[level-membership-for-pediatrics-category]
Chapter 629 Hearing Loss
Etiology
SNHL may be congenital or acquired. Acquired SNHL may be caused by genetic, infectious, autoimmune, anatomic, traumatic, ototoxic, and idiopathic factors (Tables 629-1, 629-2, 629-3, and 629-4). The recognized risk factors account for about 50% of cases of moderate to profound SNHL.
Table 629-1 INDICATORS ASSOCIATED WITH HEARING LOSS
INDICATORS ASSOCIATED WITH SENSORINEURAL AND/OR CONDUCTIVE HEARING LOSS
Neonates (Birth to 28 Days) When Universal Screening Is Not Available
Infants and Toddlers (Age 29 Days to 2 Yr) When Certain Health Conditions Develop that Require Rescreening
Infants and Toddlers (Age 29 Days to 3 Yr) Who Require Periodic Monitoring of Hearing
INDICATORS ASSOCIATED WITH DELAYED-ONSET SENSORINEURAL HEARING LOSS
INDICATORS ASSOCIATED WITH CONDUCTIVE HEARING LOSS
Note: At all ages, parents’ concern about hearing loss must be taken seriously even in the absence of risk factors.
ECMO, extracorporeal membrane oxygenation.
Adapted from American Academy of Pediatrics, Joint Committee on Infant Hearing: Joint Committee on Infant Hearing 1994 position statement, Pediatrics 95:152, 1995.
Table 629-2 COMMON TYPES OF HEREDITARY NONSYNDROMIC SENSORINEURAL HEARING LOSS
| LOCUS | GENE | AUDIO PHENOTYPE |
|---|---|---|
| DFN3 | POU3F4 | Conductive hearing loss due to stapes fixation mimicking otosclerosis; superimposed progressive SNHL |
| DFNA1 | DIAPH1 | Low-frequency loss beginning in the 1st decade and progressing to all frequencies to produce a flat audio profile with profound losses throughout the auditory range |
| DFNA2 | KCNQ4 | Symmetrical high-frequency sensorineural loss beginning in the 1st decade and progressing over all frequencies |
| GJB3 | Symmetrical high-frequency sensorineural loss beginning in the 3rd decade | |
| DFNA 6/14/38 | WFS1 | Early-onset low-frequency sensorinerual loss; about 75% of families dominantly segregating this audio profile carry missense mutations in the C-terminal domain of wolframin. |
| DFNA10 | EYA4 | Progressive loss beginning in the 2nd decade as a flat to gently sloping audio profile that becomes steeply sloping with age |
| DFNA13 | COL11A2 | Congenital mid-frequency sensorineural loss that shows age-related progression across the auditory range |
| DFNA15 | POU4F3 | Bilateral progressive sensorineural loss beginning in the 2nd decade |
| DFNA20/26 | ACTG1 | Bilateral progressive sensorineural loss beginning in the 2nd decade; with age, the loss increases with threshold shifts in all frequencies, although a sloping configuration is maintained in most cases |
| DFNB1 | GJB2, GJB6 | Hearing loss varies from mild to profound. The most common genotype, 35delG/35delG, is associated with severe to profound SNHL in about 90% of affected children; severe to profound deafness is observed in only 60% of children who are compound heterozygotes carrying 1 35delG allele and any other GJB2 SNHL-causing allele variant; in children carrying 2 GJB2 SNHL-causing missense mutations, severe to profound deafness is not observed. |
| DFNB4 | SLC26A4 | DFNB4 and Pendred syndrome (see Table 629-3) are allelic. DFNB4 hearing loss is associated with dilatation of the vestibular aqueduct and can be unilateral or bilateral. In the high frequencies, the loss is severe to profound; in the low frequencies, the degree of loss varies widely. Onset can be congenital (prelingual), but progressive postlingual loss also is common. |
| mtDNA 1555A > G |
12S rRNA | Degree of hearing loss varies from mild to profound but usually is symmetrical; high frequencies are preferentially affected; precipitous loss in hearing can occur after aminoglycoside therapy. |
From Smith RJH, Bale JF Jr, White KR: Sensorineural hearing loss in children, Lancet 365:879–890, 2005.
SNHL, sensorineural hearing loss.
