Head Injury
Perspective
The devastating consequences of head injury have been recorded since ancient times. Early neurosurgical records were primarily observational, with very few suggestions for treatment (Fig. 41-1). Despite centuries of investigation and the development of new and better intensive care, we still have not discovered effective therapies that can be applied after the injury to reverse most pathologic aspects of traumatic brain injury (TBI).

In 1996, Congress approved the Traumatic Brain Injury Act (Public Law 104-166), which charged the Centers for Disease Control and Prevention (CDC) with “determining the incidence and prevalence of traumatic brain injury in all age groups in the general population of the United States.”1
Each year, an estimated 1.7 million people sustain head injury in the United States.1
Of these, 1.36 million (1.4% of all emergency department [ED] visits) undergo emergency evaluation, including approximately 470,000 children younger than age 14 years. TBI accounts for 15.1% of all injury-related hospitalizations in the United States. Overall, 80% sustain minor head trauma (Glasgow Coma Scale [GCS] score of 14 or 15), 10% have moderate head injuries (GCS score of 9-13), and 10% have severe head injuries (GCS score of 8). Almost 20% of all head-injured patients are hospitalized, and approximately 52,000 patients die each year from TBI (Table 41-1).
Table 41-1
Traumatic Brain Injury (TBI) as a Portion of All Injuries and Emergency Department (ED) Visits
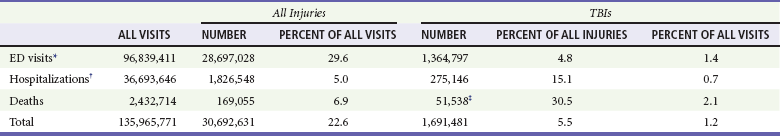
*Persons who were hospitalized, died, or were transferred to another facility were excluded.
†In-hospital deaths and patients who transferred from another hospital were excluded.
‡128 mortality records (from 2002-2006) were omitted because of missing age information.
(Adapted from Faul M XL, Wald MM, Coronado VG: Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002-2006. Atlanta, Centers for Disease Control and Prevention, National Center for Injury Prevention and Control, 2010, pp 1-74.)
The leading causes of head injury in the civilian population are falls (43.7%) and motor vehicle collisions (MVCs) (21.5%).1 TBI caused by blasts has been called the signature injury of the wars in Iraq and Afghanistan: TBI of any severity is estimated to affect as many as 10 to 20% of wartime service members.2–4 Head injury is the leading cause of traumatic death in patients younger than 25 years and accounts for nearly one third of all trauma deaths.1 Head injury from child abuse is common.5 New data suggest that with children younger than 12 months of age, TBIs and/or fractures attributable to abuse occurred in 1 of 2000 infants.6 The CDC estimates that there are at least 5.3 million Americans currently experiencing some degree of disability as a result of TBI. As veterans return to the United States, the number of patients experiencing the consequences of TBI has increased markedly.
Principles of Disease
Scalp and Cranium
The skull comprises the frontal, ethmoid, sphenoid, and occipital bones and two parietal and two temporal bones. The unique layered architecture of the bones of the skull enhances its strength. Each bone consists of solid inner and outer layers separated by a layer of cancellous bone tissue (the diploë). In adults the bones of the skull average 2 to 6 mm in thickness; the bones in the temporal region are usually the thinnest of the skull.7 The cranial bones form a smooth outer surface of the skull, but within the cranial vault are many bone protrusions and ridges. Contrecoup injuries and contusions far from the site of head impact often occur as the accelerating brain strikes against these uneven bone surfaces.
The cranial vault is rigid and nonexpandable, with an average volume in adults of approximately 1900 mL.8 Cranial contents exit or enter the skull through many foramina. The largest, the foramen magnum, is the site of exit of the brainstem and spinal cord from the cranium.
Brain and Cerebrospinal Fluid
Blood-Brain Barrier.: The blood-brain barrier (BBB) maintains the microenvironment of the brain tissue. Extracellular ion and neurotransmitter concentrations are regulated by movement across this barrier. When the BBB is intact, the ability of neuroactive drugs to penetrate into the brain tissue usually depends on their lipid solubility. Post-traumatic cerebral edema and possibly the biomechanics of the injury itself can cause a prolonged disruption of the BBB for up to several hours after trauma.9,10 Prolonged disruption of the BBB contributes to the development of post-traumatic vasogenic cerebral edema.
