[level-membership-for-pediatrics-category]
Head Injury and Facial Trauma
Head Injury
Traumatic brain injury (TBI) is an important cause of injury-related death and disability in children.1 In the USA, there are an estimated 1.7 million people with TBI annually, with a reported 52,000 deaths and 275,000 hospitalizations. Children ages 0 to 4 years and 15 to 19 years are most likely to suffer TBI. Nearly half a million emergency department visits annually are for children less than 14 years with TBI. Males, age 0 to 4 years, have the highest rates of TBI-related emergency department visits, hospitalizations, and deaths combined.2
Head trauma from child abuse remains a significant concern. In children younger than age 1 year, nonaccidental trauma (NAT) is the most common cause of fatal TBI.3,4 Head trauma associated with NAT has a higher rate of subdural hematomas, subarachnoid hemorrhages, and retinal hemorrhages when compared with nonabusive head trauma. Public health campaigns have brought attention to the dangers of shaking a young child and have provided resources for parents and caretakers who may be at risk of harming a child.5
Although the reasons are not entirely clear, there has been a general trend in improved outcomes after severe TBI in children.6 The decline in morbidity and mortality are likely due to improved prehospital care, regionalization of pediatric trauma care, adherence to evidence-based practice guidelines, more aggressive care (such as intracranial pressure [ICP] monitoring and early surgical evacuation of mass lesions), improved diagnostic imaging (computed tomography [CT], magnetic resonance imaging [MRI]), and advances in intensive care. In January 2012, the second edition of evidence-based practice guidelines was published for the acute medical management of severe TBI in infants, children, and adolescents.7 These guidelines have allowed a decrease in variability in care across centers, but there is a striking lack of data from well-designed, randomized controlled trials.
Pathophysiology
The immature brain has some structural differences from its adult counterpart that may explain the different responses to injury often seen in children after TBI. The brain doubles in size in the first 6 months of life and reaches approximately 80% of adult size by age 2. The developing brain has a higher water content and incomplete neuronal synapse formation and arborization. In addition, incomplete myelinization and neurochemical changes result in neuronal plasticity after birth. The subarachnoid space is generally smaller and offers less protection than the mature brain, owing to less buoyancy, and thereby provides less protection to the brain parenchyma during changes in head momentum. The result is a higher incidence of diffuse cerebral edema and parenchymal injuries in children. In children with distensible skulls, some argue that the open fontanelle allows for expansion of the intracranial contents and therefore affords increased protection from elevation of ICP. However, studies have indicated that the smaller neural axis of infants and young children results in a less compliant pressure–volume relationship with an increased risk of intracranial hypertension.8,9
The Monro–Kellie doctrine is an important concept relating to the understanding of ICP dynamics (Fig. 17-1). The Monro–Kellie doctrine uses a simple hydraulic approach to the cerebral circulation. Given that the cranium is a rigid, nonexpansile container, it states that the total volume of the intracranial contents must remain constant and any increase in the volume of one component must be at the expense of the others, assuming the intracranial volume remains constant. Thus, very early after injury, a mass such as an expanding hematoma may be enlarging while the ICP remains normal. Once the limit of displacement of CSF and intravascular blood has been reached, ICP rapidly increases (Fig. 17-2). Further work has shown that the relationship between ICP and cerebral blood flow (CBF) is much more complex and variable. Whereas simultaneous measurement of ICP and CBF would be most helpful in optimizing therapeutic strategies, the Monro–Kellie doctrine provides a reasonable basic explanation of intracranial dynamics.
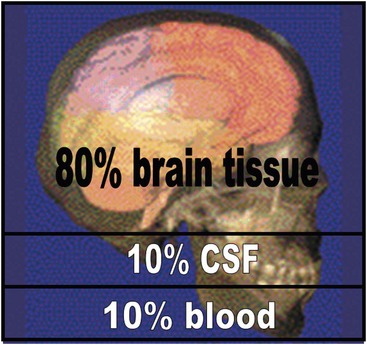
FIGURE 17-1 Monro–Kellie doctrine. The Monro–Kellie doctrine states that the cranial compartment is incompressible and that the volume inside the cranium is a fixed volume. The cranium and its constituents (blood, cerebrospinal fluid [CSF], and brain tissue) create a state of volume equilibrium such that any increase in volume of one of the cranial constituents must be compensated by a decrease in volume of another.
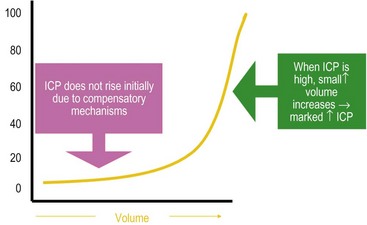
FIGURE 17-2 Intracranial pressure (ICP)–volume relationship. Small increases in brain volume do not lead to immediate increase in ICP because of the ability of the cerebrospinal fluid to be displaced into the spinal canal as well as the ability the falx cerebri to stretch slightly between the hemispheres and the tentorium between the hemispheres and the cerebellum. However, once the ICP has reached around 25 mmHg, small increases in brain volume can lead to marked elevations in ICP.
CBF is defined as the velocity of blood through the cerebral circulation. In adults, the normal CBF is 50–55 mL/100g of brain tissue/minute. In children, CBF may be much higher depending on their age. At 1 year of age, it approximates adult levels, but at 5 years of age, normal CBF is approximately 90 mL/100 g/min and then gradually declines to adult levels by the mid-late teens. Brain injury severe enough to cause coma can result in a 50% reduction in CBF during the first 6 to 12 hours after injury.10,11 It usually increases over the next 2 to 3 days, but for those patients who remain comatose, CBF remains below normal for days or weeks after injury. There is increasing evidence that such low levels of CBF are inadequate to meet the metabolic demands of the brain early after injury and that regional, if not global, cerebral ischemia results.12–14
Cerebral perfusion pressure (CPP) is the differential pressure of arterial flow into and venous flow out of the brain. CPP may be defined by the difference between mean arterial pressure (MAP) and ICP. CPP is considered the transmural pressure gradient that is ultimately the driving force required for supplying cerebral metabolic needs. As ICP increases following head injury, CPP decreases and blood flow to the brain eventually declines. At a CPP of 10 mmHg, blood vessels collapse and blood flow ceases.
Current techniques available to measure CBF, such as transcranial Doppler and Xenon-enhanced CT imaging, are still considered experimental in the management of severe TBI. Because CPP is easily determined by ICP monitoring, it has become a critical parameter for defining treatment options. Studies have shown a good correlation between CPP and CBF in patients with intact cerebral autoregulation.13 However, cerebral autoregulation is often disrupted after severe TBI and measures of cerebral vascular resistance may be more useful in guiding therapy.15,16
Cerebral autoregulation refers to a homeostatic process that allows CBF to remain constant over a wide range of MAPs. Arterial vessels can dilate or constrict in response to various physiologic changes, including ICP and systemic arterial pressure, to maintain normal flow and normal brain metabolism. In healthy adult patients, CBF remains constant with a MAP between 60–160 mmHg, or a CPP between 50–150 mmHg.17 Normally, with elevated systemic blood pressure, reflexive vasoconstriction will occur to prevent intracranial hypertension. In contrast, a moderate decrease in systemic blood pressure will paradoxically result in increased ICP because compensatory reflex vasodilatation will occur. When perfusion pressure falls below 50 mmHg, cerebral ischemia develops and compensatory cerebral arteriole vasodilatation is exhausted. When perfusion pressure exceeds 150 mmHg, cerebral arteriolar impedance is overcome, the affected vessels passively dilate, and fluid is forced through a damaged endothelium into the brain, causing diffuse vasogenic edema. Impaired cerebral autoregulation after TBI and age-related changes in CBF make the immature brain susceptible to secondary injury, both from diminished and excess CBF, and are both associated with a poor neurologic outcome.18
Historically, treatment protocols were principally directed toward reducing ICP. Hyperventilation and fluid restriction were important components in these older protocols. Sustained elevations in ICP above 20 mmHg are poorly tolerated by the injured brain and have been associated with poor neurologic outcome and increased mortality in pediatric patients.19 Sustained elevation in ICP may result in cerebral ischemia if cerebral perfusion is impaired, and ultimately may result in cerebral herniation. Current treatment strategies seek to optimize CPP while reducing ICP, with little reliance on hyperventilation or fluid restriction. CPP is likely an age-related continuum, thus making it problematic to develop treatment protocols based on a single number for all age groups. To date, no study has demonstrated that active maintenance of CPP above any target threshold in infants and children following TBI improves mortality or morbidity. However, there seems to be a threshold of less than 40 mmHg that is associated with increased mortality; therefore, most treatment guidelines recommend a minimum CPP of 40 mmHg.