Table 629-3 COMMON TYPES OF SYNDROMIC SENSORINEURAL HEARING LOSS
| SYNDROME | GENE | PHENOTYPE |
|---|---|---|
| DOMINANT | ||
| Waardenberg (WS1) | PAX3 | Major diagnostic criteria include dystopia canthorum, congenital hearing loss, heterochromic irises, white forelock, and an affected first-degree relative. About 60% of affected children have congenital hearing loss; in 90%, the loss is bilateral. |
| Waardenberg (WS2) | MITF, others | Major diagnostic criteria are as for WS1 but without dystopia canthorum. About 80% of affected children have congenital hearing loss; in 90%, the loss is bilateral. |
| Branchio-otorenal | EYA1 | Diagnostic criteria include hearing loss (98%), preauricular pits (85%), and branchial (70%), renal (40%), and external-ear (30%) abnormalities. The hearing loss can be conductive, sensorineural, or mixed, and mild to profound in degree. |
| RECESSIVE | ||
| Pendred syndrome | SLC26A4 | Diagnostic criteria include sensorineural hearing loss that is congenital, nonprogressive, and severe to profound in many cases, but can be late-onset and progressive; bilateral dilation of the vestibular aqueduct with or without cochlear hypoplasia; and an abnormal perchlorate discharge test or goiter. |
| Usher syndrome type 1 (USH1) | USH1A, MYO7A, USH1C, CDH23, USH1E, PCDH15, USH1G | Diagnostic criteria include congenital, bilateral, and profound hearing loss, vestibular areflexia, and retinitis pigmentosa (commonly not diagnosed until tunnel vision and nyctalopia become severe enough to be noticeable). |
| Usher syndrome type 2 (USH2) | USH2A, USH2B, USH2C, others | Diagnostic criteria include mild to severe, congenital, bilateral hearing loss and retinitis pigmentosa; hearing loss may be perceived as progressing over time because speech perception decreases as diminishing vision interferes with subconscious lip reading. |
| Usher syndrome type 3 (USH3) | USH3 | Diagnostic criteria include postlingual, progressive sensorineural hearing loss, late-onset retinitis pigmentosa, and variable impairment of vestibular function. |
From Smith RJH, Bale JF Jr, White KR: Sensorineural hearing loss in children, Lancet 365:879–890, 2005.
Table 629-4 INFECTIOUS PATHOGENS IMPLICATED IN SENSORINEURAL HEARING LOSS IN CHILDREN
CONGENITAL INFECTIONS
ACQUIRED INFECTIONS
From Smith RJH, Bale JF Jr, White KR: Sensorineural hearing loss in children, Lancet 365:879–890, 2005.
Infectious Causes
The most common infectious cause of congenital SNHL is cytomegalovirus (CMV), which infects 1/100 newborns in the USA (Chapters 247 and 630). Of these, 6,000-8,000 infants each year have clinical manifestations, including approximately 75% with SNHL. Congenital CMV warrants special attention because it is associated with hearing loss in its symptomatic and asymptomatic forms, and the hearing loss may be progressive. Some children with congenital CMV have suddenly lost residual hearing at 4-5 yr of age. Much less common congenital infectious causes of SNHL include toxoplasmosis and syphilis. Congenital CMV, toxoplasmosis, and syphilis also can manifest with delayed onset of SNHL months to years after birth. Rubella, once the most common viral cause of congenital SNHL, is very uncommon because of effective vaccination programs. In utero infection with herpes simplex virus is rare, and hearing loss is not an isolated manifestation.
Genetic Causes
Genetic causes of SNHL probably are responsible for as many as 50% of SNHL cases (see Tables 629-2 and 629-3). These disorders may be associated with other abnormalities, may be part of a named syndrome, or can exist in isolation. SNHL often occurs with abnormalities of the ear and eye and with disorders of the metabolic, musculoskeletal, integumentary, renal, and nervous systems.
Unlike children with an easily identified syndrome or with anomalies of the outer ear, who may be identified as being at risk for hearing loss and consequently monitored adequately, children with nonsyndromic hearing loss present greater diagnostic difficulty. Mutations of the connexin-26 and -30 genes have been identified in autosomal recessive (DNFB 1) and autosomal dominant (DNFA 3) SNHL and in sporadic patients with nonsyndromic SNHL; up to 50% of nonsyndromic SNHL may be related to a mutation of connexin-26. Mutations of the GJB2 gene co-localize with DFNA 3 and DFNB 1 loci on chromosome 13, are associated with autosomal nonsyndromic susceptibility to deafness, and are associated with as many as 30% of cases of sporadic severe to profound congenital deafness and 50% of cases of autosomal recessive nonsyndromic deafness. Sex-linked disorders associated with SNHL, thought to account for 1-2% of SNHL, include Norrie disease, the otopalatal digital syndrome, Nance deafness, and Alport syndrome. Chromosomal abnormalities such as trisomy 13-15, trisomy 18, and trisomy 21 also can be accompanied by hearing impairment. Patients with Turner syndrome have monosomy for all or part of 1 X chromosome and can have CHL, SNHL, or mixed hearing loss. The hearing loss may be progressive. Mitochondrial genetic abnormalities also can result in SNHL (see Table 629-2).
Physical Causes
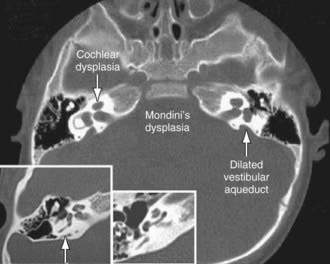
Agenesis or malformation of cochlear structures including the Scheibe, Mondini (Fig. 629-1), Alexander, and Michel anomalies; enlarged vestibular aqueducts (which may be associated with Pendred Syndrome); and semicircular canal anomalies may be genetic. These anomalies probably occur before the 8th wk of gestation and result from arrest in normal development, aberrant development, or both. Many of these anomalies also have been described in association with other congenital conditions such as intrauterine CMV and rubella infections. These abnormalities are quite common; in as many as 20% of children with SNHL, obvious or subtle temporal bone abnormalities are seen on high-resolution CT scanning or MRI.