The brain has an extremely high metabolic rate, using approximately 20% of the entire oxygen volume consumed by the body. To provide for its high metabolic demands, the brain requires approximately 15% of the total cardiac output. Optimal regional CBF is maintained by the ability of cerebral vessels to alter their diameter in response to changing physiologic conditions.10 Hypertension, alkalosis, and hypocarbia promote cerebral vasoconstriction; hypotension, acidosis, and hypercarbia cause cerebral vasodilation. In the normal brain, CBF is maintained at constant levels with a mean arterial pressure (MAP) of 60 to 150 mm Hg. This is referred to as autoregulation. Outside this range, the CBF varies linearly with MAP.
Cerebral vasoactivity is very sensitive to changes in systemic carbon dioxide and oxygen partial pressures (PCO2 and PO2, respectively). The response to changes in PCO2 is nearly linear between PCO2 values of 20 and 60 mm Hg.11 In this range, lowering PCO2 by as little as 1 mm Hg decreases the diameter of cerebral vessels by 2 or 3%, which corresponds to an overall change in CBF of 1.1 mL per 100 g of tissue per minute. The physiologic response of blood vessels to PCO2 is the rationale for the acute use of brief hyperventilation to control increased intracranial pressure (ICP) after head injury. As PCO2 decreases with hyperventilation, cerebral vasoconstriction occurs. As a result, the volume of blood per unit area of brain tissue decreases. This decrease (even if small) may buffer the effects of increasing edema or an expanding hematoma within the rigid cranial vault. The vasoconstriction produced by extreme changes in PCO2 (20 mm Hg or less) can be so pronounced that some areas of brain experience ischemia; subsequently, tissue hypoxia can occur.10,12,13 Therefore hyperventilation is controlled and monitored, with a goal of maintaining the PCO2 between 30 and 35 mm Hg during prolonged hyperventilation, and is reserved for patients who are showing signs of acute herniation.12,13 Over 12 to 24 hours, injured vessels may lose their responsiveness to hyperventilation-induced hypocarbia and become vasodilated. Blood may then be shunted to the injured area, resulting in increased brain swelling and mass effect. Prolonged (i.e., beyond the acute resuscitation) or prophylactic hyperventilation is therefore not recommended as a treatment for increased ICP, and hyperventilation is not used for the routine management of head-injured patients with no signs of increased ICP.12,13 The neurologic effects of hypocapnia are illustrated in Figure 41-2.
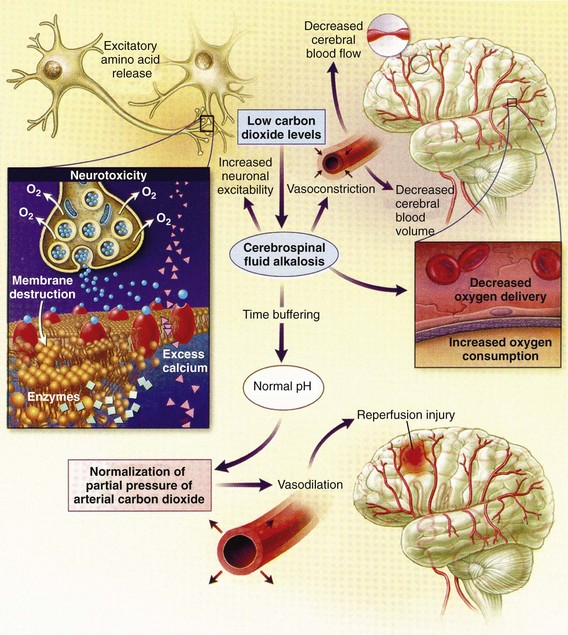
The cerebral vessels also respond to changes in PO2. As PO2 declines, cerebral vessels dilate to ensure adequate oxygen delivery to brain tissue. When brain injury has occurred, increased CBF in the presence of a disrupted BBB can promote the formation of vasogenic edema. Avoiding or reversing hypoxia is therefore an essential goal in the acute management of the head-injured patient.14 The responses of the cerebral vasculature to changing physiologic conditions protect the brain by increasing the delivery of oxygen to tissue, enhancing the removal of metabolic end products, and allowing nearly instantaneous adjustments of regional blood flow to meet the changing metabolic demands.
