in Helicobacter pylori transmission: A review of the evidence. Epidemiol Infect. 2006;134:439-49.
18. Weyermann M, Adler G, Brenner H, Rothenbacher D. The mother as source of Helicobacter pylori infection. Epidemiology. 2006;17:332-4.
19. Parsonnet J, Shmuely H, Haggerty T. Fecal and oral shedding of Helicobacter pylori from healthy infected adults [In process citation]. JAMA. 1999;282:2240-5.
20. Perry S, de la Luz SM, Yang S, et al. Gastroenteritis and transmission of Helicobacter pylori infection in households. Emerg Infect Dis. 2006;12:1701-8.
21. Burgers R, Schneider-Brachert W, Reischl U, et al. Helicobacter pylori in human oral cavity and stomach. Eur J Oral Sci. 2008;116:297-304.
22. Lin SK, Lambert JR, Schembri MA, et al. The prevalence of Helicobacter pylori in practising dental staff and dental students. Aust Dent J. 1998;43:35-9.
23. De Schryver AA, Van Hooste WL, Van Winckel MA, Van Sprundel MP. Helicobacter pylori infection: A global occupational risk for healthcare workers? Int J Occup Environ Health. 2004;10:428-32.
24. Dore MP, Malaty HM, Graham DY, et al. Risk factors associated with Helicobacter pylori infection among children in a defined geographic area. Clin Infect Dis. 2002;35:240-5.
25. Lee A. The Helicobacter pylori genome—New insights into pathogenesis and therapeutics. N Engl J Med. 1998;338:832-3.
26. Blaser MJ, Atherton JC. Helicobacter pylori persistence: Biology and disease. J Clin Invest. 2004;113:321-33.
27. Graham JE, Peek RM, Krishna U, Cover TL. Global analysis of Helicobacter pylori gene expression in human gastric mucosa. Gastroenterology. 2002;123:1637-48.
28. Merrell DS, Goodrich ML, Otto G, et al. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect Immun. 2003;71:3529-39.
29. Mobley HLT, Hu LT, Foxall PA. Helicobacter pylori urease: Properties and role in pathogenesis. Scand J Gastroenterol. 1991;187:39-46.
30. Kawakubo M, Ito Y, Okimura Y, et al. Natural antibiotic function of a human gastric mucin against Helicobacter pylori infection. Science. 2004;305:1003-6.
31. Wex T, Treiber G, Nilius M, et al. Helicobacter pylori–mediated gastritis induces local downregulation of secretory leukocyte protease inhibitor in the antrum. Infect Immun. 2004;72:2383-5.
32. Boren T, Falk P, Roth KA, et al. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892-5.
33. Ilver D, Arnqvist A, Ogren J, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373-7.
34. Linden S, Nordman H, Hedenbro J, et al. Strain- and blood group-dependent binding of Helicobacter pylori to human gastric MUC5AC glycoforms. Gastroenterology. 2002;123:1923-30.
35. Mizushima T, Sugiyama T, Komatsu Y, et al. Clinical relevance of the babA2 genotype of Helicobacter pylori in Japanese clinical isolates. J Clin Microbiol. 2001;39:2463-5.
36. Fan XJ, Crowe SE, Behar S, et al. The effect of class II MHC expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: A mechanism for Th1 cell-mediated damage. J Exp Med. 1998;187:1659-69.
37. Fan X, Gunasena H, Cheng Z, et al. Helicobacter pylori urease binds to class II MHC on gastric epithelial cells and induces their apoptosis. J Immunol. 2000;165:1918-24.
38. Clyne M, Dillon P, Daly S, et al. Helicobacter pylori interacts with the human single-domain trefoil protein TFF1. Proc Natl Acad Sci U S A. 2004;101:7409-14.
39. Lefebvre O, Chenard MP, Masson R, et al. Gastric mucosa abnormalities and tumorigenesis in mice lacking the pS2 trefoil protein. Science. 1996;274:259-62.
40. Tebbutt NC, Giraud AS, Inglese M, et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nat Med. 2002;8:1089-97.
41. Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135-45.
42. Su B, Ceponis PJ, Lebel S, et al. Helicobacter pylori activates Toll-like receptor 4 expression in gastrointestinal epithelial cells. Infect Immun. 2003;71:3496-502.
43. Smith MF, Mitchell AJr, Li G, et al. TLR2 and TLR5, but not TLR4, are required for Helicobacter pylori–induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem. 2003;278:32552-60.
44. Mandell L, Moran AP, Cocchiarella A, et al. Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun. 2004;72:6446-54.
45. Gewirtz AT, Yu Y, Krishna US, et al. Helicobacter pylori flagellin evades toll-like receptor 5-mediated innate immunity. J Infect Dis. 2004;189:1914-20.
46. Naumann M, Crabtree JE. Helicobacter pylori–induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 2004;12:29-36.
47. Elliott SN, Ernst PB, Kelly CP. The year in Helicobacter pylori 2001: Molecular inflammation. Curr Opin Gastroenterol. 2002;17:s12-18.
48. Israel DA, Salama N, Arnold CN, et al. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest. 2001;107:611-20.
49. Ogura K, Maeda S, Nakao M, et al. Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J Exp Med. 2000;192:1601-10.
50. Day AS, Jones NL, Lynett JT, et al. cagE is a virulence factor associated with Helicobacter pylori–induced duodenal ulceration in children. J Infect Dis. 2000;181:1370-5.
51. Yahiro K, Wada A, Nakayama M, et al. Protein-tyrosine phosphatase alpha, RPTP alpha, is a Helicobacter pylori VacA receptor. J Biol Chem. 2003;278:19183-9.
52. Cover TL. The vacuolating cytotoxin of Helicobacter pylori. Mol Microbiol. 1996;20:241-6.
53. van Doorn LJ, Figueiredo C, Megraud F, et al. Geographic distribution of vacA allelic types of Helicobacter pylori. Gastroenterology. 1999;116:823-30.
54. Fujikawa A, Shirasaka D, Yamamoto S, et al. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat Genet. 2003;33:375-81.
55. Atherton JC, Cao P, Peek RM, et al. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771-7.
56. Cover TL, Krishna US, Israel DA, Peek RM. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951-7.
57. Macarthur M, Hold GL, El-Omar EM. Inflammation and cancer II. Role of chronic inflammation and cytokine gene polymorphisms in the pathogenesis of gastrointestinal malignancy. Am J Physiol Gastrointest Liver Physiol. 2004;286:G515-20.
58. El-Omar E, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398-402.
59. Furuta T, El-Omar EM, Xiao F, et al. Interleukin 1beta polymorphisms increase risk of hypochlorhydria and atrophic gastritis and reduce risk of duodenal ulcer recurrence in Japan. Gastroenterology. 2002;123:92-105.