Effects of Hearing Impairment
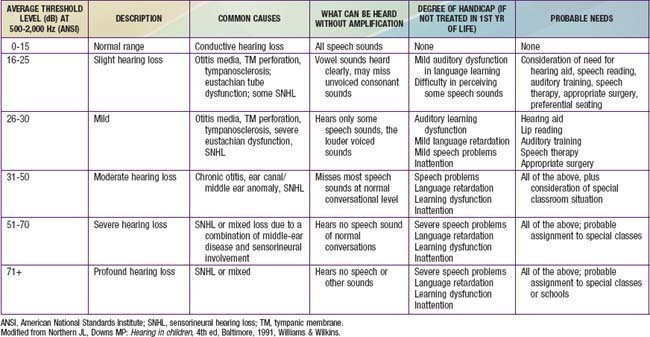
Even mild or unilateral hearing loss can have a detrimental effect on the development of a young child and on school performance. Children with such hearing impairments have greater difficulty when listening conditions are unfavorable (e.g., background noise and poor acoustics), as can occur in a classroom. The fact that schools are auditory-verbal environments is unappreciated by those who minimize the impact of hearing impairment on learning. Hearing loss should be considered in any child with speech and language difficulties or below-par performance, poor behavior, or inattention in school (Table 629-5).
Hearing Screening
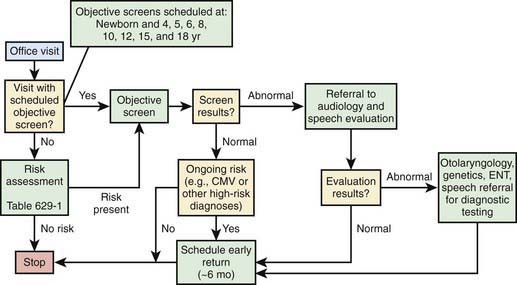
Until mandated screening programs are established universally, many hospitals will continue to use other criteria to screen for hearing loss. Some use the high-risk criteria (see Table 629-1) to decide which infants to screen; some screen all infants who require intensive care; and some do both. The problem with using high-risk criteria to screen is that 50% of cases of hearing impairment will be missed, either because the infants are hearing impaired but do not meet any of the high-risk criteria or because they develop hearing loss after the neonatal period.
Many children become hearing impaired after the neonatal period and therefore are not identified by newborn-screening programs. Often it is not until children are in preschool or kindergarten that further hearing screening takes place. Primary care physicians and pediatricians should be alert to the signs and symptoms of childhood hearing impairment, so that children with hearing impairment who have not been screened formally can be identified as early as possible. Recommendations for postneonatal screening are noted in Figure 629-2.
Identification of Hearing Impairment
The impact of hearing impairment is greatest on an infant who has yet to develop language; therefore, identification, diagnosis, description, and treatment should begin as soon as possible. In general, infants with a prenatal or perinatal history that puts them at risk (see Table 629-2) or those who have failed a formal hearing screening should be monitored closely by an experienced clinical audiologist until a reliable assessment of auditory function has been obtained. Pediatricians should encourage families to cooperate with the follow-up plan. Infants who are born at risk but who were not screened as neonates (often because of transfer from one hospital to another) should have a hearing screening by age 3 mo.
Hearing-impaired infants, who are born at risk or are screened for hearing loss in a neonatal hearing screening program, account for only a portion of hearing-impaired children. Children who are congenitally deaf because of autosomal recessive inheritance or subclinical congenital infection often are not identified until 1-3 yr of age. Usually, those with more severe hearing loss are identified at an earlier age, but identification often occurs later than the age at which intervention can provide an optimal outcome. Children who hear normally develop an extensive language by 3-4 yr of age (Table 629-6) and exhibit behavior reflecting normal auditory function (Table 629-7). Failure to fulfill these criteria should be the reason for an audiologic evaluation. Parents’ concern about hearing and any delayed development of speech and language should alert the pediatrician, because parents’ concern usually precedes formal identification and diagnosis of hearing impairment by 6 mo to 1 yr of age.
| AGE (mo) | REFERRAL GUIDELINES FOR CHILDREN WITH “SPEECH” DELAY |
|---|---|
| 12 | No differentiated babbling or vocal imitation |
| 18 | No use of single words |
| 24 | Single-word vocabulary of ≤10 words |
| 30 | <100 words; no evidence of 2-word combinations; unintelligible |
| 36 | <200 words; no use of telegraphic sentences; clarity <50% |
| 48 | <600 words; no use of simple sentences clarity ≤80% |
From Matkin ND: Early recognition and referral of hearing-impaired children, Pediatr Rev 6:151–156, 1984. Reproduced by permission of Pediatrics.