Cerebral Perfusion Pressure.: CBF also depends on cerebral perfusion pressure (CPP), which is the pressure gradient across the brain. The determinants of CPP are MAP and the resistance to CBF produced by mean systemic venous pressure and ICP. Because ICP is higher than mean systemic venous pressure, ICP effects predominate. Therefore CPP is estimated as
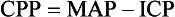
Direct Injury.: Direct impact head injury occurs when the head is struck by an object or its motion is arrested by another object. The resulting damage depends on the consistency, mass, surface area, and velocity of the object striking the head. Direct injury can also be caused by compression of the head. External signs of trauma are frequently noted at the site of application of the impact or compression force. The skull initially bends inward at the point of contact. If the force is sufficient, a skull fracture can occur. The cranium absorbs some of the applied energy, and some energy is transmitted to the brain by shock waves that travel distant to the site of impact or compression. These shock waves distort and disrupt intracranial contents and temporarily alter regional ICP as they propagate. In general, the more rapidly a force is applied, the greater the damage it causes. The extent of direct injury depends on the vasoelastic properties of the underlying region of brain tissue, the duration of the force applied, the magnitude of the force reaching the brain tissue, and the surface area of the brain that is affected by the application of the force. In cases of penetrating trauma, the mass, shape, direction, and speed of the penetrating object also affect the extent of direct injury.
Indirect Injury.: In indirect brain injury, the cranial contents are set into motion by forces other than the direct contact of the skull with another object. A common example is acceleration-deceleration injury, such as the shaken impact syndrome.15,16 No direct mechanical impact is sustained, but the cranial contents are set into vigorous motion. The brain moves within the skull, and bridging subdural vessels are strained. Subdural hematomas (SDHs) may result. Differential acceleration of the cranial contents occurs, depending on the physical characteristics of the brain region. As one brain region slides past another, shear and strain injuries are produced. This movement results in diffuse injuries, such as diffuse axonal injury (DAI) or concussion. Additional injury occurs as the movement of the intracranial contents is abruptly arrested and the brain strikes the skull or a dural structure. Contrecoup contusions are an example of the injury produced in this manner. In penetrating injury the traversal of the object produces pressure waves that can strike structures distal to the path of the missile.
Brain Cellular Damage and Death
Primary and Secondary Brain Injuries
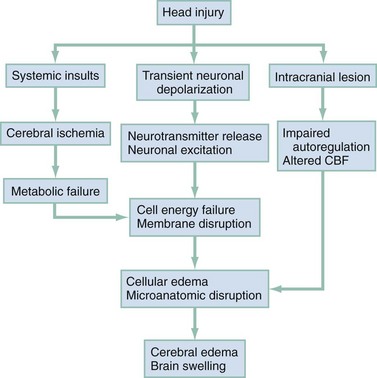
The circumstances and extent of the primary injury are not the only contributors to the final neurologic outcome after head injury. The traumatic event also produces injury at the functional and anatomic cellular level, which begins soon after the impact and continues for several hours and even days after injury. Secondary brain injury results from intracellular and extracellular derangements that are probably initiated at the time of trauma by a massive depolarization of brain cells and subsequent ionic shifts.17 Animal studies have revealed a complicated series of neurochemical, neuroanatomic, and neurophysiologic reactions after head injury (Fig. 41-3). The cell has some compensatory mechanisms to protect itself from widespread damage, such as endogenous free radical scavengers and antioxidants. With significant trauma, however, these systems are quickly overwhelmed, and the functional and structural integrity of the cell is threatened. Human studies document similar changes. The relative importance and contribution of each adverse reaction to the final functional status of the damaged cell are uncertain, as are the rate and duration of each detrimental event. All currently used acute therapies for TBI are directed at reversing or preventing secondary injury. Experimental evidence for many investigational agents aimed at specific steps in the destructive processes suggests that some aspects of secondary brain injury may be reversed or modified. Multiple ongoing head injury trials have been performed with numerous investigational therapeutic interventions; to date, none have proved useful in the clinical setting.18,19
Secondary Systemic Insults
The final neurologic outcome after head trauma is influenced by the extent and degree of secondary brain injury. In turn, the amount of secondary brain injury depends on certain premorbid and comorbid conditions, such as the age of the patient and trauma-related systemic events.19–21 A primary goal in the emergency care of the head-injured patient is prevention or reduction of systemic conditions that are known to worsen outcome after TBI.