60. El-Omar EM, Rabkin CS, Gammon MD, et al. Chow. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193-201.
61. Machado JC, Figueiredo C, Canedo P, et al. A proinflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology. 2003;125:364-71.
62. Segal ED, Cha J, Lo J, et al. Altered states: Involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proc Natl Acad Sci U S A. 1999;96:14559-64.
63. Amieva MR, Vogelmann R, Covacci A, et al. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300:1430-4.
64. Maeda S, Yoshida H, Ogura K, et al. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology. 2000;119:97-108.
65. Keates S, Keates AC, Warny M, et al. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- Helicobacter pylori. J Immunol. 1999;163:5552-9.
66. Naumann M, Wessler S, Bartsch C, et al. Activation of activator protein 1 and stress response kinases in epithelial cells colonized by Helicobacter pylori encoding the cag pathogenicity island. J Biol Chem. 1999;274:31655-62.
67. Takahashi S, Keto Y, Fujita T, et al. FR167653, a p38 mitogen-activated protein kinase inhibitor, prevents Helicobacter pylori–induced gastritis in Mongolian gerbils. J Pharmacol Exp Ther. 2001;296:48-56.
68. Ding SZ, OHara AM, Denning TL, et al. Helicobacter pylori and H2O2 increases AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology. 2004;127:845-58.
69. O’Hara AM, Bhattacharyya A, Mifflin RC, et al. Interleukin-8 induction by Helicobacter pylori in human gastric epithelial cells is dependent on apurinic/apyrimidinic endonuclease-2/redox factor-1. J Immuol. 2006;177:7990-9.
70. Serafini M, Bellocco R, Wolk A, Ekstrom AM. Total antioxidant potential of fruit and vegetables and risk of gastric cancer. Gastroenterology. 2002;123:985-91.
71. Fahey JW, Haristoy X, Dolan PM, et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci U S A. 2002;99:7610-15.
72. Stein M, Rappuoli R, Covacci A. Tyrosine phosphorylation of the Helicobacter pylori CagA antigen after cag-driven host cell translocation. Proc Natl Acad Sci U S A. 2000;97:1263-8.
73. Higashi H, Tsutsumi R, Muto S, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295:683-6.
74. Selbach M, Moese S, Hauck CR, et al. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem. 2002;277:6775-8.
75. Tanaka J, Suzuki T, Mimuro H, Sasakawa C. Structural definition on the surface of Helicobacter pylori type IV secretion apparatus. Cell Microbiol. 2003;5:395-404.
76. Tsutsumi R, Higashi H, Higuchi M, et al. Attenuation of Helicobacter pylori CagA x SHP-2 signaling by interaction between CagA and C-terminal Src kinase. J Biol Chem. 2003;278:3664-70.
77. Higashi H, Tsutsumi R, Fujita A, et al. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci U S A. 2002;99:14428-33.
78. Higashi H, Nakaya A, Tsutsumi R, et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205-16.
79. Hirata Y, Maeda S, Mitsuno Y, et al. Helicobacter pylori CagA protein activates serum response element-driven transcription independently of tyrosine phosphorylation. Gastroenterology. 2002;123:1962-71.
80. El Etr SH, Mueller A, Tompkins LS, et al. Phosphorylation-independent effects of CagA during interaction between Helicobacter pylori and T84 polarized monolayers. J Infect Dis. 2004;190:1516-23.
81. Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci U S A. 2000;97:7533-8.
82. Yamaoka Y, Kikuchi S, El Zimaity HM, et al. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology. 2002;123:414-24.
83. Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166-74.
84. Satin B, Del Giudice G, Della B, et al. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a protective antigen and a major virulence factor. J Exp Med. 2000;191:1467-76.
85. Rieder G, Einsiedl W, Hatz RA, et al. Comparison of CXC chemokines ENA-78 and interleukin-8 expression in Helicobacter pylori–associated gastritis. Infect Immun. 2001;69:81-8.
86. Eck M, Schmausser B, Scheller K, et al. CXC chemokines Gro(alpha)/IL-8 and IP-10/MIG in Helicobacter pylori gastritis. Clin Exp Immunol. 2000;122:192-9.
87. Harris PR, Mobley HLT, Perez-Perez GI, et al. Helicobacter pylori urease is a potent stimulus of mononuclear phagocyte activation and inflammatory cytokine production. Gastroenterology. 1996;111:419-25.
88. Gobert AP, Bambou JC, Werts C, et al. Helicobacter pylori heat shock protein 60 mediates interleukin-6 production by macrophages via a toll-like receptor (TLR)-2-, TLR-4-, and myeloid differentiation factor 88-independent mechanism. J Biol Chem. 2004;279:245-50.
89. Shimoyama T, Everett SM, Dixon MF, et al. Chemokine mRNA expression in gastric mucosa is associated with Helicobacter pylori cagA positivity and severity of gastritis. J Clin Pathol. 1998;51:765-70.
90. Haeberle HA, Kubin M, Bamford KB, et al. Differential stimulation of interleukin-12 (IL-12) and IL-10 by live and killed Helicobacter pylori in vitro and association of IL-12 production with gamma interferon-producing T cells in the human gastric mucosa. Infect Immun. 1997;65:4229-35.
91. Hafsi N, Voland P, Schwendy S, et al. Human dendritic cells respond to Helicobacter pylori, promoting NK cell and Th1-effector responses in vitro. J Immunol. 2004;173:1249-57.
92. Tomita T, Jackson AM, Hida N, et al. Expression of interleukin-18, a Th1 cytokine, in human gastric mucosa is increased in Helicobacter pylori infection. J Infect Dis. 2001;183:620-7.
93. Ceponis PJ, McKay DM, Menaker RJ, et al. Helicobacter pylori infection interferes with epithelial Stat6-mediated interleukin-4 signal transduction independent of cagA, cagE, or VacA. J Immunol. 2003;171:2035-41.
94. Luzza F, Parrello T, Monteleone G, et al. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori–infected human gastric mucosa. J Immunol. 2000;165:5332-7.
95. Sebkova L, Pellicano A, Monteleone G, et al. Extracellular signal-regulated protein kinase mediates interleukin 17 (IL-17)-induced IL-8 secretion in Helicobacter pylori–infected human gastric epithelial cells. Infect Immun. 2004;72:5019-26.
96. Lehmann FS, Terracciano L, Carena I, et al. In situ correlation of cytokine secretion and apoptosis in Helicobacter pylori–associated gastritis. Am J Physiol Gastrointest Liver Physiol. 2002;283:G481-8.
97. Zavros Y, Rathinavelu S, Kao JY, et al. Treatment of Helicobacter gastritis with interleukin-4 requires somatostatin. Proc Natl Acad Sci. 2003;100:12944-9.
