Chapter 3 Heart failure and cardiomyopathy
Introduction
Heart failure is a complex syndrome in which the heart functions inadequately as a pump to support a physiological circulation resulting in symptoms such as breathlessness and fatigue and signs such as fluid retention. It is associated with high morbidity and mortality and represents a major burden for healthcare services.
The cardiomyopathies are diseases of the myocardium associated with cardiac dysfunction and are subdivided on the basis of morphology into hypertrophic, dilated, restrictive and ARVC (Figure 3.1). Many cardiomyopathies are inherited through a single gene abnormality. Some cardiomyopathies have an extended phenotype with conduction disease, whilst other genetic cardiac diseases have conduction disease (such as long/short QT and the Brugada syndrome—the channelopathies) without ‘cardiomyopathy’. At least nine different sarcomeric genes can cause the same phenotype of hypertrophic cardiomyopathy (HCM) and yet different mutations in some of these nine genes can also cause DCM. Approximately half of all HCM patients have been shown to have a genetic mutation and many individuals carry mutations without phenotypic manifestation, and the impact of environment, genetic modifiers and patient age are important. There are also disease mimics known as phenocopies, such as the glycogen storage diseases, which appear phenotypically like the genetic diseases.
CMR protocol
A CMR heart failure and cardiomyopathy protocol typically obtains information on cardiac morphology, cardiac function and myocardial tissue characterization. The scan report should comment on these three areas in general terms such as:
Specific features for reporting are highlighted after discussion of the specific cardiomyopathies.
Cardiac function
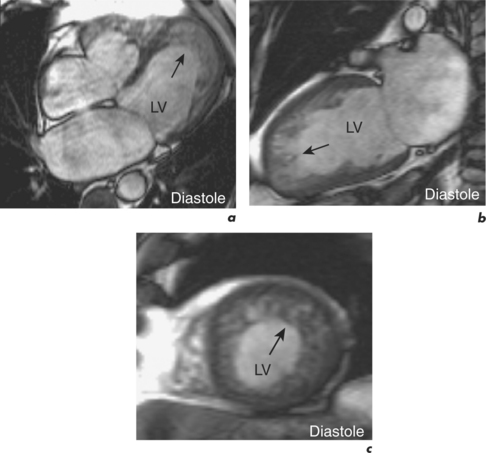
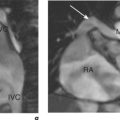
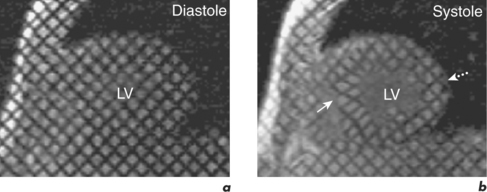
CMR is the gold standard technique for the assessment of left and right ventricular volumes and mass due to a combination of advantages (Table 3.1). CMR measurements are more reproducible than echocardiography, the apex and RV are no longer blind spots (Figure 3.2), and the technique is not window dependent (Figure 3.3). For heart failure, the SSFP GE cine sequences have major advantages because the blood to myocardium contrast is dependent not on blood flow but on intrinsic magnetic differences between blood and myocardium. This means that the blood pool is more clearly distinguished from myocardium even in slow-flow areas compared to that using older white-blood imaging methods. The SSFP sequences can also be sped up using parallel imaging acquisition techniques to minimize breath-holding times in symptomatic heart failure patients.
Table 3.1 Advantages of CMR for measuring ventricular function
| Precise and reproducible piloting |
| No problems with acoustic windows |
| No blind spots—image quality the same throughout |
| High blood–myocardial tissue contrast (even in heart failure) |
| True 3D assessment with no geometric assumptions |
| RV imaged as well as the LV |
| Any view or plane can be imaged as necessary |
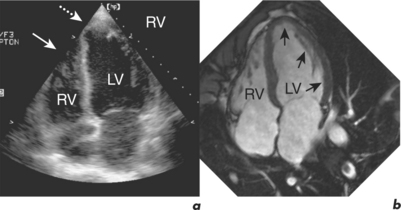
An emerging technique for functional assessment is myocardial tagging. In this technique, a grid of tag lines is laid over the heart magnetically in diastole and then tracked as they distort with myocardial contraction through systole (Figure 3.4). The tag line deformation can be analysed like tissue Doppler imaging, but rather than being limited to strain in line with the Doppler beam, tagging can be analysed in any direction simultaneously, including circumferentially, at any point in the myocardium, and is able to compare epicardial and endocardial function.