Primary Brain Injury
Skull fractures occur commonly with head injury and are readily diagnosed with CT. In children, a skull fracture should prompt an evaluation of the underlying brain parenchyma given the significant impact it takes to injure the skull. Fractures of the skull vault can occur in either a linear or a stellate fashion. Fractures involving the skull base are typically associated with a greater force than simple cranial vault fractures. The classic signs of basilar skull fractures include Battle’s sign (ecchymoses over the mastoid process associated with an ipsilateral skull fracture), raccoon eyes and CSF rhinorrhea (associated with a cribriform plate fracture), and otorrhea (associated with fracture of the mastoid air cells or temporal bone fracture). Meningitis associated with a basilar skull fracture occurs in up to 10% of patients.20 Despite the risk of infection, the routine use of prophylactic antibiotics is not recommended as they have not been shown to prevent meningitis from occurring, and tend to select out for resistant organisms.21–23 Vaccination against Streptococcus pneumoniae should be considered for all patients with a basilar skull fracture and CSF leak due to the increased risk of pneumococcal-associated meningitis.24
Post-traumatic intracranial hemorrhage includes epidural hematomas, subdural hematomas, and subarachnoid hemorrhages. Epidural hematomas usually occur in the middle fossa and are often associated with an injury to the middle meningeal artery, although they can occur in the anterior or posterior fossa. The classic CT description is a lenticular hematoma, bound by suture lines, because of the tightly bound dura (Fig. 17-3). Clot formation under the calvaria compresses the dura and can cause rapid neurologic deterioration as the brain becomes further displaced. Skull fractures overlying the epidural hematoma are common. The classic presentation of a patient with a lucid interval followed by clinical deterioration is rare in children. Only after the hematoma enlarges is clinical evidence of elevated ICP noted. Typical symptoms include headache, lethargy, emesis, irritability, confusion, and decreased level of consciousness. Progressive deterioration results in seizures, changes in vital signs with hypertension and respiratory instability, pupillary changes, posturing, and cardiovascular compromise. Prompt neurosurgical evacuation is imperative for patient survival and good outcome. Evacuation of extremely large clots (>40 mL) in children often results in very good long-term results, provided that operative intervention is timely.
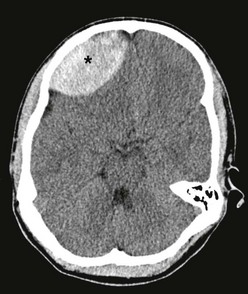
FIGURE 17-3 CT scan shows an epidural hematoma (asterisk) in a 14-year-old patient. (Courtesy of Dr Lisa Lowe.)
Subdural hemorrhages are classified as acute (<3 days old), subacute (3-10 days old), and chronic (>10 days old). Acute and subacute subdural hemorrhages are not infrequent in infants, and often the result of birth injury or NAT (Fig. 17-4). They usually result from lacerated bridging veins, or from associated contusions hemorrhaging into the subdural space. The superficial cortical veins in small children lack any reinforcement from arachnoidal trabeculae and are susceptible to inertial loading. Subdural hematomas tend to follow the convexities of the brain and cover the entire hemisphere. The cranial CT demonstrates hyperdense crescent-shaped blood collections at the surface of the brain, often associated with mass effect and cortical edema. Occasionally, and particularly when anemia is present, the CT findings of an acute subdural hematoma may have an isodense appearance that belies the actual hemorrhagic character later found at the time of operation.
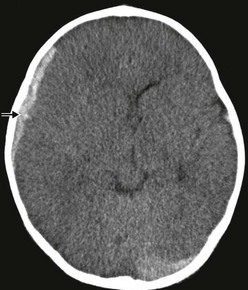
FIGURE 17-4 Hyperacute subdural bleeding (arrow) is seen on this cranial CT scan of a 9-month-old patient. (Courtesy of Dr Lisa Lowe.)
Subarachnoid hemorrhage (SAH) in acutely traumatized children is common and is rarely the result of aneurysmal bleeding (Fig. 17-5). Subarachnoid bleeding occurs from disruption of the fragile pia-arachnoidal vasculature. This often occurs over the convexities of the brain affected by coup type injuries or the frontotemporal poles affected by contrecoup injuries. Traumatic SAH can also occur in the interhemispheric fissure and in the basilar cisterns. When SAH is an isolated finding following minor trauma, no specific therapy is indicated except symptomatic amelioration of chemical meningitis, meningismus, and photophobia. SAH can result in hydrocephalus and may require ventricular shunting to relieve the increased ICP. In patients with severe TBI, SAH is associated with a poor outcome and may also be associated with cerebral vasospasm. Transcranial Doppler imaging can be utilized to identify vasospasm. Therapy for vasospasm in adults include calcium channel blockers and neurointerventional techniques; however, these are not well studied in children and not commonly used.25,26
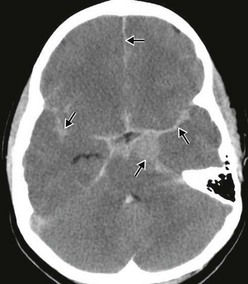
Secondary Brain Injury
Edema of the brain is an important marker for injury and is also a cause of secondary injury. In early (<24 hours) fatal closed-head injuries in children, CT scans often demonstrate little or no significant parenchymal bleeding. However, in children, rapid development of edema is commonly seen on serial CT scans and the diffuse brain swelling causes the obliteration of the ventricles and loss of the basilar cisterns and subarachnoid space. As swelling progresses and the compensatory mechanisms of the brain are exhausted, ICP increases markedly with small changes in intracranial volume (see Fig. 17-2). Cerebral edema typically develops early after injury, peaking at 72 to 96 hours, and then gradually resolves over the next week in survivors.27
Studies in adults utilizing xenon CT have shown a reduction in CBF early after severe TBI.10,12 This hypoperfusion may be further exacerbated by hypotension and hypoxia. It is clear that this early hypoperfusion or ischemia after severe TBI is associated with a poor outcome.28,29 Proposed mediators involved in early post-traumatic ischemia include direct vascular disruption, production of endothelin-1 (a potent vasoconstrictor), loss of endogenous vasodilators (endogenous nitric oxide synthase), and likely many other complex, interrelated, cellular and metabolic events.
The release of excitatory amino acids, such as glutamate, results in neuronal injury after TBI. Glutamate is the most abundant neurotransmitter in the brain. However, toxic levels cause neuronal cell death.30,31 After TBI, glutamate and other excitatory amino acids are released, resulting in neuronal swelling, calcium influx, and release of cytotoxic enzymes leading to cell death. Studies have failed to demonstrate efficacy of anti-excitotoxic therapies, perhaps owing to their application in all patients with TBI rather than those with excitotoxicity, and because treatment may have been initiated too late.32
Initial Evaluation and Management of Head Injury
As with any trauma, the initial management and resuscitation begins with an assessment of circulation, airway and breathing (CAB). Ensuring adequate oxygenation and ventilation and promptly addressing sources of ongoing blood loss serve as the basic principles in the management of persons suspected or confirmed to have head injury. Hypotension and hypoxia in the field are proven secondary insults that are associated with poor outcomes, with hypotension being considerably more detrimental than hypoxia.28,33 There has been no documented advantage to endotracheal intubation over effective bag-valve-mask ventilation in the field.34,35 Providers inexperienced in pediatric airway management should defer endotracheal intubation. Efforts should be made to control bleeding, including scalp lacerations, which may be a source of shock in the child. The administration of isotonic crystalloid solution should be utilized to promptly restore the intravascular volume. Colloid solutions should be avoided. The withholding of fluid because of concerns of concomitant head injury is unjustified and may contribute to secondary brain injury with ineffective CBF. Although young children are prone to hypoglycemia, hypoglycemia occurring early after trauma is rare. Hyperglycemia may be detrimental during periods of cellular hypoxia owing to a shift to anaerobic metabolism and lactate production. Therefore, unless there is documented hypoglycemia, dextrose-containing fluids should be avoided in the early phases of resuscitation.