98. Wang J, Fan XJ, Lindholm C, et al. Helicobacter pylori modulates lymphoepithelial cell interactions leading to epithelial cell damage through Fas/Fas Ligand interactions. Infect. Immun. 2000;68:4303-11.
99. Rudi J, Kuck D, Strand S, et al. Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori–induced gastric epithelial apoptosis. J Clin Invest. 1998;102:1506-14.
100. D’Elios MM, Bergman MP, Azzurri A, et al. H(+),K(+)-atpase (proton pump) is the target autoantigen of Th1-type cytotoxic T cells in autoimmune gastritis. Gastroenterology. 2001;120:377-386.
101. Fox JG, Beck P, Dangler CA, et al. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces Helicobacter-induced gastric atrophy. Nat Med. 2000;6:536-42.
102. Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161:72-87.
103. Okazaki K, Ohana M, Oshima C, et al. Interaction of Helicobacter pylori–induced follicular gastritis and autoimmune gastritis in BALB/c mice with post-thymectomy autoimmune gastritis. J Gastroenterol. 2003;38:1131-7.
104. Lundgren A, Suri-Payer E, Enarsson K, et al. Helicobacter pylori–specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun. 2003;71:1755-62.
105. Negrini R, Lisato L, Zanella I, et al. Helicobacter pylori infection induces antibodies cross-reacting with human gastric mucosa. Gastroenterology. 1991;101:437-45.
106. Appelmelk BJ, Simoons-Smit I, Negrini R, et al. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect Immun. 1996;64:2031-40.
107. Yokota K, Kobayashi K, Kawahara Y, et al. Gastric ulcers in SCID mice induced by Helicobacter pylori infection after transplanting lymphocytes from patients with gastric lymphoma. Gastroenterology. 1999;117:893-9.
108. Gobert AP, McGee DJ, Akhtar M, et al. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A. 2001;98:13844-9.
109. Byrd JC, Yunker CK, Xu QS, et al. Inhibition of gastric mucin synthesis by Helicobacter pylori. Gastroenterology. 2000;118:1072-9.
110. Allen LA, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med. 2000;191:115-28.
111. Molinari M, Salio M, Galli C, et al. Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin VacA. J Exp Med. 1998;187:135-40.
112. Koyama S. Apoptotic depletion of infiltrating mucosal lymphocytes associated with Fas ligand expression by Helicobacter pylori–infected gastric mucosal epithelium: human glandular stomach as a site of immune privilege. Dig Dis Sci. 2000;45:773-80.
113. Betten A, Bylund J, Cristophe T, et al. A proinflammatory peptide from Helicobacter pylori activates monocytes to induce lymphocyte dysfunction and apoptosis. J Clin Invest. 2001;108:1221-8.
114. Wang J, Brooks EG, Bamford KB, et al. Negative selection of T cells by Helicobacter pylori as a model for bacterial strain selection by immune evasion. J Immunol. 2001;167:926-34.
115. Gebert B, Fischer W, Weiss E, et al. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099-102.
116. Boncristiano M, Paccani SR, Barone S, et al. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med. 2003;198:1887-97.
117. Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci U S A. 2004;101:7727-32.
118. Zabaleta J, McGee DJ, Zea AH, et al. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR zeta-chain (CD3zeta). J Immunol. 2004;173:586-93.
119. Correa P, Houghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology. 2007;133:659-72.
120. Fennerty MB. Gastric intestinal metaplasia on routine endoscopic biopsy. Gastroenterology. 2003;125:586-90.
121. Malfertheiner P, Peitz U. The interplay between Helicobacter pylori, gastro-oesophageal reflux disease, and intestinal metaplasia. Gut. 2005;54(Suppl 1):i13-20.
122. McColl KE. Helicobacter pylori and oesophageal cancer—Not always protective. Gut. 2007;56:457-9.
123. Huang JQ, Zheng GF, Sumanac K, et al. Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology. 2003;125:1636-44.
124. Goodman KJ, Joyce SL, Ismond KP. Extragastric diseases associated with Helicobacter pylori infection. Curr Gastroenterol Rep. 2006;8:458-64.
125. Franchini M, Veneri D. Helicobacter pylori–associated immune thrombocytopenia. Platelets. 2006;17:71-7.
126. Asahi A, Nishimoto T, Okazaki Y, et al. Helicobacter pylori eradication shifts monocyte Fcgamma receptor balance toward inhibitory FcgammaRIIB in immune thrombocytopenia purpura patients. J Clin Invest. 2008;118:2939-49.
127. DuBois S, Kearney DJ. Iron-deficiency anemia and Helicobacter pylori infection: a review of the evidence. Am J Gastroenterol. 2005;100:453-9.
128. NIH Consensus Conference. Helicobacter pylori in peptic ulcer disease. NIH Consensus Development Panel on Helicobacter pylori in Peptic Ulcer Disease. JAMA. 1994;272:65-9.
129. Malfertheiner P, Megraud F, O’Morain C, et al. Current concepts in the management of Helicobacter pylori infection: The Maastricht III Consensus Report. Gut. 2007;56:772-81.
130. Fujioka T, Yoshiiwa A, Okimoto T, et al. Guidelines for the management of Helicobacter pylori infection in Japan: Current status and future prospects. J Gastroenterol. 2007;17:3-6.
131. Chey WD, Wong BC. American College of Gastroenterology guideline on the management of Helicobacter pylori infection. Am J Gastroenterol. 2007;102:1808-25.
132. Talley NJ, Vakil N. Guidelines for the management of dyspepsia. Am J Gastroenterol. 2005;100:2324-37.
133. Moayyedi P, Deeks J, Talley NJ, et al. An update of the Cochrane systematic review of Helicobacter pylori eradication therapy in nonulcer dyspepsia: Resolving the discrepancy between systematic reviews. Am J Gastroenterol. 2003;98:2621-6.
134. Chey WD, Moayyedi P. Review article: Uninvestigated dyspepsia and non-ulcer dyspepsia—the use of endoscopy and the roles of Helicobacter pylori eradication and antisecretory therapy. Aliment Pharmacol Ther. 2004;1:1-8.
135. Malfertheiner P, Sipponen P, Naumann M, et al. Helicobacter pylori eradication has the potential to prevent gastric cancer: A state-of-the-art critique. Am J Gastroenterol. 2005;100:2100-15.
136. Rostami N, Keshtkar-Jahromi M, Rahnavardi M, et al. Effect of eradication of Helicobacter pylori on platelet recovery in patients with chronic idiopathic thrombocytopenic purpura: A controlled trial. Am J Hematol. 2008;83:376-81.
