CHAPTER 55 HEADACHE PATHOGENESIS
Headache is a broad term that encompasses most of the pain syndromes that involve the head and upper neck. Headache is much more than migraine alone, and facial pain is often included in the discussion of headache. The common feature of all these pain syndromes is that they involve the regions of the body innervated by the trigeminal nerve and the upper cervical nerves, as opposed to nonheadache pain syndromes, which involve the spinal nerves. This chapter discusses the pathogenesis of headache. We discuss the tissues in the periphery and the regions of the brain that are involved in generating and maintaining headache pain, with an emphasis on the pathophysiology of migraine. This emphasis on migraine is due to the advances that have been made in animal models of headache and functional imaging studies of patients with migraine.
Migraine is a primary headache disorder arising from a brain dysfunction that leads to activation of the trigeminal vascular system. This review discusses developments in the understanding of the cellular and molecular aspects of migraine. Migraine is more than a pain disorder—there are multiple stages of a migraine attack. The prodrome may begin as early as 3 days before an attack; in some patients (≈14%) this leads to a visual or sensory aura. The prodrome phase is followed by the pain phase, which, untreated, can last from 4 to 72 hours. This is followed by the resolution phase, which is characterized by alleviation of the pain and a return to normal activity. Even during this interictal phase, auditory habituation1 and threshold measurements of magnetic stimulation of the cortex2 are different in migraineurs compared with healthy subjects; the blink reflex, however, is not different.3
ANATOMY OF HEADACHE
Most animal research into migraine has focused on activation of dural nociceptors using electrical current or proinflammatory chemicals. Dural blood vessels and the dura itself are innervated by sensory neurons whose cell bodies are in the trigeminal ganglion. A single neurite emerges from the cell bodies of the sensory neurons in the trigeminal ganglion. This neurite bifurcates and projects out to the periphery and into the dorsal horn of the midbrain. Because sensory neurons in the trigeminal ganglion have only one neurite, both projections are classified as axons. Normally, we associate the direction of information transmission along this neuron from the periphery to the central nervous system, but glutamate and neuropeptides can also be released in the periphery. This can contribute to sensory trigeminal activation, blood vessel vasodilation, and plasma protein extravasation.4 Glutamate and neuropeptides, such as calcitonin gene–related peptide, can also be released in the trigeminal ganglion itself.5–7 These sites of release are important for the strong sensory activation required for the initiation of the migraine attack and as potential targets of migraine headache treatments. Significant in headache, the large blood vessels of the meninges are mainly innervated by Aδ and C type sensory fibers, which contain glutamate as well as neuropeptides.
The fifth cranial nerve arises from the trigeminal ganglion and transmits all the somatic sensory information from the head, face, and dura to the trigeminal nucleus caudalis in the central nervous system. The trigeminal nerve has three divisions: ophthalmic, mandibular, and maxillary. Anterior structures of the head and face are innervated by the ophthalmic (first) division. Posterior regions of the head and neck are innervated by the upper cervical nerves. The trigeminal nerve enters the brainstem at the pontine level of the hindbrain and terminates in the trigeminal nucleus of the brainstem. Nociceptive input from the cervical nerves also activates the neurons in the trigeminal nucleus caudalis, which extends to the C2 spinal segment.8
The trigeminal nucleus is composed of the principal trigeminal nuclei and spinal trigeminal nuclei (subdivided into three regions: the nucleus oralis, the interpolaris, and the nucleus caudalis).9 The dorsal horn of the brainstem spinal trigeminal nucleus has a laminar structure homologous to the dorsal horn of the spinal cord. The primary afferents terminate in laminae I and II and, to a lesser extent, the deeper laminae III to V. The neurons of the deeper lamina project to the thalamus in the trigeminothalamic tract, which is homologous to the spinothalamic tract. Second-order neurons from the trigeminal spinal nuclei form the trigeminothalamic tract and project to other midbrain structures, such as the periaquaductal gray, as well as to the thalamus and hypothalamus (Fig. 55-1).