Common secondary systemic insults in trauma patients include hypotension, hypoxia, and anemia and hyperpyrexia. Hypotension, defined as a systolic blood pressure less than 90 mm Hg, has been found to have negative impact on severe head injury outcome.18,19,21,22 Systemic hypotension reduces cerebral perfusion, thereby potentiating ischemia and infarction. The presence of hypotension nearly doubles the mortality from head injury and worsens the outcome of the patients who survive.17,21–23
Hyperpyrexia (core body temperature of 38.5° C) is also correlated with worsened outcomes after TBI, and both its magnitude and its duration seem to contribute. The exact mechanism by which it causes damage is yet to be determined but likely involves stimulation of metabolism in injured areas of the brain, thus recruiting blood flow with a resultant increase in ICP.18,21
Hypoxia, defined as a PO2 less than 60 mm Hg, probably occurs often in the head-injured patient. Causes include (1) transient or prolonged apnea caused by brainstem compression or injury after the traumatic event; (2) partial airway obstruction caused by blood, vomitus, or other debris in the airway of the traumatized patient; (3) injury to the chest wall that interferes with normal respiratory excursion; (4) pulmonary injury that reduces effective oxygenation; and (5) ineffective airway management, such as the inability to bag-valve-mask or intubate the patient in an effective or timely manner. The exact incidence of hypoxia in the head-injured patient is difficult to estimate because it is often unnoticed or undocumented in the out-of-hospital setting. When its occurrence is documented, the overall mortality from severe head injury may double.18,22–24 Increased recognition of the potentially devastating consequences of hypoxia and associated hypercarbia has led to more vigilance in out-of-hospital and emergency settings.
Anemia caused by blood loss can be detrimental to the head-injured patient by reducing the oxygen-carrying capacity of the blood, thus reducing the amount of necessary substrate delivered to the injured brain tissue. When anemia (hematocrit 30%) occurs in patients with severe head injury, the mortality rate increases.18,21 Other potential reversible causes of systemic insult in head injury include hypercarbia, coagulopathy, and seizures.
Pathophysiology
Increased Intracranial Pressure
ICP represents a balance of the pressures exerted by the contents of the cranial cavity. This relationship is explained by the Monro-Kellie doctrine.25,26 Because the craniospinal intradural space is almost nonexpandable, the sum of the volume of brain, CSF, and blood within the cranium will remain constant. If the volume of any of these components increases, the volume of another must decrease for a constant ICP to be maintained. Increased ICP is defined as CSF pressure greater than 15 mm Hg (or 195 mm H2O) and is a frequent consequence of severe head injury. Initially, as ICP increases as a result of a traumatic mass lesion or edema formation, the CSF is displaced from the cranial vault to the spinal canal, offsetting the increased blood or brain volume. When this compensatory mechanism is overwhelmed, the elastic properties of the brain substance allow tissue compression to provide buffering for the increasing pressure. Depending on the location and the rate of expansion of the traumatic mass lesion and the rate of cerebral edema formation, the intracranial compensatory mechanisms can accommodate an increased volume of 50 to 100 mL. Beyond that, even small additional changes in intracranial volume relationships, such as those caused by vasodilation, CSF obstruction, or small areas of focal edema, cause a dramatic increase in ICP. If ICP increases to the point at which CPP is compromised, vasoparalysis occurs and autoregulation is lost. The CBF then depends directly on the systemic MAP. With the loss of autoregulation, massive cerebral vasodilation occurs. Systemic pressure is transmitted to the capillaries, and the outpouring of fluids into the extravascular space can contribute to vasogenic edema and thus further increase ICP. If ICP rises to the level of the systemic arterial pressure, CBF ceases and brain death occurs.
