47 Heart-Lung Interactions
Ventilation can profoundly alter cardiovascular function. The boundaries of the cardiovascular unit’s responsiveness are defined by both cardiovascular and pulmonary factors. These limitations include myocardial reserve, circulating blood volume, blood flow distribution, autonomic tone, endocrinological responses, lung volume, intrathoracic pressure (ITP) and surrounding pressures for the remainder of the circulation. That positive-pressure ventilation may influence cardiovascular function in ways not seen during spontaneous ventilation was appreciated when positive-pressure ventilation was first introduced over 50 years ago1 and still results in new perspectives today.2
 Airway Pressure, Intrathoracic Pressure, and Lung Volume Relationships
Airway Pressure, Intrathoracic Pressure, and Lung Volume Relationships
Since positive-pressure ventilation was introduced, the concept of relating hemodynamic consequences to airway pressure was widely accepted.3,4 This oversimplification has been the source of much of the confusion in the clinical literature. A major source of such confusion rests in equating changes in airway pressure (Paw) with changes in both pleural pressure (Ppl) and lung volume. Physicians often equate Paw with the hemodynamic effects seen because (1) Paw can be measured easily at the bedside in patients receiving mechanical ventilation, (2) mean Paw reflects mean alveolar pressure, and (3) increases in Paw qualitatively reflect increases in both lung volume and Ppl. However, the association between Paw and other hemodynamically relevant factors (1) is highly variable as ventilatory patterns, airway resistance, and lung compliance change, (2) does not accurately reflect changes in pericardial pressure (Ppc), which is a primary determinant of transmural left ventricular (LV) pressure, and (3) may mislead the caregiver at the bedside into altering therapy based on these wrong assumptions. Numerous studies have demonstrated that the primary determinants of hemodynamic responses to ventilation are due to changes in intrathoracic pressure and lung volume, and not Paw.5 Thus, prior to examining heart-lung interactions, we shall address the relation between Paw, Ppl, Ppc, and lung volume. To simplify the discussion, we shall use the term intrathoracic pressure (ITP) to refer to nonspecific intrathoracic surface pressure. When specific intrapleural surface pressures are identified, they will be referred to either as the lateral chest wall, diaphragm, and juxtacardiac pleural pressures or pericardial pressure where appropriate.
Airway Pressure, Lung Volume, and Regional Pleural Pressures
During positive-pressure inspiration, increases in Paw parallel increases in lung volume. In the sedated and paralyzed patient at end-inspiration, only lung and thoracic compliance determine the relationship between Paw and lung volume. However, if ventilated patients actively resist lung inflation or sustain expiratory muscle activity at end-inspiration, then end-inspiratory Paw will exceed resting Paw for that lung volume. Similarly, if patient activity prevents full exhalation by expiratory braking, then for the same end-expiratory airway pressure (often measured as positive end-expiratory pressure [PEEP]), lung volume may be higher than predicted from end-expiratory Paw values alone. Finally, even if inspiration is passive and no increased airway resistance is present, Paw may rapidly increase as chest wall compliance decreases, especially as chest wall compliance includes diaphragmatic dissention. If intraabdominal pressure were to increase then end-expiratory Paw must also increase for a constant tidal volume. In acute respiratory distress syndrome (ARDS), increases in intraabdominal pressure can occur with gut wall swelling (third-spacing) and gut distention.6
As the lung expands, it pushes on surrounding structures, distorting them and causing their surface pressures to increase. Thus lung expansion induces an increase in lateral wall, diaphragmatic, and juxtacardiac Ppl as well as Ppc. The degree of increase in each of these surface pressures will be a function of the compliance and inertance of their opposing structures. These interactions were described by Novak et al.,6 who demonstrated that changes in Ppl induced by positive-pressure ventilation are not similar in all regions of the thorax and increase differently as inspiratory flow rate and frequency increase. Pleural pressure on the diaphragm increases least during inspiration, and juxtacardiac Ppl increases most. Since the diaphragm is very compliant, it seems reasonable that diaphragmatic ITP should increase less than lateral chest wall Ppl to sudden increases in lung volume. However, if abdominal distention develops, as commonly occurs in the setting of sepsis, the diaphragm will become relatively noncompliant because of the increase in abdominal pressure. Under these conditions, ITP tends to increase similarly across the thorax, so one may incorrectly assume with abdominal distention that the lung is injured and becoming stiffer. In fact, lung compliance may be normal, but chest wall compliance is restricting expansion.6 This distinction is important because increasing Paw to overcome chest wall stiffness should increase ITP more with greater hemodynamic consequences but should not improve gas exchange, since the alveoli are not damaged. If lung compliance is reduced, as in ALI, similar increases in Paw should not increase ITP as much but should also recruit collapsed and injured alveolar units, improving gas exchange but having smaller hemodynamic effects.
A hydrostatic pressure gradient exists in the pleural space. Dependent regions have a higher baseline pressure than nondependent regions in proportion to their height above or below the heart in centimeters and equate to an equal cm H2O pressure difference. In the supine subject, steady state apneic Ppl along the horizontal plane from the apex of the lung to the diaphragm are similar, whereas anterior Ppl is less, and posterior gutter Ppl is greater (Figure 47-1).
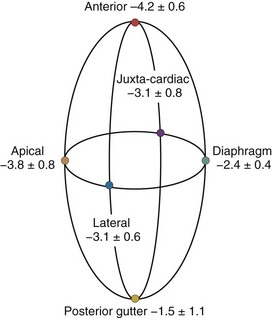
Care must be taken to determine not only what types of ventilation are being compared but also how and where estimates of Ppl and Ppc are made. For example, if estimates of transpulmonary pressure are needed to define lung compliance and its change with recruitment maneuvers, lateral chest wall Ppl appears to more accurately reflect the pressure volume characteristics of the intact lung.6 Similarly, if diaphragmatic work is to be monitored, either esophageal or diaphragmatic Ppl should be used. Finally, if heart-lung interactions are being examined, juxtacardiac Ppl is the most accurate measure of Ppl, and increases during positive-pressure inspiration will be underestimated by esophageal pressure. Since the heart is fixed within the cardiac fossa, juxtacardiac Ppl increases more than lateral chest wall or diaphragmatic Ppl. Pinsky and Guimond7 demonstrated that heart failure was associated with a greater increase in Ppc than juxtacardiac Ppl, presumably because of pericardial restraint. Importantly, with progressive increases in PEEP, juxtacardiac Ppl increased toward levels of Ppc found without PEEP, whereas Ppc initially remained constant. Once these two surface pressures became equal, further increases in PEEP increased both juxtacardiac Ppl and Ppc in parallel. If pericardial volume restraint exists, juxtacardiac Ppl will underestimate Ppc, but with sustained lung compression of the heart overriding tamponade, juxtacardiac Ppl and Ppc will become similar.
Pleural Pressure and Lung Volume in Acute Lung Injury
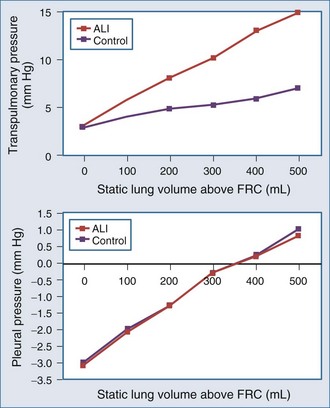
The interaction of Paw, lung volume, and ITP in the setting of lung disease is complex and can be different for the same pathologic setting depending on the tidal volume, inspiratory flow rate, ventilatory frequency, and body position. The presence of parenchymal disease, airflow obstruction, and extrapulmonary processes that directly alter chest wall–diaphragmatic contraction also profoundly alter these interactions. Static lung expansion occurs as Paw increases because the transpulmonary pressure (Paw relative to ITP) increases. If lung injury induces alveolar flooding or increased pulmonary parenchyma stiffness, greater increases in Paw will be required to distend the lungs to a constant end-inspiratory volume. Romand et al.8 demonstrated that Paw increased more during ALI than in control conditions for a constant tidal volume, whereas lateral chest wall Ppl and Ppc increased similarly between both conditions if tidal volume was held constant (Figure 47-2). These data agree with the studies of O’Quinn et al.9 that the primary determinant of the increase in Ppl and Ppc during positive-pressure ventilation is lung volume change, not Paw change. Data from Romand et al.8 demonstrated that the increase in ITP during sustained increases in lung volume is greater than the increase in Ppc. Presumably, Ppc does not increase as much as ITP because increasing lung volume also reduces filling of the ventricles, thereby reducing their size inside the cardiac fossa. To summarize, for a constant increase in lung volume, ITP will increase similarly despite drastic changes in lung compliance and airway resistance.
Relation Between Airway, Pleural, and Pericardial Pressures
Since the distribution of alveolar collapse and alterations in lung compliance in ARDS and ALI is non-homogeneous, lung distention during positive-pressure ventilation must reflect overdistention of some regions of the lung at the expense of noncompliant or poorly compliant regions. Accordingly, Paw will reflect distention of lung units that were aerated prior to inspiration but may not reflect the degree of lung inflation of nonaerated lung units. Pressure-limited ventilation assumes this is the case and aims to limit Paw in ALI so as to prevent overdistention of aerated lung units, with the understanding that tidal volume, and thus minute ventilation, must decrease. Therefore, pressure-limited ventilation will hypoventilate the lungs, leading to “permissive” hypercapnia. It is not surprising that in an animal model of ALI in which tidal volume was either kept constant at pre-injury levels or reduced to match pre-injury plateau Paw (pressure-limited ventilation), both Ppl and Ppc increased less compared to both pre–lung injury states or in ALI when tidal volume remained at pre-injury levels.8 These points underlie the fundamental hemodynamic differences seen when different modes of mechanical ventilatory support are compared to each other.
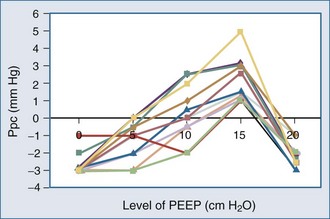
Because ALI is often non-homogeneous, with aerated areas of the lung displaying normal compliance, large increases in Paw can overdistend aerated lung units.10 Vascular structures that are distended will have a greater increase in their surrounding pressure than collapsible structures that do not distend.11 However, Romand et al.8 and Scharf and Ingram12 demonstrated that despite this non-homogeneous pattern of alveolar distention, if tidal volume is kept constant, Ppl increases in a homogeneous manner independent of the mechanical properties of the lung. Under constant tidal volume conditions, changes in peak and mean Paw will reflect changes in the mechanical properties of the lungs and patient coordination but may not reflect changes in ITP. Similarly, changes in Paw may not alter global cardiovascular dynamics. Underscoring this limitation of Paw to reflect either ITP or Ppc, Pinsky et al.13 demonstrated in postoperative patients that the percentage of Paw increase that will be transmitted to the pericardial surface is not constant from one subject to the next as PEEP is increased (Figure 47-3). Thus, one cannot predict the amount of increase in Ppc or Ppl that will occur in patients as PEEP is increased. Accordingly, assuming some constant fraction of Paw transmission to the pleural surface as a means of calculating the effect of increasing Paw on Ppl is inaccurate and potentially dangerous to patient management.
Although it may be difficult to know the actual Ppl, it is possible to determine the ventilation-induced change in Ppl. Since during airway occlusion maneuvers lung volume does not change, transpulmonary pressure is also constant so that the change in ITP is equal to the change in Paw.14 Accordingly, an increase in Paw of 20 mm Hg during a Valsalva maneuver will reflect an increase in ITP of 20 mm Hg, and a decrease in Paw to −20 mm Hg during a Mueller maneuver will reflect a decrease in ITP of 20 mm Hg below atmospheric pressure.
Clinically, esophageal pressure is often used as a surrogate for Ppc and ITP. Two limitations to the use of esophageal pressure (Pes) in estimating Ppc and ITP exist. First, Ppc and ITP may not be similar nor increase by similar amounts with the application of positive Paw if the pericardium becomes a limiting membrane.15,16 Operationally, this equates to Ppc exceeding juxtacardiac Ppl by the degree to which the pericardium limits biventricular dilation. Thus, estimates of Ppc made by using ITP measures may underestimate Ppc and overestimate the increase in Ppc as Paw is increased. Second, although esophageal pressure is often used clinically to estimate swings in both Ppl and Ppc so as to calculate the work cost of breathing, esophageal pressure is only accurate at reflecting negative swings in Ppl during spontaneous inspiration in upright seated individuals3 and in recumbent dogs in the left lateral position.17 Esophageal pressure changes underestimate both the positive swings in Ppl and the mean increase in Ppl seen with increases in lung volume during positive-pressure ventilation. During Mueller and Valsalva maneuvers, however, because lung volume does not change, swings in esophageal pressure will accurately reflect swings in ITP.3 In fact, documenting that airway and esophageal pressure swings are identical in magnitude is how esophageal manometers are validated at the bedside.
 Hemodynamic Effects of Ventilation
Hemodynamic Effects of Ventilation
Lung volume increases during both spontaneous and positive-pressure ventilation, but ITP decreases during spontaneous inspiration and increases during positive-pressure inspiration. Changes in ITP and the metabolic demand needed to create these changes represent the primary determinants of the hemodynamic differences between spontaneous and positive-pressure ventilation.18,19
Ventilation As Exercise
Spontaneous ventilatory efforts are produced by respiratory muscle contraction. Blood flow to these muscles is derived from several arterial circuits whose absolute flow is believed to exceed the highest metabolic demand of maximally exercising skeletal muscle.20 Thus, under normal cardiovascular conditions, blood flow is not the limiting factor determining maximal ventilatory effort. Although ventilation normally requires less than 5% of total O2 delivery to meet its demand,20 in lung disease states where the work of breathing is increased (e.g., pulmonary edema, bronchospasm), the work cost of breathing can increase metabolic demand for O2 to 25% or 30% of total O2 delivery.20–23 Furthermore, if cardiac output is limited, blood flow to other organs and to the respiratory muscles may be compromised, inducing both tissue hypoperfusion and lactic acidosis.24–27 Starting mechanical ventilation may reduce metabolic demand, increasing SvO2 for a constant cardiac output and CaO2. Intubation and mechanical ventilation, when adjusted to the metabolic demands of the patient, may dramatically decrease the work of breathing, resulting in increased O2 delivery to other vital organs and decreased serum lactic acid levels. These cardiovascular benefits can also be realized with effective use of noninvasive ventilation mask continuous positive airway pressure (CPAP).28 The obligatory increase in SvO2 will result in an increase in the PaO2 if fixed right-to-left shunts exist, even if mechanical ventilation does not alter the ratio of shunt blood flow to cardiac output. Finally, if cardiac output is severely limited, respiratory muscle failure develops despite high central neuronal drive such that many heart failure patients experience respiratory arrest prior to cardiovascular standstill.29
Ventilator-dependent patients who fail to wean from mechanical ventilation may occasionally have impaired baseline cardiovascular performance30 but routinely develop overt signs of heart failure during weaning, including pulmonary edema,30,31 myocardial ischemia,32–35 tachycardia, and gut ischemia.36 Jubran et al.37 demonstrated that although all subjects increase their cardiac outputs in response to a weaning trial, those who subsequently fail to wean demonstrate a reduction in mixed venous O2 saturation, consistent with a failing cardiovascular response to increased metabolic demand. Importantly, the increased work of breathing may come from endotracheal tube flow resistance.38 Thus, weaning from mechanical ventilatory support can be considered a cardiovascular stress test. Again, investigators have documented weaning-associated ECG and thallium cardiac blood flow scan-related signs of ischemia in subjects with known coronary artery disease32 and in otherwise normal patients.34,35 Placing patients with severe heart failure and/or ischemia on ventilatory support by either intubation and ventilation39 or noninvasive CPAP40 can reverse myocardial ischemia.
