[level-membership-for-critical-care-medicine-category]
178 Heavy Metals
 Arsenic
Arsenic
Background
Enormous achievements and incredible misfortune mark arsenic’s history. Dating back to 400 BC, Greek and Roman physicians included arsenic in their medical armamentarium.1 Today, arsenic continues to be found in treatments offered by practitioners of Indian folk medicine and traditional Chinese medicine.2,3 In addition, in Western medicine, arsenic trioxide (Trisenox) and melarsoprol are used to treat promyelocytic leukemia4 and late-stage African trypanosomiasis.5
Arsenic’s therapeutic usefulness is based on its ability to poison cells, and it is best known as a poison. While arsenic has been noted to produce a garlic odor and possess a characteristically sweet flavor, most arsenical compounds have no perceptible smell or taste.6 This has made the detection of arsenic difficult when employed as a homicidal agent. Historically, this has afforded arsenic the illustrious title of “Poison of Kings and the King of Poisons.”7
Toxicity
Environmental arsenic comes from both natural (volcanic eruption, water runoff) and manmade sources (mining, smelting, combustion of fossil fuels, and pesticide use).7,8 Food, predominantly seafood, represents the principal route for human exposure; however, rice, mushrooms, and poultry also contribute to exposure.6 Alternative sources include air and water, particularly in Bangladesh where tube-wells supply millions with arsenic-contaminated drinking water.9 In the United States, the average person consumes 50 µg of arsenic daily, with inorganic arsenic accounting for 3.5 µg of this total. Certain occupations such as metal working, electronics manufacturing, and glass manufacturing also increase arsenic exposure.6
Arsenic is found in nature in several different forms: combined with carbon and other elements in organic compounds, in inorganic compounds, in gaseous compounds, and in the nontoxic elemental form. Arsenobetaine, an organic arsenic compound found in fish and shellfish, possesses a low risk for human toxicity.6 Inorganic arsenicals, found as trivalent (As3+, arsenite, more toxic) or pentavalent (As5+, arsenate, less toxic) compounds, account for the majority of human toxicity. Arsenite compounds exhibit high affinity for binding to proteins, whereas the majority of arsenate compounds remain unbound. Weak protein binding allows arsenate compounds to be freely excreted. Protein-bound arsenite functions as both a storage depot and a target action site.10 Alternating hepatic reduction and methylation reactions convert inorganic arsenic to an organic form. This transformation detoxifies the parent compound but also increases arsenic’s carcinogenicity.7,10,11
The mechanism accounting for arsenic’s ability to disrupt cellular function, and that which results in acute toxicity, differs depending on the form of arsenic responsible for the exposure. By disrupting the activity of key enzymes by binding to critical sulfhydryl groups, arsenite impairs both oxidative phosphorylation and gluconeogenesis. Especially important in this regard is arsenite’s ability to inhibit the enzyme, pyruvate dehydrogenase (PDH), which catalyzes the first and rate-limiting step in the tricarboxylic acid (TCA) cycle. Inhibition of PDH hinders the production of acetyl-CoA from pyruvate, limiting cellular ATP production. In addition, arsenate can substitute for phosphate and become incorporated into arsenate analogs of glucose-6-phosphate, 6-phosphogluconate, and adenosine triphosphate (ATP).6,12,13 These arsenate analogs are less stable than their phosphate-containing counterparts, and their formation can lead to uncoupling of oxidative phosphorylation.6,13–16
Arsine gas (AsH3) is released when many arsenic-containing compounds come in contact with an acid or when metallic arsenic comes into contact with water. Workers involved with lead plating, soldering, etching, smelting, and galvanizing are at risk for exposure to arsine gas.17 Being colorless and nonirritating, arsine gas is particularly difficult to detect following industrial exposure. As with arsenic, arsine gas has been noted to possess a slight garlic odor; also like arsenic, however, arsine’s odor is not always detectable.18
Exposure to arsine causes acute hemolysis. The exact mechanism for this hemolysis is not completely understood and is likely multifactorial, involving both binding of arsine to hemoglobin and perhaps inhibition of sulfhydryl-containing enzymes.19–22 One complication of arsine exposure is acute renal failure. Arsine-induced renal failure most likely is secondary to direct nephrotoxic effects of arsine as well as the renal toxicity caused by the release of hemoglobin from lysed red blood cells.23 Although chronic renal dysfunction has been described after arsine exposure, recovery is possible.22,24
Clinical Presentation
A patient’s clinical presentation after arsenic exposure is influenced by four factors: the arsenic species, the amount, the route, and the duration of exposure. In addition, the symptoms of acute arsenic toxicity differ depending on whether exposure was oral or inhalational. Gastrointestinal symptoms are a reliable finding in cases of acute arsenic poisoning. Shortly after exposure, patients typically experience abdominal discomfort, nausea, emesis, and profuse diarrhea.7,25,26 Severe gastroenteritis and hemorrhage may develop. Furthermore, arsenic induces capillary dilation, third-spacing of fluid, ventricular arrhythmias, and cardiomyopathy. Collectively these changes can result in pulmonary edema, hypotension, congestive heart failure, and shock.7,20 Cardiac abnormalities following arsenic exposure can include QTc prolongation, T-wave abnormalities, second-degree heart block, QRS widening, nonconducted P-waves, nonsustained ventricular tachycardia, torsades de pointes, complete heart block, asystole, pericardial effusion, and serositis.27–31 Cardiac conduction abnormalities have been described following therapeutic use of arsenic trioxide. These conduction abnormalities may be a consequence of arsenic trioxide–induced electrolyte abnormalities (hypokalemia or hypomagnesemia).29 Alternatively, these changes in cardiac conduction might reflect the cardiotoxic effects of other cancer chemotherapeutic agents. Additionally, arsenic-induced blockade of IKr and IKs channels with activation of IK-ATP channels has been demonstrated.32 The variability seen in arsenic-induced QTc prolongation likely occurs secondary to the combined effects of this activation and blockade.
Neurologic symptoms are also associated with arsenic toxicity and typically include altered mental status and confusion.20,25 Seizures have been linked to arsenic-induced arrhythmias,27,33 and if related to hypoxemia may signal a terminal stage in cases of arsenic poisoning. While typically delayed 2 to 8 weeks after exposure, peripheral neuropathy from large arsenic exposures sometimes can become apparent within a few hours.7,34 Patients describe pain, numbness, and paresthesias in a “stocking/glove” sort of distribution.20,25,35 The symmetric sensorimotor neuropathy seen following arsenic exposure can be misdiagnosed as Guillain-Barré.7,35 Marked abnormalities in sensory and mixed nerve conduction in conjunction with moderate motor conduction abnormalities can be seen on electrophysiologic testing. These results are consistent with axonal degeneration, which is also apparent histologically in nerve biopsies.36
Symptoms of chronic arsenic toxicity differ from those of acute toxicity. Patients demonstrate hematologic abnormalities including pancytopenia, anemia, and macrocytosis. Dermal manifestations include Mees lines (transverse white striae on the fingernails), hyperkeratotic extremity lesions, and hyperpigmented melanosis. Gastrointestinal symptoms as well as liver disease are also described.7,26,37,38 Additionally, patients may note a metallic taste.7,38,39
Inhalational exposure to arsenic in the form of arsine gas produces symptoms that differ from those seen with oral arsenic exposure. Classically, exposure is characterized by the development of a triad of symptoms: abdominal pain, hematuria, and jaundice. Within 2 to 24 hours of exposure, patients develop headache, malaise, abdominal discomfort, nausea, and emesis. The variability in the onset of symptoms is influenced by the duration of exposure and the concentration of the gas.17 Hemolysis with subsequent gross hematuria and renal failure often follow. Additionally, patients may develop scleral icterus and a bronze skin discoloration.17,24 As with exposure to other arsenic compounds, exposure to arsine gas can lead to development of peripheral neuropathy.40
Diagnosis
The potential for arsenic toxicity can be determined by combining the patient’s clinical presentation with the likelihood of exposure. Blood and urine testing can confirm the diagnosis. Blood arsenic clearance occurs in three phases: Phase 1 takes place 2 to 3 hours following intravenous (IV) administration and is associated with a half-life (t1/2) of about 2 hours; phase 2 occurs 3 hours to 7 days following administration and is associated with a t1/2 of about 27 hours; and phase 3 occurs 10 or more days following administration and is associated with a t1/2 of about 230 hours.41 Arsenic is rapidly cleared from the blood during phases 1 and 2; thus, the reliability of blood arsenic testing is limited to the early stages of acute toxicity.33 Following IV administration of radioactive arsenic, urinary recovery has been demonstrated at 18% to 30% after 1 hour, 36% to 56% at 4 hours, 57% to 90% by 9 days, and 96.6% by 18 days.41 A positive urine arsenic level should be followed up by arsenic speciation to distinguish nontoxic organic arsenic (commonly found in seafood) from the toxic inorganic form. A random urine arsenic level greater than 50 µg/L, or a 24-hour urine sample demonstrating more than 100 µg of arsenic, can confirm the diagnosis of acute arsenic toxicity. However, this diagnosis should be questioned if there is a history of recent seafood ingestion or the sample was not collected in a metal-free container. Hair and nail samples may be used to confirm exposure when chronic toxicity is suspected.7 Hair testing can also be performed to estimate an approximate exposure date based on the rate of hair growth (0.4 mm/d) and the distance of the root from the arsenic peak.42 Additionally, an electrocardiogram (ECG) should be obtained in cases of potential arsenic toxicity; T-wave abnormalities, QTc prolongation, and torsades de pointes have been described following arsenic exposure.27,31
Following possible exposure to arsine gas, laboratory evaluation should include complete blood count, measurement of circulating lactate dehydrogenase and other liver enzyme concentrations, measurement of serum bilirubin concentration, Coombs testing, and monitoring of serum electrolyte levels and renal function. Patients will demonstrate a Coombs-negative hemolytic anemia with elevated circulating lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and bilirubin levels, renal dysfunction, and subsequent hyperkalemia. Hemoglobinuria, albuminuria, and occasional erythrocyte and hemoglobin tubular casts are commonly seen on urinalysis. A peripheral blood smear can show signs of red blood cell damage including erythrocyte fragments, basophilic stippling, anisocytosis, poikilocytosis, and Heinz bodies.17,18,22,43 Methemoglobinemia also has been described.43
Treatment
Initial treatment of arsenic toxicity must include prevention of further exposure to the poison, careful monitoring of the cardiovascular system, and judicious repletion of intravascular fluid deficits. Hyperkalemia, which can be clinically significant, should be treated. Despite a lack of supporting data, gastric lavage and administration of activated charcoal should be considered following oral exposure to a large of dose of arsenic in an attempt to limit absorption of the toxic compound(s).7 Patients presenting with renal failure following arsenic exposure may need renal replacement therapy until renal function recovers.44 Additionally, patients demonstrating ECG abnormalities, particularly QTc prolongation, should have close monitoring of their electrolytes, with replacement as indicated. Antiarrhythmic therapy should be instituted as needed.