137. Versalovic J. Helicobacter pylori. Pathology and diagnostic strategies. Am J Clin Pathol. 2003;119:403-12.
138. Megraud F, Lehours P. Helicobacter pylori detection and antimicrobial susceptibility testing. Clin Microbiol Rev. 2007;20:280-322.
139. Midolo P, Marshall BJ. Accurate diagnosis of Helicobacter pylori. Urease tests. Gastroenterol Clin North Am. 2000;29:871-8.
140. Gisbert JP, Abraira V. Accuracy of Helicobacter pylori diagnostic tests in patients with bleeding peptic ulcer: A systematic review and meta-analysis. Am J Gastroenterol. 2006;101:848-63.
141. Gatta L, Vakil N, Ricci C, et al. Effect of proton pump inhibitors and antacid therapy on 13C urea breath tests and stool test for Helicobacter pylori infection. Am J Gastroenterol. 2004;99:823-9.
142. el-Zimaity HM. Accurate diagnosis of Helicobacter pylori with biopsy. Gastroenterol Clin North Am. 2000;29:863-9.
143. Wright CL, Kelly JK. The use of routine special stains for upper gastrointestinal biopsies. Am J Surg Pathol. 2006;30:357-61.
144. Gerrits MM, van Vliet AH, Kuipers EJ, Kusters JG. Helicobacter pylori and antimicrobial resistance: Molecular mechanisms and clinical implications. Lancet Infect Dis. 2006;6:699-709.
145. Marchildon P, Balaban DH, Sue M, et al. Usefulness of serological IgG antibody determinations for confirming eradication of Helicobacter pylori infection. Am J Gastroenterol. 1999;94:2105-8.
146. Quesada M, Calvet X, Dosal A, et al. Evaluation of four different fecal tests for determination of cure after Helicobacter pylori treatment. J Clin Gastroenterol. 2006;40:790-4.
147. Chisholm SA, Owen RJ. Application of polymerase chain reaction-based assays for rapid identification and antibiotic resistance screening of Helicobacter pylori in gastric biopsies. Diagn Microbiol Infect Dis. 2008;61:67-71.
148. Vakil N, Megraud F. Eradication therapy for Helicobacter pylori. Gastroenterology. 2007;133:985-1001.
149. Megraud F. Hpylori antibiotic resistance: Prevalence, importance, and advances in testing. Gut. 2004;53:1374-84.
150. Peura DA. Treatment of Helicobacter pylori infection. In: Wolfe MM, editor. Therapy of digestive disorders. Philadelphia: Elsevier; 2006:277.
151. Jodlowski TZ, Lam S, Ashby CR. Emerging therapies for the treatment of Helicobacter pylori infections. Ann Pharmacother. 2008;42:1621-39.
152. Gisbert JP, Castro-Fernandez M, Bermejo F, et al. Third-line rescue therapy with levofloxacin after two H. pylori treatment failures. Am J Gastroenterol. 2006;101:243-7.
153. Gisbert JP, Gonzalez L, Calvet X, et al. Proton pump inhibitor, clarithromycin and either amoxycillin or nitroimidazole: A meta-analysis of eradication of helicobacter pylori [In process citation]. Aliment Pharmacol Ther. 2000;14:1319-28.
154. Vaira D, Zullo A, Vakil N, et al. Sequential therapy versus standard triple-drug therapy for Helicobacter pylori eradication: A randomized trial. Ann Intern Med. 2007;146:556-63.
155. Jafri NS, Hornung CA, Howden CW. Meta-analysis: Sequential therapy appears superior to standard therapy for Helicobacter pylori infection in patients naive to treatment. Ann Intern Med. 2008;148:923-31.
156. Laine L, Hunt R, El-Zimaity H, et al. Bismuth-based quadruple therapy using a single capsule of bismuth biskalcitrate, metronidazole, and tetracycline given with omeprazole versus omeprazole, amoxicillin, and clarithromycin for eradication of Helicobacter pylori in duodenal ulcer patients: A prospective, randomized, multicenter, North American trial. Am J Gastroenterol. 2003;98:562-7.
157. Lara LF, Cisneros G, Gurney M, et al. One-day quadruple therapy compared with 7-day triple therapy for Helicobacter pylori infection. Arch Intern Med. 2003;163:2079-84.
158. Broutet N, Tchamgoue S, Pereira E, et al. Risk factors for failure of Helicobacter pylori therapy—Results of an individual data analysis of 2751 patients. Aliment Pharmacol Ther. 2003;17:99-109.
159. Suzuki T, Matsuo K, Ito H, et al. Smoking increases the treatment failure for Helicobacter pylori eradication. Am J Med. 2006;119:217-24.
160. Kearney DJ. Retreatment of Helicobacter pylori infection after initial treatment failure. Am J Gastroenterol. 2001;96:1335-9.
161. Osato MS, Reddy R, Reddy SG, et al. Pattern of primary resistance of Helicobacter pylori to metronidazole or clarithromycin in the United States. Arch Intern Med. 2001;161:1217-20.
162. Meyer JM, Silliman NP, Wang W, et al. Risk factors for Helicobacter pylori resistance in the United States: the surveillance of H. pylori antimicrobial resistance partnership (SHARP) study, 1993-1999. Ann Intern Med. 2002;136:13-24.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 50 Helicobacter pylori
Helicobacter pylori are unique bacteria ideally suited to live in the acidic environment of the human stomach. Their spiral shape and multiple unipolar flagella allow them to move freely through the gastric mucous layer, where they remain protected from low gastric pH.1 Organisms produce large amounts of urease, an enzyme that hydrolyzes urea to alkaline ammonia and CO2. This permits the bacteria to further control the pH of their microenvironment. Urease is also the basis of clinical diagnostic tests (urea breath test and rapid urea biopsy tests) for infection. H. pylori remain difficult and tedious to culture because they grow slowly and require specialized culture media and a controlled microaerophilic environment.