Hemodynamic Effects of Changes in Lung Volume
Autonomic Tone
The lungs are richly enervated with integrated somatic and autonomic fibers that originate, traverse through, and end in the thorax. These neuronal networks mediate multiple homeostatic processes through the autonomic nervous system that alter both instantaneous cardiovascular function (e.g., respiratory sinus arrhythmia) and steady state cardiovascular status (e.g., ADH-induced fluid retention). Numerous cardiovascular reflexes are centered within this network. Inflation induces immediate changes in autonomic output. The most commonly described inflation-chronotropic responses act through vagal-mediated reflex arcs.41,42 Lung inflation to normal tidal volumes (<10 mL/kg) increases heart rate via parasympathetic tone withdrawal. Inspiration-associated cardioacceleration is referred to as respiratory sinus arrhythmia (RSA)43 and denotes normal autonomic tone.44 Loss of RSA is associated with dysautonomia, and its reappearance precedes the return of peripheral autonomic control in diabetics with peripheral neuropathy.45 However, some degree of respiratory-associated heart rate change is intrinsic to the heart itself. For example, in denervated human hearts (transplants), a small degree of ventilation-associated changes in heart rate persists,46 suggesting that mechanoreceptors in the right atrium can alter sinoatrial tone. Bernardi et al.46 demonstrated that this heart rate variability in cardiac transplant recipients had a periodicity twice that of the ventilatory cycle, suggesting that both increases and decreases in venous return and ventricular loading impart some RSA. In support of this concept, Pinna et al. documented that most of the heart rate and arterial pressure changes seen during breathing in patients with severe congestive heart failure (CHF) are more reflective of changes in intrathoracic blood volume than of alterations in autonomic input.47
Lung inflation to larger tidal volumes (>15 mL/kg) decreases heart rate. Pulmonary vasoconstriction may occur through vagal reflex arcs48 but does not appear to induce significant hemodynamic effects. Reflex arterial vasodilatation can also occur with lung hyperinflation.41,49–53 This inflation-vasodilatation response appears to be mediated by afferent vagal fibers, because it is abolished by selective vagotomy. Interestingly, blocking sympathetic afferent fibers also blocks this reflex,51,54 presumably by withdrawing central sympathetic tone. Although this inflation-vasodilatation response induces expiration-associated reductions in LV contractility in healthy volunteers55 and in ventilator-dependent patients with the initiation of high-frequency ventilation41 or hyperinflation,51 its clinical significance in other patient groups is unknown. Since patients with ALI often ventilate only a relatively small amount of their lungs, the potential exists that these patients may experience regional hyperinflation and may develop reflex cardiovascular depression. Interestingly, several studies comparing larger tidal volume ventilation with pressure-limited ventilation document better hemodynamic status with pressure-limited ventilation. Similarly, although humoral factors (including compounds whose production is dependent on cyclooxygenase activation56) released from pulmonary endothelial cells during lung inflation may also induce this depressor response,57–59 these interactions do not appear to grossly alter cardiovascular status.60 In fact, unilateral lung hyperinflation (unilateral PEEP) does not appear to influence systemic hemodynamics.61 Interestingly, increased levels of nitric oxide (NO) in the exhaled gas of rabbits ventilated at increasing tidal volumes have been reported.48 Importantly, for the same decrease in cardiac output, heart rate increases less with the application of PEEP than with hemorrhage.48 The reasons for this difference are unknown but may reflect PEEP-induced sympatholytic actions and increased arterial pressure minimizing baroreceptor stimulation.
Both positive-pressure ventilation and sustained hyperinflation stimulate endocrinological responses that induce fluid retention via right atrial stretch receptors. Plasma norepinephrine, plasma renin activity,62,63 and atrial natriuretic peptide (ANP)64 increase during positive-pressure ventilation with or without PEEP. Interestingly, when subjects with CHF are given nasal CPAP, plasma ANP activity decreases in parallel with improvements in blood flow.65,66
Determinants of Pulmonary Vascular Resistance
Changes in lung volume are caused by changes in transpulmonary distending pressure, the pressure difference between alveolar pressure and ITP. Since pulmonary tissue pressure and ITP are nearly identical, increasing lung volume increases the difference between alveolar and tissue pressures, making pulmonary vascular resistance increase independent of any effect of volume change on humoral or autonomic responses.18,67–72
Lung inflation primarily affects cardiac function and cardiac output by altering right ventricle (RV) preload and afterload.72 RV afterload is the maximal RV systolic wall stress during contraction73 which, by Laplace’s law, equals the product of the RV radius of curvature (a function of end-diastolic volume) and transmural pressure (a function of systolic RV pressure).74 Changes in ITP that occur without changes in lung volume, as may occur with obstructive inspiratory efforts, will not alter the pressure gradient between the RV and pulmonary artery nor result in change of pulmonary vascular resistance. Thus, neither straining at stools (Valsalva maneuver) nor obstructive inspiratory efforts (Mueller maneuver) primarily affect RV afterload. Although obstructive inspiratory efforts are usually associated with increased RV afterload, the reason for this effect is backward LV failure and reactive hypoxic pulmonary vasoconstriction.75,76
Systolic RV pressure approximates transmural systolic pulmonary artery pressure (Ppa) when no pulmonary stenosis is present. Transmural Ppa can increase by one of two mechanisms: (1) an increase in pulmonary arterial pressure without change in pulmonary vasomotor tone, as occurs with increases in blood flow (exercise) or passive increases in outflow pressure (LV failure), or (2) an increase in pulmonary vascular resistance. Usually any increase in transmural Ppa during positive-pressure ventilation is due to an increase in pulmonary vascular resistance, because neither instantaneous cardiac output77 nor LV filling14 changes. Increases in transmural Ppa impedes RV ejection,78 decreasing RV stroke volume79 and causing RV dilation and passive obstruction to venous return,56,58 which may rapidly progress to acute cor pulmonale.80 If RV dilation and pressure overload persist, RV free wall ischemia and infarction can develop.81 Importantly, rapid fluid challenges in the setting of acute cor pulmonale can precipitate profound cardiovascular collapse due to excessive RV dilation, RV ischemia, and compromised LV filling through the process of ventricular interdependence. During normal end-inspiration, mild hypoxemia (PaO2 > 65 mm Hg) and low levels of PEEP (<7.5 cm H2O) should minimally increase transmural Ppa. If slight increases in transmural Ppa are sustained, however, fluid retention occurs, either by intrinsic humoral mechanisms (increased ANP secretion) or by therapeutic intravascular volume infusion,82 resulting in an increase in RV end-diastolic volume maintaining cardiac output.74,83
The mechanism by which ventilation alters pulmonary vasomotor tone is complex. If regional alveolar PO2 (PAO2) decreases below 60 mm Hg, local pulmonary vasomotor tone increases, reducing local blood flow.84 This process of hypoxic pulmonary vasoconstriction is mediated in part by variations in the synthesis and release of NO by pulmonary vascular endothelial cells. The pulmonary endothelium normally synthesizes a basal low amount of NO, a potent vasodilator, using endothelial nitric oxide synthase. This basal NO release is highly regulated and dependent on O2 and is inhibited by both hypoxia and acidosis. If O2 becomes scarce, NO is not made, and pulmonary vasomotor tone increases to the level that would exist if no NO were present.
Many pulmonary pathologic processes are associated with regional reductions in PAO2, such as atelectasis, airway obstruction, and ventilation/perfusion mismatching. Hypoxic pulmonary vasoconstriction, by reducing pulmonary blood flow to those hypoxic regions, optimizes ventilation/perfusion matching. However, if alveolar hypoxia occurs throughout the lungs, overall pulmonary vasomotor tone increases, raising pulmonary vascular resistance and impeding RV ejection.73 At low lung volumes, alveoli spontaneously collapse as a result of loss of interstitial traction and closure of the terminal airways, causing alveolar hypoxia. Patients with acute hypoxemic respiratory failure have small lung volumes.85,86 Therefore, pulmonary vascular resistance is often increased in these patients owing to alveolar collapse and resultant hypoxic pulmonary vasoconstriction.
Mechanical Ventilation-Induced Changes in Pulmonary Vascular Resistance
Mechanical ventilation may reduce active pulmonary vasomotor tone by one of several related processes. Hypoxic pulmonary vasoconstrictor tone may be decreased by increasing global PAO2 by enriching alveolar gas O2,87–90 reexpansion of collapsed alveolar units thereby increasing PAO2 in those local alveoli,5,91–93 increased alveolar ventilation and reversal of acute respiratory acidosis,90 or merely through decreasing central sympathetic output by allowing the patient in ALI to not fight for every breath.94,95 Similarly, these effects need not require positive-pressure breaths as much as expansion of collapsed alveoli.96 Such recruitment of lung units is usually accomplished by the addition of PEEP or CPAP. Thus, if PEEP opens collapsed lung units and replenishes alveolar gas with O2, hypoxic pulmonary vasoconstriction will be reduced, pulmonary vascular resistance will decrease, and RV ejection will improve.
Changes in lung volume can also profoundly alter pulmonary vasomotor tone by passively compressing the alveolar vessels.85,92,93 Pulmonary circulation can be separated into two groups of blood vessels depending on what pressure surrounds them92 (Figure 47-4). The small pulmonary arterioles, venules, and alveolar capillaries sense alveolar pressure as their surrounding pressure and are referred to as alveolar vessels. The large pulmonary arteries and veins, as well as the heart and intrathoracic great vessels of the systemic circulation, sense interstitial pressure or ITP as their surrounding pressure and can be called extraalveolar vessels. Alveolar pressure minus ITP is the transpulmonary pressure. Increasing lung volume requires a rise in transpulmonary pressure, so the extravascular pressure gradient between alveolar and extraalveolar vessels varies proportionally with changes in lung volume. Importantly, the radial interstitial forces of the lung that keep the airways patent91,97,98 also act upon the extraalveolar vessels. As lung volume increases, the radial interstitial forces increase, increasing the diameter of both extraalveolar vessels and airways and resulting in a reduction in airway resistance at higher lung volumes, as well as increased extraalveolar vessel diameter and capacitance.99 This tethering effect is lost with lung deflation, thereby increasing pulmonary vascular resistance.88,91 The collapse of small airways also induces alveolar hypoxia. Thus, at small lung volumes, pulmonary vascular resistance is increased owing to the combined effect of hypoxic pulmonary vasoconstriction and extraalveolar vessel collapse.
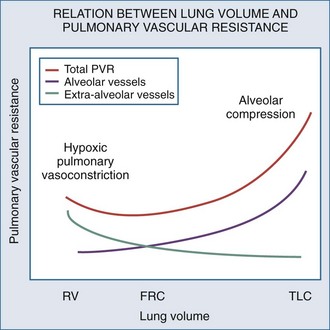
Increases in lung volume progressively raise alveolar vessel resistance, becoming noticeable above resting lung volume or functional residual capacity (FRC).88,100 There are two causes of the increased alveolar vessel resistance. First, the heart and extraalveolar vessels sense ITP as their surrounding pressure, whereas the alveolar vessels sense alveolar pressure as their surrounding pressure, so an extralumenal transpulmonary pressure gradient exists between extraalveolar and alveolar vessels. As lung volume increases, the extralumenal pressure difference increases as well. If transpulmonary pressure increases enough to exceed intralumenal vascular pressure, the pulmonary vasculature will collapse where extraalveolar vessels pass into alveolar loci, reducing the vasculature cross-sectional area and increasing pulmonary vascular resistance. Similarly, increasing lung volume by stretching and distending the alveolar septa may also compress alveolar capillaries, although this mechanism is less well substantiated. Hyperinflation can create significant pulmonary hypertension and may precipitate acute RV failure (acute cor pulmonale)101 and RV ischemia.81 Thus PEEP may increase pulmonary vascular resistance if it induces overdistention of the lung above its normal FRC. Recently the effect of inflation on RV input impedance was validated in humans, using echocardiographic techniques.102 Similarly, if lung volumes are reduced, then increasing lung volume back to baseline levels by the use of PEEP will decrease pulmonary vascular resistance by reversing hypoxic pulmonary vasoconstriction.103
Ventricular Interdependence
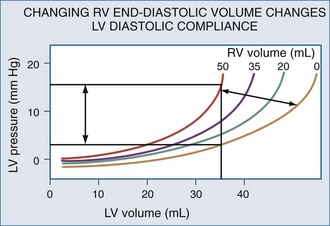
Changes in RV output must invariably alter LV filling because the two ventricles are serially linked through the pulmonary vasculature. However, LV preload can also be directly altered by changes in RV end-diastolic volume.104 If RV volume increases, LV diastolic compliance will decrease by the mechanism of ventricular interdependence.105 Ventricular interdependence functions through two separate processes. First, increasing RV end-diastolic volume will induce a shift of the intraventricular septum into the LV, thereby decreasing LV diastolic compliance106 (Figure 47-5). For the same LV filling pressure, RV dilation will decrease LV end-diastolic volume and therefore cardiac output. This interaction is believed to be the major determinant of the phasic changes in arterial pulse pressure and stroke volume seen in tamponade, referred to as pulsus paradoxus. Spontaneous inspiration increases venous return, causing RV dilation and decreasing LV end-diastolic compliance. Maintaining a relatively constant rate of venous return, either by volume resuscitation107 or vasopressor infusion,4 will minimize this effect. Thus, the presence of pulse paradoxus can be used as a marker of functional hypovolemia, even if actual intravascular volume status is not reduced.
Mechanical Heart-Lung Interactions
With hyperinflation, the heart may be compressed between the two expanding lungs,108 increasing juxtacardiac ITP. The chest wall and diaphragm can move away from the expanding lungs, whereas the heart is trapped within its cardiac fossa, so juxtacardiac ITP may increase more than lateral chest wall or diaphragmatic ITP.6,16 This compressive effect of the inflated lung can be seen with either spontaneous109 or positive pressure–induced hyperinflation.97,98 This decrease in “apparent” LV diastolic compliance106 was previously misinterpreted as impaired LV contractility, because LV stroke work for a given LV end-diastolic pressure or pulmonary artery occlusion pressure is decreased.110,111 However, numerous studies have shown that when patients are fluid resuscitated to return LV end-diastolic volume to its original level, both LV stroke work and cardiac output also returned to their original levels69,107 despite the continued application of PEEP.112
Takata et al.113 proposed a novel approach to understanding tamponade that lends itself to mechanical heart-lung interactions. Hyperinflation directly alters biventricular filling, whereas inspiration primarily alters RV filling and only indirectly affects LV volumes through changes in diastolic compliance. Thus, hyperinflation—as occurs in severe asthma and with the use of excessive amounts of PEEP—would produce a clinical picture indistinguishable from tamponade. Indeed, Rebuck and Read114 made this same observation in their analysis of the hemodynamic effects of severe asthma over 30 years ago, although they did not postulate a specific mechanism to explain this phenomenon. Presumably, the shift from “uncoupled” to “coupled” cardiac fossal restraint would occur as absolute lung volume increased, biventricular volume increased, or both. If cardiac volumes are small, and lung inflation does not overdistend the chest, RV filling will be primarily impeded. In contrast, in CHF states and with marked lung overdistention, both RV and LV filling may be compromised by ventilation.