Methods to reduce elevated ICP include hyperventilation, use of osmotic and diuretic agents, and CSF drainage. Uncontrollable increased ICP is defined as an ICP of 20 mm Hg or higher refractory to treatment. If ICP is not controlled, herniation syndromes can occur, resulting in brainstem compression and subsequent cardiorespiratory arrest. In the United States, the use of ICP monitoring and control has become standard in cases of moderate and severe TBI despite the lack of prospective controlled research showing clear efficacy as an individual patient treatment modality.18
Brain Swelling and Cerebral Edema
Two primary types of brain swelling occur after head injury: congestive brain swelling and cerebral edema. Congestive brain swelling results from an increased intracranial blood volume. Hyperemia occurs early after trauma and can persist for the first few days after injury.23,27 It is especially common in children. The increased blood volume is most likely caused by vasodilation, which occurs as a compensatory mechanism to maintain optimal CBF in the presence of increased metabolic needs of the damaged brain tissue.
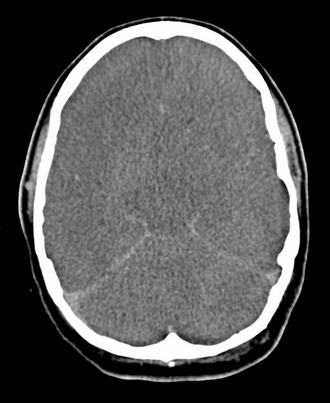
Cerebral edema is an increase in brain volume caused by an absolute increase in cerebral tissue water content. Diffuse cerebral edema may develop soon after head injury; however, its presence and extent do not always correlate with the severity of head injury. On computed tomography (CT) scans, diffuse edema manifests as bilateral compression of the ventricles, loss of definition of the cortical sulci, or effacement of the basal cisterns (Fig. 41-4). Focal edema adjacent to traumatic mass lesions demonstrates decreased density on CT scans compared with normal tissue. CT can also detect a mass effect, caused by edema surrounding a traumatic lesion.
Both vasogenic and cytotoxic cerebral edema occur in the setting of trauma; the incidence and onset of each relative to the other depend on the nature of the injury. Vasogenic edema arises from transvascular leakage caused by mechanical failure of the tight endothelial junctions of the BBB.23,27 Vasogenic edema accumulates preferentially in white matter and can become widespread. It is frequently associated with focal contusions or hematomas. Vasogenic edema eventually resolves as edema fluid is reabsorbed into the vascular space or the ventricular system.
Cytotoxic edema is an intracellular process that results from membrane pump failure. It is common after head injury and is frequently associated with post-traumatic ischemia and tissue hypoxia. Normal membrane pump activity depends on adequate CBF to ensure adequate substrate and oxygen delivery to brain tissue. If the CBF is reduced to 40% or less of baseline, cytotoxic edema begins to develop. If CBF drops to 25% of baseline, membrane pumps fail and cells begin to die. Congestive brain swelling can contribute to cytotoxic edema if it becomes severe enough to increase ICP and reduce CPP so that cerebral circulation cannot be maintained. Recent work suggests that cytotoxic cerebral edema is the predominant form of edema in patients who have experienced TBI.23,28
Cushing’s Reflex
Progressive hypertension associated with bradycardia and diminished respiratory effort is a specific response to acute, potentially lethal increases in ICP. This response is called Cushing’s reflex or Cushing‘s phenomenon, and its occurrence indicates that the ICP has reached life-threatening levels. Cushing’s reflex can occur whenever ICP is increased, regardless of the cause. The full triad of hypertension, bradycardia, and respiratory irregularity is seen in only one third of cases of life-threatening increased ICP.23,29
Cerebral herniation occurs when increasing cranial volume and ICP overwhelm the natural compensatory capacities of the CNS (Fig. 41-5). Increased ICP may be the result of post-traumatic brain swelling, edema formation, traumatic mass lesion expansion, or any combination of the three. When increasing ICP cannot be controlled, the intracranial contents shift and herniate through the cranial foramen. Herniation can occur within minutes or up to days after TBI. When the signs of herniation syndrome are present, however, mortality approaches 100% without rapid implementation of temporizing emergency measures and definitive neurosurgical therapy.