When they gain access to a human host, H. pylori recognize and attach to various gastric epithelial surface receptors, thereby chronically colonizing the mucosa, disrupting cell function, inciting an intense local inflammatory and systemic immune response, and altering acid secretory physiology.2,3 The ultimate clinical manifestations of H. pylori infection include gastric and duodenal ulcer, gastric mucosa–associated lymphoid tissue (MALT) lymphoma, and adenocarcinoma; yet most infected individuals remain asymptomatic for life despite developing chronic histologic gastritis.1,2,4,5 What factors determine why some develop disease in response to infection and others do not remains a mystery, but host genetics, bacterial characteristics, and environmental features undoubtedly can influence clinical outcome.5,6 Research focusing on epidemiology, pathogenesis, management, and prevention of H. pylori infection and associated clinical conditions continues to be fueled by the tremendous worldwide prevalence of infection (especially in less-developed countries), the huge health and economic burden imposed by ulcer disease and gastric cancer, and heightened prominence and awareness of the bacteria accompanying Drs. Robin Warren and Barry Marshall’s receipt of the 2005 Nobel Prize in Physiology or Medicine for seminal contributions to the field.7
EPIDEMIOLOGY
Helicobacter pylori infection remains one of the most common chronic bacterial infections in humans. Estimates suggest that more than 50% of the world’s population is infected with the bacterium and genetic sequence analysis proposes that humans have been infected for more than 58,000 years at a time when they first migrated from Africa.8
While H. pylori have been demonstrated worldwide in individuals of all ages, infection is more common and acquired at an earlier age in developing countries compared with industrialized nations.9,10 In developing nations, the majority of children become infected before the age of 10, and during early childhood spontaneous elimination of bacteria and subsequent reinfection is quite common. Infection persists in older children and adults so that in the developing areas of the world H. pylori prevalence can reach more than 80% by age 50. In developed countries, such as the United States, young children can also acquire H. pylori, but usually before age 5.11 Spontaneous clearance often occurs and there is less chance of reinfection; thus, persistent childhood infection is much less frequently seen than in less-developed countries.9 In fact, serologic evidence of H. pylori is uncommon in children before age 10, but rises to 10% in adults between 18 and 30 years of age and further increases to 50% in those 60 or older.9 This increased prevalence of infection with age was initially thought to represent continuing acquisition throughout adult life. However, new adult infection and reinfection are quite uncommon, especially in developed countries. Epidemiologic evidence supports childhood-acquired infection even in developed nations, so the frequency of H. pylori infection for any age group in any locality reflects that particular birth cohort’s rate of bacterial acquisition early in life.9 In the United States, within any age group, infection appears to be more common in blacks than whites.12 Also Hispanic immigrants and their first-generation children are more likely to harbor H. pylori than their second-generation relatives.13 These differences probably relate to factors early in life that are linked to acquiring infection.
The risk of acquiring H. pylori is associated with living conditions and the family’s socioeconomic status during one’s childhood.9 Housing density, crowded conditions in the home, number of siblings, sharing a bed, and lack of hot or running water have been linked to higher rates of infection. In Japan, the rapidly declining prevalence of H. pylori appears to parallel the nation’s postwar economic progress and improvement in hygiene and sanitation. Of the Japanese born before 1950, more than 70% are infected compared with 45% born between 1950 and 1960 and 25% born between 1960 and 1970.14 Presently, childhood infection in Japan is rare. Predictions based on a similar declining prevalence in the United States suggest that the organism could eventually become extinct here and in other areas of the world, which would affect infection-related illness.15
Twin studies support genetic susceptibility to H. pylori infection because monozygotic twins who were raised in different households have a greater concordance of infection than dizygotic twins also raised separately.16 However, twins growing up together have a higher concordance of H. pylori status than twins growing up separately, suggesting childhood environmental factors are also important for acquisition.
Humans appear to be the major reservoir of H. pylori. However, domestic cats and captive primates and sheep can harbor these organisms,10 although it is possible that these animals actually acquired H. pylori from a human source. In the case of cats, isolation of viable bacteria from saliva and gastric juice suggests at least the possibility of transmission to humans.10
Especially in developing countries, contaminated water might serve as an environmental source of bacteria because the organism can remain viable for several days in water.17 Bacterial deoxyribonucleic acid (DNA) can be found in samples of municipal water from endemic areas of infection but whether viable H. pylori are present remains to be proven.5 In countries where infection is common, children who drink untreated stream water, eat uncooked vegetables, or swim in rivers and streams are more likely to harbor the bacteria, providing further indirect evidence of an environmental source of organisms.
Person-to-person transmission of bacteria from fecal-oral, oral-oral, or gastro-oral exposure seems the most probable explanation for infection.5,10 Within-family clustering of infection (often with genetically identical strains of H. pylori) supports person-to-person transmission.9 Also infected individuals more often have infected spouses or children than uninfected individuals. Support for sibling-to-sibling transmission comes from studies reporting that likelihood of infection correlated with number of children in the household and that younger children were more apt to be infected if older siblings were also infected.9 Mother-to-child transmission is also quite likely.11,18
Fecal-oral transmission of bacteria is a possible mechanism by which H. pylori gain access to the human host. The bacterium can be cultured from diarrheal stools and vomitus, suggesting the potential for transmission.19 Exposure to an infected family member during an acute gastrointestinal illness, especially with vomiting, appears to be a risk factor for subsequent infection.20
How frequently bacteria are transmitted through oral-oral contact is not known. Although organisms can be identified in dental plaque and saliva, the prevalence is low and it is questionable if the mouth serves as a source or reservoir for H. pylori.21 Also dentists and oral hygienists who continually have occupational exposure to dental plaque and oral secretions do not have a higher prevalence of H. pylori.22 In developed countries, spousal transmission of infection also appears to be uncommon.
Infected gastric secretions can serve as a source of bacterial transmission. Iatrogenic infection has occurred during the use of a variety of inadequately disinfected gastric devices, endoscopes, and endoscopic accessories.10 Also gastroenterologists and nurses appear to be at greater risk for acquiring H. pylori, presumably due to occupational contact with infected gastric secretions.23 Mandated universal precautions, standardized equipment disinfection, and use of video-endoscopes that reposition the instrument channel away from the mouth should reduce such iatrogenic and occupational transmission. Natural transmission could occur through contact with infected vomitus during an acute illness20 or with regurgitated material from an infected child. Such contact could explain the higher concordance of maternal/child H. pylori infection and the presumed child-to-child transmission that occurs in an infant daycare setting.24
COLONIZATION AND VIRULENCE FACTORS
One of the interesting aspects of this pathogen is how it confers disease when the organism resides, for the most part, in the lumen. Studies describing the genome of two distinct strains of H. pylori have helped to advance our understanding of the ecology of the organism and the potential gene expression patterns that can affect the pathogenesis of disease.25,26 Importantly, bacterial genes expressed in gastric mucosa differ from the pattern observed in vitro,27 whereas exposure of the bacterium to low pH increases its expression of genes encoding proteins involved in the motility apparatus and of genes encoding urease.28 The urease helps the organism adapt to the gastric milieu as it retains optimal function at two different pH values: usually pH 7.2 and pH 3.29 H. pylori is further adapted to the gastric pH by producing the molecular machinery required to migrate rapidly to a more favorable environment below the mucus layer. Motility is one of the few H. pylori characteristics shown to be necessary for successful colonization of the host.
H. pylori show a strict tropism for the gastric mucosa or intestinal sites in which there is gastric metaplasia. The corollary is also true, as H. pylori do not colonize epithelium in the stomach that has undergone intestinal metaplastic change, possibly due to the production of antimicrobial factors that select against colonization. This possibility is supported by the fact that H. pylori rarely colonize the deeper portions of the gastric glandular mucosa, where O-glycans that impair H. pylori growth are found.30 This concept is also supported by another study that shows H. pylori decreases the expression of the antibacterial molecule, secretory leukocyte protease inhibitor,31 thereby removing an element of the host response that would be detrimental to the persistent infection.