Hemodynamic Effects of Changes in Intrathoracic Pressure
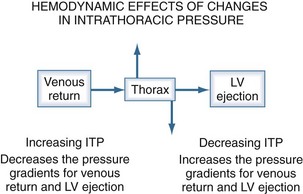
The heart within the thorax is a pressure chamber within a pressure chamber. Therefore, changes in ITP will affect the pressure gradients for both systemic venous return to the RV and systemic outflow from the LV, independent of the heart itself (Figure 47-6). Increases in ITP, by increasing right atrial pressure and decreasing transmural LV systolic pressure, will reduce the pressure gradients for venous return and LV ejection, thereby decreasing intrathoracic blood volume. Using the same argument, decreases in ITP will augment venous return and impede LV ejection and increase intrathoracic blood volume.
Systemic Venous Return
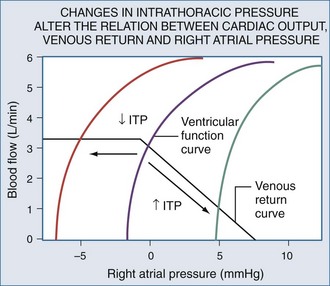
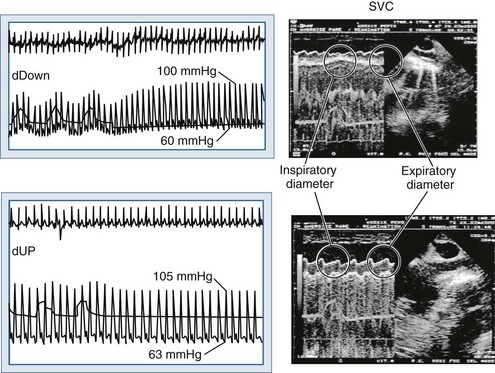
Blood flows back to the heart from the periphery through low-pressure, low-resistance venous conduits. Guyton et al. characterized venous flow from the venous reservoirs into the right atrium.115 As downstream right atrial pressure varies, as occurs with ventilation, the rate of venous return inversely changes. Pressure in the upstream venous reservoirs is called mean systemic pressure. Mean systemic pressure is a function of blood volume, peripheral vasomotor tone, and the distribution of blood within the vasculature.116 Mean systemic pressure does not change rapidly during the ventilatory cycle, whereas right atrial pressure does owing to concomitant changes in ITP. Accordingly, variations in right atrial pressure represent the major factor determining the fluctuation in pressure gradient for systemic venous return during ventilation.77,117 Positive-pressure inspiration increases ITP and right atrial pressure, decreasing the pressure gradient for venous return and RV filling,79 and consequently, RV stroke volume.77,79,118–125 These physiologic effects have recently been validated in humans, using minimally invasive echocardiographic techniques wherein vena caval flow varies with the phase of the ventilatory cycle25,126 (Figure 47-7). During normal spontaneous inspiration, the converse occurs: with decreases in ITP, right atrial pressure decreases, accelerating venous blood flow and increasing RV filling and RV stroke volume* (Figure 47-8).
The decrease in venous return during positive-pressure ventilation is often lower than one might expect based on the increase in right atrial pressure. Fessler et al.129 and Takata and Robotham130 demonstrated in dogs that PEEP increases intraabdominal pressure by causing the diaphragm to descend, thereby increasing the pressure surrounding the intraabdominal vasculature. Because a large proportion of venous blood is in the abdomen, the net effect of PEEP is to increase mean systemic pressure and right atrial pressure. Accordingly, the pressure gradient for venous return may not be reduced by PEEP, especially in patients with hypervolemia. In fact, abdominal pressurization by diaphragmatic descent may be the major mechanism by which the decrease in venous return is minimized during positive-pressure ventilation.131–135 Furthermore, Matuschak et al.131 found that although PEEP decreased blood flow to the liver in proportion to the induced decrease in cardiac output in normovolemic dogs, the liver’s ability to clear hepatocytic-specific compounds, such as indocyanine green, was unaltered. Finally, when cardiac output is restored to pre-PEEP levels by fluid resuscitation131,136 while PEEP is maintained, liver clearance mechanisms increase above pre-PEEP levels.136–139 These data are consistent with a PEEP-induced alteration in intrahepatic blood flow distribution. Thus, ventilation may have less of an effect on venous return than originally postulated, but the effect may be more complicated than we have imagined. Van den Berg et al.140 examined the effects of varying levels of CPAP on right atrial pressure, intraabdominal pressure, and cardiac output in 42 postoperative cardiac surgery patients. Up to 20 cm H2O CPAP did not significantly decrease cardiac output, as measured 30 seconds into an inspiratory hold maneuver. The reason for this apparent paradoxical effect became obvious when they compared the associated changes in right atrial pressure, abdominal pressure, and RV end-diastolic volume (Figure 47-9). What they documented was that only 30% of the increased airway pressure was transmitted to the right atrium. Perhaps more importantly, most of the increase in right atrial pressure was also realized by an increase in intraabdominal pressure, so it was not surprising that RV end-diastolic volume fell by less than 8% from pre-CPAP values. These data demonstrate that in the fluid-resuscitated patient, institution of positive-pressure ventilation may not result in a decrease in blood flow. However, if intraabdominal pressure is allowed to decrease, as would occur with an open laparotomy and decompression of tense ascites, a marked preload-responsive effect of positive-pressure ventilation can occur.
With exaggerated swings in ITP, as occur with obstructed inspiratory efforts, venous return behaves as if abdominal pressure is additive to mean systemic pressure in defining total venous blood flow.141–144 Recent interest in inverse ratio ventilation has raised questions as to its hemodynamic effect because its application includes a large component of hyperinflation. However, Mang et al.145 demonstrated in an animal model of ALI that if total PEEP (intrinsic PEEP plus extra extrinsic PEEP) was similar, no hemodynamic difference between conventional ventilation and inverse ratio ventilation was seen.
Right Ventricular Filling
Under normal conditions, it is difficult to document any relation between RV filling pressure and volume. When RV filling pressure, defined as right atrial pressure minus Ppc, was directly measured in patients undergoing open chest operations, RV filling pressure was unaltered by acute volume loading.146 Although right atrial pressure increased, Ppc also increased such that RV filling pressure remained unchanged. Similar data were seen when RV volumes are reduced by the application of PEEP in postoperative cardiac patients.147 These findings suggest that under normal conditions, RV diastolic compliance is very high, and most of the increase in right atrial pressure seen during volume loading reflects pericardial compliance and cardiac fossa stiffness, more than changes in RV distending pressure. These data also imply that with RV filling, right heart sarcomere length remains constant. Presumably, conformational changes in the RV more than wall stretch are responsible for RV enlargement.15 Accordingly, changes in right atrial pressure do not follow changes in RV end-diastolic volume. When cardiac contractility is reduced and intravascular volume is expanded, RV filling pressure increases as a result of decreased RV diastolic compliance, increased pericardial compliance, increased end-diastolic volume, or a combination of all three. In support of this hypothesis, RV filling pressure does not increase until RV volume exceeds a certain threshold value.105 Furthermore, in dogs with acute ventricular failure, volume loading increases Ppc more than ITP, consistent with pericardial rather than cardiac fossal restraint. If PEEP is increased in this setting, ITP but not Ppc selectively increases until ITP equals Ppc, then both ITP and Ppc increase equally if PEEP is increased further.7 In postoperative cardiac surgery patients,13,22,148 PEEP—and by extension lung expansion—compresses the heart within the cardiac fossa in a fashion analogous to pericardial tamponade.
Venous return is the primary determinant of cardiac output.116 Since right atrial pressure is the backpressure to venous return, venous return is maintained near maximal levels at rest51,125,126 because RV filling occurs with minimal changes in filling pressure.148 The closer right atrial pressure remains to zero relative to atmospheric pressure, the greater the pressure gradient for systemic venous blood flow.115,121 For this mechanism to operate efficiently, RV output must equal venous return, otherwise sustained increases in venous blood flow would overdistend the RV, increasing right atrial pressure. Fortunately, under normal conditions of spontaneous ventilation, the increase in venous return is in phase with inspiration, decreasing again during expiration as ITP increases.77 Likewise, the pulmonary arterial inflow circuit is highly compliant and can accept large increases in RV stroke volume without changing pressure.79,83 Thus, any increase in venous return is proportionally delivered to the pulmonary circuit without forcing the RV to increase its force of contraction or myocardial oxygen demand. Importantly, this compensatory system will rapidly become dysfunctional if RV diastolic compliance decreases or if right atrial pressure increases independent of changes in RV end-diastolic volume. An example of decreased RV diastolic compliance is acute RV dilation or cor pulmonale (pulmonary embolism, hyperinflation, and RV infarction) that induce profound decreases in cardiac output not responsive to fluid resuscitation. Dissociation between right atrial pressure and RV end-diastolic volume occurs during either tamponade or positive-pressure ventilation, because right atrial pressure is artificially increased by the increasing ITP. Accordingly, positive-pressure ventilation impairs normal circulatory adaptive processes. Furthermore, even if one restores the coupling of right atrial pressure and RV volume by using partial ventilatory support modes of ventilation, cardiac output will increase only if the RV can transduce the associated increase in venous return to forward blood flow. Thus, during weaning from mechanical ventilation, occult RV failure may be exposed and will manifest as a rapid rise in right atrial pressure and a fall in cardiac output. Since the primary effect of any form of ventilation on cardiovascular function in normal subjects is to alter RV preload via altering venous blood flow, the detrimental effect of positive-pressure ventilation on cardiac output can be minimized by either fluid resuscitation to increase mean systemic pressure4,118,140,141 or by keeping both mean ITP and swings in lung volume as low as possible. Accordingly, prolonging expiratory time, decreasing tidal volume, and avoiding PEEP all minimize the decrease in systemic venous return to the RV.1,21,77,120–124,149
Since spontaneous inspiratory efforts increase lung volume by decreasing ITP, one sees an increase in venous return with spontaneous inspiration owing to the fall in right atrial pressure.19,67,121–123 However, this augmentation of venous return is limited142,143 because if ITP decreases below atmospheric pressure, venous return becomes flow-limited as the large systemic veins collapse as they enter the thorax.115 This flow limitation is a safety valve for the heart because ITP can decrease greatly with obstructive inspiratory efforts,52 and if not flow-limited, the RV could become overdistended and fail150 (see Figure 47-8). Still, in patients with decreased RV compliance, negative swings in ITP can augment RV filling. Interestingly, negative pressure ventilation, by augmenting venous return, was shown to increase cardiac output by 39% in intubated children following repair of tetralogy of Fallot.151 In this condition, impaired RV filling secondary to RV hypertrophy and reduced RV chamber size are the primary factors limiting cardiac output.
Left Ventricular Preload and Ventricular Interdependence
Changes in venous return must eventually result in directionally similar changes in LV preload, because the two ventricles are linked in series. For example, during a Valsalva maneuver, initially RV filling is reduced, but LV filling is unaltered.14 Then, as the strain is sustained, LV filling and cardiac output both begin to decrease.108,152 This phase delay in changes from the RV to the LV is exaggerated if tidal volume or respiratory rate are increased and in the setting of hypovolemia.* Independent of this series interaction, direct ventricular interdependence can also occur and be clinically significant. Increasing RV volume shifts the intraventricular septum into the LV and simultaneously decreases LV diastolic compliance. During positive-pressure ventilation, RV volumes are usually decreased, minimizing ventricular interdependence.105,155–158 Echocardiographic studies document that although PEEP results in some degree of right-to-left intraventricular septal shift, the shift is small.69,70 In fact, increases in lung volume during positive-pressure ventilation primarily compress the two ventricles into each other, decreasing biventricular volumes.159 The decrease in cardiac output commonly seen during PEEP is due to a decrease in LV end-diastolic volume; in this setting, both LV end-diastolic volume and cardiac output are restored by fluid resuscitation160,161 without any measurable change in LV diastolic compliance.106
During spontaneous inspiration, RV volumes increase transiently, shifting the intraventricular septum into the LV,106 decreasing LV diastolic compliance and LV end-diastolic volume.103,158,162 This transient RV dilation–induced septal shift is the primary cause of inspiration-associated decreases in arterial pulse pressure; if greater than 10 mm Hg or 10% of the mean pulse pressure, the phenomenon is referred to as pulsus paradoxus.19 Since spontaneous inspiratory efforts can occur during positive-pressure ventilation and especially during partial ventilatory assist modalities, pulsus paradoxus can also be seen in mechanically ventilated patients.
Left Ventricular Afterload
Left ventricular afterload can be equated to systolic wall tension which, by the Laplace equation, is proportional to the product of transmural LV pressure and the radius of curvature of the LV, which itself is proportional to LV volume. Maximal LV wall tension normally occurs at the end of isometric contraction, reflecting both a maximal product of the LV radius of curvature (end-diastolic volume) and aortic pressure (diastolic pressure). Under normal conditions during LV ejection, LV afterload progressively decreases because LV volumes decrease markedly despite the small increase in ejection pressure. Importantly, when LV dilation exists, as in CHF, maximal LV wall stress occurs during LV ejection, since the maximal product of these two variables occurs at this time. Accordingly, LV afterload varies on the baseline level of cardiac contractility, arterial pressure, and intravascular volume. LV ejection pressure is the transmural LV systolic pressure, which can be approximated as transmural arterial pressure. Since normal baroreceptor mechanisms located in the carotid body tend to maintain arterial pressure constant with respect to atmosphere, if arterial pressure were to remain constant as ITP increased, LV wall tension would decrease as well. Similarly, if transmural arterial pressure were to remain constant as ITP increased, but LV end-diastolic volume decreased because of the increased ITP-induced decrease in systemic venous return, LV wall tension would also decrease.163 Thus, by either mechanism, increases in ITP decrease LV afterload. Similarly, decreases in ITP with a constant arterial pressure will increase LV transmural pressure, increasing LV afterload.14,164
Any process associated with marked decreases in ITP must also be associated with increased LV afterload and myocardial O2 consumption (MVO2). In fact, decreasing ITP, a common occurrence during spontaneous inspiratory efforts with bronchospasm and obstructive breathing, reflects an important cardiac stress inducing CHF. Furthermore, since weaning from positive-pressure ventilation to spontaneous ventilation may reflect dramatic changes in ITP swings and the energy requirements of the respiratory muscles, weaning from mechanical ventilation is a cardiovascular stress test.163,165,166 Interestingly, Jabran et al.37 demonstrated that all ventilator-dependent patients increased their cardiac outputs during weaning, but in those that failed to be liberated from mechanical ventilation, SvO2 decreased, consistent with cardiovascular compromise. A similar argument can be given for the observed improvement in LV systolic function in patients with severe LV failure when placed on mechanical ventilation.166 Interestingly, similar “auto-EPAP” effects of expiratory grunting have been reported in infants during crying71 and in an adult with severe LV failure.167 It may well be that our failure to define specific physiologic parameters to describe subjects who may successfully be liberated from mechanical ventilation is because we do not include in these analyses measures of cardiovascular reserve.