Table 629-7 GUIDELINES FOR REFERRAL OF CHILDREN WITH SUSPECTED HEARING LOSS
| AGE (mo) | NORMAL DEVELOPMENT |
|---|---|
| 0-4 | Should startle to loud sounds, quiet to mother’s voice, momentarily cease activity when sound is presented at a conversational level |
| 5-6 | Should correctly localize to sound presented in a horizontal plane, begin to imitate sounds in own speech repertoire or at least reciprocally vocalize with an adult |
| 7-12 | Should correctly localize to sound presented in any plane Should respond to name, even when spoken quietly |
| 13-15 | Should point toward an unexpected sound or to familiar objects or persons when asked |
| 16-18 | Should follow simple directions without gestural or other visual cues; can be trained to reach toward an interesting toy at midline when a sound is presented |
| 19-24 | Should point to body parts when asked; by 21-24 mo, can be trained to perform play audiometry |
From Matkin ND: Early recognition and referral of hearing-impaired children, Pediatr Rev 6:151–156, 1984. Reproduced by permission of Pediatrics.
Clinical Audiologic Evaluation
Audiometry
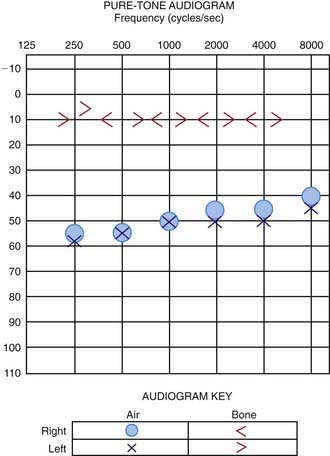
The technique of the audiologic evaluation varies as a function of the age or developmental level of the child, the reason for the evaluation, and the child’s otologic condition or history. An audiogram provides the fundamental description of hearing sensitivity (Fig. 629-3). Hearing thresholds are assessed as a function of frequency using pure tones (sine waves) at octave intervals from 250-8,000 Hz. Earphones typically are used when age-appropriate, and hearing is assessed independently for each ear. Air-conducted signals are presented through earphones (or loudspeakers) and are used to provide information about the sensitivity of the auditory system. These same test sounds can be delivered to the ear through an oscillator that is placed on the head, usually on the mastoid. Such signals are considered bone-conducted because the bones of the skull transmit vibrations as sound energy directly to the inner ear, essentially bypassing the outer and middle ears. In a normal ear, and also in children with SNHL, the air- and bone-conduction thresholds are the same. In those with CHL, the air- and bone-conduction thresholds differ. This is called the air-bone gap, which indicates the amount of hearing loss attributable to dysfunction in the outer and/or middle ear. With mixed hearing loss, both the bone- and air-conduction thresholds are abnormal, and there is an air-bone gap.
Acoustic Immittance Testing
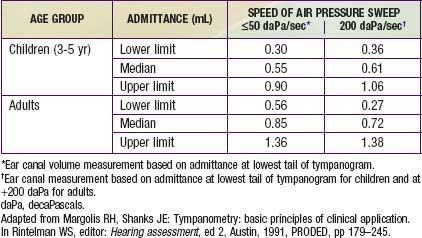
Tympanometry
When air pressure in the ear canal is equal to that in the middle ear, the middle-ear system is functioning optimally. Therefore, the ear canal pressure at which there is the greatest flow of energy (admittance) should be a reasonable estimate of the air pressure in the middle-ear space. This pressure is determined by finding the maximum or peak admittance on the tympanogram and obtaining its value on the x-axis. The value on the y-axis at the tympanogram peak is an estimate of peak admittance based on admittance tympanometry (Table 629-8). This peak measure sometimes is referred to as static acoustic admittance, even though it is estimated from a dynamic measure (Fig. 632-4A).
Tympanometry in Otitis Media with Effusion
Children who have OM with effusion often have reduced peak admittance or high negative tympanometric peak pressures (see Fig. 632-4C). However, in the diagnosis of effusion, the tympanometric measure with the greatest sensitivity and specificity is the shape of the tympanogram rather than its peak pressure or admittance. This shape sometimes is referred to as the tympanometric gradient or width; it measures the degree of roundness or peakedness of the tympanogram. The more rounded the peak (or, in an absent peak, a flat tympanogram), the higher is the probability that an effusion is present (see Fig. 632-4B). It is important to know which instrument is used, because some compute gradient automatically but others do not.
Treatment
Infants and young children with profound congenital or prelingual onset of deafness have benefited from multichannel cochlear implants (see Fig. 629-4). These implants bypass injury to the organ of Corti and provide neural stimulation by way of an external microphone and a signal processor that digitizes auditory stimuli into digital radiofrequency impulses. Cochlear implantation before age 2 yr (and even 1 yr) improves hearing and speech, enabling more than 90% of children to be in mainstream education. Most develop age-appropriate auditory perception and oral language skills.