Myocardial tissue characterization
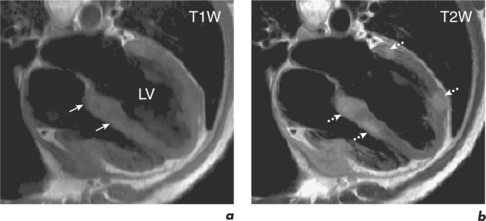
Normal and abnormal myocardium can have intrinsic contrast differences in heart failure that can be visualized by CMR. For example, fatty infiltration of the RV may be seen in ARVC using FSE sequences with and without the addition of a specially applied fat saturation pulse, which eliminates only fat from the images. Also, tissues which have increased water content, such as in myocardial oedema from acute infiltration or infarction, can be differentiated with T2W sequences (Figure 3.5). Extrinsic contrast can be created by LGE, which uses an inversion recovery sequence and a gadolinium contrast agent (see Chapter 2). Gadolinium highlights areas of myocardium with expanded interstitium, such as fibrosis, necrosis or infiltration. Different patterns of fibrosis exist in different aetiologies of heart failure and this can be used to distinguish them (Figure 3.6). MI leads to patterns of LGE which spread from the subendocardium outward to the subepicardium in the territory of a coronary artery. However, changes in myocarditis are the reverse and start at the subepicardium and can later become mid-myocardial, typically at the lateral wall. EGE in combination with relevant cines allows identification of thrombi. This technique has been discussed in the previous chapter and will also be covered in Chapter 5 on cardiac masses. Coronary CMR assessment of the proximal coronary arterial tree can be combined with myocardial tissue characterization but this technique is not robust—it is further discussed in Chapter 9.
CMR in selected cardiomyopathies
Hypertrophic cardiomyopathy
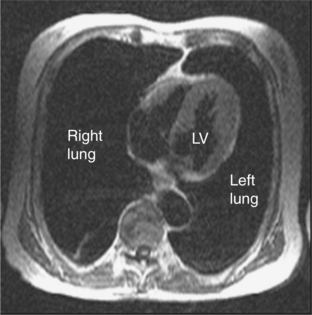
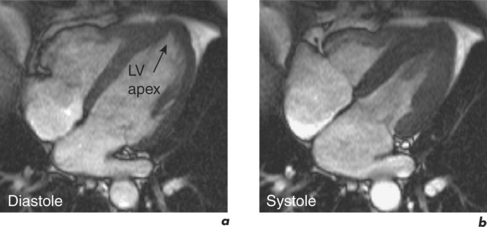
HCM strictly includes the sarcomeric variants but the phenocopies are also discussed here since the differentiation of these two groups is an important role of CMR (Table 3.2). The HCM phenotype shows high variability and also changes throughout life. Expression of early familial disease may be with an abnormal ECG and normal echocardiogram, fulfilling proposed familial criteria for HCM. In a proportion of these, CMR can detect early hypertrophy overlooked at echocardiography. CMR is especially useful in disease which is limited to the LV apex (Figure 3.7). Indicators of apical HCM in the context of non-diagnostic echocardiography include giant negative T waves on the ECG and subtle echocardiographic abnormalities such as diastolic dysfunction and apical akinesis.