Pulsus paradoxus occurs during spontaneous inspiration under conditions of marked pericardial restraint. This may occur because of pericardial limitations such as tamponade or constrictive pericarditis, as well as during loaded spontaneous ventilatory efforts when RV volumes swell and ITP decreases. In both cases, LV stroke volume decreases.168–172 Perhaps the most prominent mechanism creating an inspiratory decrease in both LV stroke volume and systolic arterial pressure is the increased venous return–induced transient decrease in LV diastolic compliance that then results in decreased LV end-diastolic volume. The negative swings in ITP also increase LV ejection pressure (LV pressure minus ITP), increasing LV end-systolic volume.14 Other influences on LV systolic function can also occur during loaded inspiratory efforts, such as with obstructive sleep apnea. These include an increase in aortic input impedance,173 altered synchrony of contraction of the global LV myocardium,174 and hypoxemia-induced decreased contractility.175 Hypoxia has the added detriment of also directly reducing LV diastolic compliance as well as decreasing myocardial contractile function.176
Experimental repetitive periodic airway obstructions induce pulmonary edema in normal animals.75,76 Furthermore, removing the negative swings in ITP by applying nasal CPAP results in improved global LV performance in patients with combined obstructive sleep apnea and CHF.176
If ITP were to increase rapidly, as during a cough, arterial pressure would also increase by a similar amount such that both arterial pressure relative to ITP (transmural arterial pressure or LV ejection pressure)14,177 and aortic blood flow108 would remain constant. However, sustained increases in ITP must eventually decrease aortic blood flow and arterial pressure as a result of the associated decrease in venous return.14 Since normal baroreceptor-based homeostatic mechanisms tend to sustain a constant arterial pressure so as to maintain organ perfusion constant,51 if ITP increased arterial pressure without changing transmural arterial pressure, the periphery would reflexively vasodilate to maintain a constant extrathoracic arterial pressure-flow relation.153 Since coronary perfusion pressure reflects the intrathoracic pressure gradient for blood flow, it is not increased by ITP-induced increases in arterial pressure. However, compression of the coronaries by the expanding lungs may obstruct coronary blood flow. Thus, the combined decrease in coronary blood flow may induce myocardial ischemia.178–180
The effect of removing large negative levels of ITP is not similar to adding positive ITP on venous return. Relative increases in ITP from very negative values to zero, relative to atmosphere, will minimally alter venous return, whereas increases in ITP above atmosphere will impede venous return by increasing right atrial pressure. Once right atrial pressure becomes negative, venous return becomes flow limited. Very negative swings in ITP, as seen during vigorous inspiratory efforts in the setting of airway obstruction (asthma, upper airway obstruction, vocal cord paralysis) or stiff lungs (interstitial lung disease, pulmonary edema, and ALI), will selectively increase LV afterload. Such spontaneous inspiratory efforts may be the cause of LV failure and pulmonary edema that is often seen in these conditions,18,52,75,76 especially if LV systolic function is already compromised30,181 (see Figure 47-8). Similarly, removing large negative swings in ITP by either bypassing upper airway obstruction (endotracheal intubation) or through the institution of mechanical ventilation or PEEP-induced loss of spontaneous inspiratory efforts should selectively reduce LV afterload without significantly decreasing either venous return or cardiac output.* Reversing this argument, weaning from mechanical ventilation, with its associated increase in both metabolic demand and LV afterload, is a form of cardiac stress testing.
Using Changes in Intrathoracic Pressure to Define Cardiovascular Performance
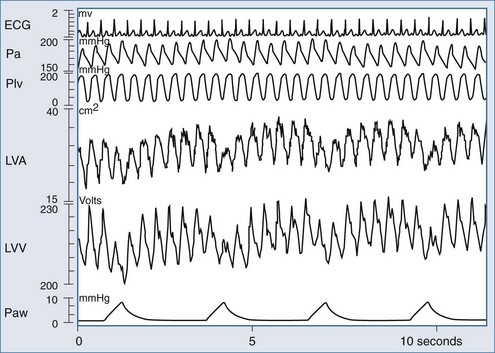
Renewed interest in using the cardiovascular response to either spontaneous or positive-pressure ventilation as a means to diagnose cardiovascular responsiveness has recently occurred. However, sustained increases in airway pressure can also be used to measure cardiac contractility, as defined by the end-systolic pressure-volume relation (ESPVR).183 Traditionally, LV ESPVR is generated by rapidly reducing LV preload by transient inferior vena caval occlusion. In the preload-dependent patient, passive inspiratory hold maneuvers that increase airway pressure by 5 to 10 cm H2O (e.g., CPAP) will selectively decrease venous return without substantially altering either pulmonary vascular resistance or LV afterload, because the changes in lung volume and ITP are relatively small. Using this approach, Haney et al.184,185 generated LV ESPVR similar to that created by transient inferior vena caval occlusion in a canine model. Since it is much easier to give 5 to 10 cm H2O CPAP to patients than to pass an intravascular balloon into the inferior vena cava to induce transient vascular obstruction, this technique has promise as a means of selectively altering venous return in the bedside assessment of LV contractility. Denault et al., using the same logic, documented in mechanically ventilated patients during cardiac surgery that LV performance could be measured during ventilation using combined estimates of LV volume and ejection pressure.186 As shown in Figure 47-10, ventilation induces profound dynamic changes in LV volumes consistent with rapidly varying changes in LV filling.
Clinical Application of Heart-Lung Interaction Physiologic Concepts: Predicting Preload Responsiveness
One of the fundamental uses of hemodynamic monitoring is to access intravascular volume status and predict the cardiac output response to volume loading.187 Regrettably, measures of ventricular filling pressure and volumes, though useful in assessing cardiac performance, are very poor predictors of volume responsiveness.188 Traditionally, the bedside clinician simply gave a rapid infusion of volume (e.g., 500 mL saline over 15 minutes) and noted whether cardiac output increased or not. Although effective at defining volume responsiveness, a volume challenge is time consuming and may delay appropriate treatment if there is no increase in cardiac output in response to this fluid challenge. Importantly, fully 50% of all hemodynamically unstable patients are not volume responsive188; thus giving volume to all unstable patients will not benefit half. Consequently, interest in using the responses of the cardiovascular system to ventilation as a surrogate for a transient volume challenge emerged. Based on the previously described physiologic principles of heart-lung interaction, we know that positive-pressure inspiration will decrease venous return to the right ventricle, which will decrease filling of the left ventricle in 2 to 3 beats, resulting in a decrease in LV stroke volume if both ventricles are volume responsive. Under such conditions, LV stroke volume will decrease from apneic baseline values proportional to the IPPV-induced decrease in venous return and the slope of the LV function curve. Since arterial pulse pressure (diastolic to systolic arterial pressure) changes directly with LV stroke volume, one can use either stroke volume or pulse pressure variation to define volume responsiveness. Analysis of the arterial pressure waveform and its phase relationship to inspiration yield powerful tools to understanding the stroke volume and pulse pressure variations with inspiration. These variations are the basis for the prediction of preload responsiveness, and thus the application of the previously described physiologic considerations to the practical management of ventilator-dependent patients. A pulse pressure variation more than 13% or a stroke volume variation more than 10% on positive-pressure ventilation is highly predictive of volume responsiveness.189
One of the most central aspects of cardiovascular homeostasis is the preload-dependent nature of LV performance. Documenting that LV EDV is above some minimal value, despite cardiac output and stroke work both being depressed, is essential for the diagnosis of cardiac pump dysfunction. Similarly, demonstrating that LV EDV is decreasing in the setting of hemodynamic instability presumes the diagnosis of inadequate circulating blood volume as the most likely cause of the hemodynamic instability, even though other etiologies (e.g., tamponade, cor pulmonale, restrictive cardiomyopathies) can coexist and require different treatments. However, knowing absolute LV EDV does not predict whether LV stroke volume will increase in response to volume loading.188 Knowing the preload of a patient is not the same as knowing if the patient will be preload responsive. In addition, being preload responsive is not necessarily indicative of fluid requirement. Just knowing that a patient is volume responsive does not equate to the need for fluid resuscitation. One must also presume or document that inadequate blood flow exists, because normal subjects who are otherwise healthy may also be volume responsive but do not need fluid resuscitation.
Assessing Fluid Responsiveness During Positive-Pressure Ventilation
For either pulse pressure variation (PPV, ratio of maximal minus minimal pulse pressure to mean pulse pressure over 5 or more breaths) or stroke volume variation (SVV, ratio of maximal minus minimal stroke volume to mean stroke volume over 5 or more breaths) to reflect preload responsiveness, the tidal volume must be fixed, the sequential R-R intervals must be constant (i.e., no arrhythmias), and both parameters should be measured during unassisted positive-pressure breathing with a tidal volume (VT) of 8 mL/kg or more. Several studies have been conducted to assess the reliability of this technique in the assessment of volume responsiveness. Marik et al. summarized the published data in the last 10 years in a systematic review of the literature specifically “to determine the ability of dynamic changes in arterial waveform-derived variables to predict fluid responsiveness and compare these with static indices of fluid responsiveness.”190 Twenty-nine studies, which enrolled 685 patients, were selected. The authors concluded that PPV, SVV, and systolic pressure variation (SPV) consistently predicted increments in stroke volume with fluid challenges. They also reported that the threshold values for such prediction of volume responsiveness were remarkably consistent throughout the studies and pointed to being between 11% and 13%. The diagnostic accuracy of these variables, as judged by area under the curve, was excellent (PPV 0.94, SPV 0.86, and SVV 0.84), with PPV significantly better than either SVV or SPV (P < 0.001). The efficacy of using either SVV or PPV metrics to predict fluid responsiveness has been validated in different clinical scenarios and populations, such as cardiac surgery,191 orthotopic liver transplantation,192 sepsis,193 ARDS,194–197 and critically ill children.198
Effect of Tidal Volume on Pulse Pressure Variation and Stroke Volume Variation
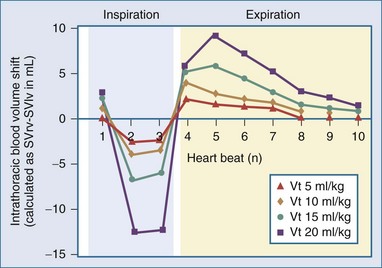
Changes in tidal volume also alter the phasic swings in ITP and thus the dynamic changes in venous return. The greater the tidal volume, the greater the cycle-specific changes in venous return augmenting PPV and SVV at the same volume status. This is not surprising given that PPV is generated by the pressure transmitted from the airways to the pleural and pericardial spaces and thus will theoretically be decreased with low tidal volume ventilation and low pulmonary compliance. These interactions were reported by three separate groups of investigators. DeBacker et al.,199 in a simple and elegant clinical description of the impact of changing tidal volume on PPV, showed that PPV varied directly with tidal volume. Renner et al.200 demonstrated that SVV also varied directly with tidal volume. Finally, Kim and Pinsky201 showed that in anesthetized ventilated dogs, PPV decreased from 20.1 ± 10.8 to 9.5 ± 5.4% when tidal volume was decreased from 20 to 5 mL/kg. This can be conceptually described as the change in intrathoracic blood volume on a beat-to-beat basis as the difference between SVRV and SVIV associated with increasing tidal volume during intermittent positive-pressure ventilation. The greater the tidal volume the greater dynamic change in intrathoracic blood volume (Figure 47-11).
PPV is generated by the pressure transmitted from the airways to the pleural and pericardial spaces, and thus will theoretically be decreased with low tidal volume ventilation and low pulmonary compliance. In addition, Muller et al.202 showed that PPV failed to predict fluid responsiveness (defined as an increase of stroke index > 15%) after a fluid challenge (normal saline or hydroxy-ethyl-starch 6% 500 mL) in patients with low airway driving pressure (PPlat − PEEP < 20 cm H2O). In that study, PPV over 13% or even over 7% was highly predictive of fluid responsiveness, but fluid responsiveness could not be ruled out in patients with lower PPV values.
Vistisen et al. have suggested that PPV should be indexed to tidal volume.203 In their study, they showed that PPV increased significantly with increments in tidal volume in three different states: hypovolemia, normovolemia, and hypervolemia. This approach is appealing because as Romand et al. previously showed,8 it is the change in lung volume, not in airway pressure, that determines the ITP change during positive-pressure breathing, and changes in ITP create the changes in venous return.
Effect of Positive End-Expiratory Pressure on Pulse Pressure Variation and Stroke Volume Variation
Since the primary effect of PEEP is to distend the lungs and increase ITP, PEEP normally reduces venous return and creates a functional hypovolemic state. Accordingly, Kubitz et al.204 showed in a porcine model that increasing PEEP levels increased both PPV and SVV. Interestingly, they also saw that this effect persisted in an open chest condition, albeit to a lesser degree. Potentially one could use the emergence of an increasing PPV during positive-pressure ventilation as a sign that lung recruitment has occurred and may have started to create hyperinflation.
Limits on the Ability of Arterial Pulse Contour to Assess Stroke Volume Variation
DeCastro et al.205,206 validated the finding that stroke volume calculated by the PiCCO arterial pulse contour technique closely tracks steady state arterial pressure but does not track dynamic SVV. These data agree with a canine study by Gunn et al.207 that examined how accurately this same device tracked SVV as vasomotor tone was pharmacologically varied. These findings are important because they illustrate that the technique and device used to estimate SVV will have limitations minimizing its clinical utility. To reduce this inherent measurement bias across monitoring devices, clinicians should use only one minimally invasive device for the continuous measuring of SVV in the same patient over time.
Fluid Responsiveness Assessment During Spontaneous Ventilation
In spontaneously breathing patients and those with arrhythmias, the mean increase in flow 20 seconds after passive leg raising to 30 to 45 degrees gives a predictive value similar to PPV/SVV during positive-pressure ventilation. However, in this setting, the variable to measure is not PPV/SVV but the change in flow of greater than 10%. Passive leg raising (PLR) is a suitable test, given that it induces a gravitational transfer of blood from the periphery to the central circulatory compartment sufficient to significantly modify the preload of the LV and cause a change in flow or cardiac output.208
The best hemodynamic marker of passive leg raising as a predictor of volume responsiveness is a change in stroke volume, given that the test is transient in nature. Monnet et al.209 demonstrated that a change in descending aortic blood flow (measured by esophageal Doppler) more than 10% after passive leg raising was predictive of volume responsiveness in spontaneously breathing patients, with or without arrhythmia, and in sedated patients with sinus rhythm. Furthermore, Lamia et al.,210 using transthoracic echocardiography, studied the stroke volume response to passive leg raising in patients breathing spontaneously. They showed that an increment in stroke volume over 12.5% was predictive of fluid responsiveness, with a sensitivity of 77% and a specificity of 100%.
 Hemodynamic Effects of Ventilation Based on Cardiopulmonary Status
Hemodynamic Effects of Ventilation Based on Cardiopulmonary Status
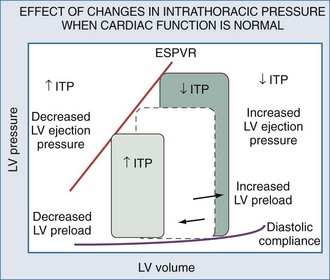
Spontaneous and positive-pressure ventilation may have profound hemodynamic consequences. Furthermore, the same ventilatory maneuver (initiation or withdrawal from mechanical ventilation) can have the opposite effects on cardiovascular stability in differing patient populations. Schematic examples of how increasing or decreasing ITP will alter the LV pressure-volume relation are depicted for conditions in which LV function is normal (Figure 47-12) and depressed (Figure 47-13). Since the hemodynamic responses to ventilation are highly dependent on existing cardiovascular state, specific responses to defined ventilatory maneuvers not only define the baseline cardiovascular state but also allow for accurate predictions about what hemodynamic effects will occur.