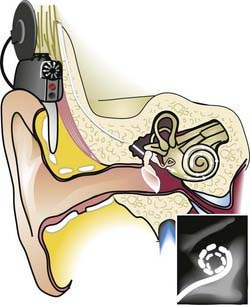
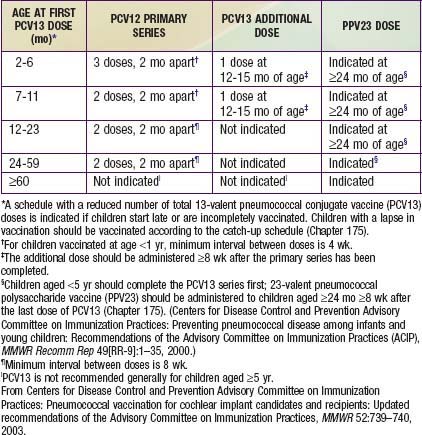
A serious complication of cochlear implants is an excessively high incidence of pneumococcal meningitis. All children receiving a cochlear implant must be vaccinated with the PCV-13 vaccine (Table 629-9).
Ahmed ZM, Masmoudi S, Kalay E, et al. Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nat Genet. 2008;40:1335-1340.
Baranano CF, Sweitzer RS, Mahalak ML, et al. The management of myringotomy tubes in pediatric cochlear implant recipients. Arch Otolaryngol Head Neck Surg. 2010;136(6):557-560.
Biernath KR, Reefhuis J, Whitney CG, et al. Bacterial meningitis among children with cochlear implants beyond 24 months after implantation. Pediatrics. 2006;117:284-289.
Bitner-Glindzicz M, Rahman S. Ototoxicity caused by aminoglycosides. BMJ. 2007;335:784-785.
Brownstein Z, Avraham KB. Deafness genes in Israel: implications for diagnostics in the clinic. Pediatr Res. 2009;66:128-134.
Capaccio P, Ottaviani F, Cuccarini V, et al. Sudden hearing loss and MTHFR 677C>T/1298A>C gene polymorphisms. Genet Med. 2005;7:206-208.
Centers for Disease Control and Prevention. Identifying infants with hearing loss—United States, 1999–2007. MMWR. 2010;59:220-223.
Choi KY, Schimmenti LA, Jurek AM, et al. Detection of cytomegalovirus DNA in dried blood spots of Minnesota infants who do not pass newborn hearing screening. Pediatr Infect Dis J. 2009;28:1095-1098.
Declau F, Boudewyns A, Van den Ende J, et al. Etiologic and audiologic evaluations after universal neonatal hearing screening: analysis of 170 referred neonates. Pediatrics. 2008;121:1119-1126.
Fugazzola L, Cerutti N, Mannavola D, et al. Differential diagnoses between Pendred and pseudo-Pendred syndromes: clinical, radiologic, and molecular studies. Pediatr Res. 2002;51:479-484.
Gates GA, Miyamoto RT. Cochlear implants. N Engl J Med. 2003;349:421-423.
Harlor ADJr, Bower C. Clinical report—hearing assessment in infants and children: recommendations beyond neonatal screening. Pediatrics. 2009;124:1252-1263.
Hinson JT, Fantin VR, Schönberger J, et al. Missense mutations in the BCS1L gene as a cause of Björnstad syndrome. N Engl J Med. 2007;356:809-819.
Huang XY, Tay GS, Wansaicheong GKL, Low WK. Preauricular sinus. Arch Otolaryngol Head Neck Surg. 2007;133:65-68.
Katbamna B, Flamme GA. Acquired hearing loss in adolescents. Pediatr Clin North Am. 2008;55:1391-1402.
Kennedy C, McCann D, Campbell MJ, et al. Universal newborn screening for permanent childhood hearing impairment: An 8-year follow-up of a controlled trial. Lancet. 2005;366:660-662.
Kimani JW, Buchman CA, Booker JK, et al. Sensorineural hearing loss in a pediatric population. Arch Otolaryngol Head Neck Surg. 2010;136(10):999-1004.
Korver AMH, Konings S, Dekker FW, et al. Newborn hearing screening vs later hearing screening and developmental outcomes in children with permanent childhood hearing impairment. JAMA. 2010;304(15):1701-1708.
Krai A, O’Donoghue GM. Profound deafness in childhood. N Engl J Med. 2010;363(15):1438-1450.
Lovett RES, Kitterick PT, Hewitt CE, Summerfield AQ. Bilateral or unilateral cochlear implantation for deaf children: an observational study. Arch Dis Child. 2010;95:107-112.
Mac Ardle B, Bitner-Glindzicz M. Investigation of the child with permanent hearing impairment. Arch Dis Child Educ Pract Ed. 2010;95:14-23.
Marti R, Nascomento A, Colomer J, et al. Hearing loss in a patient with the myopathic form of mitochondrial DNA depletion syndrome and a novel mutation in the TK2 gene. Pediatr Res. 2010;68:151-154.