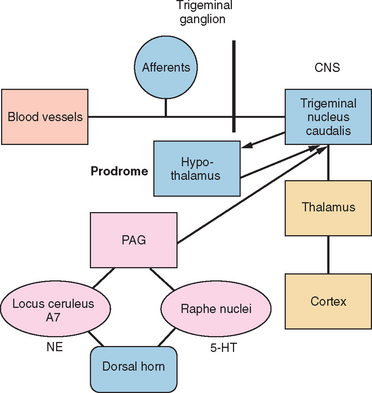
HEADACHE PAIN
Migraine pain is caused by trigeminal neurovascular activation.10 Neural events result in activation of nociceptive afferents and dilation of blood vessels on the dura. In patients with migraine accompanied by aura, cortical spreading depression is the neural event that activated the nociceptors on the dura. Cortical spreading depression is really a spreading wave of cortical spreading excitation followed by long-lasting depression. In migraine patients without aura, the mechanism for the activation of the dural afferents is unknown.
Studies in humans and research on headache models in rats have elucidated three main components involved in headache pain: (1) the cranial blood vessels, (2) the trigeminal innervation of the vessels, and (3) the reflex connections of the trigeminal system with the cranial parasympathetic system.11 The key pathway for headache pain is trigeminovascular input from the meningeal vessels and the meninges.12 Brain imaging studies suggest that important modulation of the trigeminovascular nociceptive input stems from the dorsal raphe nucleus, the locus ceruleus, and the nucleus raphe magnus.13,14
PERIPHERAL MECHANISMS AND NOCICEPTOR ACTIVATION
Before the contribution of neurogenic inflammation during the pain phase of migraine was demonstrated, the predominant theory of migraine was Harold Wolff’s vascular theory. Wolff postulated that migraine was a vascular disorder caused by a tightening (constriction) and sudden opening (dilation) of the blood vessels in the head. Research has shown that changes in blood flow are due to activation of afferents and the brain. This led to the neurogenic theory of migraine. During neurogenic inflammation, trigeminal afferent activation is accompanied by the release of vasoactive neuropeptides, including calcitonin gene–related peptide, substance P, and neurokinin A from the nerve terminals.10,15,16 These mediators produce vasodilation, sensitization of the nerve terminals, and extravasation of fluid into the perivascular space around the dural blood vessels (plasma protein extravasation) (Fig. 55-2). The nerve terminals become further excited due to a positive feedback loop. The afferents excite the blood vessels and other trigeminal nerve endings through glutamate and peptide release, and then the blood vessels further excite the sensitized afferent terminals. These afferents project in the central nervous system and terminate in the upper lamina of the dorsal horn. Some project to the deeper lamina.17,18 This strong sensory stimulation causes the induction of c-fos (an immediate-early gene product) in the secondary sensory neurons of the trigeminal nucleus caudalis in the brainstem.19 Substance P and calcitonin gene–related peptide further amplify the trigeminal terminal sensitivity by stimulating the release of bradykinin and other inflammatory mediators from mast cells. Prostaglandins and nitric oxide (a diffusible gas that acts as a neurotransmitter) are both endogenous neuromodulators that can sensitize nociceptors and the secondary sensory neurons in the trigeminal nucleus caudalis. There is evidence that inflammatory mediators, such as prostaglandin release in the central nervous system by microglia, may be more important than its release on the dura.20 Microglia are important modulators on sustained pain in chronic pain conditions.21
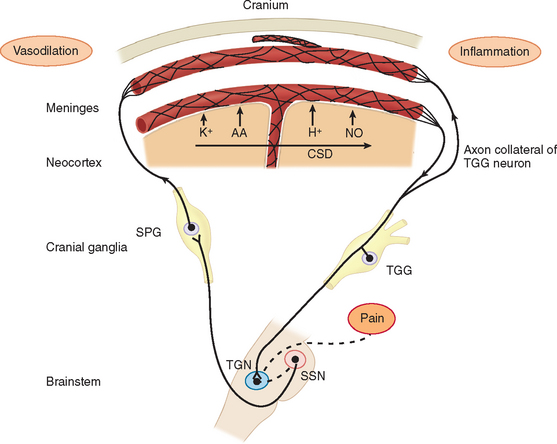
Cortical spreading depression (the mechanism of the visual aura) can also activate nociceptors of the trigeminal system through local changes in extracellular ion concentrations.22 Although the brain itself is largely insensate, pain can be generated by large cranial vessels and the dura mater, which receive nociceptor innervation. The involvement of the ophthalmic division of the trigeminal nerve and its overlap of structures innervated by branches of the C2 nerve roots explain the typical distribution of migraine pain over the frontal and temporal regions and the referral of pain to the parietal, occipital, and high cervical regions.