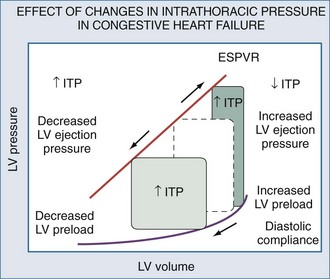
In patients with cardiovascular insufficiency due to impaired LV ejection and/or volume overload, the institution of mechanical ventilatory support can be lifesaving because of its ability to support the cardiovascular system while decreasing global O2 demand. In patients who are predominately preload dependent or hypovolemic (hemorrhagic shock, loss of vasomotor tone) and those who may develop RV failure with hyperinflation (anterior chest trauma, spinal cord shock, severe obstructive lung disease), the institution of positive-pressure ventilation must be done with caution; profound cardiovascular insufficiency may rapidly develop during the course of intubation and initiation of mechanical ventilation. Similarly, withdrawal of ventilatory support can be considered an exercise stress test such that if patients have limited cardiovascular reserve, they may not be weaned even if their traditional weaning parameter values are acceptable.30,181
Mechanical Ventilation
The hemodynamic differences between different modes of total mechanical ventilation at a constant airway pressure and PEEP can be explained by their differential effects on lung volume and ITP.211 Importantly, when two different modes of total or partial ventilatory support have similar changes in ITP and ventilatory effort, their hemodynamic effects are also similar despite markedly different airway waveforms. Partial ventilatory support with either intermittent mandatory ventilation or pressure support ventilation gives similar hemodynamic responses when matched for similar tidal volumes.212 Sternberg and Sahebjami213 demonstrated similar tissue oxygenation in stable ventilator-dependent patients when they were switched from assist-control intermittent mandatory ventilation to pressure-support ventilation with matched tidal volumes. Finally, high-frequency jet ventilation delivered at low levels results in a constant cardiac output in patients with heart failure as compared to conventional.214
Acute Lung Injury
Patients with ALI often require PEEP to maintain alveolar distention and arterial oxygenation. Positive-pressure ventilation decreases intrathoracic blood volume,121 and PEEP decreases it even more146,147 without altering LV contractile function.215 However, increases in airway pressure may not reflect increases in ITP because patients with ALI have varying degrees of increased lung stiffness and decreased chest wall compliance. Furthermore, it is the increase in lung volume, not airway pressure, that defines the degree of increase of ITP during positive-pressure ventilation.8 Lessard et al.216 saw no significant hemodynamic differences between volume-controlled, pressure-controlled, and pressure-controlled inverse-ratio ventilation adjusted to keep total PEEP and tidal volume consistent between treatment arms in patients with ARDS. Chan and Abraham217 saw similar results in patients with ARDS matched for comparable tidal volumes and total PEEP. However, when pressure control with a smaller tidal volume was compared to volume control, both Abraham and Yoshihara218 and Poelaert et al.219 found that pressure control was associated with a higher cardiac output. Davis et al.220 studied the hemodynamic effects of volume control versus pressure-controlled ventilation in 25 patients with ALI. When matched for the same mean airway pressure, both methods gave the same cardiac outputs. However, when airway pressure was increased during volume-controlled ventilation by sign wave to square wave flow pattern, cardiac output fell. Furthermore, Kiehl et al.221 found that cardiac output was better with biphasic positive airway pressure than with volume-controlled ventilation, leading to an increased SvO2 and indirectly increasing PaO2. Singer et al.222 showed in ventilator-dependent but hemodynamically stable patients that the degree of hyperinflation, not the airway pressure, determined the decrease in cardiac output. Different modes of mechanical ventilation will affect cardiac output to a similar extent for similar increases in lung volume.134,217,223 Most of the decrement in cardiac output can be reversed by fluid resuscitation that restores intrathoracic blood volume to pre-PEEP levels, as assessed by noninvasive blood pool scanning215 or echocardiography224–226; in addition, cardiac output also returned to its basal level despite the continued application of PEEP. That the PEEP-induced decrease in cardiac output was due to a decreased pressure gradient for venous return was elegantly shown by Gunter et al.,227 who minimized the decrease in cardiac output in ventilator-dependent septic patients by lowering body compression. Importantly, if cardiac output does not increase with fluid resuscitation, then other processes (e.g., cor pulmonale, increased pulmonary vascular resistance, cardiac compression) may also be inducing this cardiovascular depression.228
Congestive Heart Failure
Increases in cardiac output with increases in airway pressure suggest the presence of CHF.40,229 Grace and Greenbaum230 noted that adding PEEP to patients with heart failure did not decrease cardiac output, and actually increased cardiac output if pulmonary artery occlusion pressure exceeded 18 mm Hg. Similarly, Calvin et al.231 noted that patients with cardiogenic pulmonary edema had no decrease in cardiac output when given PEEP.232 Unfortunately, PEEP may be detrimental in patients with combined heart failure and ALI. PEEP can result in increased leukocyte retention in human lungs.233 Rasanen et al. documented that decreasing levels of ventilatory support in patients with myocardial ischemia and acute LV failure worsened ischemia40,233 and could be minimized by preventing spontaneous inspiratory effort–induced negative swings in ITP.39 Since weaning from mechanical ventilatory support is a form of exercise stress test, withdrawal of ventilatory support can unmask cardiac failure in otherwise stable patients with acute respiratory failure.30 Such patients may not be “weanable” from mechanical ventilatory support unless supplemented by positive inotropes.181
The cardiovascular benefits of positive airway pressure can be seen by withdrawing negative swings in ITP by using increasing levels of CPAP.234,235 Even CPAP levels as low as 5 cm H2O can increase cardiac output in CHF patients, whereas cardiac output decreases with similar levels of CPAP in both normal subjects and in heart failure patients without volume overload; these hemodynamic effects of increased airway pressure do not require endotracheal intubation. Patients with CHF, but in whom forced diuresis has induced a relative hypovolemic state (as manifested by a pulmonary artery occlusion pressure ≤12 mm Hg), decreased their cardiac outputs equally whether they received CPAP or BiPAP at the same mean airway pressure.236 Nasal CPAP can also accomplish the same results in patients with obstructive sleep apnea and heart failure,237 although the benefits do not appear to be related to changes in the obstructive breathing pattern.238 Prolonged nighttime nasal CPAP can selectively improve respiratory muscle strength as well as LV contractile function in patients with preexistent heart failure.239,240 These benefits are associated with reductions of serum catecholamine levels.241
If positive airway pressure augments LV ejection in heart failure states, systolic arterial pressure should not decrease but actually increase during inspiration, so-called reverse pulsus paradoxus. This was what Abel et al.207 found in post–cardiac surgery patients. Perel et al.242–244 suggested that the relation between ventilatory efforts and systolic arterial pressure may be used to identify which patients may benefit from cardiac assist maneuvers. Patients who increase their systolic arterial pressure during ventilation relative to an apneic baseline tend to have a greater degree of volume overload 243 and heart failure,242 whereas those subjects in whom systolic arterial pressure decreases tend to be volume responsive. This logic has been recently taken to be used as a hemodynamic test, and arterial pulse pressure substituted for systolic pressure. Michard et al.245 found in a series of ventilator-dependent septic patients that the greater the degree of arterial pulse pressure variation during positive-pressure ventilation, the greater the subsequent increase in cardiac output in response to volume expansion therapy.
Chronic Obstructive Pulmonary Disease
The primary hemodynamic problem seen in patients with COPD is related to hyperinflation, either due to bronchospasm, loss of lung parenchyma, or dynamic hyperinflation. In each case, the lungs expand, compressing the heart, increasing pulmonary vascular resistance, and impeding RV filling. Dynamic hyperinflation is also referred to as intrinsic PEEP. Intrinsic PEEP will alter hemodynamic function in patients in a fashion similar to extrinsic PEEP. Matching intrinsic PEEP with externally applied PEEP has no measurable detrimental hemodynamic effect,246–248 although such matching decreases the work cost of spontaneous breathing. Furthermore, CPAP, like PEEP, has little detrimental effect in these patients when delivered below the intrinsic PEEP level.249 There is little hemodynamic difference between increasing airway pressure to generate a breath and decreasing extrathoracic pressure (as with iron lung negative-pressure ventilation). Ambrosino et al.250 used negative-pressure ventilation to augment ventilation in COPD patients and found no differences in hemodynamic response with similar levels of tidal volume.
Weaning of patients with COPD will tax the cardiovascular system. Patients with severe COPD but adequate ventilatory weaning parameters may go into cardiogenic pulmonary edema during weaning.30 This probably reflects combined volume overload and increased LV failure, because LV ejection fraction decreases during such trials251; following diuresis, many of these patients can be subsequently weaned. The difficulty bedside clinicians have in predicting weaning from mechanical ventilation using simple measures of ventilatory reserve, airflow, and gas exchange parameters may reflect lack of insight into the patient’s cardiovascular reserve and the exercise load spontaneous breathing places on the rest of the circulation. Mohsenifar et al. assessed the effect of weaning on gastric intramucosal pH (pHi), as a marker of splanchnic blood flow, in ventilated patients deemed ready for weaning.36 Patients who could not be weaned demonstrated substantially reduced gastric pHi, from 7.36 during intermittent positive-pressure ventilation to 7.09 during weaning. Patients who were successfully weaned showed no change in pHi (7.45 to 7.46). Jabran et al.37 demonstrated that although all ventilator-dependent COPD patients increased their cardiac outputs during weaning trials, those who failed to wean also decreased the SvO2, consistent with an increased metabolic demand in excess of the cardiovascular reserve. Thus, occult cardiovascular insufficiency may play a major role in the development of failure to wean in critically ill patients.252 However, this assumption, though attractive, has not been proven conclusively, only suggested by this one clinical trial.
Holt JP. The effect of positive and negative intrathoracic pressure on cardiac output and venous return in the dog. Am J Physiol. 1944;142(4):594-603.
Sharpey-Schaffer EP. Effects of Valsalva’s manoeuver on the normal and failing circulation. Br Med J. 1955;1(4915):693-699.
Marini JJ, Culver BN, Butler J. Mechanical effect of lung distention with positive pressure on cardiac function. Am Rev Respir Dis. 1980;124(4):382-386.
Jardin F, Farcot JC, Boisante L, et al. Influence of positive end-expiratory pressure on left ventricular performance. N Engl J Med. 1981;304(7):387-392.
Van den Berg P, Jansen JRC, Pinsky MR. The effect of positive-pressure inspiration on venous return in volume loaded post-operative cardiac surgical patients. J Appl Physiol. 2002;92(3):1223-1231.
Jardin F, Vieillard-Baron A. Right ventricular function and positive-pressure ventilation in clinical practice: from hemodynamic subsets to respirator settings. Intensive Care Med. 2003;29(9):1426-1434.
Ventilation and Left Ventricular Performance
Buda AJ, Pinsky MR, Ingels NB, et al. Effect of intrathoracic pressure on left ventricular performance. N Engl J Med. 1979;301(9):453-459.
Calvin JE, Driedger AA, Sibbald WJ. Positive end-expiratory pressure (PEEP) does not depress left ventricular function in patients with pulmonary edema. Am Rev Respir Dis. 1981;124(2):121-128.
Rasanen J, Nikki P, Heikkila J. Acute myocardial infarction complicated by respiratory failure. The effects of mechanical ventilation. Chest. 1984;85(1):21-28.
Pinsky MR, Matuschak GM, Klain M. Determinants of cardiac augmentation by increases in intrathoracic pressure. J Appl Physiol. 1985;58(4):1189-1198.
Lemaire F, Teboul JL, Cinoti L, et al. Acute left ventricular dysfunction during unsuccessful weaning from mechanical ventilation. Anesthesiology. 1988;69(2):171-179.
Denault AY, Gorcsan J3rd, Pinsky MR. Dynamic effects of positive-pressure ventilation on canine left ventricular pressure-volume relations. J Appl Physiol. 2001;91(1):298-308.
Kaneko Y, Floras JS, Usui K, et al. Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med. 2003;348(13):1233-1241.
Kim HK, Pinsky MR. Effect of tidal volume, sampling duration and cardiac contractility on pulse pressure and stroke volume variation during positive-pressure ventilation. Crit Care Med. 2008;36(10):2858-2862.
Monnet X, Rienzo M, Osman D, et al. Passive leg raising predicts fluid responsiveness in the critically ill. Crit Care Med. 2006;34(5):1402-1407.
Pinsky MR. Hemodynamic evaluation and monitoring in the ICU. Chest. 2007;132(6):2020-2029.
Marik PE, Cavallazzi R, Vasu T, Hirani A. Dynamic changes in arterial waveform derived variables and fluid responsiveness in mechanically ventilated patients: a systematic review of the literature. Crit Care Med. 2009;37(9):2642-2647.
1 Cournaud A, Motley HL, Werko L, et al. Physiologic studies of the effect of intermittent positive pressure breathing on cardiac output in man. Am J Physiol. 1948;152:162-174.
2 Tyberg JV, Grant DA, Kingma I, et al. Effects of positive intrathoracic pressure on pulmonary and systemic hemodynamics. Respir Physiol. 2000;119:171-179.
3 Milic-Emili J, Mead J, Turner JM. Improved method for assessing the validity of the esophageal balloon technique. J Appl Physiol. 1964;19:207-211.
4 Braunwald E, Binion JT, Morgan WL, Sarnoff SJ. Alterations in central blood volume and cardiac output induced by positive pressure breathing and counteracted by metaraminol (Aramine). Circ Res. 1957;5:670-675.
5 Whittenberger JL, McGregor M, Berglund E, et al. Influence of state of inflation of the lung on pulmonary vascular resistance. J Appl Physiol. 1960;15:878-882.
6 Novak RA, Matuschak GM, Pinsky MR. Effect of ventilatory frequency on regional pleural pressure. J Appl Physiol. 1988;65:1314-1323.
7 Pinsky MR, Guimond JG. The effects of positive end- expiratory pressure on heart-lung interactions. J Crit Care. 1991;6:1-11.
8 Romand JA, Shi W, Pinsky MR. Cardiopulmonary effects of positive pressure ventilation during acute lung injury. Chest. 1995;108:1041-1048.
9 O’Quinn RJ, Marini JJ, Culver BH, et al. Transmission of airway pressure to pleural pressure during lung edema and chest wall restriction. J Appl Physiol. 1985;59:1171-1177.
10 Gattinoni L, Mascheroni D, Torresin A, et al. Morphological response to positive end-expiratory pressure in acute respiratory failure. Intensive Care Med. 1986;12:137-142.
11 Globits S, Burghuber OC, Koller J, et al. Effect of lung transplantation on right and left ventricular volumes and function measured by magnetic resonance imaging. Am J Respir Crit Care Med. 1994;149:1000-1004.
12 Scharf SM, Ingram RHJr. Effects of decreasing lung compliance with oleic acid on the cardiovascular response to PEEP. Am J Physiol. 1977;233:H635-H641.
13 Pinsky MR, Vincent JL, DeSmet JM. Estimating left ventricular filling pressure during positive end-expiratory pressure in humans. Am Rev Respir Dis. 1991;143:25-31.
14 Buda AJ, Pinsky MR, Ingels NB, et al. Effect of intrathoracic pressure on left ventricular performance. N Engl J Med. 1979;301:453-459.