McClay JE, Booth TN, Parry DA, et al. Evaluation of pediatric sensorineural hearing loss with magnetic resonance imaging? Arch Otolaryngol Head Neck Surg. 2008;134:945-952.
Mehra S, Eavey RD, Keamy DGJr. The epidemiology of hearing impairment in the United States: newborns, children, and adolescents. Otolaryngol Head Neck Surg. 2009;140:461-472.
Mencía A, Modamio-Høybjør S, Redshaw N, et al. Mutation in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet. 2009;41:609-613.
Moeller MP. Early intervention and language development in children who are deaf and hard of hearing. Pediatrics. 2000;106:E43.
Morton CC, Nance WE. Newborn hearing screening—a silent revolution. N Engl J Med. 2006;354:2151-2164.
Nikolopoulos TP, Archbold SM, O’Donoghue GM. Does cause of deafness influence outcome after cochlear implantation in children? Pediatrics. 2006;118:1350-1356.
Nikolopoulos TP, Dyar D, Archbold S, et al. Development of spoken language grammar following cochlear implantation in perlingually deaf children. Arch Otolaryngol Head Neck Surg. 2004;130:629-633.
Niparko JK, Tobey EA, Thal DJ, et al. Spoken language development in children following cochlear implantation. JAMA. 2010;303:1498-1506.
Olusanya BO, Somefun AO, Swanepoel de W. The need for standardization of methods for worldwide infant hearing screening: a systematic review. Laryngoscope. 2008;118:1830-1836.
Pirozzo S, Papinczak T, Glasziou P. Whispered voice test for screening for hearing impairment in adults and children: systematic review. BMJ. 2003;327:967-970.
Quintanilla-Dieck MD, Artunduaga M, Eavey RD. Intentional exposure to loud music: the second MTV.com survey reveals an opportunity to educate. J Pediatr. 2009;155:550-555.
Rabinowitz PM. Hearing loss and personal music players. BMJ. 2010;341:57-58.
Ramsden RT. Prognosis after cochlear implantation. BMJ. 2004;328:419-420.
Reefhuis J, Honein MA, Whitney CG, et al. Risk of bacterial meningitis in children with cochlear implants. N Engl J Med. 2003;349:435-445.
Roth D, Hildesheimer M, Bardenstein S, et al. Preauricular skin tags and ear pits are associated with permanent hearing impairment in newborns. Pediatrics. 2008;122:e884-e890.
Rubin LG, Papsin B, Committee on Infectious Diseases and Section on Otolaryngology—Head and Neck Surgery. Policy statement—cochlear implants in children: surgical site infections and prevention and treatment of acute otitis media and meningitis. Pediatrics. 2010;126:381-391.
Schreiber BE, Agrup C, Haskard DO, et al. Sudden sensorineural hearing loss. Lancet. 2010;375:1203-1210.
Schultz JM, Yang Y, Caride AJ, et al. Modification of human hearing loss by plasma-membrane calcium pump PMCA2. N Engl J Med. 2005;352:1557-1564.
Shargorodsky J, Curhan SG, Curhan GC, et al. Change in prevalence of hearing loss in US adolescents. JAMA. 2010;304:772-778.
Sharma A, Ruscetta MN, Chi DH. Ophthalmologic findings in children with sensorineural hearing loss. Arch Otolaryngol Head Neck Surg. 2009;135:119-123.
Smith RJH. Deafness: from bedside to bench and back. Lancet. 2002;360:656-657.
Smith RJH, Bale JFJr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879-890.
Thomas MA, Der Kaloustian VM, Tewfik TL. Connexin mutation testing of children with nonsyndromic, autosomal recessive sensorineural hearing loss. J Otolaryngol. 2004;33:189-192.
Thompson DC, McPhillips H, Davis RL, et al. Universal newborn hearing screening: summary of evidence. JAMA. 2001;286:2000-2010.
US Preventive Services Task Force. Universal screening for hearing loss in newborns: US Preventive Services Task Force recommendation statement. Pediatrics. 2008;122:143-148.
Venail F, Sicard M, Piron JP, et al. Reliability and complications of 500 consecutive cochlear implantations. Arch Otolaryngol Head Neck Surg. 2008;134:1276-1281.
Venail F, Vieu A, Artieres F, et al. Educational and employment achievements in perlingually deaf children who receive cochlear implants. Arch Otolaryngol Head Neck Surg. 2010;136:366-372.
Vogel I, Brug J, van der Ploeg CPB, et al. Strategies for the prevention of MP3-induced hearing loss among adolescents: expert opinions from a Delphi study. Pediatrics. 2009;123:1257-1262.
Waltzman SB, Roland TJr. Cochlear implantation in children younger than 12 months. Pediatrics. 2005;16:487-493.
Watkin PM, Baldwin M. Identifying deafness in early childhood: requirements after the newborn hearing screen. Arch Dis Child. 2011;96:62-66.
Willems PJ. Genetic causes of hearing loss. N Engl J Med. 2000;342:1101-1109.