SENSITIZATION IN MIGRAINE
Sensory sensitization is manifested in patients in two ways: hyperalgesia or allodynia. Hyperalgesia is present when lightly painful stimuli, such as a soft pinch, are perceived as very painful by the patient. Allodynia is the phenomenon wherein pain is perceived following a nonpainful stimulus. Clinicians have observed that during migraine attacks patients complain of increased pain with stimuli that would ordinarily be nonnociceptive.23 These stimuli include hair brushing, wearing a hat, showering, and resting the head on a pillow. Burstein and colleagues24 explored the development of allodynia in migraineurs. They measured pain thresholds for hot, cold, and pressure stimuli, both within the region of spontaneous pain and outside it. They found that as an attack progressed, cutaneous allodynia developed in the region of pain and then outside it at extracephalic locations in many migraine sufferers. Sensitization in migraine is important because patients with allodynia often fail to respond to triptans.25 Allodynia can be used as an outcome measure to evaluate the efficacy of migraine treatments.
Sensitization of nociceptors, secondary sensory neurons in the trigeminal nucleus caudalis, or even the projection neurons in the thalamus are responsible for initiation and maintenance of the clinical symptoms of allodynia. The afferent or central neurons processing the sensory information may have an increased spontaneous discharge rate or increased responsiveness to both painful and nonpainful stimuli. The receptive fields of these neurons can expand, resulting in pain being felt over a greater part of the dermatome. This results in hyperalgesia (increased sensitivity to pain) and cutaneous allodynia (pain perceived in response to nonpainful stimuli). Peripheral sensitization is measured in minutes, up to 1 hour, whereas central sensitization is measured in hours to days. Peripheral sensitization produces an increase in pain sensitivity that is restricted to the site of inflammation—in the case of migraine, this is the dura. This results in the throbbing quality of migraine pain and its activation by movement.26 Sensitization of these neurons reduces their threshold to a level where blood vessel and cerebrospinal fluid pulsations are painful.
Central sensitization is an activity-dependent increase in the excitability of neurons responsive to nociceptor inputs in the dorsal horn of the spinal cord due to nociceptor input. The increase in activity outlasts the initial afferent stimulation. Central sensitization is initiated by nociceptor afferent activation and is characterized by a reduction in activation threshold, increases in the magnitude of responsiveness, and an increase in receptive field. The prevailing theory is that most inputs received by dorsal horn neurons are subthreshold.27 The synaptic strength is too weak to evoke an action potential output. After a strong activation of nociceptive afferents in the periphery and a cascade of events associated with this activation, central sensitization is induced in the neuron of the deep lamina of the dorsal horn (laminae III to V), so that this input is now about threshold. These subthreshold inputs may be from Aδ or C fibers. The change in synaptic strength of the C fibers leads to the clinical symptom of hyperalgesia and an increase in the efficacy of Aδ fibers leads to allodynia. This has important implications for migraine treatment. Acute compounds must prevent central sensitization if they are to work early in an attack, and they have to reverse central sensitization if they are to be useful after allodynia has developed.
PAIN MODULATION
The nervous system contains networks that modulate nociceptive transmission.28 The trigeminal brainstem nuclear complex receives monoaminergic, enkephalinergic, and peptidergic projections from regions that are important in the modulation of nociceptive systems. A descending inhibitory neuronal network extends from the frontal cortex and hypothalamus through the periaquaductal gray to the rostral ventromedial medulla and the medullary and spinal dorsal horn. The rostral ventromedial medulla includes the raphe nuclei and the adjacent reticular formation and projects to the outer laminae of the spinal and medullary dorsal horn. Electrical stimulation or injection of opioids into the periaquaductal gray or rostral ventromedial medulla inhibits neuron activity in the dorsal horn. The periaquaductal gray receives projections from the insular cortex and the amygdala.