15 Kingma I, Smiseth OA, Frais MA, Smith ER, Tyberg JV. Left ventricular external constraint: relationship between pericardial, pleural and esophageal pressures during positive end-expiratory pressure and volume loading in dogs. Ann Biomed Eng. 1987;15:331-346.
16 Tsitlik JE, Halperin HR, Guerci AD, et al. Augmentation of pressure in a vessel indenting the surface of the lung. Ann Biomed Eng. 1987;15:259-284.
17 Marini JJ, Rodriguez RM, Lamb V. The inspiratory workload of patient-initiated mechanical ventilation. Am Rev Respir Dis. 1986;134:902-909.
18 Bromberger-Barnea B. Mechanical effects of inspiration on heart functions: A review. Fed Proc. 1981;40:2172-2177.
19 Wise RA, Robotham JL, Summer WR. Effects of spontaneous ventilation on the circulation. Lung. 1981;159:175-192.
20 Roussos C, Macklem PT. The respiratory muscles. N Engl J Med. 1982;307:786-797.
21 Grenvik A. Respiratory, circulatory and metabolic effects of respiratory treatment. Acta Anaesth Scand. Suppl, 1966.
22 Shuey CB, Pierce AK, Johnson RL. An evaluation of exercise tests in chronic obstructive lung disease. J Appl Physiol. 1969;27:256-261.
23 Stock MC, David DW, Manning JW, Ryan ML. Lung mechanics and oxygen consumption during spontaneous ventilation and severe heart failure. Chest. 1992;102:279-283.
24 Kawagoe Y, Permutt S, Fessler HE. Hyperinflation with intrinsic PEEP and respiratory muscle blood flow. J Appl Physiol. 1994;77:2440-2448.
25 Aubier M, Vires N, Sillye G, Mozes R, Roussos C. Respiratory muscle contribution to lactic acidosis in low cardiac output. Am Rev Respir Dis. 1982;126:648-652.
26 Frazier SK, Stone KS, Schertel ER, Moser DK, Pratt JW. A comparison of hemodynamic changes during the transition from mechanical ventilation to T-piece, pressure support, and continuous positive airway pressure in canines. Biol Res Nurs. 2000;1:253-264.
27 Magder S, Erian R, Roussos C. Respiratory muscle blood flow in oleic acid-induced pulmonary edema. J Appl Physiol. 1986;60:1849-1856.
28 Baratz DM, Westbrook PR, Shah K, Mohsenifar Z. Effects of nasal continuous positive airway pressure on cardiac output and oxygen delivery in patients with congestive heart failure. Chest. 1992;102:1397-1401.
29 Vires N, Sillye G, Rassidakis A, et al. Effect of mechanical ventilation on respiratory muscle blood flow during shock. Physiologist. 1980;23:1-8.
30 Lemaire F, Teboul JL, Cinoti L, et al. Acute left ventricular dysfunction during unsuccessful weaning from mechanical ventilation. Anesthesiology. 1988;69:171-179.
31 Richard C, Teboul JL, Archambaud F, Hebert JL, Michaut P, Auzepy P. Left ventricular dysfunction during weaning in patients with chronic obstructive pulmonary disease. Intensive Care Med. 1994;20:171-172.
32 Hurford WE, Lynch KE, Strauss HW, Lowenstein E, Zapol WM. Myocardial perfusion as assessed by thallium-201 scintigraphy during the discontinuation of mechanical ventilation in ventilator-dependent patients. Anesthesiology. 1991;74:1007-1016.
33 Abalos A, Leibowitz AB, Distefano D, Halpern N, Iberti TJ. Myocardial ischemia during the weaning period. Am J Crit Care. 1992;1:32-36.
34 Chatila W, Ani S, Guaglianone D, Jacob B, Amoateng-Adjepong Y, Manthous CA. Cardiac ischemia during weaning from mechanical ventilation. Chest. 1996;109:1421-1422.
35 Srivastava S, Chatila W, Amoateng-Adjepong Y, et al. Myocardial ischemia and weaning failure in patients with coronary disease: an update. Crit Care Med. 1999;27:2109-2112.
36 Mohsenifar Z, Hay A, Hay J, Lewis MI, Koerner SK. Gastric intramural pH as a predictor of success or failure in weaning patients from mechanical ventilation. Ann Intern Med. 1993;119:794-798.
37 Jabran A, Mathru M, Dries D, Tobin MJ. Continuous recordings of mixed venous oxygen saturation during weaning from mechanical ventilation and the ramifications thereof. Am J Respir Crit Care Med. 1998;158:1763-1769.
38 Straus C, Lewis B, Isebey D, Lemaire F, Harf A, Brochard L. Contribution of the endotracheal tube and the upper airway to breathing workload. Am J Respir Crit Care Med. 1998;157:23-30.
39 Rasanen J, Nikki P, Heikkila J. Acute myocardial infarction complicated by respiratory failure. The effects of mechanical ventilation. Chest. 1984;85:21-28.
40 Rasanen J, Vaisanen IT, Heikkila J, et al. Acute myocardial infarction complicated by left ventricular dysfunction and respiratory failure. The effects of continuous positive airway pressure. Chest. 1985;87:156-162.
41 Glick G, Wechsler AS, Epstein DE. Reflex cardiovascular depression produced by stimulation of pulmonary stretch receptors in the dog. J Clin Invest. 1969;48:467-472.
42 Painal AS. Vagal sensory receptors and their reflex effects. Physiol Rev. 1973;53:59-88.
43 Anrep GV, Pascual W, Rossler R. Respiratory variations in the heart rate. I The reflex mechanism of the respiratory arrhythmia. Proc R Soc Lond B Biol Sci. 1936;119:191-217.
44 Taha BH, Simon PM, Dempsey JA, Skatrud JB, Iber C. Respiratory sinus arrhythmia in humans: an obligatory role for Vagal feedback from the lungs. J Appl Physiol. 1995;78:638-645.
45 Bernardi L, Calciati A, Gratarola A, Battistin I, Fratino P, Finardi G. Heart rate-respiration relationship: computerized method for early detection of cardiac autonomic damage in diabetic patients. Acta Cardiol. 1986;41:197-206.
46 Bernardi L, Keller F, Sanders M, et al. Respiratory sinus arrhythmia in the totally denervated human heart. J Appl Physiol. 1989;67:1447-1455.
47 Pinna GD, Maestri R, Mortara A, La Rovere MT. Cardiovascular interactions during periodic breathing in awake chronic heart failure patients. Am J Physiol. 2000;278:H932-H941.
48 Persson MG, Lonnqvist PA, Gustafsson LE. Positive end-expiratory pressure ventilation elicits increases in endogenously formed nitric oxide as detected in air exhaled by rabbits. Anesthesiology. 1995;82:969-974.
49 Cassidy SS, Eschenbacher WI, Johnson RLJr. Reflex cardiovascular depression during unilateral lung hyperinflation in the dog. J Clin Invest. 1979;64:620-626.
50 Daly MB, Hazzledine JL, Ungar A. The reflex effects of alterations in lung volume on systemic vascular resistance in the dog. J Physiol (London). 1967;188:331-351.
51 Shepherd JT. The lungs as receptor sites for cardiovascular regulation. Circulation. 1981;63:1-10.
52 Stalcup SA, Mellins RB. Mechanical forces producing pulmonary edema in acute asthma. N Engl J Med. 1977;297:592-596.
53 Vatner SF, Rutherford JD. Control of the myocardial contractile state by carotid chemo- and baroreceptor and pulmonary inflation reflexes in conscious dogs. J Clin Invest. 1978;63:1593-1601.
54 Pick RA, Handler JB, Murata GH, Friedman AS. The cardiovascular effects of positive end-expiratory pressure. Chest. 1982;82:345-350.
55 Karlocai K, Jokkel G, Kollai M. Changes in left ventricular contractility with the phase of respiration. J Auton Nerv Syst. 1998;73:86-92.
56 Said SI, Kitamura S, Vreim C. Prostaglandins: Release from the lung during mechanical ventilation at large tidal ventilation. J Clin Invest. 1972;51:83a.
57 Bedetti C, Del Basso P, Argiolas C, Carpi A. Arachidonic acid and pulmonary function in a heart-lung preparation of guinea-pig. Modulation by PCO2. Arch Int Pharmacodyn Ther. 1987;285:98-116.
58 Berend N, Christopher KL, Voelkel NF. Effect of positive end- expiratory pressure on functional residual capacity: Role of prostaglandin production. Am Rev Respir Dis. 1982;126:641-647.
59 Pattern MY, Liebman PR, Hetchman HG. Humorally mediated decreases in cardiac output associated with positive end- expiratory pressure. Microvasc Res. 1977;13:137-144.
60 Berglund JE, Halden E, Jakobson S, Svensson J. PEEP ventilation does not cause humorally mediated cardiac output depression in pigs. Intensive Care Med. 1994;20:360-364.
61 Fuhrman BP, Everitt J, Lock JE. Cardiopulmonary effects of unilateral airway pressure changes in intact infant lambs. J Appl Physiol. 1984;56:1439-1448.
62 Payen DM, Brun-Buisson CJL, Carli PA, et al. Hemodynamic, gas exchange, and hormonal consequences of LBPP during PEEP ventilation. J Appl Physiol. 1987;62:61-70.
63 Frage D, de la Coussaye JE, Beloucif S, Fratacci MD, Payen DM. Interactions between hormonal modifications during Peep-Induced Antidiuresis and Antinatriuresis. Chest. 1995;107:1095-1100.
64 Frass M, Watschinger B, Traindl O, et al. Atrial natriuretic peptide release in response to different positive end-expiratory pressure levels. Crit Care Med. 1993;21:343-347.
65 Wilkins MA, Su XL, Palayew MD, et al. The effects of posture change and continuous positive airway pressure on cardiac natriuretic peptides in congestive heart failure. Chest. 1995;107:909-915.
66 Shirakami G, Magaribuchi T, Shingu K, et al. Positive end-expiratory pressure ventilation decreases plasma atrial and brain natriuretic peptide levels in humans. Anesth Analg. 1993;77:1116-1121.
67 Brecher GA, Hubay CA. Pulmonary blood flow and venous return during spontaneous respiration. Circ Res. 1955;3:40-214.
68 Goldstein JA, Vlahakes GJ, Verrier ED. The role of right ventricular systolic dysfunction and elevated intrapericardial pressures in the genesis of low output in experimental right ventricular infarction. Circulation. 1982;65:513-520.
69 Jardin F, Farcot JC, Boisante L. Influence of positive end-expiratory pressure on left ventricular performance. N Engl J Med. 1981;304:387-392.
70 Jardin FF, Farcot JC, Gueret P, Prost JF, Ozier Y, Bourdarias JP. Echocardiographic evaluation of ventricles during continuous positive pressure breathing. J Appl Physiol. 1984;56:619-627.
71 Prec KJ, Cassels DE. Oximeter studies in newborn infants during crying. Pediatr. 1952;9:756-761.
72 Luce JM. The cardiovascular effects of mechanical ventilation and positive end-expiratory pressure. JAMA. 1984;252:807-811.
73 Maughan WL, Shoukas AA, Sagawa K, Weisfeldt ML. Instantaneous pressure-volume relationships of the canine right ventricle. Circ Res. 1979;44:309-315.
74 Sibbald WJ, Driedger AA. Right ventricular function in disease states: Pathophysiologic considerations. Crit Care Med. 1983;11:339.
75 Fletcher EC, Proctor M, Yu J, et al. Pulmonary edema develops after recurrent obstructive apneas. Am J Respir Crit Care Med. 1999;160:1688-1696.
76 Chen L, Shi Q, Scharf SM. Hemodynamic effects of periodic obstructive apneas in sedated pigs with congestive heart failure. J Appl Physiol. 2000;88:1051-1060.
77 Pinsky MR. Instantaneous venous return curves in an intact canine preparation. J Appl Physiol. 1984;56:765-771.
78 Piene H, Sund T. Does pulmonary impedance constitute the optimal load for the right ventricle? Am J Physiol. 1982;242:H154-H160.
79 Pinsky MR. Determinants of pulmonary arterial flow variation during respiration. J Appl Physiol. 1984;56:1237-1245.
80 Theres H, Binkau J, Laule M, et al. Phase-related changes in right ventricular cardiac output under volume-controlled mechanical ventilation with positive end-expiratory pressure. Crit Care Med. 1999;27:953-958.
81 Johnston WE, Vinten-Johansen J, Shugart HE, Santamore WP. Positive end-expiratory pressure potentates the severity of canine right ventricular ischemia-reperfusion injury. Am J Physiol. 1992;262:H168-H176.
82 Sibbald WJ, Calvin JE, Holliday RL, et al. Concepts in the pharmacologic support of cardiovascular function in critically ill surgical patients. Surg Clin North Am. 1983;63:455-466.
83 Sibbald WH, Calvin J, Driedger AA. Right and left ventricular preload, and diastolic ventricular compliance: Implications of therapy in critically ill patients. Critical Care State of the Art. Fullerton, Calif. Society of Critical Care. 3, 1982.
84 Madden JA, Dawson CA, Harder DR. Hypoxia-induced activation in small isolated pulmonary arteries from the cat. J Appl Physiol. 1985;59:113-118.
85 Hakim TS, Michel RP, Chang HK. Effect of lung inflation on pulmonary vascular resistance by arterial and venous occlusion. J Appl Physiol. 1982;53:1110-1115.
86 Quebbeman EJ, Dawson CA. Influence of inflation and atelectasis on the hypoxic pressure response in isolated dog lung lobes. Cardiovasc Res. 1976;10:672-677.
87 Brower RG, Gottlieb J, Wise RA, Permutt W, Sylvester JT. Locus of hypoxic vasoconstriction in isolated ferret lungs. J Appl Physiol. 1987;63:58-65.
88 Hakim TS, Michel RP, Minami H, Chang K. Site of pulmonary hypoxic vasoconstriction studied with arterial and venous occlusion. J Appl Physiol. 1983;54:1298-1302.
89 Marshall BE, Marshall C. A model for hypoxic constriction of the pulmonary circulation. J Appl Physiol. 1988;64:68-77.
90 Marshall BE, Marshall C. Continuity of response to hypoxic pulmonary vasoconstriction. J Appl Physiol. 1980;49:189-196.
91 Dawson CA, Grimm DJ, Linehan JH. Lung inflation and longitudinal distribution of pulmonary vascular resistance during hypoxia. J Appl Physiol. 1979;47:532-536.
92 Howell JBL, Permutt S, Proctor DF, et al. Effect of inflation of the lung on different parts of the pulmonary vascular bed. J Appl Physiol. 1961;16:71-76.
93 West JB, Dollery CT, Naimark A. Distribution of blood flow in isolated lung; relation to vascular and alveolar pressures. J Appl Physiol. 1964;19:713-724.
94 Fuhrman BP, Everitt J, Lock JE. Cardiopulmonary effects of unilateral airway pressure changes in intact infant lambs. J Appl Physiol. 1984;56:1439-1448.
95 Fuhrman BP, Smith-Wright DL, Kulik TJ, Lock JE. Effects of static and fluctuating airway pressure on the intact, immature pulmonary circulation. J Appl Physiol. 1986;60:114-122.
96 Thorvalson J, Ilebekk A, Kiil F. Determinants of pulmonary blood volume. Effects of acute changes in airway pressure. Acta Physiol Scand. 1985;125:471-479.
97 Hoffman EA, Ritman EL. Heart-lung interaction: effect on regional lung air content and total heart volume. Ann Biomed Eng. 1987;15:241-257.