Chelators are an additional treatment to be considered. Dimercaprol, a parenteral chelating agent which functions intracellularly as well as extracellularly, is the first line of therapy for patients experiencing abdominal symptoms following acute arsenic exposure.33 Dimercaprol should be administered intramuscularly (IM) at doses of 3 to 4 mg/kg every 4 to 12 hours.20 In cases of subacute or chronic exposure when the patient can take oral medications, succimer (DMSA or 2,3-dimercaptosuccinic acid) is the chelator of choice. Succimer should be administered orally (10 mg/kg every 8 hours for 5 days). Following the initial 5 days of administration, succimer dosing is adjusted to every 12 hours.33 Clinical course determines the duration of chelator therapy.20 Urinary arsenic levels can be used to calibrate duration of treatment, with therapy being discontinued when 24-hour urinary arsenic levels are below 50 µg/L.33 Despite its recommended use, succimer has not consistently demonstrated increased urinary arsenic excretion following administration, and neuropathy can progress despite therapy.25,42 Succimer is not currently approved by the U.S. Food and Drug Administration (FDA) for treatment of arsenic toxicity.45
Treatment of arsine gas exposure represents a special circumstance. As with other forms of arsenic exposure, initial therapy requires elimination of the source. Caution should be taken to prevent additional casualties as first responders enter the exposure site. Since arsine toxicity is associated with hemolysis, exchange transfusion can be used to replenish red blood cell mass and remove both the toxic complexes formed from the arsine/hemoglobin interaction as well as the released hemoglobin pigment.40,46 Patients with renal failure should receive renal replacement therapy as indicated by clinical findings.17,18 The use of chelation therapy to treat patients acutely poisoned by arsine is controversial and may not affect the clinical course.18
 Mercury
Mercury
Background
Mercury is a naturally occurring metal found in elemental, inorganic salt, and organic forms. The use of mercury dates back to before 1500 BC. Throughout history, mercury has been employed as a cosmetic component, decoration, and even medicine. Mercury is also a natural part of our atmosphere, with 30,000 to 50,000 tons degassing from the earth’s surface annually; human activity adds another 20,000 tons each year.47 Toxicity from mercury can occur as a result of occupational, environmental, or medical exposure. Each year, mercury exposure accounts for over 6000 cases of toxicity reported to U.S. poison centers.48 The dose, length of exposure, and form of mercury can cause wide variation in the clinical presentation and ultimate outcome.49–52
Toxicity
Elemental mercury can be found in barometers, dental amalgams, electronics, thermostats, thermometers, and batteries. It is also found in some folk remedies. Elemental mercury is a liquid at room temperature. Even without heating, elemental mercury releases sufficient mercury in the gas phase to cause toxicity. Most problems from elemental mercury are the result of vapor inhalation, with nearly 80% of inhaled vapor being absorbed by the alveoli and transferred into circulating red blood cells. Most absorbed mercury is converted to its divalent (mercuric) form, thus decreasing its lipid solubility. If the patient is exposed to a very high dose of mercury vapor, a small amount of gaseous mercury can remain in the bloodstream, leading to penetration of the blood-brain barrier and central nervous system (CNS) injury.8 By comparison, the gastrointestinal tract takes up less than 2% of ingested elemental mercury53; however, in patients with mucosal disruption, transmucosal absorption of mercury can be markedly increased.54 Absorption of mercury across intact skin is minimal,55 but subcutaneous injection can cause an increase in urinary mercury concentrations. Intravenous injection of mercury can cause both mercury toxicity and mechanical obstruction of pulmonary blood flow.56,57 Elimination of inhaled vapor is via the urinary tract, and the elimination t1/2 of absorbed elemental mercury is about 60 days.56 Elemental mercury when orally ingested is typically nontoxic owing to minimal absorption across intact gastric and intestinal mucosa.58
Elemental mercury primarily targets the lungs and brain, but the poison also can cause renal and gastrointestinal injury.5,14,15 By forming complexes with sulfhydryl groups, mercury interferes with protein and nucleic acid synthesis, protein phosphorylation, and calcium homeostasis. It can also cause oxidant stress via this mechanism.59,60 Inhaled mercury vapor has a corrosive effect on the lungs and is capable of producing acute inflammation of the bronchi and bronchioles. This may result in a fatal interstitial pneumonitis.60
Elemental mercury poisoning can be misdiagnosed as pheochromocytoma. This can occur as mercury inactivates the coenzyme, S-adenosylmethionine, which inhibits catechol-O-methyltransferase (COMT). As a result, catecholamine breakdown decreases and adrenergic symptoms such as hypertension and diaphoresis develop.18–20 A constellation of personality changes in affected individuals has come to be known as erethism. The symptoms include the following61:
There has been a reported association between mercury exposure, erethism, and the development of parkinsonism61–63; however, the evidence for this association comes from only two studies64,65 and two case reports.21,22
Inorganic mercury is most commonly found in nature as cinnabar (mercury [II] sulfide). In humans, however, exposure to inorganic mercury compounds comes from germicides, pesticides, and mercury-containing antiseptics.61 Of interest, there have been several recent reports of inorganic mercury intoxication due to the use of skin-lightening beauty creams.66,67 Inorganic mercury can be absorbed through the skin, via the lungs, and via the gastrointestinal tract. In blood, inorganic mercury has a t1/2 of about 24 to 40 hours, and clearance of mercury by the kidneys is responsible for the toxic effects on the distal portion of the proximal convoluted tubules.68,69
More corrosive to the gut than elemental mercury,61,70 ingestion of mercury salts commonly causes nausea, vomiting, abdominal pain, and hematemesis. Ingestion of relatively large doses of mercury salts can lead to colitis with necrosis and mucosal sloughing, resulting in massive fluid losses.14,29,30
Along with gastrointestinal symptoms, the other notable acute effect of inorganic mercury is development of acute renal failure. Potentially reversible, renal injury can occur within hours to days of an acute exposure.71,72 Membranous glomerulonephritis and nephrotic syndrome also may occur after chronic exposure. Termination of exposure may lead to resolution of nephrotic syndrome.50,73
With prolonged skin exposure, gray-brown hyperpigmentation of skin folds of the neck and face can occur. Used as an analgesic for teething in the 19th century, dental application of calomel (mercuric chloride) can cause loose teeth, blue discoloration of the gingiva, and systemic toxicity.14,31 As with elemental mercury, acrodynia and erethism have been reported with inorganic mercury exposure.50,61
Organic mercury compounds are found in fungicides, antiseptics such as merthiolate and mercurochrome, preservatives including thimerosal, and as a contaminant of predatory fish including tuna and swordfish. As much as 90% of ingested organic mercury is absorbed by the gastrointestinal tract.74 Pulmonary absorption of organic compounds such as methylmercury vapor, approaches 80%, depending on particle size. Absorption across intact skin also can occur.47 Methylmercury readily crosses the blood-brain barrier, achieving CNS levels that are three to six times those in blood.75
Perhaps the best-known mass poisonings of mercury involved methylmercury. During the 1950s and 1960s, a chemical company in the Japanese fishing village of Minamata dumped wastewater containing mercury into Hyakken Harbor on Minamata Bay. Aquatic organisms converted the inorganic waste to an organic form (methylmercury) that was then passed up the food chain. The mercury eventually was concentrated in larger fish, which were then consumed by residents of the area.76 Over time, more than 2265 patients developed ataxia, sensory disturbances, constriction of visual fields, dysarthria, auditory disturbances, and tremor. Children exposed in utero developed congenital Minamata disease, which was characterized by seizures, spasticity, deafness, and severe mental deficiency.77 All these children had mental retardation, cerebellar ataxia, limb deformities, primitive reflexes, and dysarthria. Hypersalivation and chorea were seen in 95% and microcephaly in 60% of affected children.77–79
In a second event in 1971, 6500 Iraqis suffered symptoms similar to the Minamata patients after eating bread baked with flour made from grain intended for use as seed. The grain had been treated with a fungicide containing methylmercury.80
These two events demonstrated that organic mercury targets the CNS and that fetal brain tissue is more susceptible than the adult brain to the toxic effects of organic mercury. Postmortem findings have shown damage to gray matter of the cerebral and cerebellar cortex. The temporal cortex and calcarine region of the occipital lobe are most affected.81,82 Pathologic changes in adults and children include cortical atrophy, hypoplasia of the corpus callosum, hypoplasia of the granular cell layer of the cerebellum, and demyelination of the pyramidal tracts.78
Diagnostic Tests
Urine testing (24-hour collection) is most useful for confirming exposure/toxicity to either elemental or inorganic mercury. Collection containers are frequently washed with nitric acid, but laboratory practices may vary. Specimens should be refrigerated to decrease bacterial reduction of mercury to volatile elemental mercury.83 Samples should not be collected within 48 hours of gadolinium administration (as with magnetic resonance imaging) or within 72 hours of consuming predatory fish (e.g., tuna or swordfish).
Because approximately 90% of methylmercury is bound to red blood cells74 and very little is excreted via the kidneys, the preferred test to determine organic mercury is a whole-blood mercury level. While most persons have whole-blood mercury concentrations of less than 6 ng/mL, diets rich in predatory fish can elevate levels to 200 ng/mL or higher.84 For that reason, patients should avoid consumption of fish for at least 72 hours prior to blood testing.
Treatment
The first and most important step in treatment of mercury toxicity is to avoid further exposure of the patient to the toxin.50,61,85,86 Whereas removal of clothing and decontamination of skin may be helpful, gastrointestinal and pulmonary decontamination are of little use. Chelation is often considered the cornerstone of therapy, although the benefit remains somewhat controversial. Chelating agents bind metal ions to form a complex that can be excreted, thereby reducing the body burden of the offending metal. Dimercaptosuccinic acid (DMSA, Chemet, Succimer) is currently the favored agent. Because DMSA is available only in an oral form, dimercaprol (British antilewisite [BAL]) is used in patients unable to take oral medications. D-penicillamine is an alternate but less effective choice. Chelation therapy may take several months to eliminate the body burden of heavy metal, and clear evidence for long-term benefit is lacking.86,87 Patients with renal failure may require renal replacement therapy. Aggressive fluid replacement may be necessary to correct large losses of fluid from the gastrointestinal tract, particularly in cases of inorganic mercury exposure.
 Lead
Lead
Background
Mined by the ancient Egyptians, Phoenicians, Greeks, and Romans, lead and its toxicity have had a long, storied history. There is evidence that some of the leaders of ancient Rome suffered neurotoxicity and sterility due to lead poisoning.88 In 1763, Benjamin Franklin described abdominal pain and peripheral neuropathy associated with lead poisoning.89 With the onset of the Industrial Revolution, lead increasingly was used in machinery and manufacturing. In the United States in the 20th century, lead-containing paint and leaded gasoline became prominent sources of lead exposure in the population. It was not until 1978 that residential use of lead-based paint was banned. Leaded gasoline was not banned until the 1990s.88 With the recognition of lead toxicity and its removal from households and many industrial processes, the incidence of cases of lead poisoning has steadily declined since the 1970s.90,91 Nevertheless, lead products—both new and old—continue to be sources of exposure and toxicity.
Toxicity
Lead is still used in a variety of products. It is sometimes found in paints, plumbing pipes, gasoline, solder, batteries, bullets, toys, moonshine, traditional folk remedies, curtain weights, necklace charms, and food containers. Today the most common source of exposure is from lead-based paints and lead-contaminated soils.92,93 Many occupations including battery plant worker, metal welder, painter, construction worker, lead miner, firing range worker, glass blower, and ship builder increase the risk of lead exposure.