Current recommendations do not support ICP-directed measures in the field with the possible exception of hyperventilation and hyperosmolar therapy for signs of herniation.7,36 Every effort should be made to transport the patient to a pediatric trauma center, or to an adult center equipped to manage pediatric trauma patients. Careful attention should be paid to cervical spine immobilization as there is an increased risk of spinal cord injury with TBI.
On arrival in the emergency department (ED), the Advanced Trauma Life Support (ATLS) protocol of the American College of Surgeons should be followed.37 Early hospital evaluation and management is the same as that described for the prehospital setting. Endotracheal intubation should be performed in the initial hospital management, if needed. After initial stabilization and fluid resuscitation, a brief neurologic examination should be performed, preferably before the administration of sedation and neuromuscular blockade. Adjunctive diagnostic studies should be performed only after the initial resuscitation phase.
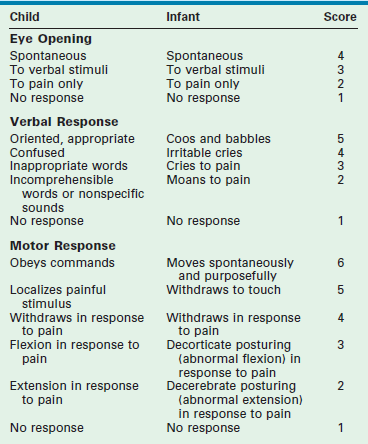
The severity of brain injury is often classified using the modified Glasgow Coma Scale (GCS) score.38 Even with modifications, the GCS score has limitations in its use for infants and very young children (Table 17-1). In addition, the use of sedation and pharmacologic paralysis, orbital swelling impairing eye opening, endotracheal intubation making verbal responses impossible, and preexisting conditions such as alterations in motor function further limit the use of the GCS score. Nonetheless, it is the best tool currently available to classify TBI and to share information between treating physicians.
Severe TBI: GCS 3–8
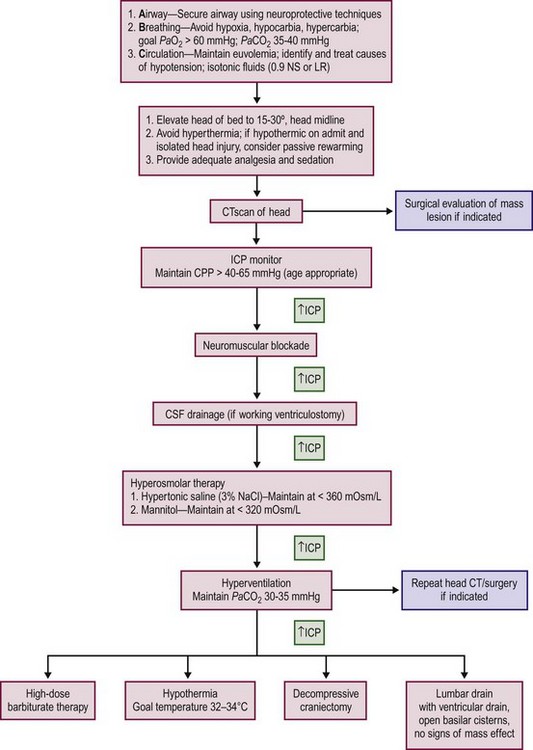
The treatment of severe TBI begins with the fundamentals of resuscitation previously described including restoration of an adequate circulating blood volume, blood pressure support, appropriate ventilation, and adequate oxygenation. Patients with severe head injury often have multisystem injuries and require a multidisciplinary team to aggressively identify and treat all injuries. An evidence-based approach to resuscitation and treatment of patients with severe brain injury is presented in Figure 17-6.
Respiratory Monitoring and Management
Ensuring adequate oxygenation to avoid ongoing and worsening neuronal ischemia is essential. The airway should be secured with special attention for avoidance of hypoxia, hypercarbia, or hypocarbia. The goals of ventilation should include a target PaCO2 of 35–40 mmHg. In the head-injured patient, hypercapnia (PaCO2 >45 mmHg) can cause significant elevation in ICP because of CO2-induced dilatation of the cerebral vasculature resulting in increased cerebral blood volume and flow. The immediate effect of hyperventilation is a reduction of ICP, although this response is neither universal nor sustained. Hyperventilation reduces ICP by causing cerebral vasoconstriction, with a subsequent reduction in CBF in reactive vascular beds. Hyperventilation should be reserved for those patients with obvious signs of brain stem herniation, often heralded by Cushing’s triad (abnormal respiratory pattern, hypertension, and bradycardia), and nonreactive, dilated pupils.
Sedation and Analgesia
Sedation and analgesia are commonly used in the management of TBI. The maintenance of the airway, placement of invasive catheters or other monitoring devices, and the safe transport of the patient for diagnostic or therapeutic procedures are examples of some aspects of patient care facilitated by these medications. Sedation and analgesia may also be useful in maintaining or decreasing ICP by decreasing the metabolic rate and thereby decreasing CBF. Studies have demonstrated a two- to threefold increase in basal metabolic rate resulting from painful or stressful stimuli. Noxious stimuli, such as suctioning, can increase ICP. Painful and noxious stimuli and stress can also increase sympathetic tone, resulting in hypertension and bleeding from operative sites.39,40 However, these medications must be used with caution, because sedative or narcotic related vasodilation may decrease CBF with resulting hypotension, or conversely may increase CBF by causing cerebral vasodilatation.
Intracranial Pressure Monitoring and Management
Despite the absence of prospective, randomized clinical trials to establish efficacy in improving outcome, the use of ICP devices has become standard in the USA for the treatment of severe head injury. There are two lines of thinking to support the use of ICP monitoring in the pediatric TBI patient. First, there is strong evidence that supports the association between intracranial hypertension and poor neurologic outcome.41 Second, ICP monitoring and aggressive treatment of intracranial hypertension are associated with the best reported clinical outcomes.42 The presence of open fontanelles and/or sutures in an infant does not preclude the development of intracranial hypertension or negate the utility of ICP monitoring.7
The treatment threshold for intracranial hypertension is typically defined as an ICP greater than 20 mmHg. Age-specific and injury mechanism–specific ICP thresholds have yet to be defined. In more recent years, the focus has switched from ICP-directed therapy to multimodal-directed therapy. Support for this change has been bolstered by a recent multicenter randomized prospective trial comparing two groups of patients ages 13 years and older. Patients were randomized into a guidelines-based management in which a protocol for monitoring intraparenchymal intracranial pressure was used (pressure-monitoring group) or a protocol in which treatment was based on imaging and clinical examination (imaging-clinical examination group).43 The study failed to find superiority with either method in the both short and long-term outcomes of patients with severe TBI. The study authors clearly state that this study does not argue against the use of ICP monitoring, but rather indicates that more work needs to be done on interpreting and utilizing this tool in the treatment and management of TBI.