Another important factor that controls colonization is the expression of receptors on host cells that allow H. pylori to bind. Lewis (Le) antigens expressed by host cells serve as a receptor for bacterial binding.32 Specific bacterial gene products, most notably BabA, act as the bacterial ligand for the Leb receptor.33 The Leb receptor may be located in the gastric mucus because strains bearing BabA also bind to the mucin MUC5AC.34 Some studies suggest that the babA2 genotype is more frequently associated with inflammation, duodenal ulcer and gastric cancer.2 However, observations that binding of H. pylori to epithelial cells freshly isolated from human gastric biopsy specimens is unaffected by the expression of Le antigen, that infection is not increased in subjects with the Leb phenotype, and that individuals who do not express Leb can clearly be infected with H. pylori cast doubt on the true role of Leb and BabA.2 Additionally, in one report, the majority of strains infecting the individuals studied did not induce ulcers or cancer, despite expressing the babA2 gene.35
H. pylori also bind to the molecular complex of invariant chain and class II human leukocyte antigens (HLAs) expressed on the surface of gastric epithelial cells.36 Class II major histocompatibility complex (MHC) molecules, with their expression increased by infection, were the first epithelial cell receptor for H. pylori demonstrated to directly affect signaling in host cells. Binding of urease to epithelial cells via class II MHC was sufficient to induce apoptosis.37 More recently, the gastric trefoil protein TFF1 was shown to serve as a receptor for H. pylori.38 This molecule is predominantly expressed in the gastric mucosa and found in association with gastric mucus. In genetically engineered mice with TFF1 deficiency, a spontaneous, antral adenoma develops, suggesting that this molecule provides an element of control over gastric epithelial cell growth.39,40
The pathogen-associated molecular receptors (PAMPS) have also been examined for their role in binding of H. pylori to the host epithelial cells. The Toll-like receptors (TLRs) are a family of PAMPS, with an apparent different specificity for various bacterial molecules.41 For example, TLR4 is able to recognize the LPS of many bacteria, with cytokines and H. pylori particles increasing the expression of TLR4.42 H. pylori LPS stimulates monocytes and gastric epithelial cell responses via TLR4.2 Other PAMPs, including TLR2, are also activated by highly purified H. pylori LPS and have even been described as being more important than TLR4.43,44 These receptors may bind bacterial products, and thereby enhance bacterial binding and cell signaling.42 TLR5 binds bacterial flagellins and, similar to TLR2, induces a signaling response that can trigger acute inflammation. H. pylori produce flagellin that binds TLR5 and activates a response in vitro.43 There is disagreement on which TLR is stimulated by H. pylori LPS or whether H. pylori flagellin is ever able to bind TLR5,45 and this may reflect the different conditions used for bacterial culture and LPS or flagellin purification. Further studies are needed to establish the significance of H. pylori binding to TLRs in the pathogenesis of infection.
After H. pylori migrate to the gastric epithelium, the organism attaches to host cells and may damage them in order to obtain nutrients from the subsequent inflammatory exudate or transudate. A key interaction between the bacteria and gastric epithelium involves a segment of bacterial DNA referred to as the cag pathogenicity island (cag PAI). Genes within the cag PAI encode proteins that provide a type IV secretion apparatus (i.e., cagE) that allows bacterial macromolecules to translocate into the host cell (i.e., cagA).26,46 cag PAI plays an important role in the pathogenesis of gastritis in humans26,47 because H. pylori bearing the cag PAI are associated with increased interleukin-8 (IL-8) expression and inflammation in gastric mucosal biopsy specimens and increased IL-8 expression and apoptosis in vitro.48 Infecting gerbils with mutated strains lacking cagE reduces the severity of gastritis and the development of gastric ulcers, intestinal metaplasia, and gastric cancer compared with gerbils infected with the wild-type strain.48,49 Human studies in which duodenal ulceration occurred more frequently in children carrying strains expressing cagE associated with higher levels of gastric IL-850 corroborate animal and in vitro studies.
All strains of H. pylori possess the vacA gene, with more than half expressing the vacuolating cytotoxin (VacA), which attaches to epithelial cells via an interaction with protein-tyrosine phosphatases.51 Although the majority of the VacA is secreted, some may remain on the surface of the bacteria and serve as a ligand for bacterial attachment via this protein-tyrosine phosphatase receptor. Several studies have examined the structure and function of VacA and its association with disease.26,52,53 For example, mice deficient in protein-tyrosine phosphatase beta do not develop ulceration when exposed to VacA.54 Different vacA alleles have been classified in the 5′ signal region (s-region) and the middle region (m-region) of the vacA gene.52 The s-region is present as s1 (which can be further distinguished as s1a, s1b, s1c) or s2, whereas the m-region is present as m1 or m2. Production of VacA is designated by the allelic combination s1/m1 and s1/m2. Specific vacA alleles (s1 and m1) are associated with disease55 and the induction of epithelial cell apoptosis.56 The interaction between VacA and its receptor(s) appears to be important in the pathogenesis of gastroduodenal disease, whether it serves as a ligand for bacterial attachment or as a secreted virulence factor.
HOST RESPONSE TO INFECTION
Increasing evidence suggests that the host response to H. pylori infection is an intrinsic component of the pathogenesis of gastrointestinal disease (Chapters 29, 52, and 54). The possibility that the host response may play a direct role in gastric cancer is supported by the observation that heterogeneity in the regions of the genome that control the magnitude of inflammation is associated with cancer linked to H. pylori infection.57 Polymorphisms in the regions controlling IL-1β58 were shown to be associated with an increased incidence of hypochlorhydria and gastric cancer. This seminal observation has been replicated in other studies in which IL-1β polymorphisms were associated not only with gastric cancer but also a decrease in recurrence of duodenal ulcer.59 An increase in IL-1 may not only drive inflammation but also lead to a physiologic state known to precede gastric cancer development because IL-1 potently inhibits gastric acid secretion. Increased gastric IL-1, more severe gastritis, gastric atrophy, and greater colonization with H. pylori strains have been associated with gastric cancer.2 Other genes that regulate the magnitude of the inflammatory response, including IL-10, tumor necrosis factor-α (TNF-α), and IL-8, have also been associated with the sequence of events leading to cancer.60,61
It is apparent that epithelial cells play an integral part in the host response to H. pylori infection as well as being the target of infection. The epithelial response to infection is complex, as it is driven by several variables: bacterial virulence factors; the signaling linked to specific receptors that recognize the bacterial components; and the local milieu of hormones, neurotransmitters, immune or inflammatory cytokines and mediators, as well as stromal factors. The epithelial cell responses include changes in epithelial cell morphology (the hummingbird phenotype),62 disruption of the tight junctional complexes,63 the production of cytokines,46 increased epithelial cell proliferation, increased rates of epithelial cell death via apoptosis, and the induction of numerous genes associated with the stress encountered in response to infection.2 The best overview of the epithelial cell response can be ascertained from the broad changes in the expression of hundreds of genes demonstrated with high-throughput gene expression systems.2 Detailed analyses of the effects of infection, different cell lines and various inflammatory mediators have yet to be carried out over the wide range of time points that would be relevant to the conditions epithelial cells face in vivo.