In the pediatric population, the most common route of exposure is ingestion, especially among children between the ages of 18 and 36 months. Compared to adults, children are also more prone to toxicity from lead ingestion because of increased gastrointestinal absorption of lead, immaturity of the blood-brain barrier, ongoing development of organ systems, and frequent concomitant iron deficiency.94
Inhalational exposure is the most common cause of lead exposure in adults. Exposure to lead among adults is often related to occupational exposure or exposure from a hobby. Ingestion of lead-contaminated moonshine also has resulted in “outbreaks” of adult lead poisoning.95,96
Lead interferes with numerous enzymatic pathways, including those that utilize sulfhydryl-containing enzymes, calcium, zinc, and iron. Within the CNS, lead poisoning can increase the permeability of the blood-brain barrier, resulting in increased intracranial fluid and pressure. Lead also has been shown to increase the spontaneous firing of neurons which utilize acetylcholine, dopamine, or γ-aminobutyric acid (GABA) as neurotransmitters within the CNS. This change in neuronal signaling can result in the erroneous enhancement of unnecessary neural pathways and destruction of necessary neural pathways. Children are particularly susceptible to this effect from lead poisoning, because the peak of fortification and elimination of neural pathways occurs around 2 years of age.94,97
Lead inhibits several enzymes necessary for the synthesis of heme. These enzymes include aminolevulinic acid (ALA) synthetase, δ-ALA dehydratase, coproporphyrinogen decarboxylase, and ferrochelatase. By interfering with erythrocyte membrane formation, lead poisoning shortens the lifespan of erythrocytes.98,99 Basophilic stippling is sometimes seen with lead toxicity and is a result of the inhibition of pyrimidine-5-nucleotidase.100 Hematologic consequences of lead poisoning can be compounded by poor nutritional status, especially iron deficiency.101
Lead is deposited in the proximal tubules of the kidney as a lead-protein complex, where it can interfere with mitochondrial function. In animal models, exposure to high doses of lead results in renal failure with the nonspecific findings of tubular atrophy, interstitial fibrosis, and glomerular sclerosis.102
Chronic lead exposure has been associated with hypertension. Lead can affect vascular smooth muscle by decreasing Na+/K+-ATPase function, resulting in increased calcium-mediated contractility. Lead also can increase protein kinase C activity, further altering vascular smooth muscle activity.103
The t1/2 of lead in blood is 4 to 6 weeks. However, in bone, which is the main reservoir for lead in the body, the t1/2 is considerably longer, ranging from 5 to 19 years.104 This lead depot in bone can be mobilized and consequently reexpose vital tissues to lead during times of increased bone turnover such as growth spurts, pregnancy, and after fractures.105 In the pediatric population, lead causes increased density of metaphyses through increased calcification of the bone metaphysis.106 These “lead lines” on radiographs may be the first manifestation of chronic lead exposure in children.
Clinical Presentation
In the pediatric population, lead-induced encephalopathy is the most critical presentation of plumbism. The peak incidence occurs between 18 and 36 months of age. Typically patients with encephalopathy caused by lead have circulating levels ≥70 µg/dL. The reverse is not true, however. Patients with circulating lead levels ≥70 µg/dL do not always have encephalopathy and may have no symptoms at all. Patients with lead encephalopathy may present with coma, seizures, failure to meet developmental milestones, ataxia, visual changes, and lethargy. Patients with acute-on-chronic or acute lead ingestion are more likely to present with severe neurologic symptoms. Children with blood lead levels between 50 and 100 µg/dL can have more subtle neurologic signs including irritability, hyperactivity, and developmental delay.94,97 There is continued controversy regarding the effects of chronic low-level lead exposure and neurodevelopment, particularly with blood lead levels less than 10 µg/dL.107–109
Adults also can present with encephalopathy after lead poisoning. Brain edema, coma, seizures, and lethargy are potential symptoms when blood lead levels are over 150 µg/dL. Personality changes, insomnia, and memory deficiencies have been reported with levels above 80 µg/dL, and less obvious symptoms such as mood changes can occur with levels in the 40 to 70 µg/dL range.91,110,111 Another neurologic sign, often described with plumbism, is predominantly motor neuropathy with clinical signs of foot and wrist drop. This neuropathy can be seen with blood lead levels as low as 40 µg/dL and is more common with long-term exposure in adults and in children with sickle cell disease.112
Lead poisoning can cause gastrointestinal symptoms including abdominal pain, anorexia, vomiting, pancreatitis, and hepatotoxicity.91
The anemia of plumbism typically does not cause clinical symptoms requiring hospitalization, but anemia can serve as a clue to the diagnosis in patients with signs of other organ system damage after lead poisoning. Lead poisoning can cause normocytic or microcytic anemia. Decreased red cell mass is generally seen in children with blood lead levels higher than 40 µg/dL and in adults with levels above 50 µg/dL.99 Basophilic stippling is sometimes observed in cases of plumbism but also has been described with arsenic poisoning, other disease states, and in healthy individuals.100
Chronic lead exposure has been associated with nephrotoxicity.102 Adult plumbism can cause renal changes consistent with a Fanconi-like syndrome characterized by aminoaciduria, phosphaturia, and glycosuria. Another complication of renal toxicity from lead is decreased uric acid clearance, leading to flares of gout aptly named “saturnine gout.”
Hypertension has been associated with chronic lead poisoning, although the strength of this association is controversial.113,114
Poisoning as a result of exposure to organic lead compounds primarily results in neurologic and gastrointestinal symptoms; hematologic toxicity is not always present. The neurologic sequelae of organic lead intoxication can include personality changes, hallucinations, lethargy, and coma.115 Patients with organic lead poisoning also can exhibit nausea and vomiting as well as liver or kidney damage.
Diagnosis
The best laboratory test to confirm lead exposure is measurement of lead concentration in a whole-blood sample obtained by venipuncture. This test should be sent to the laboratory in a lead-free tube. Blood lead levels are less useful in cases of organic lead toxicity, as they do not correlate well with expected neurotoxicity. A disadvantage to sending a whole-blood lead level is that most laboratories are unable to report results the same day the sample is received. Capillary blood lead levels can be used as a screening test, but they are less reliable than whole-blood levels and may be falsely elevated if contaminated with lead on the skin.116 Protoporphyrin levels in red blood cells are elevated when heme synthesis is inhibited, as occurs in cases of lead poisoning. However, elevated protoporphyrin levels are also observed in other conditions, such as iron deficiency, sickle cell disease, and vanadium toxicity. Elevated protoporphyrin is more likely to occur in chronic plumbism than with acute lead exposure.117 A laboratory test that detects urine lead concentration after a dose of CaNa2EDTA is available but has limited clinical utility and would not be used to guide management. A test that utilizes x-ray fluorescence to determine lead burden in bone is available in some research centers but would not be useful in the acutely ill patient.118
Radiographic imaging may help the clinician determine the etiology and proper treatment of a patient with lead poisoning. A head CT scan should be obtained in anyone with CNS findings and evidence of lead exposure. A reported finding on head CT in patients with lead encephalopathy is evidence of cerebral edema. Anyone with an elevated lead level from an undetermined source should have an abdominal radiograph to look for ingested lead objects. Long-bone radiographs of children with plumbism may show increased densities at the metaphyses (lead lines). Lead lines are a nonspecific finding and can also be seen with bismuth, phosphate, and fluoride toxicity.106
Treatment
An important intervention in the treatment of lead toxicity is removal of the source of exposure. This often requires changes in the patient’s living environment or occupation. The source of lead may also require direct medical intervention for removal. Patients who have ingested smaller lead objects may benefit from polyethylene glycol administration; radiographs should be followed to make sure this treatment is effective. A surgeon or gastroenterologist may have to become involved to remove larger lead objects found in the gastrointestinal tract.119 Lead bullets, especially ones that are in contact with synovial fluid, should be surgically removed.120
Children with blood lead levels greater than 69 µg/dL or who have symptoms of plumbism should be hospitalized and started on parenteral chelation therapy. Pediatric patients with levels between 45 and 69 µg/dL who are asymptomatic should receive oral chelation therapy. Children who are asymptomatic and have blood lead levels below 45 µg/dL do not require chelation therapy. This recommendation is based on results from a double-blind randomized clinical trial that showed no benefit from chelation therapy among children with lead levels equal to 20 to 44 µg/dL.122 The local health department should be notified to help with environmental measures and prevention education with any child with a blood lead level above 20 µg/dL or a child with more than one level equal to 15 to 19 µg/dL.121 The effectiveness of environmental cleanup and educational initiatives recently has come under question after the publication of a Cochrane review that indicated that 12 published studies have not shown a clear benefit to these interventions.123
Adults with severe symptoms such as lead-induced encephalopathy or with blood lead levels over 100 µg/dL should be admitted to the hospital and started on parenteral chelation therapy. Oral chelation therapy is recommended for adults with mild symptoms or levels equal to 70 to 100 µg/dL. In general, chelation is unnecessary for adults who are asymptomatic and have levels below 70 µg/dL.91
In the United States, there are several options for lead chelation: edetate calcium disodium, dimercaprol, and succimer. Edetate calcium disodium is a parenteral chelator approved by the FDA for lead toxicity in all age groups. It is widely available and can be administered IV or IM. The recommended IV dose in adults for severe lead poisoning is 1 to 1.5 g/m2/d infused over 8 to 12 hours for a total of 5 days; after 2 days, a repeat 5-day course can be administered if indicated. The recommended pediatric IV dose for severe lead poisoning is 1 to 1.5 g/m2/d divided into equal doses infused every 8 or 12 hours; an additional 5-day course can be given after 2 days if needed. Serious side effects of treatment with edetate calcium disodium include fever, hypersensitivity immune reactions, hypotension, nephrotoxicity, and thrombophlebitis. Patients with renal failure are at greater risk for toxicity from edetate calcium disodium, and in these patients the dose may have to be adjusted or an alternative chelator selected. Because edetate calcium disodium can increase intracranial pressure in patients with cerebral edema, the manufacturer recommends using the IM route, or alternatively using the IV route with a slow infusion rate in these cases. Edetate calcium disodium can exacerbate symptoms when given as the sole chelator to patients with high blood lead levels124; therefore, dimercaprol should be given in conjunction with edetate calcium disodium inpatients with symptomatic lead poisoning. Edetate disodium without calcium should never be used because of the risk of fatal hypocalcemia.125,126
Another FDA-approved chelator for lead toxicity is dimercaprol. It is administered by deep IM injection. In severe plumbism, dimercaprol is administered at a dose of 4 mg/kg IM every 4 hours for 2 to 7 days in both pediatric and adult patients. For mild lead poisoning, the recommended dose is 4 mg/kg IM for the first dose followed by 3 mg/kg IM every 4 hours for 2 to 7 days. Adverse reactions with dimercaprol are common and include fever, hypertension, tachycardia, and injection-site abscesses. Dimercaprol is administered in a peanut oil vehicle and should be avoided in patients with peanut allergies.127
Succimer is an oral chelator that is FDA approved for lead poisoning. The recommendation for pediatric dosing of succimer is 10 mg/kg/dose every 8 hours for 5 days, followed by 10 mg/kg/dose every 12 hours for 14 days. Succimer is not officially approved for adults, but similar doses to those approved in children have been used. Adverse reactions to succimer include neutropenia, hemolytic anemia, and transient elevations in circulating liver transaminase levels.128
There are other lead toxicity treatment options available that are not approved by the FDA, not as widely studied, or not available within the United States. Penicillamine is not approved by the FDA for lead poisoning and should only be considered in cases of serious lead poisoning when the use of other chelators was associated with unacceptable side effects. Penicillamine itself can cause a life-threatening side effect, agranulocytosis, and can also can be associated with serious dermatologic and renal complications.129 Exchange transfusions have been used in conjunction with chelation in rare cases of neonatal plumbism. Unithiol is a chelator available for oral or parenteral administration that is used mainly in Europe. Side effects include fever and allergic dermatologic reactions, as well as hypotension with the IV formulation.130
 Thallium
Thallium
Background
Discovered at a time when new techniques in spectrochemical analysis led to a race among scientists to discover the next element in the periodic table,131 thallium has had a notorious past. Shortly after discovery of the element in 1861 by Sir Edmund Crookes, the toxicity of thallium salts was recognized.132 One of the earliest recognized manifestations of thallium toxicity was hair loss; this discovery led to the use of thallium as a depilatory agent.133 It was the use of thallium compounds to treat ringworm in children that first led to medical reports of systemic toxicity. After several reported deaths, thallium as a therapeutic agent was largely abandoned.134 Although not effective as a medical treatment, the recognized toxicity of thallium, along with its lack of taste and odor, led to its widespread use as a rodenticide. Although effective, its toxicity to unsuspecting children and pets eventually resulted in it being pulled from the U.S. marketplace.135 Its only clinical use today is as a radiocontrast agent for cardiac disease, where it is used in an extremely low, nontoxic concentrations.136 Today, cases of thallium poisoning typically are the result of its malicious use. Because of its toxicity, ease of administration, and difficulty in detection, thallium has long been an agent of choice among those seeking to harm themselves or others.137 Despite its difficulty in detection, a clinician knowledgeable in the early signs and symptoms of thallium poisoning can improve their patients’ outcomes by instituting early antidotal therapy.