Although there is no single ICP monitoring device that is superior to the others, intraventricular devices are effective monitors that allow CSF drainage as part of the treatment for elevated ICP. However, if not inserted early after injury, placement may be difficult when damaged brain and cerebral edema have effaced the normal ventricle. Intraparenchymal monitors have fiberoptic or strain-gauge transducers that provide continuous pressure readings, and can be placed in any region of the brain. Newer monitors can measure brain tissue oxygen levels and permit manipulations to optimize regional oxygenation, including adjustments in the fraction of inspired oxygen (FiO2) and blood transfusion. Coagulation abnormalities are a relative contraindication to the insertion of these devices. Therefore, prompt correction using plasma or activated factor VIIa should be considered before insertion.44
The optimal CPP for a given individual is not clear. Global and/or regional cerebral ischemia commonly complicates brain injury. Studies have consistently shown that a CPP less than 40 mmHg is associated with increased mortality.41,45 A CPP between 40–65 mmHg likely represents an age-related continuum with inter-individual variability. Studies have suggested that avoidance of hypotension, rather than elevation of CPP, is more beneficial.41 Efforts to increase CPP in adults to a supernormal level with the use of fluids and vasoactive infusions are associated with significant risks, including pulmonary complications, fluid overload, pressor toxicity, and renal insufficiency. Furthermore, in cases of disrupted autoregulation, exacerbation of intracranial hypertension can result.
Neuromuscular Blockade
Neuromuscular blockade is commonly used as a treatment strategy for patients with increased ICP. Neuromuscular blocking agents should be used only in those patients with a secure airway, and who are mechanically ventilated and adequately sedated. The proposed mechanisms of neuromuscular blocking agents in reducing ICP include a reduction in airway and intrathoracic pressure via facilitation of cerebral venous outflow and prevention of shivering, posturing, or breathing against the ventilator.46 These medications have been associated with prolonged ICU stays and increased risk of nosocomial infections. Therefore, neuromuscular blockade should be reserved for the patient with increased ICP who is unresponsive to other methods such as increased sedation and/or analgesia.
Hyperosmolar Therapy
The use of hyperosmolar agents to decrease ICP was first described in the early 1900s. Hypertonic saline and mannitol were among the first agents trialed. Mannitol has become the mainstay of therapy over the past several decades. The mechanism of action of mannitol has been well studied. However, its effect on survival and functional outcome after TBI has not been well demonstrated. Mannitol requires the presence of an intact blood–brain barrier to exert its effect. Mannitol has two primary mechanisms of action. The first is an initial rheologic effect, resulting in a decrease in blood viscosity that occurs within minutes of administration. A decrease in blood vessel diameter occurs from autoregulation after reflex vasoconstriction, and leads to a decrease in cerebral blood volume and ICP.47 Second, mannitol acts as a potent osmotic diuretic, thus pulling fluid from the interstitial space into the intravascular space. Because of its diuretic effect, intravascular volume can be depleted with the risk of decreasing cerebral perfusion, resulting in secondary brain ischemia. Careful attention must be paid to promptly replenishing intravascular volume. Finally, mannitol therapy has been associated with acute tubular necrosis. Therefore, monitoring of serum osmolarity is recommended when using mannitol. Osmolar levels greater than 320 mOsm/L should be avoided. However, most of the reports describing the development of renal damage occurred in the era of dehydration therapy for cerebral edema. It is unclear whether the same osmolar thresholds are valid in the current era of maintaining normal intravascular volume. The beneficial effects of mannitol are best achieved with rapid bolus administration, with dosage ranging from 0.25–1.0 g/kg. Chronic administration of mannitol has been associated with a reverse osmotic shift, resulting in rebound cerebral edema and disruption of the blood–brain barrier.
More recently, hypertonic saline has re-emerged as a hyperosmolar agent and is the preferred agent in the TBI patient with hypovolemia.48,49 Hypertonic saline includes concentrations ranging from 3 to 23.4%, with 3% hypertonic saline being the most studied in children. There is no evidence that one concentration is more effective than another for reducing intracranial hypertension. Similar to mannitol, hypertonic saline requires the presence of an intact blood-brain barrier to exert its effect. Hypertonic saline is an osmolar agent and it pulls fluid from the interstitium into the intravascular space without a strong diuretic effect, thus maintaining blood volume and cerebral perfusion. Hypertonic saline can be used as a bolus (6.5–10 mL/kg) or continuous infusion (0.1–1.0 mL/kg/h titrated to the minimum dose needed to achieve a reduction in ICP). In addition to the hyperosmolar effect, hypertonic saline has been reported to have several other potentially beneficial effects for the trauma patient, including vasoregulatory, hemodynamic, neurochemical, and immunologic properties. Serum osmolarity of 360 mOsm/L has been reported with the use of hypertonic saline and has been well tolerated in the pediatric patient with a head injury.48 The primary theoretical concerns associated with the risk of hypertonic saline include the development of central pontine myelinolysis (rapid shrinking of the brain associated with mechanical tearing of bridging vessels leading to subarachnoid hemorrhage), renal failure, and rebound intracranial hypertension (Table 17-2).
TABLE 17-2
A Comparison of Mannitol and Hypertonic Saline (For Hyperosmolar Therapy)
| Mannitol | 3% Hypertonic Saline | |
| Bolus dosing guidelines | 0.25–1.0 g/kg rapid bolus | 3–5 mL/kg |
| Infusion guidelines | None | 0.1–1.0 mL/kg/h |
| Effectiveness | May wane with repeated administration | Effective after repeated administration; effective when mannitol efficacy has waned |
| Augmentation of MAP | Moderate | Greater, more prolonged |
| Rheologic properties | Yes | Yes |
| Diuretic effect | Osmotic diuretic, may necessitate volume replacement to avoid hypovolemia | Diuresis through action of atrial natriuretic peptide |
| Maximum serum osmolarity | 320 mOsm/L | 360 mOsm/L |
| Adverse effects | Renal failure, hypotension, rebound elevation in ICP | Rebound elevation in ICP, central pontine myelinolysis, bleeding, electrolyte abnormal |
| Proposed beneficial effects | Antioxidant effects | Restoration of resting membrane potential and cell volume, inhibition of inflammation |
ICP, intracranial pressure; MAP, mean arterial pressure.
From Knapp JM. Hyperosmolar therapy in the treatment of severe head injury in children: Mannitol and hypertonic saline. AACN Clin Issues 2005;16:199–211. Reprinted with permission.
Prophylactic administration of mannitol or other hyperosmolar therapy either in the field or in the hospital is no longer routinely recommended. Hyperosmolar therapy should be reserved for those patients with documented intracranial hypertension or those with signs of impending herniation to avoid secondary injury and complications associated with hyperosmolar therapy.
Medically Refractory Intracranial Hypertension
It is estimated that 21–42% of children with severe TBI will develop refractory intracranial hypertension despite medical and surgical management.7 In these patients, therapies and interventions with higher risk profiles may need to be considered. Decompressive craniectomy, high-dose barbiturate therapy, hyperventilation, lumbar drain placement, and the use of moderate hypothermia should be considered in the patient with medically refractory intracranial hypertension.
Decompressive Craniectomy
Children are more likely than adults to have diffuse brain swelling after TBI and may be more amenable to a treatment strategy utilizing early decompressive craniectomy. Decompressive craniectomy should be considered in children with severe TBI and infants with abusive head trauma and medically refractory intracranial hypertension. The main goal of decompressive craniectomy is to control ICP, thereby maintaining CPP and cerebral oxygenation. Improved outcomes have been demonstrated in several small single-center studies, and appears to be most effective when done early, before the development of extensive secondary brain injury.50–52
Barbiturate Therapy
High doses of barbiturates are known to reduce ICP and have been used in the management of increased ICP for decades. Their side effects limit their current use to those patients with injuries refractory to first-line therapies. Barbiturates are effective in lowering ICP by suppressing brain metabolism and altering vascular tone. In addition to the ICP-lowering benefits, barbiturates also inhibit free radical–mediated lipid peroxidation and have membrane-stabilizing effects. Small case series of children with severe TBI suggest that barbiturates may be effective in lowering ICP in the setting of refractory intracranial hypertension.53,54 Their use is associated with myocardial depression, increased risk of hypotension, and the need for blood pressure support with intravascular fluids and inotropic infusions. It is important that barbiturates are used in the setting of systemic monitoring with the ability to rapidly detect hemodynamic instability.