The expression of genes in epithelial cells stimulated with H. pylori is regulated by transcription factors that are controlled by a series of signaling mechanisms. While many transcription factors are likely activated by infection, the most studied are nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1) which regulate the expression of a wide variety of proinflammatory cytokines and cellular adhesion molecules in response to infection or the local cytokine milieu. H pylori activates NF-κB in gastric epithelial cells, both in vitro and in vivo in patients with H. pylori gastritis; gastric epithelial cell NF-κB activity is markedly enhanced, correlating with the intensity of neutrophil infiltration and IL-8 protein levels.46 This pathway is of particular interest given the recent report that polymorphisms in the IL-8 gene lead to increased mucosal IL-8 expression, inflammation, and other premalignant changes associated with gastric cancer. H. pylori infection appears to activate NF-κB and AP-1 in gastric epithelial cell lines through various signaling mechanisms including mitogen-activated protein (MAP) kinases.47,64 The MAP kinase cascades regulate a wide range of cell functions, including proliferation, inflammatory responses, and cell survival. For example, cag PAI-positive H. pylori activate the ERK, JNK, and p38 MAP kinase pathways, and ERK and p38 regulate IL-8 production in gastric epithelial cells.65,66 An inhibitor of p38α MAP kinase, FR167653, reduced both neutrophil infiltration and gastric mucosal injury in H. pylori–infected Mongolian gerbils.67
One of the novel concepts emerging is the role of oxidative stress in regulating gene expression during H. pylori infection. Apurinic-apyrimidinic endonuclease-1 (also referred to as redox factor-1) plays a key role in the regulation of redox-sensitive signaling and is expressed in gastric epithelial cells during infection with H. pylori,68 contributing to activation of AP-1 and NF-κB required for the host response to infection, including IL-8 production.69 However, the role it and other redox-regulatory molecules play in the pathogenesis of diseases associated with H. pylori has yet to be clearly defined. Oxidation of DNA by reactive oxygen species such as hydroxyl radicals are thought to play a causal role in malignant transformation through the induction of DNA damage. Oxidative DNA damage is increased in gastric epithelial cells by H. pylori infection. There is growing interest in the role of antioxidants in disease prevention or treatment because infection is associated with decreased levels of a tissue antioxidant scavenger, vitamin C. Moreover, there is evidence that diets high in antioxidants70 or “nutraceuticals” of the isothiocyanate group, such as sulforaphane,71 can antagonize oxidative stress and protect the host from gastric cancer, perhaps by decreasing inflammation and attenuating bacterial load.
CagA protein translocates into the host cell cytoplasm where it is tyrosine phosphorylated by host Src kinases, and through other interactions, regulates epithelial cell morphology.72–76 CagA in strains from distinct geographic populations appears to be phosphorylated in a different manner resulting in different effects on intracellular signaling.77,78 It is intriguing to speculate that heterogeneity in the CagA protein may lead to distinct effects on the host response that account for some of the geographic differences in disease. Although phosphorylation of the CagA protein may be important, it is not the only mechanism whereby this molecule regulates the host response. A phosphorylation-independent effect on gene transcription, which is also attributed to the CagA protein, also has been reported.79 Moreover, greater than 30% of host gene expression is altered independently of the phosphorylation of CagA, at least in intestinal epithelial cell lines.80
Outer inflammatory protein A (OipA) is another bacterial product that induces epithelial cell IL-8 production81 and strains that express OipA are associated with increased bacterial density, mucosal IL-8 levels, and neutrophil infiltration, as well as more severe clinical consequences.82 Peptidoglycan has been reported to translocate into gastric epithelial cells via the type IV secretion system encoded by the cag PAI. Once inside the cell these bacterial compounds are recognized by nucleotide-binding oligomerization domain-1 (NOD1), thereby providing a novel mechanism of bacterial sensing.83 Binding to NOD1 can lead to activation of NF-κB and the subsequent expression of various host genes encoding proinflammatory molecules.
As discussed in Chapter 49, gastric acid secretion is a major function of the gastric mucosa that is regulated by a variety of neural, endocrine, and immune factors.3 Elevated fasting and meal- or hormone-stimulated levels of gastrin are well documented in H. pylori infection, and there is evidence that gastrin expression is regulated by bacterial factors and cytokines. Expression of somatostatin, an acid-inhibitory peptide, is diminished in infected individuals as is duodenal bicarbonate secretion. The net effect of H. pylori infection on acid secretion is complex and varies depending on the duration and distribution of infection and presence of mucosal atrophy. Secretion of mucus is also affected by H. pylori infection with decreased amounts of mucus and gastric mucosal hydrophobicity; these abnormalities reverse after eradication of infection. Epithelial barrier function is altered during H. pylori infection as a consequence of both direct effects of H. pylori and the accompanying inflammatory response that collectively increase epithelial cell proliferation and programmed cell death.2
Infection with H. pylori results in a unique inflammatory response in which infection persists despite the recruitment and activation of T and B lymphocytes, phagocytic cells, and other immune cell populations. Whereas several epithelial cell responses to H. pylori have been described earlier, they do not appear to fully account for the magnitude of the inflammatory response to an organism that resides predominantly in the lumen. Some bacteria may infect epithelial cells, and significant amounts of bacterial material may “leak” around epithelial cells and reach the lamina propria, where it can activate underlying phagocytes, including neutrophils and macrophages. One bacterial factor is the H. pylori neutrophil-activating protein (HP-NAP). This 150-kd decamer protein promotes neutrophil adhesion to endothelial cells and stimulates chemotaxis of monocytes and neutrophils, NADPH oxidase complex assembly at the plasma membrane, and the subsequent production of reactive oxygen intermediates (ROIs).84 In the inflammatory environment present during H. pylori gastritis, TNF-α and interferon-γ (IFN-γ) can prime neutrophils and potentate the effects of HP-NAP.