98 Olson LE, Hoffman EA. Heart-lung interactions determined by electron beam x-ray CT in laterally recumbent rabbits. J Appl Physiol. 1995;78:417-427.
99 Grant BJB, Lieber BB. Compliance of the main pulmonary artery during the ventilatory cycle. J Appl Physiol. 1992;72:535-542.
100 Lopez-Muniz R, Stephens NL, Bromberger-Barnea B, Permutt S, Riley RL. Critical closure of pulmonary vessels analyzed in terms of Starling resistor model. J Appl Physiol. 1968;24:625-635.
101 Block AJ, Boyson PG, Wynne JW. The origins of cor pulmonale, a hypothesis. Chest. 1979;75:109-114.
102 Vieillard-Baron A, Loubieres Y, Schmitt JM, Page B, Dubourg O, Jardin F. Cyclic changes in right ventricular output impedance during mechanical ventilation. J Appl Physiol. 1999;87:1644-1650.
103 Canada E, Benumnof JL, Tousdale FR. Pulmonary vascular resistance correlated in intact normal and abnormal canine lungs. Crit Care Med. 1982;10:719-723.
104 Mitchell JR, Whitelaw WA, Sas R, Smith ER, Tyberg TV, Belenkie I. RV filling modulates left ventricular function by direct ventricular interaction during mechanical ventilation. Am J Physiol. 2005;289:H549-H557.
105 Taylor RR, Corell JW, Sonnenblick EH, Ross J.Jr. Dependence of ventricular distensibility on filling the opposite ventricle. Am J Physiol. 1967;213:711-718.
106 Brinker JA, Weiss I, Lappe DL, et al. Leftward septal displacement during right ventricular loading in man. Circulation. 1980;61:626-633.
107 Marini JJ, Culver BN, Butler J. Mechanical effect of lung distention with positive pressure on cardiac function. Am Rev Respir Dis. 1980;124:382-386.
108 Butler J. The heart is in good hands. Circulation. 1983;67:1163-1168.
109 Cassidy SS, Wead WB, Seibert GB, Ramanathan M. Changes in left ventricular geometry during spontaneous breathing. J Appl Physiol. 1987;63:803-811.
110 Cassidy SS, Robertson CH, Pierce AK, et al. Cardiovascular effects of positive end-expiratory pressure in dogs. J Appl Physiol. 1978;4:743-749.
111 Conway CM. Hemodynamic effects of pulmonary ventilation. Br J Anaesth. 1975;47:761-766.
112 Berglund JE, Halden E, Jakobson S, Landelius J. Echocardiographic analysis of cardiac function during high PEEP ventilation. Intensive Care Med. 1994;20:174-180.
113 Takata M, Harasawa Y, Beloucif S, Robotham JL. Coupled vs. Uncoupled pericardial restraint: effects on cardiac chamber interactions. J Appl Physiol. 1997;83:1799-1813.
114 Rebuck AS, Read J. Assessment and management of severe asthma. Am J Med. 1971;51:788-792.
115 Guyton AC, Lindsey AW, Abernathy B, et al. Venous return at various right atrial pressures and the normal venous return curve. Am J Physiol. 1957;189:609-615.
116 Goldberg HS, Rabson J. Control of cardiac output by systemic vessels: Circulatory adjustments of acute and chronic respiratory failure and the effects of therapeutic interventions. Am J Cardiol. 1981;47:696.
117 Kilburn KH. Cardiorespiratory effects of large pneumothorax in conscious and anesthetized dogs. J Appl Physiol. 1963;18:279-283.
118 Chevalier PA, Weber KC, Engle JC, et al. Direct measurement of right and left heart outputs in Valsalva-like maneuver in dogs. Proc. Soc Exper Biol Med. 1972;139:1429-1437.
119 Guntheroth WC, Gould R, Butler J, et al. Pulsatile flow in pulmonary artery, capillary and vein in the dog. Cardiovascular Res. 1974;8:330-337.
120 Guntheroth WG, Morgan BC, Mullins GL. Effect of respiration on venous return and stroke volume in cardiac tamponade. Mechanism of pulsus paradoxus. Circ Res. 1967;20:381-390.
121 Guyton AC. Effect of cardiac output by respiration, opening the chest, and cardiac tamponade. In: Circulatory Physiology: Cardiac Output and Its Regulation. Philadelphia, PA: Saunders; 1963:378-386.
122 Holt JP. The effect of positive and negative intrathoracic pressure on cardiac output and venous return in the dog. Am J Physiol. 1944;142:594-603.
123 Morgan BC, Abel FL, Mullins GL, et al. Flow patterns in cavae, pulmonary artery, pulmonary vein and aorta in intact dogs. Am J Physiol. 1966;210:903-909.
124 Morgan BC, Martin WE, Hornbein TF, et al. Hemodynamic effects of intermittent positive pressure respiration. Anesthesiology. 1960;27:584-590.
125 Scharf SM, Brown R, Saunders N, Green LH. Hemodynamic effects of positive pressure inflation. J Appl Physiol. 1980;49:124-131.
126 Jardin F, Vieillard-Baron A. Right ventricular function and positive-pressure ventilation in clinical practice: from hemodynamic subsets to respirator settings. Intensive Care Med. 2003;29:1426-1434.
127 Scharf SM, Brown R, Saunders N, et al. Effects of normal and loaded spontaneous inspiration on cardiovascular function. J Appl Physiol. 1979;47:582-590.
128 Groeneveld AB, Berendsen RR, Schneider AJ, Pneumatikos IA, Stokkel LA, Thijs LG. Effect of the mechanical ventilatory cycle on thermodilution right ventricular volumes and cardiac output. J Appl Physiol. 2000;89:89-96.
129 Fessler HE, Brower RG, Wise RA, Permutt S. Effects of positive end-expiratory pressure on the canine venous return curve. Am Rev Respir Dis. 1992;146:4-10.
130 Takata M, Robotham JL. Effects of inspiratory diaphragmatic descent on inferior vena caval venous return. J Appl Physiol. 1992;72:597-607.
131 Matuschak GM, Pinsky MR, Rogers RM. Effects of positive end-expiratory pressure on hepatic blood flow and hepatic performance. J Appl Physiol. 1987;62:1377-1383.
132 Chihara E, Hasimoto S, Kinoshita T, Hirpose M, Tanaka Y, Morimoto T. Elevated mean systemic filling pressure due to intermittent positive-pressure ventilation. Am J Physiol. 1992;262:H1116-H1121.
133 Takata M, Wise RA, Robotham JL. Effects of abdominal pressure on venous return: abdominal vascular zone conditions. J Appl Physiol. 1990;69:1961-1972.
134 Barnes GE, Laine GA, Giam PY, Smith EE, Granger HJ. Cardiovascular responses to elevation of intra-abdominal hydrostatic pressure. Am J Physiol. 1985;248:R208-R213.
135 Lichtwarck-Aschoff M, Zeravik J, Pfeiffer UJ. Intrathoracic blood volume accurately reflects circulatory volume status in critically ill patients with mechanical ventilation. Intensive Care Med. 1992;18:142-145.
136 Brienza N, Revelly JP, Ayuse T, Robotham JL. Effect of PEEP on liver arterial and venous blood flows. Am J Respir Crit Care Med. 1995;152:504-510.
137 Sha M, Saito Y, Yokoyama K, Sawa T, Amaha K. Effects of continuous positive-pressure ventilation on hepatic blood flow and intrahepatic oxygen delivery in dogs. Crit Care Med. 1987;15:1040-1417.
138 Richard C, Berdeaux A, Delion F, et al. Effect of mechanical ventilation on hepatic drug pharmacokinetics. Chest. 1986;90:837-842.
139 Dorinsky PM, Hamlin RL, Gadek JE. Alterations in regional blood flow during positive end-expiratory pressure ventilation. Crit Care Med. 1987;15:106-115.
140 Van den Berg P, Jansen JRC, Pinsky MR. The effect of positive-pressure inspiration on venous return in volume loaded post-operative cardiac surgical patients. J Appl Physiol. 2002;92:1223-1231.
141 Magder S, Georgiadis G, Cheong T. Respiratory variation in right atrial pressure predict the response to fluid challenge. J Crit Care. 1992;7:76-85.
142 Terada N, Takeuchi T. Postural changes in venous pressure gradients in anesthetized monkeys. Am J Physiol. 1993;264:H21-H25.
143 Scharf S, Tow DE, Miller MJ, Brown R, McIntyre K, Dilts C. Influence of posture and abdominal pressure on the hemodynamic effects of Mueller’s maneuver. J Crit Care. 1989;4:26-34.
144 Tarasiuk A, Scharf SM. Effects of periodic obstructive apneas on venous return in closed-chest dogs. Am Rev Respir Dis. 1993;148:323-329.
145 Mang H, Kacmarek RM, Ritz R, Wilson RS, Kimball WP. Cardiorespiratory effects of volume- and pressure-controlled ventilation at various I/E ratios in an acute lung injury model. Am J Respir Crit Care Med. 1995;151:731-736.
146 Tyberg JV, Taichman GC, Smith ER, Douglas NWS, Smiseth OA, Keon WJ. The relationship between pericardial pressure and right atrial pressure: An intraoperative study. Circulation. 1986;73:428-432.
147 Pinsky MR, Vincent JL, DeSmet JM. Effect of positive end-expiratory pressure on right ventricular function in man. Am Rev Respir Dis. 1992;146:681-687.
148 Jayaweera AR, Ehrlich W. Changes of phasic pleural pressure in awake dogs during exercise: potential effects on cardiac output. Ann Biomed Eng. 1987;15:311-318.
149 Harken AH, Brennan MF, Smith N, Barsamian EM. The hemodynamic response to positive end-expiratory ventilation in hypovolemic patients. Surgery. 1974;76:786-793.
150 Lores ME, Keagy BA, Vassiliades T, Henry GW, Lucas CL, Wilcox BR. Cardiovascular effects of positive end-expiratory pressure (PEEP) after pneumonectomy in dogs. Ann Thorac Surg. 1985;40:464-473.
151 Shekerdemian LS, Bush A, Shore DF, Lincoln C, Redington AN. Cardiorespiratory responses to negative pressure ventilation after Tetralogy of Fallot repair: a hemodynamic tool for patients with low-output state. J Am Coll Cardiol. 1999;33:549-555.
152 Sharpey-Schaffer EP. Effects of Valsalva maneuver on the normal and failing circulation. Br Med J. 1955;1:693-699.
153 Peters J, Kindred MK, Robotham JL. Transient analysis of cardiopulmonary interactions II. Systolic events. J Appl Physiol. 1988;64:1518-1526.
154 Pinsky MR, Matuschak GM, Klain M. Determinants of cardiac augmentation by increases in intrathoracic pressure. J Appl Physiol. 1985;58:1189-1198.
155 Rankin JS, Olsen CO, Arentzen CE, et al. The effects of airway pressure on cardiac function in intact dogs and man. Circulation. 1982;66:108-120.
156 Robotham JL, Rabson J, Permutt S, Bromberger-Barnea B. Left ventricular hemodynamics during respiration. J Appl Physiol. 1979;47:1295-1303.
157 Ruskin J, Bache RJ, Rembert JC, GreenfieldJr. Pressure-flow studies in man: Effect of respiration on left ventricular stroke volume. Circulation. 1973;48:79-85.
158 Olsen CO, Tyson GS, Maier GW, et al. Dynamic ventricular interaction in the conscious dog. Circ Res. 1983;52:85-104.
159 Bell RC, Robotham JL, Badke FR, Little WC, Kindred MK. Left ventricular geometry during intermittent positive pressure ventilation in dogs. J Crit Care. 1987;2:230-244.
160 Qvist J, Pontoppidan H, Wilson RS, Lowenstein E, Laver MB. Hemodynamic responses to mechanical ventilation with PEEP: the effects of hypovolemia. Anesthesiology. 1975;42:45-53.
161 Denault AY, Gorcsan JIII, Deneault LG, Pinsky MR. Effect of positive pressure ventilation on left ventricular pressure-volume relationship. Anesthesiology. 1993;79:A315.
162 Janicki JS, Weber KT. The pericardium and ventricular interaction, distensibility and function. Am J Physiol. 1980;238:H494-H503.
163 Beyar R, Goldstein Y. Model studies of the effects of the thoracic pressure on the circulation. Ann Biomed Eng. 1987;15:373-383.
164 Pinsky MR, Summer WR, Wise RA, Permutt S, Bromberger-Barnea B. Augmentation of Cardiac Function by Elevation of Intrathoracic Pressure. J Appl Physiol. 1983;54:950-955.
165 Cassidy SA, Wead WB, Seibert GB, Ramanathan M. Geometric left-ventricular responses to interactions between the lung and left ventricle: positive pressure breathing. Ann Biomed Eng. 1987;15:285-295.
166 Scharf SM, Brown R, Warner KG, Khuri S. Intrathoracic pressure and left ventricular configuration with respiratory maneuvers. J Appl Physiol. 1989;66:481-491.
167 Pinsky MR, Summer WR. Cardiac Augmentation by Phasic High Intrathoracic Support (PHIPS) in Man. Chest. 1983;84:370-375.
168 Pinsky MR, Matuschak GM, Itzkoff JM. Respiratory Augmentation of Left Ventricular Function During Spontaneous Ventilation in Severe Left Ventricular Failure by Grunting: An Auto-EPAP Effect. Chest. 1984;86:267-269.
169 Blaustein AS, Risser TA, Weiss JW, Parker JA, Holman L, McFadden ER. Mechanisms of pulsus paradoxus during resistive respiratory loading and asthma. J Am Coll Cardiol. 1986;8:529-536.
170 Strohl KP, Scharf SM, Brown R, Ingram RHJr. Cardiovascular performance during bronchospasm in dogs. Respiration. 1987;51:39-48.
171 Scharf SM, Graver LM, Balaban K. Cardiovascular effects of periodic occlusions of the upper airways in dogs. Am Rev Respir Dis. 1992;146:321-329.
172 Viola AR, Puy RJM, Goldman E. Mechanisms of pulsus paradoxus in airway obstruction. J Appl Physiol. 1990;68:1927-1931.
173 Scharf SM, Graver LM, Khilnani S, Balaban K. Respiratory phasic effects of inspiratory loading on left ventricular hemodynamics in Vagotomized dogs. J Appl Physiol. 1992;73:995-1003.
174 Latham RD, Sipkema P, Westerhof N, Rubal BJ. Aortic input impedance during Mueller maneuver: an evaluation of “effective strength. J Appl Physiol. 1988;65:1604-1610.
175 Virolainen J, Ventila M, Turto H, Kupari M. Effect of negative intrathoracic pressure on left ventricular pressure dynamics and relaxation. J Appl Physiol. 1995;79:455-460.
176 Gomez A, Mink S. Interaction between effects of hypoxia and hypercapnia on altering left ventricular relaxation and chamber stiffness in dogs. Am Rev Respir Dis. 1992;146:313-320.
177 Denault AY, Gorcsan J3rd, Pinsky MR. Dynamic effects of positive-pressure ventilation on canine left ventricular pressure-volume relations. J Appl Physiol. 2001;91:298-308.
178 Abel FL, Mihailescu LS, Lader AS, Starr RG. Effects of pericardial pressure on systemic and coronary hemodynamics in dogs. Am J Physiol. 1995;268:H1593-H1605.