Toxicity
Similar to other metals, it is not the elemental form but rather the salts of thallium that are toxic. As a constituent of the earth’s crust, thallium is found throughout the environment. In addition, thallium is emitted from the combustion of coal and in the process of iron smelting.138 Despite its widespread environmental distribution, the majority of clinical problems resulting from thallium are the result of suicide or homicide attempts. Its salts are tasteless, odorless, water soluble, and completely and rapidly absorbed by the gastrointestinal tract. In animal studies, thallium concentrations in urine and feces can be measured within 1 hour of oral administration.139 Once absorbed, thallium is widely distributed throughout the body, where it interferes with activities of several critical metabolic enzymes. Because its charge and atomic radius are similar to the potassium ion, thallous ion interferes with K+-dependent enzymatic processes, including those catalyzed by pyruvate kinase and Na+/K+-ATPase.135 It is this similarity of thallous ion and potassium ion that makes thallium a useful radionuclide for cardiac stress testing. Along with interfering with potassium-dependent processes, thallium also inhibits sulfhydryl-containing enzymes such as pyruvate dehydrogenase.135 The inhibition of potassium- and sulfhydryl-dependent enzymes results in impaired cellular energy production, which if severe enough, leads to cell death.135 Unlike other metals, the primary route of elimination for thallium is not renal but rather fecal, a result of enterohepatic circulation.139
Acute Clinical Presentation
After a toxic exposure to thallium, the earliest reliable finding is the development of a rapidly progressive painful peripheral neuropathy. Beginning typically within 2 or 3 days of exposure, symptoms begin in the feet.140 If the dose is large enough, symptoms can also involve or progress to involve the hands. The neuropathic pain resulting from thallium is often described as a “pins-and-needles” sensation and is commonly excruciating to the point where even the weight of a bedsheet is intolerable.141 In severe cases, the neuropathy can be misdiagnosed as Guillain-Barré syndrome. This diagnostic confusion is prompted by absent or diminished deep tendon reflexes in cases of thallium poisoning142,143 and symptoms that can rapidly ascend from the lower extremities to involve the respiratory muscles, necessitating in some cases the need for mechanical ventilation.141 A relatively unique feature of thallium’s neurotoxicity is that along with involvement of peripheral nerves, the cranial nerves also can be affected. All of the cranial nerves can be involved, but those innervating the eye (II, III, IV, VI) are the most commonly affected.144,145 In addition, CNS findings are also commonly seen in cases of severe thallium poisoning and can manifest as hallucinations, insomnia, acute psychosis, or coma.133,144
The best-known complication of thallium poisoning is alopecia, which begins around 5 to 14 days after exposure to the toxin.133 By 3 to 4 weeks, near-total body alopecia including loss of axillary, pubic, and the lateral eyebrow hair can be apparent.146 The medial part of the eyebrow is typically spared, as its hairs are typically in a resting phase.147 The exact cause of alopecia is unknown, but it is likely that interruption of keratin synthesis and/or metabolism in the hair matrix is responsible.146 While alopecia is the most recognizable feature of thallium poisoning, patients can die before it develops, making early diagnosis of thallium poisoning challenging. In addition, peripheral neuropathy can occur in mild cases, without the development of hair loss.148 In addition to hair changes, other cutaneous manifestations of thallium poisoning can include acneform and eczematous skin eruptions.146
Along with the neurologic and dermatologic manifestations of thallium toxicity, other less specific findings are also commonly reported. In some cases, the earliest symptoms of thallium poisoning are abdominal pain, vomiting, and diarrhea. Over days to weeks, constipation or obstipation may develop.133,147 Compared to arsenic poisoning, the gastrointestinal symptoms associated with thallium exposure are not as severe and do not dominate the early clinical picture. Other nonspecific manifestations of thallium poisoning include myalgia, pleuritic chest pain, insomnia, hypertension, nonspecific ST-T wave ECG changes, bradycardia, tachycardia, hypotension, acute respiratory distress syndrome, acute hepatitis, delayed development of nail dystrophy (Mees lines), and acute and chronic neuropsychiatric manifestations.143,148–152
Diagnostic Tests
Because many of the initial features of thallium poisoning are nonspecific, and few clinicians see many if any cases in their career, making the early diagnosis of thallium poisoning can be difficult.148 However, by being familiar with its most common clinical presentation and obtaining some readily available neurologic and clinical examination findings, diagnosis is possible early in the course of the illness when treatment is most likely to benefit the patient.
Neuropathy is universally present in severe cases of thallium poisoning, so nerve conduction studies may be helpful in diagnosing patients.148 Although the findings are not specific and may be normal early in the clinical course, electromyogram findings indicative of sensorimotor axonopathy in the setting of rapidly progressive symptoms should prompt consideration of acute thallium poisoning. Once the diagnosis of thallium poisoning is part of the differential diagnosis, visualizing a pulled hair from the patient under a low-powered light microscope and finding markedly darkened hair roots should prompt immediate chelation therapy (Figure 178-1).147,148 Blackened hair roots after thallium poisoning are secondary to accumulated gaseous inclusions.146 Darkened roots can be seen as early as 4 days after poisoning,147 and in cases of repeated poisoning, it is possible to observe several bands.147 The highest percentage of darkened roots is seen in hairs pulled from the scalp (95%), followed by hairs from the chest and legs (50% to 60%), and less commonly from eyebrows and eyelids (30%).147 The absence of darkened hair roots in persons with other signs of thallium poisoning should not dissuade clinicians from beginning treatment.
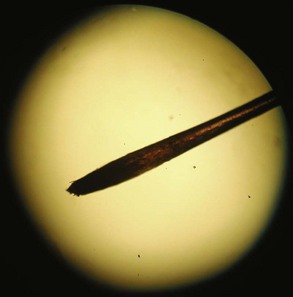
Figure 178-1 Darkened hair root of a thallium-poisoned patient seen under low-power light microscopy.
Although clinical features and hair examination may prompt treatment for a presumptive diagnosis of thallium poisoning, the definitive diagnosis requires analytic confirmation of elevated concentrations of thallium in urine or hair. A 24-hour urine specimen is considered the gold standard for laboratory confirmation of thallium poisoning. In most unexposed persons, the thallium concentration in urine should be less than the lower limit for detection. Nevertheless, depending on occupational and environmental exposure, levels ≤20 µg/specimen can be considered normal. Analysis hair of hair samples is not thought to be as reliable as urinalysis, but it has been shown to correlate with concentrations in 24-hour urine specimens.148 In patients with exposure to thallium many days or weeks prior to obtaining specimens, analyzing samples of hair and nails may be the only means to confirm the diagnosis. Hair levels below 15 ng/g are generally considered normal.135
Treatment
Once thallium poisoning is suspected or recognized, treatment should focus on decreasing circulating thallium concentrations. As mentioned previously, thallium undergoes enterohepatic circulation. The most effective agent for treating thallium poisoning is Prussian blue, a brightly colored hexacyanoferrate compound (Fe4[Fe(CN)6]3). The unabsorbed and insoluble complex binds thallium in the gut, thereby increasing fecal elimination. The FDA has approved Prussian blue, sold under the brand name Radiogardase, for treatment of both cesium and thallium poisoning. The recommended dose of Prussian blue is 3 g orally 3 times a day for adults. For children (2-12 years), the dose is 1 g 3 times a day. Numerous studies in animals demonstrate Prussian blue’s effectiveness for preventing mortality from thallium poisoning.144 Although the very nature of thallium poisoning makes a human clinical trial impossible, case reports support the safety and efficacy of Prussian blue.140,149,153 Treatment is generally recommended until 24-hour urine thallium concentrations fall below 0.5 mg/24 h.140 Although Prussian blue contains cyanide complexes, in vitro studies suggest that the small amounts of cyanide released are clinically insignificant.154 Adverse events related to the use of Prussian blue have not been reported.153
Not all hospitals stock Prussian blue in the pharmacy, so until a supply arrives, alternative treatments may be required. Activated charcoal binds thallium155,156 and has been used to treat cases of thallium poisoining.148,157 Although there are no specific dose recommendations, 50 g of charcoal twice a day has been used.148 Because thallium can promote constipation, the use of charcoal suspended in sorbitol is recommended.
Although thallium is binds sulfhydryl-containing compounds,135 studies in animals indicate that sulfhydryl-containing chelators such as BAL, D-penicillamine, DMSA, and DMPS either provide no benefit or actually increase toxicity,156,158–160 possibly by increasing the concentration of thallium within the CNS.159
Hemodialysis has been used in cases of thallium poisoning. Commonly employed along with potassium supplementation, it is unclear if the amount of thallium removed results in any clinical benefit.140,141,144,161,162 For critically ill patients with thallium toxicity, hemodialysis may be justified.
Agency for Toxic Substances and Disease Registry. Toxicological profile for arsenic. Atlanta: U.S. Department of Health and Human Services, Public Health Service; 2007.
In-depth description of arsenic toxicity, including sections geared towards patient education. It can be accessed at http://www.atsdr.cdc.gov/toxprofiles/tp2.html.
Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol. 2006;36:609-662. Comment in: Crit Rev Toxicol 2007;37:537-49; discussion 551-2
American Academy of Pediatrics Committee on Environmental Health. Lead exposure in children: prevention, detection, and management. Pediatrics. 2005;116(4):1036-1046.
A comprehensive review of pediatric lead poisoning, including treatment recommendations.
Rogan WJ, Dietrich KN, Ware JH, et alTreatment of Lead-Exposed Children Trial Group. The effect of chelation therapy with succimer on neuropsychological development in children exposed to lead. N Engl J Med. 2001;344:1421-1426.
Hoffman RS. Thallium toxicity and the role of Prussian blue in therapy. Toxicol Rev. 2003;22:29-40.
An excellent and well-researched review on the toxicity of thallium and its treatment.
1 Hunt E, Hader SL, Files D, Corey GR. Arsenic poisoning seen at Duke Hospital, 1965-1998. N C Med J. Mar-Apr 1999;60(2):70-74.
2 Lynch E, Braithwaite R. A review of the clinical and toxicological aspects of ‘traditional’ (herbal) medicines adulterated with heavy metals. Expert Opin Drug Saf. Jul 2005;4(4):769-778.
3 Wong ST, Chan HL, Teo SK. The spectrum of cutaneous and internal malignancies in chronic arsenic toxicity. Singapore Med J. Apr 1998;39(4):171-173.
4 Arsenic Trioxide (Drug Evaluation). In: DRUGDEX® System (electronic version). Thomson Reuters (Healthcare) Inc., Greenwood Village, Colorado, USA. Available at: http://www.thomsonhc.com. (cited: 4/28/2010)
5 Melarsoprol (Drug Evaluation). In: DRUGDEX® System (electronic version). Thomson Reuters (Healthcare) Inc., Greenwood Village, Colorado, USA. Available at: http://www.thomsonhc.com. (cited: 4/28/2010)
6 Agency for Toxic Substances and Disease Registry. Toxicological profile for arsenic. Atlanta: U.S. Department of Health and Human Services, Public Health Service; 2007.
7 Vahidnia A, van der Voet GB, de Wolff FA. Arsenic neurotoxicity–a review. Hum Exp Toxicol. Oct 2007;26(10):823-832.
8 Peters GR, McCurdy RF, Hindmarsh JT. Environmental aspects of arsenic toxicity. Crit Rev Clin Lab Sci. Dec 1996;33(6):457-493.
9 Jones H, Visoottiviseth P, Bux MK, et al. Case reports: arsenic pollution in Thailand, Bangladesh, and Hungary. Rev Environ Contam Toxicol. 2008;197:163-187.