These nuclei are believed to modulate the activity of the trigeminal nucleus caudalis and the dorsal horn neurons. Three classes of neurons have been identified in the rostral ventromedial medulla and periaquaductal gray. “OFF” cells pause immediately before the nociceptive reflex, whereas “ON” cells are activated.29 Neutral cells show no consistent changes in activation. Increased ON cell activity in the brainstem’s pain modulation system enhances the response to both painful and nonpainful stimuli. Headache may be caused, in part, by enhanced neuronal activity in the nucleus caudalis as a result of enhanced ON cell or decreased OFF cell activity. Other conditioned stimuli associated with pain and stress also can turn on the pain system and may account, in part, for the association between pain and stress.
Positron emission tomography and functional MRI in primary headaches, such as migraine and cluster headache, have demonstrated activations in the brain areas associated with pain, such as the cingulate cortex, the insulae, the frontal cortex, the thalamus, the basal ganglia, and the cerebellum. These areas are similarly activated when head pain is induced by injecting capsaicin into volunteers’ foreheads. In addition to the generic pain areas activated by the capsaicin injections, specific brainstem areas, such as the dorsal pons, are activated in episodic migraine.30
GENETICS AND MIGRAINE
There is mounting evidence that migraine is a genetic disorder. For other headache disorders, such as familial hemiplegic migraine (FHM), there are very good data demonstrating inherited genes responsible for the disorder.31 Genome-wide screens have shown that there are several susceptibility loci associated with migraine, for example, on chromosomes 4q24, 6p12.2-p21.1, 11q24, and 14q21.2-q22.3 and loci on 1q31 and Xq24-28.32 None of these loci has been demonstrated to cause migraine as with the loci associated with familial hemiplegic migraine. Familial hemiplegic migraine is a rare autosomal dominantly inherited subtype of migraine with aura. Mutations in at least two genes result in the disorder (FMH1 [CACNA1A at 19p13] and FMH2 [ATP1A2 at 1q23]). The success in discovering several genes responsible for familial hemiplegic migraine has led to increased work on the genetic components of migraine. These studies should lead to new discoveries in this area.
SUMMARY
Although clinical and basic science research has revealed many pieces of the puzzle of migraine pathophysiology, the full picture is still elusive. For example, we know that environmental and behavioral triggers, such as lack of sleep, too much sleep, fasting, and thirst, can initiate a migraine attack; however, the mechanism by which these triggers initiate the migraine is not clear.33 Many of these triggers are related to hypothalamic function. Most of the basic science research of migraine has focused on the pain phase of the disorder and has ignored the prodrome and resolution phases. In addition, although we have isolated genes that are associated with classic forms of inherited headache syndromes, such as familial hemiplegic migraine, we do not know how mutations in these genes lead to episodic headache disorders. Understanding these aspects of migraine and recognizing that it is more than just a pain disorder will be critical to fully elucidating its pathophysiology.
Bolay H, Reuter U, Dunn AK, et al. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136-142.
Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89:107-110.
Knight Y. Brainstem modulation of caudal trigeminal nucleus: a model for understanding migraine biology and future drug targets. Headache Curr. 2005;2:108-118.
Piovesan EJ, Kowacs PA, Oshinsky ML. Convergence of cervical and trigeminal sensory afferents. Curr Pain Headache Rep. 2003;7:377-383.
Watkins LR, Milligan ED, Maier SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol. 2003;521:1-21.
1 Ambrosini A, De PV, Afra J, et al. Reduced gating of middle-latency auditory evoked potentials (P50) in migraine patients: another indication of abnormal sensory processing? Neurosci Lett. 2001;306:132-134.
2 Young WB, Oshinsky ML, Shechter AL, et al. Consecutive transcranial magnetic stimulation: phosphene thresholds in migraineurs and controls. Headache J Head Face Pain. 2004;44:131-135.
3 Kröner-Herwig B, Ruhmland M, Zintel W, et al. Are migraineurs hypersensitive? a test of the stimulus processing disorder hypothesis. Eur J Pain. 2005;9:661-671.
4 Omote K, Kawamata T, Kawamata M, et al. Activation of peripheral NMDA-nitric oxide cascade in formalin test. Anesthesiology. 2000;93:173-178.
5 Neubert JK, Maidment NT, Matsuka Y, et al. Inflammation-induced changes in primary afferent-evoked release of substance P within trigeminal ganglia in vivo. Brain Res. 2000;871:181-191.