| Storage diseases |
| Anderson–Fabry disease Glycogen storage diseases Mucopolysaccharidoses Mitochondrial myopathies Syndromes (Noonan’s, Friedreich’s ataxia) |
| Physiological hypertrophy |
| Increased afterload (hypertension, AS, outflow tract obstruction) Athlete’s heart Afro-Caribbean Compensatory hypertrophy (after MI) |
| Infiltration |
| Amyloid Senile amyloid |
| Ageing |
| Sigmoid septum |
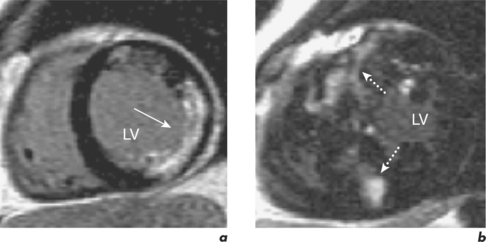
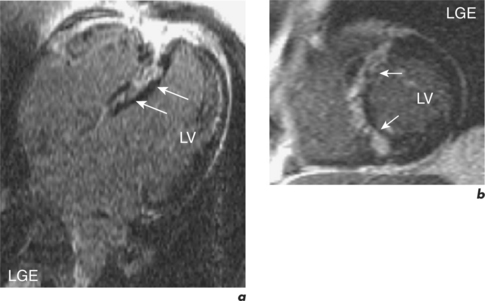
In established HCM, there is a need to identify patients at risk of sudden death and this is mainly done by assessing the presence of known risk factors such as family history of sudden death, unexplained syncope, ventricular tachycardia, abnormal exercise test, and LV wall thickness greater than 30 mm. Adding up the number of risk factors gives a reasonable indication of risk. In the absence of any risk factors, the patient is considered low risk and management is with reassurance and regular reassessment. In the presence of more than two risk factors, patients are high risk and warrant anti-arrhythmia treatment, often with an AICD. However, a third of patients have only one risk factor and are at intermediate risk. LGE CMR may help further risk stratify this group into low or high risk since myocardial fibrosis may represent the substrate for heart failure and arrhythmias (Figure 3.8). The risk of sudden death is greater in those with greater myocardial late enhancement and in the presence of progressive LV dilatation and thinning. This is especially true for the younger patient population (< 40 years old). The specific pattern of LGE may also be important and is also helpful in distinguishing HCM mimics such as Anderson–Fabry disease (AFD). AFD is an X-linked storage disease that causes left ventricular hypertrophy (LVH) and accounts for 1 to 3% of patients with phenotypic HCM. AFD should be considered in middle-aged men with suspected HCM where an auto-somal dominant family history is not present, especially if there is no evidence of LVOT obstruction. Contrast enhancement in AFD occurs predominantly at the basal inferolateral wall. Other mimics include cardiac amyloid where the heart is infiltrated with the amyloid protein. This protein accumulates preferentially throughout the subendocardium, the site of the longitudinal fibres, leading to reduced long-axis function. The myocardium becomes non-compliant and exhibits a restrictive physiology. There is symmetrical and global myocardial hypertrophy, which is readily visualized by CMR, and there may be characteristic subendocardial contrast enhancement and a black-blood pool due to rapid wash-out of gadolinium from the blood (Figure 3.9).
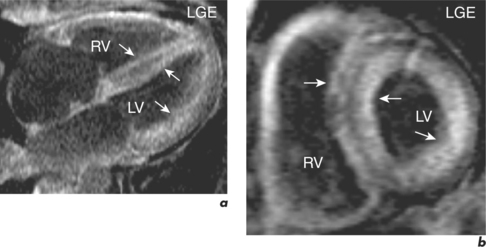
Dilated cardiomyopathy
When cardiac dilatation heart due to coronary disease is advanced, myocardial wall thinning due to infarction, remote remodelling and subendocardial infarction can make the appearance of the heart very similar to that found in DCM. LGE CMR studies in this situation demonstrate transmural or subendocardial MI. By contrast, in most cases of DCM there is either no enhancement or a characteristic mid-myocardial wall fibrosis affecting the circumferential fibres (Figure 3.10). Late enhancement in some suspected cases of DCM may represent sequelae of previous myocarditis.
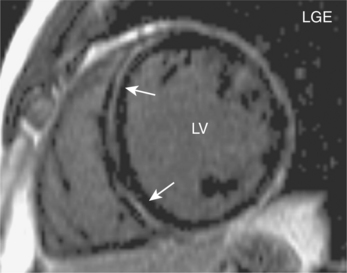
Thalassaemia major
Thalassaemia is a common genetic condition worldwide which results in anaemia secondary to defective globin synthesis. β-Thalassaemia major is the homozygous form of defective β-globin synthesis and affected individuals are blood transfusion dependent. Repeated transfusions contribute to iron overload and iron becomes deposited throughout the body, especially the heart, liver, pancreas and endocrine glands. The resulting cardiomyopathy can progress to overt cardiac failure which can be rapidly progressive. Mortality in thalassaemia major is often from heart failure, with many patients dying prematurely in their mid-thirties. Timely detection of cardiac dysfunction allows targeting of aggressive iron chelation therapy in these individuals and can reverse the cardiomyopathic process.