10 Ghosh P, Banerjee M, Giri AK, Ray K. Toxicogenomics of arsenic: classical ideas and recent advances. Mutat Res. Sep-Oct 2008;659(3):293-301.
11 Vahter M, Concha G. Role of metabolism in arsenic toxicity. Pharmacol Toxicol. Jul 2001;89(1):1-5.
12 Crane RK, Lipmann F. The effect of arsenate on aerobic phosphorylation. J Biol Chem. 1953;201(1):235-243.
13 Moore SA, Moennich DM, Gresser MJ. Synthesis and hydrolysis of ADP-arsenate by beef heart submitochondrial particles. J Biol Chem. May 25 1983;258(10):6266-6271.
14 Gresser MJ. ADP-arsenate. Formation by submitochondrial particles under phosphorylating conditions. J Biol Chem. Jun 25 1981;256(12):5981-5983.
15 ter Welle HF, Slater EC. Uncoupling of respiratory-chain phosphorylation by arsenate. Biochim Biophys Acta. Jul 5 1967;143(1):1-17.
16 Winski SL, Carter DE. Arsenate toxicity in human erythrocytes: characterization of morphologic changes and determination of the mechanism of damage. J Toxicol Environ Health A. Mar 13 1998;53(5):345-355.
17 Fowler BA, Weissberg JB. Arsine poisoning. N Engl J Med. Nov 28 1974;291(22):1171-1174.
18 Klimecki WT, Carter DE. Arsine toxicity: chemical and mechanistic implications. J Toxicol Environ Health. Dec 1995;46(4):399-409.
19 Hatlelid KM. Reactions of arsine with hemoglobin. J Toxicol Environ Health. 1996;47(2):145-157.
20 Klaassen C. Heavy Metals and Heavy-Metal Antagonists. Brunton L, editor. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill; 2006. http://online.statref.com/document.aspx?fxid=75&docid=649. Accessed 4/29/10
21 Blair PC, Thompson MB, Bechtold M, Wilson RE, Moorman MP, Fowler BA. Evidence for oxidative damage to red blood cells in mice induced by arsine gas. Toxicology. Jul 1990;63(1):25-34.
22 Teitelbaum DT, Kier LC. Arsine poisoning. Report of five cases in the petroleum industry and a discussion of the indications for exchange transfusion and hemodialysis. Arch Environ Health. Jul 1969;19(1):133-143.
23 Jones NW. Arseniureted hydrogen poisoning. With report of five cases. JAMA. 1907;XLVIII(13):1099-1105.
24 Parish GG, Glass R, Kimbrough R. Acute arsine posioning in two workers cleaning a clogged drain. Arch Environ Health. Jul-Aug 1979;34(4):224-227.
25 Vantroyen B, Heilier JF, Meulemans A, et al. Survival after a lethal dose of arsenic trioxide. J Toxicol Clin Toxicol. 2004;42(6):889-895.
26 Ratnaike RN. Acute and chronic arsenic toxicity. Postgrad Med J. Jul 2003;79(933):391-396.
27 Beckman KJ, Bauman JL, Pimental PA, Garrard C, Hariman RJ. Arsenic-induced torsades de pointes. Crit Care Med. Feb 1991;19(2):290-292.
28 Ohnishi K, Yoshida H, Shigeno K, et al. Prolongation of the QT interval and ventricular tachycardia in patients treated with arsenic trioxide for acute promyelocytic leukemia. Ann Intern Med. December 5, 2000;133(11):881-885.
29 Product Information: Trisenox (arsenic trioxide) injection. Frazer, PA: Cephalon, 2008.
30 Unnikrishnan D, Dutcher JP, Varshneya N, et al. Torsades de pointes in 3 patients with leukemia treated with arsenic trioxide. Blood. Mar 1 2001;97(5):1514-1516.
31 Glazener FS, Ellis JG, Johnson PK. Electrocardiographic findings with arsenic poisoning. Calif Med. Aug 1968;109(2):158-162.
32 Drolet B, Simard C, Roden DM. Unusual effects of a QT-prolonging drug, arsenic trioxide, on cardiac potassium currents. Circulation. Jan 6 2004;109(1):26-29.
33 Ford M. Arsenic. In: Flomenbaum NE, Goldfrank LR, Hoffman RS, Howland MA, Lewin NA, Nelson LS, editors. Goldfrank’s Toxicologic Emergencies. 8th ed. New York: McGraw-Hill; 2006:1251-1264.
34 Kishi Y, Sasaki H, Yamasaki H, Ogawa K, Nishi M, Nanjo K. An epidemic of arsenic neuropathy from a spiked curry. Neurology. May 22, 2001;56(10):1417-1418.
35 Donofrio PD, Wilbourn AJ, Albers JW, Rogers L, Salanga V, Greenberg HS. Acute arsenic intoxication presenting as Guillain-Barre-like syndrome. Muscle Nerve. Feb 1987;10(2):114-120.
36 Oh SJ. Electrophysiological profile in arsenic neuropathy. J Neurol Neurosurg Psychiatry. Dec 1991;54(12):1103-1105.
37 Alain G, Tousignant J, Rozenfarb E. Chronic arsenic toxicity. Int J Dermatol. Dec 1993;32(12):899-901.
38 Hall JC, Harruff R. Fatal cardiac arrhythmia in a patient with interstitial myocarditis related to chronic arsenic poisoning. South Med J. 1989;82(12):1557-1560.
39 Hall AH. Chronic arsenic poisoning. Toxicol Lett. Mar 10 2002;128(1-3):69-72.
40 Ibrahim D, Froberg B, Wolf A, Rusyniak DE. Heavy metal poisoning: clinical presentations and pathophysiology. Clin Lab Med. Mar 2006;26(1):67-97. viii
41 Mealey JJr, Brownell GL, Sweet WH. Radioarsenic in plasma, urine, normal tissues, and intracranial neoplasms; distribution and turnover after intravenous injection in man. AMA Arch Neurol Psychiatry. Mar 1959;81(3):310-320.
42 Stenehjem AE, Vahter M, Nermell B, et al. Slow recovery from severe inorganic arsenic poisoning despite treatment with DMSA (2.3-dimercaptosuccinic acid). Clin Toxicol (Phila). May 2007;45(4):424-428.
43 Uldall PR, Khan HA, Ennis JE, McCallum RI, Grimson TA. Renal damage from industrial arisine poisoning. Br J Ind Med. Oct 1970;27(4):372-377.
44 Vaziri ND, Upham T, Barton CH. Hemodialysis clearance of arsenic. Clin Toxicol. Oct 1980;17(3):451-456.
45 Succimer (Drug Evaluation). In: DRUGDEX® System (electronic version). Thomson Reuters (Healthcare) Inc., Greenwood Village, Colorado, USA. Available at: http://www.thomsonhc.com. (cited: 4/29/2010)
46 Danielson C, Houseworth J, Skipworth E, Smith D, McCarthy L, Nanagas K. Arsine toxicity treated with red blood cell and plasma exchanges. Transfusion (Paris). Sep 2006;46(9):1576-1579.
47 Berlin M, Zallups R, Fowler B. Mercury. In: Nordberg G, Fowler B, Nordberg M, Friberg L, editors. Handbook on the Toxicology of Metals. 3rd ed. Amsterdam: Elsevier; 2007:675-729.
48 Bronstein A, Spyker D, Cantilena LJr, Green J, Rumack B, Griffin S. 2008 Annual report of the American Association of Poison Control Centers’. National Poison Data System (NPDS) Clin Toxicol. 2009;47:911-1084.
49 Goldwater LJ. Hat industry. Mercury; a History of Quicksilver: York Press; 1955.
50 Graeme KA, Pollack CV. Heavy metal toxicity, Part I: arsenic and mercury. J Emerg Med. Jan-Feb 1998;16(1):45-56.
51 O’Shea JG. “Two minutes with Venus, two years with mercury”—mercury as an antisyphilitic chemotherapeutic agent. J R Soc Med. 1990;83:392-395.
52 Risher J, De Woskin R. Toxicological profile for mercury. Atlanta: US Department of Health and Human services, Public Health Service, ATSDR; 1999.
53 Song Y, Li A. Massive elemental mercury ingestion. Clin Toxicol. 2007;45(2):193.
54 Bredfeldt JE, Moeller DD. Systemic mercury intoxication following rupture of a Miller-Abbott tube. Am J Gastroenterol. Apr 1978;69(4):478-480.
55 Hursh JB, Clarkson TW, Miles EF, Goldsmith LA. Percutaneous absorption of mercury vapor by man. Arch Environ Health. Mar-Apr 1989;44(2):120-127.
56 dell’Omo M, Muzi G, Bernard A, Lauwerys RR, Abbritti G. Long-term toxicity of intravenous mercury injection. Lancet. Jul 6 1996;348(9019):64.
57 Kedziora A, Duflou J. Attempted suicide by intravenous injection of mercury: a rare cause of cardiac granulomas. A case report. Am J Forensic Med Pathol. Jun 1995;16(2):172-176.
58 Rusyniak DE, Nanagas KA. Conservative management of elemental mercury retained in the appendix. Clin Toxicol (Phila). Nov 2008;46(9):831-833.
59 Chang LW, Verity MA. Mercury Neurotoxicity: Effects and Mechanisms. In: Chang LW, Dyer RS, editors. Handbook of Neurotoxicology. New York: Marcel Dekker; 1995:31-59.
60 Garnier R, Fuster JM, Conso F, Dautzenberg B, Sors C, Fournier E. Acute mercury vapour poisoning (author’s transl). Toxicological European Research—Recherche Europeenne en Toxicologie. Mar 1981;3(2):77-86.
61 Boyd AS, Seger D, Vannucci S, Langley M, Abraham JL, King LEJr. Mercury exposure and cutaneous disease. J Am Acad Dermatol. Jul 2000;43(1 Pt 1):81-90.
62 Finkelstein Y, Vardi J, Kesten MM, Hod I. The enigma of parkinsonism in chronic borderline mercury intoxication, resolved by challenge with penicillamine. Neurotoxicology. 1996;17(1):291-295.
63 Miller K, Ochudlo S, Opala G, Smolicha W, Siuda J. Parkinsonism in chronic occupational metallic mercury intoxication. Neurol Neurochir Pol. 2003;37(Suppl 5)):31-38.
64 Ngim CH, Devathasan G. Epidemiologic study on the association between body burden mercury level and idiopathic Parkinson’s disease. Neuroepidemiology. 1989;8(3):128-141.
65 Ohlson CG, Hogstedt C. Parkinson’s disease and occupational exposure to organic solvents, agricultural chemicals and mercury–a case-referent study. Scand J Work Environ Health. Dec 1981;7(4):252-256.
66 Centers for Disease Control and P. Mercury poisoning associated with beauty cream–Texas, New Mexico, and California, 1995-1996. MMWR Morb Mortal Wkly Rep. 1996;45(19):400-403.
67 Mahe A, Ly F, Perret J-L. Systemic complications of the cosmetic use of skin-bleaching products. Int J Dermatol. Oct 2005;44(Suppl 1)):37-38.
68 Sanchez-Sicilia L, Seto D, Nakamoto S, Kolff W. Acute mercurial intoxication treated by hemodialysis. Ann Intern Med. 1963;59:692-706.
69 Schreiner G, Maher J. Toxic nephropathy. Am J Med. 1965;38:409-449.
70 Dargan P, Giles L, Wallace C, et al. Case report: Severe mercuric sulphate poisoning treated with 2,3-dimercaptopropane-1-sulphonate and haemodiafiltration. Critical Care. 2003;7(3):R1-R6.
71 Newton JA, House IM, Volans GN, Goodwin FJ. Plasma mercury during prolonged acute renal failure after mercuric chloride ingestion. Hum Toxicol. 1983;2(3):535-537.
72 Dargan PI, Giles LJ, Wallace CI, et al. Case report: severe mercuric sulphate poisoning treated with 2,3-dimercaptopropane-1-sulphonate and haemodiafiltration. Crit Care. Jun 2003;7(3):R1-R6.