6 Holland GR. Sympathetic-sensory axon to axon contacts in the dental pulp. Proc Finn Dent Soc. 1989;85:375-378.
7 Amir R, Devor M. Chemically mediated cross-excitation in rat dorsal root ganglia. J Neurosci. 1996;16:4733-4741.
8 Piovesan EJ, Kowacs PA, Oshinsky ML. Convergence of cervical and trigeminal sensory afferents. Curr Pain Headache Rep. 2003;7:377-383.
9 Strassman AM, Vos BP, Mineta Y, et al. Fos-like immunoreactivity in the superficial medullary dorsal horn induced by noxious and innocuous thermal stimulation of facial skin in the rat. J Neurophysiol. 1993;70:1811-1821.
10 Reuter U, Bolay H, Jansen-Olesen I, et al. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain. 2001;124:2490-2502.
11 Bolay H, Reuter U, Dunn AK, et al. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136-142.
12 Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89:107-110.
13 Hadjikhani N, Sanchez DR, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687-4692.
14 Knight Y. Brainstem modulation of caudal trigeminal nucleus: a model for understanding migraine biology and future drug targets. Headache Curr. 2005;2:108-118.
15 Goadsby PJ, MacDonald GJ. Extracranial vasodilation mediated by vasoactive intestinal polypeptide (VIP). Brain Res. 1985;329:285-288.
16 May A, Buchel C, Bahra A, et al. Intracranial vessels in trigeminal transmitted pain: a PET study. Neuroimage. 1999;9:453-460.
17 Jacquin MF, Chiaia NL, Haring JH, et al. Intersubnuclear connections within the rat trigeminal brainstem complex. Somatosens Mot Res. 1990;7:399-420.
18 Hayashi H. Morphology of terminations of small and large myelinated trigeminal primary afferent fibers in the cat. J Comp Neurol. 1985;240:71-89.
19 Mitsikostas DD, Sanchez del Rio M. Receptor systems mediating c-fos expression within trigeminal nucleus caudalis in animal models of migraine. Brain Res Brain Res Rev. 2001;35:20-35.
20 Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol. 2005;58:698-705.
21 Watkins LR, Milligan ED, Maier SF. Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol. 2003;521:1-21.
22 Wei F, Dubner R, Ren K. Nucleus reticularis gigantocellularis and nucleus raphe magnus in the brain stem exert opposite effects on behavioral hyperalgesia and spinal Fos protein expression after peripheral inflammation. Pain. 1999;80:127-141.
23 Burstein R, Cutrer MF, Yarnitsky D. The development of cutaneous allodynia during a migraine attack. Clinical evidence for the sequential recruitment of spinal and supraspinal nociceptive neurons in migraine. Brain. 2000;123:1703-1709.
24 Burstein R, Yarnitsky D, Goor-Aryeh I, et al. An association between migraine and cutaneous allodynia. Ann Neurol. 2000;47:614-624.
25 Burstein R, Levy D, Jakubowski M. Effects of sensitization of trigeminovascular neurons to triptan therapy during migraine. Rev Neurol (Paris). 2005;161:658-660.
26 Burstein R, Jakubowski M. Implications of multimechanism therapy: when to treat? Neurology. 2005;64(10 Suppl 2):S16-S20.
27 Ji RR, Kohno T, Moore KA, et al. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696-705.
28 Millan MJ. Descending control of pain. Progr Neurobiol. 2002;66:355-474.
29 Fields HL, Malick A, Burstein R. Dorsal horn projection targets of ON and OFF cells in the rostral ventromedial medulla. J Neurophysiol. 1995;74:1742-1759.
30 Goadsby PJ. Neuroimaging in headache. Microsc Res Tech. 2001;53:179-187.
31 Haan J, Kors EE, Vanmolkot KRJ, et al. Migraine genetics: an update. Curr Pain Headache Rep. 2005;9:213-220.
32 Kors EE, Haan J, Van Den Maagdenberg AMJM, et al. Recent findings in headache genetics. Curr Opin Neurol. 2004;17:283-288.
33 Burstein R, Jakubowski M. Unitary hypothesis for multiple triggers of the pain and strain of migraine. J Comp Neurol. 2005;493:9-14.