When cells undergo apoptosis and die, they are removed by phagocytes. This response occurs in the digestive tract as epithelial cells migrate toward the lumen and undergo apoptosis, providing another means whereby the host can sample the antigenic challenges facing epithelial cells. Viral antigens are presented to T cells when infected apoptotic epithelial cells overlie the Peyer’s patch. Engulfment of H. pylori infected epithelial cells by phagocytes may also be an important mechanism by which H. pylori can activate the host response, and it has been shown that macrophages bind and then engulf gastric epithelial cells that undergo apoptosis due to infection.2
Recruitment and activation of macrophages and neutrophils cause the release of other inflammatory mediators. Increased expression of inducible nitric oxide synthase (iNOS) is observed in the gastric mucosa during infection with H. pylori.2 Nitric oxide (NO) and superoxide (O2−), which may be produced by infiltrating neutrophils, react to form peroxynitrite (ONOO−), a potent oxidant and reducing agent. Although these products have potent antimicrobial effects, uncontrolled or inappropriate production could play a role in the gastric mucosal damage observed during H. pylori infection. The catabolism of urea by urease provides CO2, which rapidly neutralizes the bactericidal activity of the peroxynitrate by reacting with it to form ONO-OCO2. Urease may favor bacterial colonization by neutralizing some host responses but this also enhances the nitration potential of ONOO− and may favor mutagenesis of host cell DNA.
Cytokines secreted by epithelial cells complement those released in the lamina propria. For example, neutrophils are not only activated by IL-8 but also by chemokines such as ENA-7885 and Gro-α,86 which can derive from the epithelium, the adjacent myofibroblasts or the macrophages within the lamina propria. Cytokine induction in macrophages includes induction of TNF-α and IL-6 by urease,87 whereas heat shock protein 60 induces IL-6.88 Intact bacteria can induce the production of chemokines that recruit T cells,89 as well as IL-1290,91 and IL-18,92 two cytokines that favor the selection of Th1 cells. Thus, intact bacteria or bacterial factors trigger a broad cytokine response within the lamina propria.
As adaptive responses develop, different T lymphocyte helper (Th) cell subsets emerge, with characteristic patterns of cytokine secretion. Th1 cells promote cell-mediated immune responses through the production of IFN-γ and TNF-α, whereas Th2 cells produce IL-4, IL-5, IL-10, and transforming growth factor-β (TGF-β). Th2 cells can promote mucosal IgA or IgE responses, as well as diminish the inflammation caused by Th1 cytokines. Previous studies suggest that the infected gastric mucosa is preconditioned to favor Th1 cell development.90 One possible hypothesis to account for this tendency is that infection selectively blocks Th2 development. H. pylori can interfere with STAT6 activation by IL-4,93 which could impair Th2 development, and IL-12 and IL-18 induced in response to infection may positively select for the predominant Th1 response. Other cytokines that may enhance Th1 responses, such as IL-23 and IL-27, have yet to be studied in human tissue.
T cell activation by H. pylori infection may contribute to more severe inflammation and gastroduodenal diseases. Increased levels of biologically active IL-17, a cytokine produced by activated CD4+ T lymphocytes, are found in the mucosa of H. pylori–infected patients.94,95 IL-17, in turn, induces IL-8 expression by gastric epithelial cells, thereby enhancing neutrophil recruitment. Activation of transcription factors by IL-17 may also contribute to the increased levels of numerous other proinflammatory cytokines and enzymes observed during H. pylori infection, such as IL-1β, TNF-α, and cyclooxygenase-2 (COX-2). IFN-γ and TNF-α produced by Th1 cells can increase the expression of many genes in the epithelium, including IL-8. These cytokines also enhance bacterial binding36 and may contribute to enhanced bacterial load.96 In animal models Th1 cells increase epithelial cell apoptosis36 as well as inflammation, atrophy, and dysplasia.97 TNF-α, IFN-γ, and IL-1β up-regulate gastric mucosal Fas antigen expression.98 Since Th1 cells express higher levels of Fas ligand (FasL) than Th2 cells, the relative increase in Th1 cells during H. pylori infection may induce epithelial cell death through Fas-FasL interactions.98,99 This notion is substantiated by the observation that proton pump H+,K+-ATPase–specific Th1 cells in the gastric mucosa kill target cells via Fas-FasL interactions and may act as effector cells in autoimmune gastritis.100
Because Th1 cells cannot clear H. pylori, some other T cell subset may have to be stimulated in order to confer immunity. Studies in animal models indicate that protective immunity was induced by vaccines for Helicobacter spp. via Th cells other than Th1 cells, possibly including Th2 cells. The anti-inflammatory cytokines associated with Th2 cells or other regulatory subsets of Th cells can attenuate the pathogenic effects of Th1 cells.101 More direct evidence suggests that IL-4 can decrease gastritis, an effect that may be mediated by the release of somatostatin.97 As gastric responses can be modified by Th2 cells, the role of other T cell subsets, such as regulatory T cells (Treg), in the pathogenesis of disease associated with H. pylori infection is being addressed. Depletion of Treg in neonatal mice leads to autoimmune gastritis,102 and infection with H. pylori alleviates autoimmune gastritis induced in neonatal mice.103 This suggests that infection may stimulate a subset of anti-inflammatory T cells that impair excessive inflammation, which could otherwise lead to the spontaneous clearance of the organism, an effect that appears to occur in the human mucosa in response to H. pylori infection.104
Antibodies in the gastrointestinal tract are normally of the immunoglobulin A (IgA) isotype, which are highly adapted for mucosal protection, conferring protective immunity without activating complement and stimulating deleterious amounts of inflammation. During infection with H. pylori, the number of IgA producing cells increases. IgG and IgM are also detected, along with activated complement. It has been suggested that the level of autoantibodies in humans correlates with the severity of gastritis.2 Local immune complexes contribute to gastroduodenal inflammation and tissue damage during infection and may contribute to autoimmune gastritis. Monoclonal antibodies that recognize H. pylori cross-react with human and murine gastric epithelial cells.105,106 Adoptive transfer of these antibodies to recipient mice induces gastritis,105 as does the transfer of B cells that recognize heat shock proteins from individuals with maltoma.107 Anti-Le antibodies have been described in humans and occur independently of the Le phenotype of the host but they do not appear to be autoreactive. Autoantibodies induced in mice may recognize different targets within the gastric mucosa and even though they may cross-react with human gastric tissue, autoantibodies induced in humans may have a completely different specificity.
With few exceptions, infection with H. pylori persists for the life of the host unless there is some intervention with antibiotics. This observation has led to investigations as to whether immunity is impaired by immunologic avoidance or tolerance. Several bacterial factors, including catalase and urease, antagonize innate host responses. Production of the enzyme arginase by H. pylori inhibits NO production and may favor bacterial survival,108 whereas virulent strains of H. pylori have also been shown to alter mucus production109 and phagocytosis.110
[/not-level-membership-for-gastroenterology-and-hepatology-category]