Hyperventilation
Hyperventilation has been a mainstay in the management of severe TBI, but more recent concerns about its role in cerebral ischemia have lessened its use. As discussed previously, the cerebral vasculature is sensitive to changes in PaCO2, with hypocarbia producing cerebral vasoconstriction and a decrease in CBF. Historically, hyperemia, or excessive CBF, was thought to be the primary mechanism resulting in cerebral edema after TBI, thus making hyperventilation a reasonable approach in the management of the patient with severe TBI. More recent studies have demonstrated that hyperemia is uncommon after severe TBI. Hyperventilation may decrease cerebral oxygenation, resulting in secondary brain ischemia.55,56 The use of aggressive hyperventilation (PaCO2 <30 mmHg) should be reserved for refractory intracranial hypertension. Monitoring of CBF, jugular venous oxygen saturation, or brain tissue oxygenation may help identify cerebral ischemia in this setting.
Therapeutic Hypothermia
The avoidance of hyperthermia and use of therapeutic hypothermia after TBI is based on the rationale that temperature plays an important role in mechanisms contributing to secondary brain injury (excitotoxicity, free radical formation). Despite evidence in animal models demonstrating efficacy of therapeutic moderate hypothermia (32–34°C), large randomized clinical studies in both adults and children have not proven the effectiveness of hypothermia on improved outcomes after TBI.57–61 Studies extrapolated from the adult literature indicate that hyperthermia adversely affects outcome so it may be advisable to consider passive re-warming of the mild to moderately hypothermic trauma patient with isolated head injury.62
Facial Trauma
The same mechanisms of injury associated with TBI (sports, motor vehicle accidents, and falls) also lead to facial trauma in infants and children. The elasticity of the facial skeleton, the enhanced facial soft tissue padding, the lack of sinuses that thin the surrounding bone, and the prominence of the skull protect the younger child’s face from injury, resulting in a lower incidence of traumatic injury. As children age, develop, and become more active, the frequency of injury increases. No matter the age, the reconstructive principles remain the same: restoration of function, maximization of aesthetic results, limited visible incisions for repair, avoidance of complications, and minimal effect on later growth and development from both the trauma and the operative repair. Except for the last principle, these goals are the same as repair of facial trauma in adults. The last principle, related to later growth and development, must be understood by both the surgical team and the family as it can alter the treatment plan. Furthermore, the treatment plan can often be different for a given fracture that occurs during infancy versus childhood versus adolescence.
Evaluation and Diagnosis
Examination should begin with an overall assessment of neurologic status and airway competency. Craniofacial fractures have a high incidence of TBI. CSF leaks may occur and neurosurgical consultation is often warranted. An unstable airway can be challenging secondary to mandibular or maxillary fractures. Teeth can be loose, free-floating, or aspirated. The cervical spine is often not cleared and the child’s neck is immobilized. Intubation with a flexible endoscope may be necessary. A tracheotomy may be needed in some cases.
Mandibular examination begins by asking the patient if his or her teeth come together normally. Intraoral examination will demonstrate malocclusion. Lower lip paresthesias are indicative of fractures in the body or parasymphyseal regions of the mandible. Gingival lacerations can also expose an underlying fracture. Dental and/or alveolar ridge injuries are also common. The clinician must palpate the mandible from temporomandibular joint to the contralateral temporomandibular joint, documenting pain if present. The older the child, the more likely they are to have two concomitant mandible fractures similar to adults (Fig. 17-7).
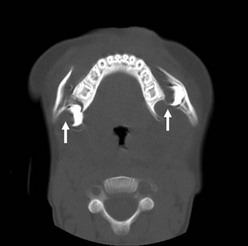
FIGURE 17-7 This 18-month-old suffered a crush injury after a swing fell on his face. This CT scan shows bilateral fractures (arrows) of the body of the mandible. He required open reduction and internal fixation with intermaxillary wiring.
Treatment
Midfacial Fractures
Isolated displaced zygoma fractures require open reduction with fixation (Fig. 17-8). As the zygoma is a bone that constitutes part of the orbital walls and floor, the orbital floor needs to be explored and repaired at the time of zygoma fracture repair. Orbital sequelae of inadequately repaired zygoma fractures include enopthalmos, orbital dystopia, and diplopia.
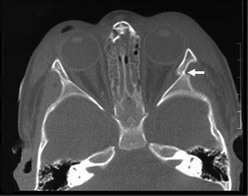
Unique Pediatric Concerns
Pediatric facial fractures and their repair pose long-term concerns distinct from adult trauma. The fractures themselves may inhibit long-term growth of the involved bony structures leading not only to distortion, but to asymmetry as well. Fracture repair techniques, including subperiosteal exposure of the involved bones, may further impede long-term growth. Bony fixation with permanent metallic plates also can result in growth restriction. The recent development of resorbable plates which function during the period of bony healing, and then resorb, is an attempt to eliminate this long-term concern. To date, these plates have shown great efficacy in nonweight-bearing bones, but are more limited in bones involved in mastication. Because of these concerns, because of the differences in anatomy including tooth buds and development, and because of bony elasticity and the absence of sinuses, pediatric fractures are different than adult fractures as are the philosophies for pediatric fracture repair. In addition, the fracture type and repair varies by age in this growing population.
References
1. Adekoya, N, Thurman, DJ, White, DD, et al. Surveillance for traumatic brain injury deaths–United States, 1989–1998. Morbidity & Mortality Weekly Report Surveillance Summaries. 2002; 51:1–14.
2. Langlois, JA, Rutland-Brown, W, Thomas, KE. The incidence of traumatic brain injury among children in the United States: Differences by race. J Head Trauma Rehab. 2005; 20:229–238.
3. Billmire, ME, Myers, PA. Serious head injury in infants: Accident or abuse? Pediatr. 1985; 75:340–342.
4. Bruce, DA, Zimmerman, RA. Shaken impact syndrome. Pediatr Ann. 1989; 18:482–484.
5. Child Maltreatment Prevention. Center for Disease Control and Prevention. [cited 2008 May 7]; Available from: http://www.cdc.gov/ncipc/dvp/CMP/CMP-prvt-strat.htm, 2008.
6. Tilford, JM, Aitken, ME, Anand, KJ, et al. Hospitalizations for critically ill children with traumatic brain injuries: A longitudinal analysis. Crit Care Med. 2005; 33:2074–2081.
7. Kochanek, PM, Carney, N, Adelson, PD, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents–second edition. Pediatric Critical Care Medicine: A Journal of the Society of Critical Care Medicine and the World Federation of Pediatric Intensive and Critical Care Societies. 2012; 13(Suppl 1):S1–82.
8. Muizelaar, JP, Marmarou, A, DeSalles, AA, et al. Cerebral blood flow and metabolism in severely head-injured children. Part 1: Relationship with GCS score, outcome, ICP, and PVI. J Neurosurg. 1989; 71:63–71.
9. Shapiro, K, Marmarou, A. Clinical applications of the pressure-volume index in treatment of pediatric head injuries. J Neurosurg. 1982; 56:819–825.
10. Bouma, GJ, Muizelaar, JP. Evaluation of regional cerebral blood flow in acute head injury by stable xenon-enhanced computerized tomography. Acta Neur S. 1993; 59:34–40.
11. Bouma, GJ, Muizelaar, JP. Cerebral blood flow in severe clinical head injury. New Horizons. 1995; 3:384–394.
12. Bouma, GJ, Muizelaar, JP, Choi, SC, et al. Cerebral circulation and metabolism after severe traumatic brain injury: the elusive role of ischemia. J Neurosurg. 1991; 75:685–693.
13. Cold, GE. Cerebral blood flow in the acute phase after head injury. Part 2: Correlation to intraventricular pressure (IVP), cerebral perfusion pressure (CPP), PaCO2, ventricular fluid lactate, lactate/pyruvate ratio and pH. Acta Anaesthesiologica Scandinavica. 1981; 25:332–335.