73 Lund A. The effect various substances on the excretion and the toxicity of thallium in the rat. Acta Pharmacol Toxicol (Copenh). 1956;12:260-268.
74 Aberg B, Ekman L, Falk R, Greitz U, Persson G, Snihs JO. Metabolism of methyl mercury (203Hg) compounds in man. Arch Environ Health. Oct 1969;19(4):478-484.
75 Berlin M, Carlson J, Norseth T. Dose-dependence of methylmercury metabolism. A study of distribution: biotransformation and excretion in the squirrel monkey. Arch Environ Health. Jun 1975;30(6):307-313.
76 Rusyniak DE, Furbee RB, Pascuzzi R. Historical neurotoxins: what we have learned from toxins of the past about diseases of the present. Neurol Clin. May 2005;23(2):337-352.
77 Harada M. Minamata disease: methylmercury poisoning in Japan caused by environmental pollution. Crit Rev Toxicol. 1995;25(1):1-24.
78 Harada M. Congenital Minamata disease: intrauterine methylmercury poisoning. Teratology. Oct 1978;18(2):285-288.
79 Kondo K. Congenital Minamata disease: warnings from Japan’s experience. J Child Neurol. Jul 2000;15(7):458-464.
80 Bakir F, Damluji SF, Amin-Zaki L, et al. Methylmercury poisoning in Iraq. Science. Jul 20 1973;181(96):230-241.
81 Eto K. Minamata disease. Neuropathology. Sep 2000;20(Suppl):S14-S19.
82 Eto K, Tokunaga H, Nagashima K, Takeuchi T. An autopsy case of minamata disease (methylmercury poisoning)–pathological viewpoints of peripheral nerves. Toxicol Pathol. Nov-Dec 2002;30(6):714-722.
83 Cornelis R, Heinzow B, Herber R, et al. Sample collection guidelines for trace elements in blood and urine. Pure Appl Chem. 1995;67(8/9):1575-1608.
84 Baselt RC. Disposition of Toxic Drugs and Chemicals in Man, 8th ed. Foster City: Biomedical Publications; 2008.
85 Clarkson TW, Magos L, Myers GJ. The toxicology of mercury–current exposures and clinical manifestations. N Engl J Med. 2003;349:1731-1737.
86 Rishler JF, Amler SN. Mercury exposure: evaluation and intervention in the inappropriate use of chelation agents in the diagnosis and treatment of putative mercury poisoning. Neurotoxicology. May 11 2005;26:691-699.
87 Kosnett MJ. Unanswered questions in metal chelation. Clin Toxicol. 1992;30(4):529-547.
88 Hernberg S. Lead poisoning in a historical perspective. Am J Ind Med. Sep 2000;38(3):244-254.
89 Felton JS. Man, medicine, and work in America: a historical series. 3. Benjamin Franklin and his awareness of lead poisoning. J Occup Med. Nov 1967;9(11):543-554.
90 Blood lead levels–United States, 1999-2002. MMWR Morb Mortal Wkly Rep. May 27 2005;54(20):513-516.
91 Henretig FM. 8th ed Lead. Goldfrank’s Toxicologic Emergencies. vol 8. New York: McGraw-Hill; 2006. p. 1315, 1318-1319
92 Jacobs DE, Clickner RP, Zhou JY, et al. The prevalence of lead-based paint hazards in U.S. housing. Environ Health Perspect. Oct 2002;110(10):A599-A606.
93 Haley VB, Talbot TO. Seasonality and trend in blood lead levels of New York State children. BMC Pediatr. Jun 4 2004;4:8.
94 Goldstein GW. Neurologic concepts of lead poisoning in children. Pediatr Ann. Jun 1992;21(6):384-388.
95 Elevated blood lead levels associated with illicitly distilled alcohol–Alabama, 1990-1991. MMWR Morb Mortal Wkly Rep. May 1 1992;41(17):294-295.
96 Holstege CP, Ferguson JD, Wolf CE, Baer AB, Poklis A. Analysis of moonshine for contaminants. J Toxicol Clin Toxicol. 2004;42(5):597-601.
97 Lidsky TI, Schneider JS. Lead neurotoxicity in children: basic mechanisms and clinical correlates. Brain. Jan 2003;126(Pt 1):5-19.
98 Piomelli S. Chemical toxicity of red cells. Environ. Health Perspect. Jun 1981;39:65-70.
99 Albahary C. Lead and hemopoiesis. The mechanism and consequences of the erythropathy of occupational lead poisoning. Am J Med. Mar 1972;52(3):367-378.
100 Cheson BD, Rom WN, Webber RC. Basophilic stippling of red blood cells: a nonspecific finding of multiple etiology. Am J Ind. Med. 1984;5(4):327-334.
101 Levander OA. Lead toxicity and nutritional deficiencies. Environ. Health Perspect. Apr 1979;29:115-125.
102 Loghman-Adham M. Renal effects of environmental and occupational lead exposure. Environ. Health Perspect. Sep 1997;105(9):928-939.
103 Prozialeck WC, Edwards JR, Nebert DW, Woods JM, Barchowsky A, Atchison WD. The vascular system as a target of metal toxicity. Toxicol Sci. Apr 2008;102(2):207-218.
104 Rabinowitz MB. Toxicokinetics of bone lead. Environ. Health Perspect. Feb 1991;91:33-37.
105 Rastogi S, Nandlike K, Fenster W. Elevated blood lead levels in pregnant women: identification of a high-risk population and interventions. J Perinat Med. 2007;35(6):492-496.
106 Sachs HK. The evolution of the radiologic lead line. Radiology. Apr 1981;139(1):81-85.
107 Canfield RL, Henderson CRJr, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 microg per deciliter. N Engl J Med. Apr 17 2003;348(16):1517-1526.
108 Pocock SJ, Smith M, Baghurst P. Environmental lead and children’s intelligence: a systematic review of the epidemiological evidence. BMJ. Nov 5 1994;309(6963):1189-1197.
109 Koller K, Brown T, Spurgeon A, Levy L. Recent developments in low-level lead exposure and intellectual impairment in children. Environ. Health Perspect. Jun 2004;112(9):987-994.
110 Cullen MR, Robins JM, Eskenazi B. Adult inorganic lead intoxication: presentation of 31 new cases and a review of recent advances in the literature. Medicine (Baltimore). Jul 1983;62(4):221-247.
111 Baker EL, Feldman RG, White RA, et al. Occupational lead neurotoxicity: a behavioural and electrophysiological evaluation. Study design and year one results. Br J Ind Med. Aug 1984;41(3):352-361.
112 Erenberg G, Rinsler SS, Fish BG. Lead neuropathy and sickle cell disease. Pediatrics. Oct 1974;54(4):438-441.
113 Navas-Acien A, Schwartz BS, Rothenberg SJ, Hu H, Silbergeld EK, Guallar E. Bone lead levels and blood pressure endpoints: a meta-analysis. Epidemiology. May 2008;19(3):496-504.
114 Nawrot TS, Thijs L, Den Hond EM, Roels HA, Staessen JA. An epidemiological re-appraisal of the association between blood pressure and blood lead: a meta-analysis. J Hum Hypertens. Feb 2002;16(2):123-131.
115 Boeckx RL, Postl B, Coodin FJ. Gasoline sniffing and tetraethyl lead poisoning in children. Pediatrics. Aug 1977;60(2):140-145.
116 Anderson MK, Amrich M, Decker KL, Mervis CA. Using state lead poisoning surveillance system data to assess false positive results of capillary testing. Matern Child Health J. Nov 2007;11(6):603-610.
117 Martin CJ, Werntz CL3rd, Ducatman AM. The interpretation of zinc protoporphyrin changes in lead intoxication: a case report and review of the literature. Occup Med (Lond). Dec 2004;54(8):587-591.
118 Hu H, Shih R, Rothenberg S, Schwartz BS. The epidemiology of lead toxicity in adults: measuring dose and consideration of other methodologic issues. Environ. Health Perspect. Mar 2007;115(3):455-462.
119 Clifton JC2nd, Sigg T, Burda AM, Leikin JB, Smith CJ, Sandler RH. Acute pediatric lead poisoning: combined whole bowel irrigation, succimer therapy, and endoscopic removal of ingested lead pellets. Pediatr Emerg Care. Jun 2002;18(3):200-202.
120 Sokolowski MJ, Sisson GJr. Systemic lead poisoning due to an intra-articular bullet. Orthopedics. Apr 2005;28(4):411-412.
121 Lead exposure in children: prevention, detection, and management. Pediatrics. Oct 2005;116(4):1036-1046.
122 Rogan WJ, Dietrich KN, Ware JH, et al. The effect of chelation therapy with succimer on neuropsychological development in children exposed to lead. N Engl J Med. May 10 2001;344(19):1421-1426.
123 Yeoh B, Woolfenden S, Wheeler D, Alperstein G, Lanphear B. Household interventions for prevention of domestic lead exposure in children. Cochrane Database Syst Rev 2008(2):CD006047.
124 Chisolm JJJr. The use of chelating agents in the treatment of acute and chronic lead intoxication in childhood. J Pediatr. 1968;73(1):1-38.
125 Edetate Calcium Disodium. http://www.thomsonhc.com. Accessed May 2, 2010
126 Brown MJ, Willis T, Omalu B, Leiker R. Deaths resulting from hypocalcemia after administration of edetate disodium: 2003-5. Pediatrics. Aug 2006;118(2):e534-e536.
127 Dimercaprol. http://www.thomsonhc.com. Accessed May 2, 2010
128 Succimer. http://www.thomsonhc.com. Accessed May 2, 2010
129 Penicillamine. http://www.thomsonhc.com. Accessed May 2, 2010
130 Unithiol. http://www.thomsonhc.com. Accessed May 2, 2010
131 James FA. Of ‘Medals and Muddles’ the context of the discovery of thallium: William Crookes’s early spectro-chemical work. Notes Rec R Soc Lond. 1984;39(1):65-90.
132 Swain RE, Bateman WG. The toxicity of Thallium Salts. J Biol Chem. January 1, 1910;7(2):137-151.
133 Prick JJG, Sillevis Smitt WG, Muller L. Thallium Poisoning. Elsevier Publishing Company; 1955.
134 Lynch GR, Lond MB. The toxicology of thallium. Lancet. 1930;Dec 20:1340-1344.
135 Mulkey JP, Oehme FW. A review of thallium toxicity. Vet Hum Toxicol. 1993;35(5):445-453.
136 Lebowitz E, Greene MW, Fairchild R, et al. Thallium-201 for Medical Use. I J Nucl Med. February 1, 1975;16(2):151-155.
137 Rusyniak DE. Thallium. In: Holstege CP, Neer T, Saathoff GB, Furbee RB, editors. Criminal Poisoning: Clinical and Forensic Perspectives. Sudbury, MA: Jones and Bartlett Learning, 2010.
138 Peter ALJ, Viraraghavan T. Thallium: a review of public health and environmental concerns. Environ Int. 2005;31(4):493-501.
139 Lund A. Distribution of thallium in the organism and its elimination. Acta Pharmacol Toxicol (Copenh). 1956;12:251-259.
140 Malbrain ML, Lambrecht GL, Zandijk E, et al. Treatment of severe thallium intoxication. J Toxicol Clin Toxicol. 1997;35(1):97-100.
141 Misra UK, Kalita J, Yadav RK, Ranjan P. Thallium poisoning: emphasis on early diagnosis and response to haemodialysis. Postgrad Med J. Feb 2003;79(928):103-105.
142 Nordentoft T, Andersen EB, Mogensen PH. Initial sensorimotor and delayed autonomic neuropathy in acute thallium poisoning. Neurotoxicology. 1998;19(3):421-426.