Wei BPC, Clark GM, O’Leary SJ, et al. Meningitis after cochlear implantation. BMJ. 2007;335:1058.
Wei B, O’Leary S, Dowell R. Cochlear implantation: one or two? Lancet. 2007;370:719-720.
Wolf B, Spencer R, Gleason T. Hearing loss is a common feature of symptomatic children with profound biotinidase deficiency. Pediatrics. 2002;140:242-246.
Yoshinaga-Itano C, Sedey AL, Coulter DK, et al. Language of early- and later-identified children with hearing loss. Pediatrics. 1998;102:1161-1171.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 629 Hearing Loss
Etiology
SNHL may be congenital or acquired. Acquired SNHL may be caused by genetic, infectious, autoimmune, anatomic, traumatic, ototoxic, and idiopathic factors (Tables 629-1, 629-2, 629-3, and 629-4). The recognized risk factors account for about 50% of cases of moderate to profound SNHL.
Table 629-1 INDICATORS ASSOCIATED WITH HEARING LOSS
INDICATORS ASSOCIATED WITH SENSORINEURAL AND/OR CONDUCTIVE HEARING LOSS
Neonates (Birth to 28 Days) When Universal Screening Is Not Available
Infants and Toddlers (Age 29 Days to 2 Yr) When Certain Health Conditions Develop that Require Rescreening
Infants and Toddlers (Age 29 Days to 3 Yr) Who Require Periodic Monitoring of Hearing
INDICATORS ASSOCIATED WITH DELAYED-ONSET SENSORINEURAL HEARING LOSS
INDICATORS ASSOCIATED WITH CONDUCTIVE HEARING LOSS
Note: At all ages, parents’ concern about hearing loss must be taken seriously even in the absence of risk factors.
ECMO, extracorporeal membrane oxygenation.
Adapted from American Academy of Pediatrics, Joint Committee on Infant Hearing: Joint Committee on Infant Hearing 1994 position statement, Pediatrics 95:152, 1995.
Table 629-2 COMMON TYPES OF HEREDITARY NONSYNDROMIC SENSORINEURAL HEARING LOSS
| LOCUS | GENE | AUDIO PHENOTYPE |
|---|---|---|
| DFN3 | POU3F4 | Conductive hearing loss due to stapes fixation mimicking otosclerosis; superimposed progressive SNHL |
| DFNA1 | DIAPH1 | Low-frequency loss beginning in the 1st decade and progressing to all frequencies to produce a flat audio profile with profound losses throughout the auditory range |
| DFNA2 | KCNQ4 | Symmetrical high-frequency sensorineural loss beginning in the 1st decade and progressing over all frequencies |
| GJB3 | Symmetrical high-frequency sensorineural loss beginning in the 3rd decade | |
| DFNA 6/14/38 | WFS1 | Early-onset low-frequency sensorinerual loss; about 75% of families dominantly segregating this audio profile carry missense mutations in the C-terminal domain of wolframin. |
| DFNA10 | EYA4 | Progressive loss beginning in the 2nd decade as a flat to gently sloping audio profile that becomes steeply sloping with age |
| DFNA13 | COL11A2 | Congenital mid-frequency sensorineural loss that shows age-related progression across the auditory range |
| DFNA15 | POU4F3 | Bilateral progressive sensorineural loss beginning in the 2nd decade |
| DFNA20/26 | ACTG1 | Bilateral progressive sensorineural loss beginning in the 2nd decade; with age, the loss increases with threshold shifts in all frequencies, although a sloping configuration is maintained in most cases |
| DFNB1 | GJB2, GJB6 | Hearing loss varies from mild to profound. The most common genotype, 35delG/35delG, is associated with severe to profound SNHL in about 90% of affected children; severe to profound deafness is observed in only 60% of children who are compound heterozygotes carrying 1 35delG allele and any other GJB2 SNHL-causing allele variant; in children carrying 2 GJB2 SNHL-causing missense mutations, severe to profound deafness is not observed. |
| DFNB4 | SLC26A4 | DFNB4 and Pendred syndrome (see Table 629-3) are allelic. DFNB4 hearing loss is associated with dilatation of the vestibular aqueduct and can be unilateral or bilateral. In the high frequencies, the loss is severe to profound; in the low frequencies, the degree of loss varies widely. Onset can be congenital (prelingual), but progressive postlingual loss also is common. |
| mtDNA 1555A > G |
12S rRNA | Degree of hearing loss varies from mild to profound but usually is symmetrical; high frequencies are preferentially affected; precipitous loss in hearing can occur after aminoglycoside therapy. |
From Smith RJH, Bale JF Jr, White KR: Sensorineural hearing loss in children, Lancet 365:879–890, 2005.
SNHL, sensorineural hearing loss.