14. Jaggi, J, Obrist, W, Gennarelli, T, et al. Relationship of early cerebral blood flow and metabolism to outcome in acute head injury. J Neurosurg. 1990; 72:176–182.
15. Cruz, J, Jaggi, JL, Hoffstad, OJ. Cerebral blood flow, vascular resistance, and oxygen metabolism in acute brain trauma: Redefining the role of cerebral perfusion pressure? Crit Care Med. 1995; 23:1412–1417.
16. Lang, EW, Lagopoulos, J, Griffith, J, et al. Cerebral vasomotor reactivity testing in head injury: The link between pressure and flow. J Neurol Psychiatry. 2003; 74:1053–1059.
17. Paulson, OB, Strandgaard, S, Edvinsson, L. Cerebral autoregulation. Cerebrovas Brain Met. 1990; 2:161–192.
18. Vavilala, MS, Muangman, S, Tontisirin, N, et al. Impaired cerebral autoregulation and 6-month outcome in children with severe traumatic brain injury: Preliminary findings. Dev Neurosci. 2006; 28:348–353.
19. Pfenninger, J, Kaiser, G, Lutschg, J, et al. Treatment and outcome of the severely head injured child. Intens Care Med. 1983; 9:13–16.
20. Dagi, TF, Meyer, FB, Poletti, CA. The incidence and prevention of meningitis after basilar skull fracture. Am J Emer Med. 1983; 1:295–298.
21. Rathore, MH. Do prophylactic antibiotics prevent meningitis after basilar skull fracture? Pediatr Infect Dis J. 1991; 10:87–88.
22. Villalobos, T, Arango, C, Kubilis, P, et al. Antibiotic prophylaxis after basilar skull fractures: A meta-analysis. Clin Infect Dis. 1998; 27:364–369.
23. O, RB, João, C, Cristina, S, et al. Antibiotic prophylaxis for preventing meningitis in patients with basilar skull fractures. John Wiley & Sons, Ltd; 2011.
24. Venetz, I, Schopfer, K, Muhlemann, K. Paediatric, invasive pneumococcal disease in Switzerland, 1985–1994. Swiss Pneumococcal Study Group. Int J Epidem. 1998; 27:1101–1104.
25. Harders, A, Kakarieka, A, Braakman, R. Traumatic subarachnoid hemorrhage and its treatment with nimodipine. German TSAH Study Group. J Neurosurg. 1996; 85:82–89.
26. Romner, B, Bellner, J, Kongstad, P, et al. Elevated transcranial Doppler flow velocities after severe head injury: Cerebral vasospasm or hyperemia? J Neurosurg. 1996; 85:90–97.
27. Bareyre, F, Wahl, F, McIntosh, TK, et al. Time course of cerebral edema aftertraumatic brain injury in rats: Effects of riluzole and mannitol. J Neurotrauma. 1997; 14:839–889.
28. Chestnut, R, Marshall, LF, Klauber, MR, et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993; 34:216–222.
29. Gopinath, SP, Robertson, CS, Contant, CF, et al. Jugular venous desaturation and outcome after head injury. J Neurol Psychiatry. 1994; 57:717–723.
30. Choi, DW. Excitotoxic cell death. J Neurobiol. 1992; 23:1261–1276.
31. Choi, DW, Maulucci-Gedde, M, Kriegstein, AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987; 7:357–368.
32. Koh, JY, Choi, DW. Selective blockade of non-NMDA receptors does not block rapidly triggered glutamate-induced neuronal death. Brain Res. 1991; 548:318–321.
33. Stocchetti, N, Furlan, A, Volta, F. Hypoxemia and arterial hypotension at the accident scene in head injury. J Trauma. 1996; 40:764–767.
34. Cooper, A, DiScala, C, Foltin, G, et al. Prehospital endotracheal intubation for severe head injury in children: A reaapraisal. Semin Pediatr Surg. 2001; 10:3–6.
35. Gausche, M, Lewis, R, Stratton, S, et al. Effect of out-of-hospital pediatric endotracheal intubation on survival and neurological outcome: A controlled clinical trial. JAMA. 2000; 283:783–790.
36. Gabriel, E, Ghajar, J, Jagoda, A, et al. Guidelines for pre-hospital management of traumatic brain injury. New York: Brain Trauma Foundation; 2000.
37. Advanced Trauma Life Support (ATLS). 7th ed, Chicago, IL: American College of Surgeons; 2004.
38. Teasdale, G, Jennett, B. Assessment of coma and impaired consciousness: A practical scale. Lancet. 1974; 2:81–84.
39. Kerr, ME, Weber, BB, Sereika, SM, et al. Effect of endotracheal suctioning on cerebral oxygenation in traumatic brain-injured patients. Crit Care Med. 1999; 27:2776–2781.
40. Raju, TN, Vidyasagar, D, Torres, C, et al. Intracranial pressure during intubation and anesthesia in infants. J Pediatr. 1980; 96:860–862.
41. Downard, C, Hulka, F, Mullins, RJ, et al. Relationship of cerebral perfusion pressure and survival in pediatric brain-injured patients. J Trauma. 2000; 49:654–658.
42. Tilford, J, Aitken, M, Anand, K, et al. Hospitalizations for critically ill children with traumatic brain injuries: A longitudinal analysis. Crit Care Med. 2005; 33:2074–2081.
43. Chestnut, RM, Temkin, N, Carney, N, et al. A trial of intracranial-pressure monitoring in traumatic brain injury. N Engl J Med. 2012; 367:2471–2481.
44. Morenski, JD, Tobias, JD, Jimenez, DF. Recombinant activated factor VII for cerebral injury-induced coagulopathy in pediatric patients. Report of three cases and review of the literature. J Neurosurg. 2003; 98:611–616.
45. Elias-Jones, AC, Punt, JA, Turnbull, AE, et al. Management and outcome of severe head injuries in the Trent region 1985–90. Arch Dis Child. 1992; 67:1430–1435.
46. Hsiang, JK, Chesnut, RM, Crisp, CB, et al. Early, routine paralysis for intracranial pressure control in severe head injury: is it necessary? Crit Care Med. 1994; 22:1471–1476.
47. Muizelaar, JP, Wei, EP, Kontos, HA, et al. Mannitol causes compensatory cerebral vasoconstriction and vasodilation in response to blood viscosity changes. J Neurosurg. 1983; 59:822–828.
48. Khanna, S, Davis, D, Peterson, B, et al. Use of hypertonic saline in the treatment of severe refractory posttraumatic intracranial hypertension in pediatric traumatic brain injury. Crit Care Med. 2000; 28:1144–1151.
49. Peterson, B, Khanna, S, Fisher, B, et al. Prolonged hypernatremia controls elevated intracranial pressure in head-injured pediatric patients. Crit Care Med. 2000; 28:1136–1143.
50. Guresir, E, Schuss, P, Seifert, V, et al. Decompressive craniectomy in children: Single-center series and systematic review; 70. Neurosurg, 2012.
51. Oluigbo, CO, Wilkinson, CC, Stence, NV, et al. Comparison of outcomes following decompressive craniectomy in children with accidental and nonaccidental blunt cranial trauma. J Neuros-Pediatr. 2012; 9:125–132.
52. Pérez Suárez, E. Decompressive craniectomy in 14 children with severe head injury: Clinical results with long-term follow-up and review of the literature. J Trauma. 2011; 71:133–140.
53. Kasoff, SS, Lansen, TA, Holder, D, et al. Aggressive physiologic monitoring of pediatric head trauma patients with elevated intracranial pressure. Pediatr Neurosci. 1988; 14:241–249.
54. Pittman, T, Bucholz, R, Williams, D. Efficacy of barbiturates in the treatment of resistant intracranial hypertension in severely head-injured children. Pediatr Neurosci. 1989; 15:13–17.