143 Bank WJ, Pleasure DE, Suzuki K, Nigro M, Katz R. Thallium poisoning. Arch Neurol. 1972;26(5):456-464.
144 Hoffman RS. Thallium toxicity and the role of Prussian blue in therapy. Toxicol Rev. 2003;22(1):29-40.
145 Tabandeh H, Crowston JG, Thompson GM. Ophthalmologic features of thallium poisoning. Am J Ophthalmol. Feb 15 1994;117(2):243-245.
146 Tromme I, Van Neste D, Dobbelaere F, et al. Skin signs in the diagnosis of thallium poisoning. Br J Dermatol. 1998;138(2):321-325.
147 Moeschlin S. Thallium poisoning. Clin Toxicol. 1980;17(1):133-146.
148 Rusyniak DE, Furbee RB, Kirk MS. Thallium and arsenic poisoning in a small Midwest town. Ann Emerg Med. 2002;39(3):307-311.
149 Meggs WJ, Hoffman RS, Shih RD, Weisman RS, Goldfrank LR. Thallium poisoning from maliciously contaminated food. J Toxicol Clin Toxicol. 1994;32(6):723-730.
150 McMillan TM, Jacobson RR, Gross M. Neuropsychology of thallium poisoning. J Neurol Neurosurg Psychiatry. 1997;63(2):247-250.
151 Reed D, Crawley J, Faro SN, Pieper SJ, Kurland LT. Thallotoxicosis. JAMA. 1963;183(7):516-522.
152 Roby DS, Fein AM, Bennett RH, Morgan LS, Zatuchni J, Lippmann ML. Cardiopulmonary effects of acute thallium poisoning. Chest. February 1984;85(2):236-240.
153 Pearce J. Studies of any toxicological effects of Prussian blue compounds in mammals–a review. Food Chem Toxicol. 1994;32(6):577-582.
154 Yang Y, Brownell C, Sadrieh N, May J, Del A. Quantitative measurement of cyanide released from Prussian blue. Clin Toxicol (Phila). Oct-Nov 2007;45(7):776-781.
155 Hoffman RS, Stringer JA, Feinberg RS, Goldfrank LR. Comparative efficacy of thallium adsorption by activated charcoal, Prussian blue, and sodium polystyrene sulfonate. J Toxicol Clin Toxicol. 1999;37(7):833-837.
156 Lund A. The effect various substances on the excretion and the toxicity of thallium in the rat. Acta Pharmacol Toxicol (Copenh). 1956;12:260-268.
157 Tsai Y-T, Huang C-C, Kuo H-C, et al. Central nervous system effects in acute thallium poisoning. Neurotoxicology. 2006;27(2):291-295.
158 Mulkey JP, Oehme FW. Are 2,3-dimercapto-1-propanesulfonic acid or Prussian blue beneficial in acute thallotoxicosis in rats? Vet Hum Toxicol. 2000;42(6):325-329.
159 Rios C, Monroy-Noyola A. D-penicillamine and Prussian blue as antidotes against thallium intoxication in rats. Toxicology. 1992;74(1):69-76.
160 Rusyniak DE, Kao LW, Nanagas KA, et al. Dimercaptosuccinic acid and Prussian blue in the treatment of acute thallium poisoning in rats. J Toxicol Clin Toxicol. 2003;41(2):137-142.
161 Pedersen RS, Olesen AS, Freund LG, Solgaard P, Larsen E. Thallium intoxication treated with long-term hemodialysis, forced diuresis and Prussian blue. Acta Med Scand. 1978;204(5):429-432.
162 Kielstein JT, Linnenweber S, Schoepke T, Fliser D. One for all–a multi-use dialysis system for effective treatment of severe thallium intoxication. Kidney Blood Press Res. 2004;27(3):197-199.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
178 Heavy Metals
Arsenic
Background
Enormous achievements and incredible misfortune mark arsenic’s history. Dating back to 400 BC, Greek and Roman physicians included arsenic in their medical armamentarium.1 Today, arsenic continues to be found in treatments offered by practitioners of Indian folk medicine and traditional Chinese medicine.2,3 In addition, in Western medicine, arsenic trioxide (Trisenox) and melarsoprol are used to treat promyelocytic leukemia4 and late-stage African trypanosomiasis.5
Arsenic’s therapeutic usefulness is based on its ability to poison cells, and it is best known as a poison. While arsenic has been noted to produce a garlic odor and possess a characteristically sweet flavor, most arsenical compounds have no perceptible smell or taste.6 This has made the detection of arsenic difficult when employed as a homicidal agent. Historically, this has afforded arsenic the illustrious title of “Poison of Kings and the King of Poisons.”7
Toxicity
Environmental arsenic comes from both natural (volcanic eruption, water runoff) and manmade sources (mining, smelting, combustion of fossil fuels, and pesticide use).7,8 Food, predominantly seafood, represents the principal route for human exposure; however, rice, mushrooms, and poultry also contribute to exposure.6 Alternative sources include air and water, particularly in Bangladesh where tube-wells supply millions with arsenic-contaminated drinking water.9 In the United States, the average person consumes 50 µg of arsenic daily, with inorganic arsenic accounting for 3.5 µg of this total. Certain occupations such as metal working, electronics manufacturing, and glass manufacturing also increase arsenic exposure.6
Arsenic is found in nature in several different forms: combined with carbon and other elements in organic compounds, in inorganic compounds, in gaseous compounds, and in the nontoxic elemental form. Arsenobetaine, an organic arsenic compound found in fish and shellfish, possesses a low risk for human toxicity.6 Inorganic arsenicals, found as trivalent (As3+, arsenite, more toxic) or pentavalent (As5+, arsenate, less toxic) compounds, account for the majority of human toxicity. Arsenite compounds exhibit high affinity for binding to proteins, whereas the majority of arsenate compounds remain unbound. Weak protein binding allows arsenate compounds to be freely excreted. Protein-bound arsenite functions as both a storage depot and a target action site.10 Alternating hepatic reduction and methylation reactions convert inorganic arsenic to an organic form. This transformation detoxifies the parent compound but also increases arsenic’s carcinogenicity.7,10,11
The mechanism accounting for arsenic’s ability to disrupt cellular function, and that which results in acute toxicity, differs depending on the form of arsenic responsible for the exposure. By disrupting the activity of key enzymes by binding to critical sulfhydryl groups, arsenite impairs both oxidative phosphorylation and gluconeogenesis. Especially important in this regard is arsenite’s ability to inhibit the enzyme, pyruvate dehydrogenase (PDH), which catalyzes the first and rate-limiting step in the tricarboxylic acid (TCA) cycle. Inhibition of PDH hinders the production of acetyl-CoA from pyruvate, limiting cellular ATP production. In addition, arsenate can substitute for phosphate and become incorporated into arsenate analogs of glucose-6-phosphate, 6-phosphogluconate, and adenosine triphosphate (ATP).6,12,13 These arsenate analogs are less stable than their phosphate-containing counterparts, and their formation can lead to uncoupling of oxidative phosphorylation.6,13–16
Arsine gas (AsH3) is released when many arsenic-containing compounds come in contact with an acid or when metallic arsenic comes into contact with water. Workers involved with lead plating, soldering, etching, smelting, and galvanizing are at risk for exposure to arsine gas.17 Being colorless and nonirritating, arsine gas is particularly difficult to detect following industrial exposure. As with arsenic, arsine gas has been noted to possess a slight garlic odor; also like arsenic, however, arsine’s odor is not always detectable.18
Exposure to arsine causes acute hemolysis. The exact mechanism for this hemolysis is not completely understood and is likely multifactorial, involving both binding of arsine to hemoglobin and perhaps inhibition of sulfhydryl-containing enzymes.19–22 One complication of arsine exposure is acute renal failure. Arsine-induced renal failure most likely is secondary to direct nephrotoxic effects of arsine as well as the renal toxicity caused by the release of hemoglobin from lysed red blood cells.23 Although chronic renal dysfunction has been described after arsine exposure, recovery is possible.22,24
Clinical Presentation
A patient’s clinical presentation after arsenic exposure is influenced by four factors: the arsenic species, the amount, the route, and the duration of exposure. In addition, the symptoms of acute arsenic toxicity differ depending on whether exposure was oral or inhalational. Gastrointestinal symptoms are a reliable finding in cases of acute arsenic poisoning. Shortly after exposure, patients typically experience abdominal discomfort, nausea, emesis, and profuse diarrhea.7,25,26 Severe gastroenteritis and hemorrhage may develop. Furthermore, arsenic induces capillary dilation, third-spacing of fluid, ventricular arrhythmias, and cardiomyopathy. Collectively these changes can result in pulmonary edema, hypotension, congestive heart failure, and shock.7,20 Cardiac abnormalities following arsenic exposure can include QTc prolongation, T-wave abnormalities, second-degree heart block, QRS widening, nonconducted P-waves, nonsustained ventricular tachycardia, torsades de pointes, complete heart block, asystole, pericardial effusion, and serositis.27–31 Cardiac conduction abnormalities have been described following therapeutic use of arsenic trioxide. These conduction abnormalities may be a consequence of arsenic trioxide–induced electrolyte abnormalities (hypokalemia or hypomagnesemia).29 Alternatively, these changes in cardiac conduction might reflect the cardiotoxic effects of other cancer chemotherapeutic agents. Additionally, arsenic-induced blockade of IKr and IKs channels with activation of IK-ATP channels has been demonstrated.32 The variability seen in arsenic-induced QTc prolongation likely occurs secondary to the combined effects of this activation and blockade.