Table 629-3 COMMON TYPES OF SYNDROMIC SENSORINEURAL HEARING LOSS
| SYNDROME | GENE | PHENOTYPE |
|---|---|---|
| DOMINANT | ||
| Waardenberg (WS1) | PAX3 | Major diagnostic criteria include dystopia canthorum, congenital hearing loss, heterochromic irises, white forelock, and an affected first-degree relative. About 60% of affected children have congenital hearing loss; in 90%, the loss is bilateral. |
| Waardenberg (WS2) | MITF, others | Major diagnostic criteria are as for WS1 but without dystopia canthorum. About 80% of affected children have congenital hearing loss; in 90%, the loss is bilateral. |
| Branchio-otorenal | EYA1 | Diagnostic criteria include hearing loss (98%), preauricular pits (85%), and branchial (70%), renal (40%), and external-ear (30%) abnormalities. The hearing loss can be conductive, sensorineural, or mixed, and mild to profound in degree. |
| RECESSIVE | ||
| Pendred syndrome | SLC26A4 | Diagnostic criteria include sensorineural hearing loss that is congenital, nonprogressive, and severe to profound in many cases, but can be late-onset and progressive; bilateral dilation of the vestibular aqueduct with or without cochlear hypoplasia; and an abnormal perchlorate discharge test or goiter. |
| Usher syndrome type 1 (USH1) | USH1A, MYO7A, USH1C, CDH23, USH1E, PCDH15, USH1G | Diagnostic criteria include congenital, bilateral, and profound hearing loss, vestibular areflexia, and retinitis pigmentosa (commonly not diagnosed until tunnel vision and nyctalopia become severe enough to be noticeable). |
| Usher syndrome type 2 (USH2) | USH2A, USH2B, USH2C, others | Diagnostic criteria include mild to severe, congenital, bilateral hearing loss and retinitis pigmentosa; hearing loss may be perceived as progressing over time because speech perception decreases as diminishing vision interferes with subconscious lip reading. |
| Usher syndrome type 3 (USH3) | USH3 | Diagnostic criteria include postlingual, progressive sensorineural hearing loss, late-onset retinitis pigmentosa, and variable impairment of vestibular function. |
From Smith RJH, Bale JF Jr, White KR: Sensorineural hearing loss in children, Lancet 365:879–890, 2005.
Table 629-4 INFECTIOUS PATHOGENS IMPLICATED IN SENSORINEURAL HEARING LOSS IN CHILDREN
CONGENITAL INFECTIONS
ACQUIRED INFECTIONS
From Smith RJH, Bale JF Jr, White KR: Sensorineural hearing loss in children, Lancet 365:879–890, 2005.
Infectious Causes
The most common infectious cause of congenital SNHL is cytomegalovirus (CMV), which infects 1/100 newborns in the USA (Chapters 247 and 630). Of these, 6,000-8,000 infants each year have clinical manifestations, including approximately 75% with SNHL. Congenital CMV warrants special attention because it is associated with hearing loss in its symptomatic and asymptomatic forms, and the hearing loss may be progressive. Some children with congenital CMV have suddenly lost residual hearing at 4-5 yr of age. Much less common congenital infectious causes of SNHL include toxoplasmosis and syphilis. Congenital CMV, toxoplasmosis, and syphilis also can manifest with delayed onset of SNHL months to years after birth. Rubella, once the most common viral cause of congenital SNHL, is very uncommon because of effective vaccination programs. In utero infection with herpes simplex virus is rare, and hearing loss is not an isolated manifestation.
Genetic Causes
Genetic causes of SNHL probably are responsible for as many as 50% of SNHL cases (see Tables 629-2 and 629-3). These disorders may be associated with other abnormalities, may be part of a named syndrome, or can exist in isolation. SNHL often occurs with abnormalities of the ear and eye and with disorders of the metabolic, musculoskeletal, integumentary, renal, and nervous systems.
Unlike children with an easily identified syndrome or with anomalies of the outer ear, who may be identified as being at risk for hearing loss and consequently monitored adequately, children with nonsyndromic hearing loss present greater diagnostic difficulty. Mutations of the connexin-26 and -30 genes have been identified in autosomal recessive (DNFB 1) and autosomal dominant (DNFA 3) SNHL and in sporadic patients with nonsyndromic SNHL; up to 50% of nonsyndromic SNHL may be related to a mutation of connexin-26. Mutations of the GJB2 gene co-localize with DFNA 3 and DFNB 1 loci on chromosome 13, are associated with autosomal nonsyndromic susceptibility to deafness, and are associated with as many as 30% of cases of sporadic severe to profound congenital deafness and 50% of cases of autosomal recessive nonsyndromic deafness. Sex-linked disorders associated with SNHL, thought to account for 1-2% of SNHL, include Norrie disease, the otopalatal digital syndrome, Nance deafness, and Alport syndrome. Chromosomal abnormalities such as trisomy 13-15, trisomy 18, and trisomy 21 also can be accompanied by hearing impairment. Patients with Turner syndrome have monosomy for all or part of 1 X chromosome and can have CHL, SNHL, or mixed hearing loss. The hearing loss may be progressive. Mitochondrial genetic abnormalities also can result in SNHL (see Table 629-2).