55. Skippen, P, Seear, M, Poskitt, K, et al. Effect of hyperventilation on regional cerebral blood flow in head-injured children. Crit Care Med. 1997; 25:1402–1409.
56. Stringer, WA, Hasso, AN, Thompson, JR, et al. Hyperventilation-induced cerebral ischemia in patients with acute brain lesions: Demonstration by xenon-enhanced CT. Am J Neuroradiol. 1993; 14:475–484.
57. Adelson, PD. Hypothermia following pediatric traumatic brain injury. J Neurotraum. 2009; 26:429–436.
58. Adelson, PD, Ragheb, J, Kanev, P, et al. Phase II clinical trial of moderate hypothermia after severe traumatic brain injury in children. Neurosurg. 2005; 56:740–754.
59. Biswas, AK, Bruce, DA, Sklar, FH, et al. Treatment of acute traumatic brain injury in children with moderate hypothermia improves intracranial hypertension. Crit Care Med. 2002; 30:2742–2751.
60. Hutchison, JS, Ward, RE, Lacroix, J, et al. Hypothermia Pediatric Head Injury Trial Investigators and the Canadian Critical Care Trials Group. Hypothermia therapy after traumatic brain injury in children. N Engl J Med. 2008; 358:2447–2456.
61. Marion, DW, Penrod, LE, Kelsey, SF, et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med. 1997; 336:540–546.
62. Jones, PA, Andrews, PJ, Midgley, S, et al. Measuring the burden of secondary insults in head-injured patients during intensive care. J Neurosurg Anesth. 1994; 6:4–14.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Head Injury and Facial Trauma
Head Injury
Traumatic brain injury (TBI) is an important cause of injury-related death and disability in children.1 In the USA, there are an estimated 1.7 million people with TBI annually, with a reported 52,000 deaths and 275,000 hospitalizations. Children ages 0 to 4 years and 15 to 19 years are most likely to suffer TBI. Nearly half a million emergency department visits annually are for children less than 14 years with TBI. Males, age 0 to 4 years, have the highest rates of TBI-related emergency department visits, hospitalizations, and deaths combined.2
Head trauma from child abuse remains a significant concern. In children younger than age 1 year, nonaccidental trauma (NAT) is the most common cause of fatal TBI.3,4 Head trauma associated with NAT has a higher rate of subdural hematomas, subarachnoid hemorrhages, and retinal hemorrhages when compared with nonabusive head trauma. Public health campaigns have brought attention to the dangers of shaking a young child and have provided resources for parents and caretakers who may be at risk of harming a child.5
Although the reasons are not entirely clear, there has been a general trend in improved outcomes after severe TBI in children.6 The decline in morbidity and mortality are likely due to improved prehospital care, regionalization of pediatric trauma care, adherence to evidence-based practice guidelines, more aggressive care (such as intracranial pressure [ICP] monitoring and early surgical evacuation of mass lesions), improved diagnostic imaging (computed tomography [CT], magnetic resonance imaging [MRI]), and advances in intensive care. In January 2012, the second edition of evidence-based practice guidelines was published for the acute medical management of severe TBI in infants, children, and adolescents.7 These guidelines have allowed a decrease in variability in care across centers, but there is a striking lack of data from well-designed, randomized controlled trials.
Pathophysiology
The immature brain has some structural differences from its adult counterpart that may explain the different responses to injury often seen in children after TBI. The brain doubles in size in the first 6 months of life and reaches approximately 80% of adult size by age 2. The developing brain has a higher water content and incomplete neuronal synapse formation and arborization. In addition, incomplete myelinization and neurochemical changes result in neuronal plasticity after birth. The subarachnoid space is generally smaller and offers less protection than the mature brain, owing to less buoyancy, and thereby provides less protection to the brain parenchyma during changes in head momentum. The result is a higher incidence of diffuse cerebral edema and parenchymal injuries in children. In children with distensible skulls, some argue that the open fontanelle allows for expansion of the intracranial contents and therefore affords increased protection from elevation of ICP. However, studies have indicated that the smaller neural axis of infants and young children results in a less compliant pressure–volume relationship with an increased risk of intracranial hypertension.8,9
The Monro–Kellie doctrine is an important concept relating to the understanding of ICP dynamics (Fig. 17-1). The Monro–Kellie doctrine uses a simple hydraulic approach to the cerebral circulation. Given that the cranium is a rigid, nonexpansile container, it states that the total volume of the intracranial contents must remain constant and any increase in the volume of one component must be at the expense of the others, assuming the intracranial volume remains constant. Thus, very early after injury, a mass such as an expanding hematoma may be enlarging while the ICP remains normal. Once the limit of displacement of CSF and intravascular blood has been reached, ICP rapidly increases (Fig. 17-2). Further work has shown that the relationship between ICP and cerebral blood flow (CBF) is much more complex and variable. Whereas simultaneous measurement of ICP and CBF would be most helpful in optimizing therapeutic strategies, the Monro–Kellie doctrine provides a reasonable basic explanation of intracranial dynamics.
FIGURE 17-1 Monro–Kellie doctrine. The Monro–Kellie doctrine states that the cranial compartment is incompressible and that the volume inside the cranium is a fixed volume. The cranium and its constituents (blood, cerebrospinal fluid [CSF], and brain tissue) create a state of volume equilibrium such that any increase in volume of one of the cranial constituents must be compensated by a decrease in volume of another.
FIGURE 17-2 Intracranial pressure (ICP)–volume relationship. Small increases in brain volume do not lead to immediate increase in ICP because of the ability of the cerebrospinal fluid to be displaced into the spinal canal as well as the ability the falx cerebri to stretch slightly between the hemispheres and the tentorium between the hemispheres and the cerebellum. However, once the ICP has reached around 25 mmHg, small increases in brain volume can lead to marked elevations in ICP.
CBF is defined as the velocity of blood through the cerebral circulation. In adults, the normal CBF is 50–55 mL/100g of brain tissue/minute. In children, CBF may be much higher depending on their age. At 1 year of age, it approximates adult levels, but at 5 years of age, normal CBF is approximately 90 mL/100 g/min and then gradually declines to adult levels by the mid-late teens. Brain injury severe enough to cause coma can result in a 50% reduction in CBF during the first 6 to 12 hours after injury.10,11 It usually increases over the next 2 to 3 days, but for those patients who remain comatose, CBF remains below normal for days or weeks after injury. There is increasing evidence that such low levels of CBF are inadequate to meet the metabolic demands of the brain early after injury and that regional, if not global, cerebral ischemia results.12–14
Cerebral perfusion pressure (CPP) is the differential pressure of arterial flow into and venous flow out of the brain. CPP may be defined by the difference between mean arterial pressure (MAP) and ICP. CPP is considered the transmural pressure gradient that is ultimately the driving force required for supplying cerebral metabolic needs. As ICP increases following head injury, CPP decreases and blood flow to the brain eventually declines. At a CPP of 10 mmHg, blood vessels collapse and blood flow ceases.