179 Khilnani S, Graver LM, Balaban K, Scharf SM. Effects of inspiratory loading on left ventricular myocardial blood flow and metabolism. J Appl Physiol. 1992;72:1488-1492.
180 Satoh S, Watanabe J, Keitoku M, Itoh N, Maruyama Y, Takishima T. Influences of pressure surrounding the heart and intracardiac pressure on the diastolic coronary pressure-flow relation in excised canine heart. Circ Res. 1988;63:788-797.
181 Beach T, Millen E, Grenvik. Hemodynamic response to discontinuance of mechanical ventilation. Crit Care Med. 1973;1:85-90.
182 Garpestad E, Parker JA, Katayama H, et al. Decrease in ventricular stroke volume at apnea termination is independent of oxygen desaturation. J Appl Physiol. 1994;77:1602-1608.
183 Suga H, Sagawa K. Instantaneous pressure-volume relationships and their ratio in the excised supported canine left ventricle. Circ Res. 1974;35:117-134.
184 Haney MF, Johansson G, Haggmark S, Biber B. Heart-lung interactions during positive-pressure ventilation: left ventricular pressure-volume momentary response to airway pressure elevation. Acta Anaesthesiol Scand. 2001;45:702-709.
185 Haney MF, Johansson G, Haggmark S, Biber B. Method of preload reduction during LVESPVR analysis of systolic function: airway pressure elevations and vena caval occlusion. Anesthesiology. 2002;97:436-446.
186 Denault AY, Gasior TA, Gorcsan J3rd, Mandarino WA, Deneault LG, Pinsky MR. Determinants of aortic pressure variation during positive-pressure ventilation in man. Chest. 1999;116:176-186.
187 Pinsky MR. Hemodynamic evaluation and monitoring in the ICU. Chest. 2007;123:2020-2029.
188 Michard F, Teboul JL. Predicting fluid responsiveness in ICU patients: a critical analysis of the evidence. Chest. 2002;121:2000-2008.
189 Michard F, Boussat S, Chemla D, et al. Relation between respiratory changes in arterial pulse pressure and fluid responsiveness in septic patients with acute circulatory failure. Am J Respir Crit Care Med. 2000;162:134-138.
190 Marik PE, Cavallazzi R, Vasu T, Hirani A. Dynamic changes in arterial waveform derived variables and fluid responsiveness in mechanically ventilated patients: A systematic review of the literature. Crit Care Med. 2009;37:2642-2647.
191 De Blasi RA, Palmisani S, Cigognetti L, et al. Effects of sternotomy on heart-lung interaction in patients undergoing cardiac surgery receiving pressure-controlled mechanical ventilation. Acta Anaesthesiol Scand.. 2007;51:441-446.
192 Biais M, Nouette-Gaulain K, Cottenceau V, Revel P, Sztark F. Uncalibrated pulse contour-derived stroke volume variation predicts fluid responsiveness in mechanically ventilated patients undergoing liver transplantation. Br J Anaesth.. 2008;101:761-768.
193 Yu YH, Dai HW, Yan ML, et al. An evaluation of stroke volume variation as a predictor of fluid responsiveness in mechanically ventilated elderly patients with severe sepsis. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2009;21:463-465. [article in Chinese] PMID: 19695166
194 Vallée1 F, Richard JCM, Mari A, et al. Pulse pressure variations adjusted by alveolar driving pressure to assess fluid responsiveness. Intensive Care Med. 2009;35:1004-1010.
195 Lefrant J-Y, De Backer D. Can we use pulse pressure variations to predict fluid responsiveness in patients with ARDS? Intensive Care Med. 2009;35:966-968.
196 Payen D, Vallée F, Mari A, Richard JC, De Backer D. Can pulse pressure variations really better predict fluid responsiveness than static indices of preload in patients with acute respiratory distress syndrome? Crit Care Med. 2009;37:1178-1182.
197 Huang Chung-Chi, Fu Jui-Ying, Hu Han-ChungMD, Kao Kuo-Chin, Chen Ning-Hung, Hsieh Meng-Jer, Tsai Ying-Huang. Prediction of fluid responsiveness in acute respiratory distress syndrome patients ventilated with low tidal volume and high positive end-expiratory pressure. Crit Care Med. 2008;36:2810-2816.
198 Durand P, Chevret L, Essouri S, Haas V, Devictor D. Respiratory variations in aortic blood flow predict fluid responsiveness in ventilated children. Intensive Care Med. 2008;34:888-894.
199 De Backer D, Heenen S, Piagnerelli M, Koch M, Vincent JL. Pulse pressure variations to predict fluid responsiveness: influence of tidal volume. Intensive Care Med. 2005;31:517-523.
200 Renner J, Cavus E, Meybohm P, et al. Stroke volume variation during hemorrhage and after fluid loading: impact of different tidal volumes. Acta Anesthesiol Scand. 2007;51:538-544.
201 Kim HK, Pinsky MR. Effect of tidal volume, sampling duration and cardiac contractility on pulse pressure and stroke volume variation during positive-pressure ventilation. Crit Care Med. 2008;36:2858-2862.
202 Muller L, Louart G, Bousquet PJ, et al. The influence of the airway driving pressure on pulsed pressure variation as a predictor of fluid responsiveness. Intensive Care Med. 2010;36:496-503.
203 Vistisen ST, Koefoed-Nielsen J, Larsson A. Should dynamic parameters for prediction of fluid responsiveness be indexed to the tidal volume? Acta Anaesthesiol Scand. 2010;54:191-198.
204 Kubitz JC, Annecke T, Kemming GI, et al. The influence of positive end-expiratory pressure on stroke volume variation and central blood volume during open and closed chest conditions. Eur J Cardiothorac Surg. 2006;30:90-95.
205 De Castro V, Goarin JP, Lhotel L, Mabrouk N, Perel A, Coriat P. Comparison of stroke volume (SV) and stroke volume respiratory variation (SVV) measured by the axillary artery pulse-contour method and by aortic Doppler echocardiography in patients undergoing aortic surgery. Br J Anesth. 2006;5:605-610.
206 Pinsky MR. Hemodynamic Evaluation and Monitoring in the ICU. Chest. 2007;132:2020-2029.
207 Gunn SR, Kim HK, Harrigan PWJ, Pinsky MR. Ability of pulse contour and esophageal Doppler to estimate rapid changes in stroke volume. Intensive Care Med. 2006;32:1537-1546.
208 Teboul JL, Monnet X. Prediction of volume responsiveness in critically ill patients with spontaneous breathing activity. Curr Opin Crit Care. 2008;14:334-339.
209 Monnet X, Rienzo M, Osman D, et al. Passive leg raising predicts fluid responsiveness in the critically ill. Crit Care Med. 2006;34:1402-1407.
210 Lamia B, Ochagavia A, Monnet X, Chemla D, Richard C, Teboul JL. Echocardiographic prediction of volume responsiveness in critically ill patients with spontaneously breathing activity. Intensive Care Med. 2007;33:1125-1132.
211 Pinsky MR, Matuschak GM, Bernardi L, Klain M. Hemodynamic effects of cardiac cycle-specific increases in intrathoracic pressure. J Appl Physiol. 1986;60:604-612.
212 Dries DJ, Kumar P, Mathru M, et al. Hemodynamic effects of pressure support ventilation in cardiac surgery patients. Am Surg. 1991;57:122-125.
213 Sternberg R, Sahebjami H. Hemodynamic and oxygen transport characteristics of common ventilatory modes. Chest. 1994;105:1798-1803.
214 Bayly R, Sladen A, Guntapalli K, Klain M. Synchronous versus nonsynchronous high-frequency jet ventilation: effects on cardio-respiratory variables and airway pressures in postoperative patients. Crit Care Med. 1987;15:915-923.
215 Dhainaut JF, Devaux JY, Monsallier JF, Brunet F, Villemant D, Huyghebaert MF. Mechanisms of decreased left ventricular preload during continuous positive pressure ventilation in ARDS. Chest. 1986;90:74-80.
216 Lessard MR, Guerot E, Lorini H, Lemaire F, Brochard L. Effects of pressure-controlled with different I:E ratios versus volume-controlled ventilation on respiratory mechanics, gas exchange and hemodynamics in patients with adult respiratory distress syndrome. Anesthesiology. 1994;80:983-991.
217 Chan K, Abraham E. Effects of inverse ratio ventilation on cardio-respiratory parameters in severe respiratory failure. Chest. 1992;102:1556-1561.
218 Abraham E, Yoshihara G. Cardiorespiratory effects of pressure controlled ventilation in severe respiratory failure. Chest. 1990;98:1445-1449.
219 Poelaert JI, Visser CA, Everaert JA, Koolen JJ, Colardyn FA. Acute hemodynamic changes of pressure-controlled inverse ration ventilation in the adult respiratory distress syndrome. A transesophageal echocardiographic and Doppler study. Chest. 1993;104:214-219.
220 Davis KJr, Branson RD, Campbell RS, Porembka DT. Comparison of volume control and pressure control ventilation: is flow waveform the difference? J Trauma. 1996;41:808-814.
221 Kiehl M, Schiele C, Stenzinger W, Kienast J. Volume-controlled versus biphasic positive airway pressure ventilation in leukopenic patients with severe respiratory failure. Crit Care Med. 1996;24:780-784.
222 Singer M, Vermaat J, Hall G, Latter G, Patel M. Hemodynamic effects of manual hyperinflation in critically ill mechanically ventilated patients. Chest. 1994;106:1182-1187.
223 Hartmann M, Rosberg B, Jonsson K. The influence of different levels of PEEP on peripheral tissue perfusion measured by subcutaneous and transcutaneous oxygen tension. Intensive Care Med. 1992;18:474-478.
224 Huemer G, Kolev N, Kurz A, Zimpfer M. Influence of positive end-expiratory pressure on right and left ventricular performance assessed by Doppler two-dimensional echocardiography. Chest. 1994;106:67-73.
225 Jardin F. PEEP and ventricular function. Intensive Care Med. 1994;20:169-170.
226 Goertz A, Heinrich H, Winter H, Deller A. Hemodynamic effects of different ventilatory patterns. A prospective clinical trial. Chest. 1991;99:1166-1171.
227 Gunter JP, deBoisblanc BP, Rust BS, Johnson WD, Summer WR. Effect of synchronized, systolic, lower body, positive pressure on hemodynamics in human septic shock: a pilot study. Am J Respir Crit Care Med. 1995;151:719-723.
228 Schuster S, Erbel R, Weilemann LS, et al. Hemodynamics during PEEP ventilation in patients with severe left ventricular failure studied by transesophageal echocardiography. Chest. 1990;97:1181-1189.
229 Abel JG, Salerno TA, Panos A, et al. Cardiovascular effects of positive pressure ventilation in humans. Ann Thorac Surg. 1987;43:36-43.
230 Grace MP, Greenbaum DM. Cardiac performance in response to PEEP in patients with cardiac dysfunction. Crit Care Med. 1982;20:358-360.
231 Calvin JE, Driedger AA, Sibbald WJ. Positive end-expiratory pressure (PEEP) does not depress left ventricular function in patients with pulmonary edema. Am Rev Respir Dis. 1981;124:121-128.
232 Loick HM, Wendt M, Rotker J Theissen JL. Ventilation with positive end-expiratory airway pressure causes leukocyte retention in human lung. J Appl Physiol. 1993;75:301-306.
233 Rasanen J. Respiratory failure in acute myocardial infarction. Appl Cardiopulm Pathophys. 1988;2:271-279.
234 DeHoyos A, Liu PP, Benard DC, Bradley TD. Haemodynamic effects of continuous positive airway pressure in humans with normal and impaired left ventricular function. Clin Sci Colch. 1995;88:173-178.
235 Naughton MT, Rahman MA, Hara K, Flora JS, Bradley TD. Effect of continuous positive airway pressure on intrathoracic and left ventricular transmural pressures in patients with congestive heart failure. Circulation. 1995;91:1725-1731.
236 Joet-Philip FF, Paganelli FF, Dutau HL, Saadjian AY. Hemodynamic effects of bi-level nasal positive airway pressure ventilation in patients with heart failure. Respiration. 1999;66:136-143.
237 Lin M, Yang Y-F ChiangH-T, Chang M-S, Chiang BN, Cheitlin MD. Reappraisal of continuous positive airway pressure therapy in acute cardiogenic pulmonary edema. Chest. 1995;107:1379-1386.
238 Buckle P, Millar T, Kryger M. The effect of short-term nasal CPAP on Cheyne-Stokes respiration in congestive heart failure. Chest. 1992;102:31-35.
239 Granton JT, Naughton MT, Benard DC, Liu PP, Goldstein RS, Bradley TD. CPAP improves inspiratory muscle strength in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med. 1996;153:277-282.
240 Kaneko Y, Floras JS, Usui K, et al. Cardiovascular effects of continuous positive airway pressure in patients with heart failure and obstructive sleep apnea. N Engl J Med. 2003;348:1233-1241.
241 Naughton MT, Benard DC, Liu PP, Rutherford R, Rankin F, Bradley TD. Effects of nasal CPAP on sympathetic activity in patients with heart failure and central sleep apnea. Am J Respir Crit Care Med. 1995;152:473-479.
242 Baeaussier M, Coriat P, Perel A, et al. Determinants of systolic pressure variation in patients ventilated after vascular surgery. J Cardiothorac Vasc Anesth. 1995;9:547-551.
243 Coriat P, Vrillon M, Perel A, et al. A comparison of systolic blood pressure variations and echocardiographic estimates of end-diastolic left ventricular size in patients after aortic surgery. Anesth Analg. 1994;78:46-53.
244 Szold A, Pizov R, Segal E, Perel A. The effect of tidal volume and intravascular volume state on systolic pressure variation in ventilated dogs. Intensive Care Med. 1989;15:368-371.
245 Michard F, Boussat S, Chemla D, et al. Relation between respiratory changes in arterial pulse pressure and fluid responsiveness in septic patients with acute circulatory failure. Am J Respir Crit Care Med. 2000;162:134-138.
246 Ranieri VM, Giuliani R, Cinnella G, et al. Physiologic effects of positive end-expiratory pressure in patients with chronic obstructive lung disease during acute ventilatory failure and controlled mechanical ventilation. Am Rev Respir Dis. 1993;147:5-13.
247 Baigorri F, De Monte A, Blanch L, et al. Hemodynamic response to external counterbalancing of auto-positive end-expiratory pressure in mechanically ventilated patients with chronic obstructive lung disease. Crit Care Med. 1994;22:1782-1791.
248 Pinsky MR. Though the past darkly: Ventilatory management of patients with chronic obstructive pulmonary disease. Crit Care Med. 1994;22:1714-1717.
249 Ambrosino N, Nava S, Torbicki A, et al. Hemodynamic effects of pressure support and PEEP ventilation by nasal route in patients with stable chronic obstructive pulmonary disease. Thorax. 1993;48:523-528.
250 Ambrosino N, Cobelli F, Torbicki A, et al. Hemodynamic effects of negative-pressure ventilation in patients with COPD. Chest. 1990;97:850-856.
251 Richard C, Teboul J-L, Archambaud F, Hebert J-L, Michaut P, Auzepy P. Left ventricular function during weaning of patients with chronic obstructive pulmonary disease. Intensive Care Med. 1994;20:181-186.
252 Brochard L, Isabey D, Piquet J, et al. Reversal of acute exacerbations of chronic obstructive lung disease by inspiratory assistance with a facemask. N Engl J Med. 1990;323:1523-1530.