Neurologic symptoms are also associated with arsenic toxicity and typically include altered mental status and confusion.20,25 Seizures have been linked to arsenic-induced arrhythmias,27,33 and if related to hypoxemia may signal a terminal stage in cases of arsenic poisoning. While typically delayed 2 to 8 weeks after exposure, peripheral neuropathy from large arsenic exposures sometimes can become apparent within a few hours.7,34 Patients describe pain, numbness, and paresthesias in a “stocking/glove” sort of distribution.20,25,35 The symmetric sensorimotor neuropathy seen following arsenic exposure can be misdiagnosed as Guillain-Barré.7,35 Marked abnormalities in sensory and mixed nerve conduction in conjunction with moderate motor conduction abnormalities can be seen on electrophysiologic testing. These results are consistent with axonal degeneration, which is also apparent histologically in nerve biopsies.36
Symptoms of chronic arsenic toxicity differ from those of acute toxicity. Patients demonstrate hematologic abnormalities including pancytopenia, anemia, and macrocytosis. Dermal manifestations include Mees lines (transverse white striae on the fingernails), hyperkeratotic extremity lesions, and hyperpigmented melanosis. Gastrointestinal symptoms as well as liver disease are also described.7,26,37,38 Additionally, patients may note a metallic taste.7,38,39
Inhalational exposure to arsenic in the form of arsine gas produces symptoms that differ from those seen with oral arsenic exposure. Classically, exposure is characterized by the development of a triad of symptoms: abdominal pain, hematuria, and jaundice. Within 2 to 24 hours of exposure, patients develop headache, malaise, abdominal discomfort, nausea, and emesis. The variability in the onset of symptoms is influenced by the duration of exposure and the concentration of the gas.17 Hemolysis with subsequent gross hematuria and renal failure often follow. Additionally, patients may develop scleral icterus and a bronze skin discoloration.17,24 As with exposure to other arsenic compounds, exposure to arsine gas can lead to development of peripheral neuropathy.40
Diagnosis
The potential for arsenic toxicity can be determined by combining the patient’s clinical presentation with the likelihood of exposure. Blood and urine testing can confirm the diagnosis. Blood arsenic clearance occurs in three phases: Phase 1 takes place 2 to 3 hours following intravenous (IV) administration and is associated with a half-life (t1/2) of about 2 hours; phase 2 occurs 3 hours to 7 days following administration and is associated with a t1/2 of about 27 hours; and phase 3 occurs 10 or more days following administration and is associated with a t1/2 of about 230 hours.41 Arsenic is rapidly cleared from the blood during phases 1 and 2; thus, the reliability of blood arsenic testing is limited to the early stages of acute toxicity.33 Following IV administration of radioactive arsenic, urinary recovery has been demonstrated at 18% to 30% after 1 hour, 36% to 56% at 4 hours, 57% to 90% by 9 days, and 96.6% by 18 days.41 A positive urine arsenic level should be followed up by arsenic speciation to distinguish nontoxic organic arsenic (commonly found in seafood) from the toxic inorganic form. A random urine arsenic level greater than 50 µg/L, or a 24-hour urine sample demonstrating more than 100 µg of arsenic, can confirm the diagnosis of acute arsenic toxicity. However, this diagnosis should be questioned if there is a history of recent seafood ingestion or the sample was not collected in a metal-free container. Hair and nail samples may be used to confirm exposure when chronic toxicity is suspected.7 Hair testing can also be performed to estimate an approximate exposure date based on the rate of hair growth (0.4 mm/d) and the distance of the root from the arsenic peak.42 Additionally, an electrocardiogram (ECG) should be obtained in cases of potential arsenic toxicity; T-wave abnormalities, QTc prolongation, and torsades de pointes have been described following arsenic exposure.27,31
Following possible exposure to arsine gas, laboratory evaluation should include complete blood count, measurement of circulating lactate dehydrogenase and other liver enzyme concentrations, measurement of serum bilirubin concentration, Coombs testing, and monitoring of serum electrolyte levels and renal function. Patients will demonstrate a Coombs-negative hemolytic anemia with elevated circulating lactate dehydrogenase, aspartate aminotransferase, alanine aminotransferase, and bilirubin levels, renal dysfunction, and subsequent hyperkalemia. Hemoglobinuria, albuminuria, and occasional erythrocyte and hemoglobin tubular casts are commonly seen on urinalysis. A peripheral blood smear can show signs of red blood cell damage including erythrocyte fragments, basophilic stippling, anisocytosis, poikilocytosis, and Heinz bodies.17,18,22,43 Methemoglobinemia also has been described.43
Treatment
Initial treatment of arsenic toxicity must include prevention of further exposure to the poison, careful monitoring of the cardiovascular system, and judicious repletion of intravascular fluid deficits. Hyperkalemia, which can be clinically significant, should be treated. Despite a lack of supporting data, gastric lavage and administration of activated charcoal should be considered following oral exposure to a large of dose of arsenic in an attempt to limit absorption of the toxic compound(s).7 Patients presenting with renal failure following arsenic exposure may need renal replacement therapy until renal function recovers.44 Additionally, patients demonstrating ECG abnormalities, particularly QTc prolongation, should have close monitoring of their electrolytes, with replacement as indicated. Antiarrhythmic therapy should be instituted as needed.
Chelators are an additional treatment to be considered. Dimercaprol, a parenteral chelating agent which functions intracellularly as well as extracellularly, is the first line of therapy for patients experiencing abdominal symptoms following acute arsenic exposure.33 Dimercaprol should be administered intramuscularly (IM) at doses of 3 to 4 mg/kg every 4 to 12 hours.20 In cases of subacute or chronic exposure when the patient can take oral medications, succimer (DMSA or 2,3-dimercaptosuccinic acid) is the chelator of choice. Succimer should be administered orally (10 mg/kg every 8 hours for 5 days). Following the initial 5 days of administration, succimer dosing is adjusted to every 12 hours.33 Clinical course determines the duration of chelator therapy.20 Urinary arsenic levels can be used to calibrate duration of treatment, with therapy being discontinued when 24-hour urinary arsenic levels are below 50 µg/L.33 Despite its recommended use, succimer has not consistently demonstrated increased urinary arsenic excretion following administration, and neuropathy can progress despite therapy.25,42 Succimer is not currently approved by the U.S. Food and Drug Administration (FDA) for treatment of arsenic toxicity.45
Treatment of arsine gas exposure represents a special circumstance. As with other forms of arsenic exposure, initial therapy requires elimination of the source. Caution should be taken to prevent additional casualties as first responders enter the exposure site. Since arsine toxicity is associated with hemolysis, exchange transfusion can be used to replenish red blood cell mass and remove both the toxic complexes formed from the arsine/hemoglobin interaction as well as the released hemoglobin pigment.40,46 Patients with renal failure should receive renal replacement therapy as indicated by clinical findings.17,18 The use of chelation therapy to treat patients acutely poisoned by arsine is controversial and may not affect the clinical course.18
Mercury
Background
Mercury is a naturally occurring metal found in elemental, inorganic salt, and organic forms. The use of mercury dates back to before 1500 BC. Throughout history, mercury has been employed as a cosmetic component, decoration, and even medicine. Mercury is also a natural part of our atmosphere, with 30,000 to 50,000 tons degassing from the earth’s surface annually; human activity adds another 20,000 tons each year.47 Toxicity from mercury can occur as a result of occupational, environmental, or medical exposure. Each year, mercury exposure accounts for over 6000 cases of toxicity reported to U.S. poison centers.48 The dose, length of exposure, and form of mercury can cause wide variation in the clinical presentation and ultimate outcome.49–52
Toxicity
Elemental mercury can be found in barometers, dental amalgams, electronics, thermostats, thermometers, and batteries. It is also found in some folk remedies. Elemental mercury is a liquid at room temperature. Even without heating, elemental mercury releases sufficient mercury in the gas phase to cause toxicity. Most problems from elemental mercury are the result of vapor inhalation, with nearly 80% of inhaled vapor being absorbed by the alveoli and transferred into circulating red blood cells. Most absorbed mercury is converted to its divalent (mercuric) form, thus decreasing its lipid solubility. If the patient is exposed to a very high dose of mercury vapor, a small amount of gaseous mercury can remain in the bloodstream, leading to penetration of the blood-brain barrier and central nervous system (CNS) injury.8 By comparison, the gastrointestinal tract takes up less than 2% of ingested elemental mercury53; however, in patients with mucosal disruption, transmucosal absorption of mercury can be markedly increased.54 Absorption of mercury across intact skin is minimal,55 but subcutaneous injection can cause an increase in urinary mercury concentrations. Intravenous injection of mercury can cause both mercury toxicity and mechanical obstruction of pulmonary blood flow.56,57 Elimination of inhaled vapor is via the urinary tract, and the elimination t1/2 of absorbed elemental mercury is about 60 days.56 Elemental mercury when orally ingested is typically nontoxic owing to minimal absorption across intact gastric and intestinal mucosa.58
Elemental mercury primarily targets the lungs and brain, but the poison also can cause renal and gastrointestinal injury.5,14,15 By forming complexes with sulfhydryl groups, mercury interferes with protein and nucleic acid synthesis, protein phosphorylation, and calcium homeostasis. It can also cause oxidant stress via this mechanism.59,60 Inhaled mercury vapor has a corrosive effect on the lungs and is capable of producing acute inflammation of the bronchi and bronchioles. This may result in a fatal interstitial pneumonitis.60
Elemental mercury poisoning can be misdiagnosed as pheochromocytoma. This can occur as mercury inactivates the coenzyme, S-adenosylmethionine, which inhibits catechol-O-methyltransferase (COMT). As a result, catecholamine breakdown decreases and adrenergic symptoms such as hypertension and diaphoresis develop.18–20 A constellation of personality changes in affected individuals has come to be known as erethism. The symptoms include the following61:
There has been a reported association between mercury exposure, erethism, and the development of parkinsonism61–63; however, the evidence for this association comes from only two studies64,65 and two case reports.21,22
Inorganic mercury is most commonly found in nature as cinnabar (mercury [II] sulfide). In humans, however, exposure to inorganic mercury compounds comes from germicides, pesticides, and mercury-containing antiseptics.61 Of interest, there have been several recent reports of inorganic mercury intoxication due to the use of skin-lightening beauty creams.66,67 Inorganic mercury can be absorbed through the skin, via the lungs, and via the gastrointestinal tract. In blood, inorganic mercury has a t1/2 of about 24 to 40 hours, and clearance of mercury by the kidneys is responsible for the toxic effects on the distal portion of the proximal convoluted tubules.68,69
More corrosive to the gut than elemental mercury,61,70 ingestion of mercury salts commonly causes nausea, vomiting, abdominal pain, and hematemesis. Ingestion of relatively large doses of mercury salts can lead to colitis with necrosis and mucosal sloughing, resulting in massive fluid losses.14,29,30
Along with gastrointestinal symptoms, the other notable acute effect of inorganic mercury is development of acute renal failure. Potentially reversible, renal injury can occur within hours to days of an acute exposure.71,72 Membranous glomerulonephritis and nephrotic syndrome also may occur after chronic exposure. Termination of exposure may lead to resolution of nephrotic syndrome.50,73
With prolonged skin exposure, gray-brown hyperpigmentation of skin folds of the neck and face can occur. Used as an analgesic for teething in the 19th century, dental application of calomel (mercuric chloride) can cause loose teeth, blue discoloration of the gingiva, and systemic toxicity.14,31 As with elemental mercury, acrodynia and erethism have been reported with inorganic mercury exposure.50,61
Organic mercury compounds are found in fungicides, antiseptics such as merthiolate and mercurochrome, preservatives including thimerosal, and as a contaminant of predatory fish including tuna and swordfish. As much as 90% of ingested organic mercury is absorbed by the gastrointestinal tract.74 Pulmonary absorption of organic compounds such as methylmercury vapor, approaches 80%, depending on particle size. Absorption across intact skin also can occur.47 Methylmercury readily crosses the blood-brain barrier, achieving CNS levels that are three to six times those in blood.75
Perhaps the best-known mass poisonings of mercury involved methylmercury. During the 1950s and 1960s, a chemical company in the Japanese fishing village of Minamata dumped wastewater containing mercury into Hyakken Harbor on Minamata Bay. Aquatic organisms converted the inorganic waste to an organic form (methylmercury) that was then passed up the food chain. The mercury eventually was concentrated in larger fish, which were then consumed by residents of the area.76 Over time, more than 2265 patients developed ataxia, sensory disturbances, constriction of visual fields, dysarthria, auditory disturbances, and tremor. Children exposed in utero developed congenital Minamata disease, which was characterized by seizures, spasticity, deafness, and severe mental deficiency.77 All these children had mental retardation, cerebellar ataxia, limb deformities, primitive reflexes, and dysarthria. Hypersalivation and chorea were seen in 95% and microcephaly in 60% of affected children.77–79
In a second event in 1971, 6500 Iraqis suffered symptoms similar to the Minamata patients after eating bread baked with flour made from grain intended for use as seed. The grain had been treated with a fungicide containing methylmercury.80
These two events demonstrated that organic mercury targets the CNS and that fetal brain tissue is more susceptible than the adult brain to the toxic effects of organic mercury. Postmortem findings have shown damage to gray matter of the cerebral and cerebellar cortex. The temporal cortex and calcarine region of the occipital lobe are most affected.81,82 Pathologic changes in adults and children include cortical atrophy, hypoplasia of the corpus callosum, hypoplasia of the granular cell layer of the cerebellum, and demyelination of the pyramidal tracts.78
Diagnostic Tests
Urine testing (24-hour collection) is most useful for confirming exposure/toxicity to either elemental or inorganic mercury. Collection containers are frequently washed with nitric acid, but laboratory practices may vary. Specimens should be refrigerated to decrease bacterial reduction of mercury to volatile elemental mercury.83 Samples should not be collected within 48 hours of gadolinium administration (as with magnetic resonance imaging) or within 72 hours of consuming predatory fish (e.g., tuna or swordfish).
Because approximately 90% of methylmercury is bound to red blood cells74 and very little is excreted via the kidneys, the preferred test to determine organic mercury is a whole-blood mercury level. While most persons have whole-blood mercury concentrations of less than 6 ng/mL, diets rich in predatory fish can elevate levels to 200 ng/mL or higher.84 For that reason, patients should avoid consumption of fish for at least 72 hours prior to blood testing.