Actual myocardial iron concentration can be more directly and better quantified using a multiecho T2* GE CMR technique which measures the myocardial relaxation parameter known as ‘T2 star’ (Figure 3.11). Myocardial T2* measurements are made using a mid-ventricular short-axis slice and normal values are approximately 40 ms, with a lower boundary of 20 ms. In thalassaemia, values greater than 20 ms suggest no significant iron loading whilst values less than 10 ms indicate severe iron loading (Table 3.3). Approximately 90% of patients presenting in heart failure have a T2* of less than 10 ms and are at high risk of death. They require urgent increases of iron chelation therapy, often with two drugs. Liver T2* measurements are made in a hepatic transaxial slice avoiding hepatic blood vessels. Values greater than 6.3 ms indicate no significant iron loading and values less than 1.4 ms indicate severe iron loading (Table 3.3). The total study duration is approximately 15 minutes and the technique can also be used in other iron overload conditions such as haemochromatosis and sickle cell anaemia.
| Iron loading | Myocardial T2* (ms) | Hepatic T2* (ms) |
|---|---|---|
| None | >20 | >6.3 |
| Mild | 14–20 | 2.7–6.3 |
| Moderate | 10–14 | 1.4–2.7 |
| Severe | <10 | <1.4 |
Arrhythmogenic right ventricular cardiomyopathy
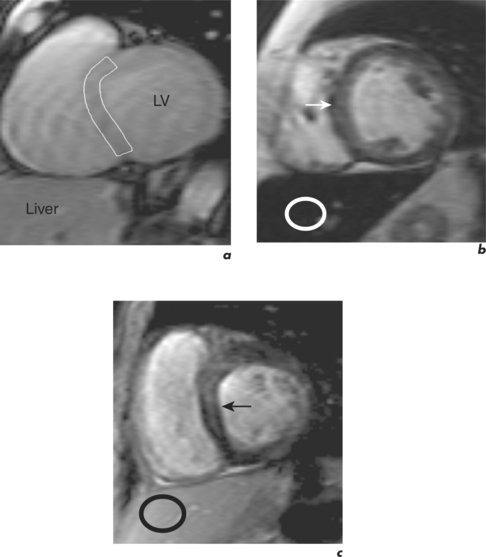
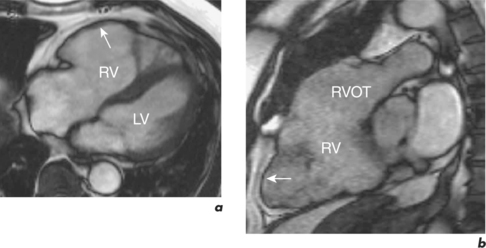
In ARVC, ventricular tachycardia arising from the RV may cause sudden death. The disease is usually autosomal dominant with abnormalities in genes coding for proteins in the cell junctions known as desmosomes. Injury results from impaired tissue integrity with fibro-fatty healing typically affecting the thinner walled RV. Affected individuals can have associated abnormalities of RV and LV structure, and the former are better delineated by CMR than TTE (Figure 3.12). Abnormalities include regional RV WMA, wall thinning, fatty infiltration and LGE. In the late stages, the RV may become poorly functioning. Early changes are difficult to detect and there are traps for the unwary: in the normal RV, the free wall at the moderator insertion may appear akinetic, normal RV wall motion is highly variable, and intra-myocardial fat must be distinguished from epicardial fat. CMR in ARVC requires experience, results must be interpreted in conjunction with the defined major and minor criteria and with other clinical tests to generate an overall probability of disease.
The specific CMR comments here reflect the underlying suspected or known diagnosis:
Other cardiomyopathies
Restrictive cardiomyopathies are well visualized with CMR and key features include reduced long-axis function and atrial dilatation. In addition, subendocardial enhancement may be seen. Examples of diseases that cause a restrictive pattern include cardiac amyloid (Figure 3.9) and eosinophilic heart disease causing endomyocardial fibrosis.
LVNC is a cardiomyopathy characterized by an altered structure of the LV myocardium with very thinned, hypokinetic segments consisting of two layers: thin, compacted myocardium on the subepicardial side, and a thicker non-compacted subendocardial layer (Figure 3.13). RV non-compaction accompanies LVNC in a significant proportion of patients. Clinical manifestations include heart failure, ventricular arrhythmias and systemic embolic events. TTE and CMR are the imaging methods of choice with the criteria for diagnosis being a ratio of non-compacted to compacted myocardial layers of greater than 2.