Current techniques available to measure CBF, such as transcranial Doppler and Xenon-enhanced CT imaging, are still considered experimental in the management of severe TBI. Because CPP is easily determined by ICP monitoring, it has become a critical parameter for defining treatment options. Studies have shown a good correlation between CPP and CBF in patients with intact cerebral autoregulation.13 However, cerebral autoregulation is often disrupted after severe TBI and measures of cerebral vascular resistance may be more useful in guiding therapy.15,16
Cerebral autoregulation refers to a homeostatic process that allows CBF to remain constant over a wide range of MAPs. Arterial vessels can dilate or constrict in response to various physiologic changes, including ICP and systemic arterial pressure, to maintain normal flow and normal brain metabolism. In healthy adult patients, CBF remains constant with a MAP between 60–160 mmHg, or a CPP between 50–150 mmHg.17 Normally, with elevated systemic blood pressure, reflexive vasoconstriction will occur to prevent intracranial hypertension. In contrast, a moderate decrease in systemic blood pressure will paradoxically result in increased ICP because compensatory reflex vasodilatation will occur. When perfusion pressure falls below 50 mmHg, cerebral ischemia develops and compensatory cerebral arteriole vasodilatation is exhausted. When perfusion pressure exceeds 150 mmHg, cerebral arteriolar impedance is overcome, the affected vessels passively dilate, and fluid is forced through a damaged endothelium into the brain, causing diffuse vasogenic edema. Impaired cerebral autoregulation after TBI and age-related changes in CBF make the immature brain susceptible to secondary injury, both from diminished and excess CBF, and are both associated with a poor neurologic outcome.18
Historically, treatment protocols were principally directed toward reducing ICP. Hyperventilation and fluid restriction were important components in these older protocols. Sustained elevations in ICP above 20 mmHg are poorly tolerated by the injured brain and have been associated with poor neurologic outcome and increased mortality in pediatric patients.19 Sustained elevation in ICP may result in cerebral ischemia if cerebral perfusion is impaired, and ultimately may result in cerebral herniation. Current treatment strategies seek to optimize CPP while reducing ICP, with little reliance on hyperventilation or fluid restriction. CPP is likely an age-related continuum, thus making it problematic to develop treatment protocols based on a single number for all age groups. To date, no study has demonstrated that active maintenance of CPP above any target threshold in infants and children following TBI improves mortality or morbidity. However, there seems to be a threshold of less than 40 mmHg that is associated with increased mortality; therefore, most treatment guidelines recommend a minimum CPP of 40 mmHg.
Primary Brain Injury
Skull fractures occur commonly with head injury and are readily diagnosed with CT. In children, a skull fracture should prompt an evaluation of the underlying brain parenchyma given the significant impact it takes to injure the skull. Fractures of the skull vault can occur in either a linear or a stellate fashion. Fractures involving the skull base are typically associated with a greater force than simple cranial vault fractures. The classic signs of basilar skull fractures include Battle’s sign (ecchymoses over the mastoid process associated with an ipsilateral skull fracture), raccoon eyes and CSF rhinorrhea (associated with a cribriform plate fracture), and otorrhea (associated with fracture of the mastoid air cells or temporal bone fracture). Meningitis associated with a basilar skull fracture occurs in up to 10% of patients.20 Despite the risk of infection, the routine use of prophylactic antibiotics is not recommended as they have not been shown to prevent meningitis from occurring, and tend to select out for resistant organisms.21–23 Vaccination against Streptococcus pneumoniae should be considered for all patients with a basilar skull fracture and CSF leak due to the increased risk of pneumococcal-associated meningitis.24
Post-traumatic intracranial hemorrhage includes epidural hematomas, subdural hematomas, and subarachnoid hemorrhages. Epidural hematomas usually occur in the middle fossa and are often associated with an injury to the middle meningeal artery, although they can occur in the anterior or posterior fossa. The classic CT description is a lenticular hematoma, bound by suture lines, because of the tightly bound dura (Fig. 17-3). Clot formation under the calvaria compresses the dura and can cause rapid neurologic deterioration as the brain becomes further displaced. Skull fractures overlying the epidural hematoma are common. The classic presentation of a patient with a lucid interval followed by clinical deterioration is rare in children. Only after the hematoma enlarges is clinical evidence of elevated ICP noted. Typical symptoms include headache, lethargy, emesis, irritability, confusion, and decreased level of consciousness. Progressive deterioration results in seizures, changes in vital signs with hypertension and respiratory instability, pupillary changes, posturing, and cardiovascular compromise. Prompt neurosurgical evacuation is imperative for patient survival and good outcome. Evacuation of extremely large clots (>40 mL) in children often results in very good long-term results, provided that operative intervention is timely.
FIGURE 17-3 CT scan shows an epidural hematoma (asterisk) in a 14-year-old patient. (Courtesy of Dr Lisa Lowe.)
Subdural hemorrhages are classified as acute (<3 days old), subacute (3-10 days old), and chronic (>10 days old). Acute and subacute subdural hemorrhages are not infrequent in infants, and often the result of birth injury or NAT (Fig. 17-4). They usually result from lacerated bridging veins, or from associated contusions hemorrhaging into the subdural space. The superficial cortical veins in small children lack any reinforcement from arachnoidal trabeculae and are susceptible to inertial loading. Subdural hematomas tend to follow the convexities of the brain and cover the entire hemisphere. The cranial CT demonstrates hyperdense crescent-shaped blood collections at the surface of the brain, often associated with mass effect and cortical edema. Occasionally, and particularly when anemia is present, the CT findings of an acute subdural hematoma may have an isodense appearance that belies the actual hemorrhagic character later found at the time of operation.
FIGURE 17-4 Hyperacute subdural bleeding (arrow) is seen on this cranial CT scan of a 9-month-old patient. (Courtesy of Dr Lisa Lowe.)
Subarachnoid hemorrhage (SAH) in acutely traumatized children is common and is rarely the result of aneurysmal bleeding (Fig. 17-5). Subarachnoid bleeding occurs from disruption of the fragile pia-arachnoidal vasculature. This often occurs over the convexities of the brain affected by coup type injuries or the frontotemporal poles affected by contrecoup injuries. Traumatic SAH can also occur in the interhemispheric fissure and in the basilar cisterns. When SAH is an isolated finding following minor trauma, no specific therapy is indicated except symptomatic amelioration of chemical meningitis, meningismus, and photophobia. SAH can result in hydrocephalus and may require ventricular shunting to relieve the increased ICP. In patients with severe TBI, SAH is associated with a poor outcome and may also be associated with cerebral vasospasm. Transcranial Doppler imaging can be utilized to identify vasospasm. Therapy for vasospasm in adults include calcium channel blockers and neurointerventional techniques; however, these are not well studied in children and not commonly used.25,26
Secondary Brain Injury
Edema of the brain is an important marker for injury and is also a cause of secondary injury. In early (<24 hours) fatal closed-head injuries in children, CT scans often demonstrate little or no significant parenchymal bleeding. However, in children, rapid development of edema is commonly seen on serial CT scans and the diffuse brain swelling causes the obliteration of the ventricles and loss of the basilar cisterns and subarachnoid space. As swelling progresses and the compensatory mechanisms of the brain are exhausted, ICP increases markedly with small changes in intracranial volume (see Fig. 17-2). Cerebral edema typically develops early after injury, peaking at 72 to 96 hours, and then gradually resolves over the next week in survivors.27
Studies in adults utilizing xenon CT have shown a reduction in CBF early after severe TBI.10,12 This hypoperfusion may be further exacerbated by hypotension and hypoxia. It is clear that this early hypoperfusion or ischemia after severe TBI is associated with a poor outcome.28,29 Proposed mediators involved in early post-traumatic ischemia include direct vascular disruption, production of endothelin-1 (a potent vasoconstrictor), loss of endogenous vasodilators (endogenous nitric oxide synthase), and likely many other complex, interrelated, cellular and metabolic events.
The release of excitatory amino acids, such as glutamate, results in neuronal injury after TBI. Glutamate is the most abundant neurotransmitter in the brain. However, toxic levels cause neuronal cell death.30,31 After TBI, glutamate and other excitatory amino acids are released, resulting in neuronal swelling, calcium influx, and release of cytotoxic enzymes leading to cell death. Studies have failed to demonstrate efficacy of anti-excitotoxic therapies, perhaps owing to their application in all patients with TBI rather than those with excitotoxicity, and because treatment may have been initiated too late.32
Initial Evaluation and Management of Head Injury
As with any trauma, the initial management and resuscitation begins with an assessment of circulation, airway and breathing (CAB). Ensuring adequate oxygenation and ventilation and promptly addressing sources of ongoing blood loss serve as the basic principles in the management of persons suspected or confirmed to have head injury. Hypotension and hypoxia in the field are proven secondary insults that are associated with poor outcomes, with hypotension being considerably more detrimental than hypoxia.28,33 There has been no documented advantage to endotracheal intubation over effective bag-valve-mask ventilation in the field.34,35
[/not-level-membership-for-pediatrics-category]