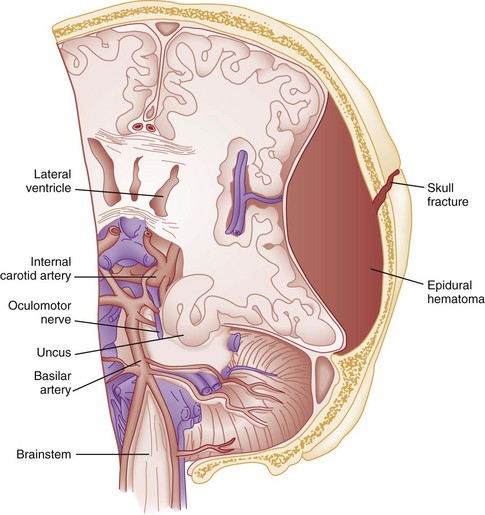
Uncal Herniation.: The most common clinically significant traumatic herniation syndrome is uncal herniation, a form of transtentorial herniation. Uncal herniation is often associated with traumatic extra-axial hematomas in the lateral middle fossa or the temporal lobe. The classic signs and symptoms are caused by compression of the ipsilateral uncus of the temporal lobe on the U-shaped edge of the tentorium cerebelli as the brain is forced through the tentorial hiatus. As compression of the uncus begins, the third cranial nerve (CN) is compressed; anisocoria, ptosis, impaired extraocular movements, and a sluggish pupillary light reflex develop on the side ipsilateral to the expanding mass lesion. This phase may last for minutes to hours, depending on how rapidly the expanding lesion is changing. As the herniation progresses, compression of the ipsilateral oculomotor nerve eventually causes ipsilateral pupillary dilation and nonreactivity.
Initially in the uncal herniation process, motor examination findings can be normal, but contralateral Babinski’s responses develop early.23 Babinski’s sign is dorsiflexion of the great toe and fanning of the other toes. Contralateral hemiparesis develops as the ipsilateral peduncle is compressed against the tentorium. With continued progression of the herniation, bilateral decerebrate posturing eventually occurs; decorticate posturing is not always seen with the uncal herniation syndrome. Pupillary dilation and nonreactivity are the most reliable physical examination findings with regard to lesion location.
Central Transtentorial Herniation.: The central transtentorial herniation syndrome is demonstrated by rostrocaudal neurologic deterioration caused by an expanding lesion at the vertex or the frontal or occipital pole of the brain. It is less common than uncal transtentorial herniation. Clinical deterioration occurs as bilateral central pressure is exerted on the brain from above. The initial clinical manifestation may be a subtle change in mental status or decreased level of consciousness, bilateral motor weakness, and pinpoint pupils (2 mm). Light reflexes are still present but are often difficult to detect. Muscle tone is increased bilaterally, and bilateral Babinski’s signs may be present. As central herniation progresses, both pupils become midpoint and lose light responsiveness. Respiratory patterns are affected, and sustained hyperventilation may occur. Motor tone increases. Decorticate posturing is elicited by noxious stimuli. This progresses to bilateral decorticate and then spontaneous decerebrate posturing. Respiratory patterns initially include yawns and sighs and progress to sustained tachypnea, followed by shallow slow and irregular breaths immediately before respiratory arrest.
Cerebellotonsillar Herniation.: Cerebellotonsillar herniation occurs when the cerebellar tonsils herniate downward through the foramen magnum. This is usually caused by a cerebellar mass or a large central vertex mass causing the rapid displacement of the entire brainstem.23 Clinically, patients demonstrate sudden respiratory and cardiovascular collapse as the medulla is affected. Pinpoint pupils are noted. Flaccid quadriplegia is the most common motor presentation because of bilateral compression of the corticospinal tracts. Mortality from cerebellar herniation approaches 70%.23
Upward Transtentorial Herniation.: Upward transtentorial herniation occasionally occurs as a result of an expanding posterior fossa lesion. Level of consciousness declines rapidly. These patients may have pinpoint pupils from compression of the pons. Downward conjugate gaze is accompanied by the absence of vertical eye movements.
Clinical Features and Diagnostic Strategies
Acute Neurologic Examination
Glasgow Coma Scale
The GCS is an objective method of following the patient’s neurologic status (Table 41-2). The GCS assesses a patient’s best eye, verbal, and motor responsiveness. It was developed for the clinical evaluation of head trauma patients at 6 hours after trauma, and all initial validation studies investigated its application at this time. It was designed for assessment of patients with isolated head trauma who were hemodynamically stable and adequately oxygenated.26 The GCS is only one aspect of the neurologic examination (e.g., the motor score reflects best limb movement, and it cannot detect subtle changes in mental status). However, because of its inter-rater reliability, reliance on objective clinical data, and ease of application, the GCS has become a standard acute measure of neurologic function in patients with altered mental status from any cause, including head trauma.
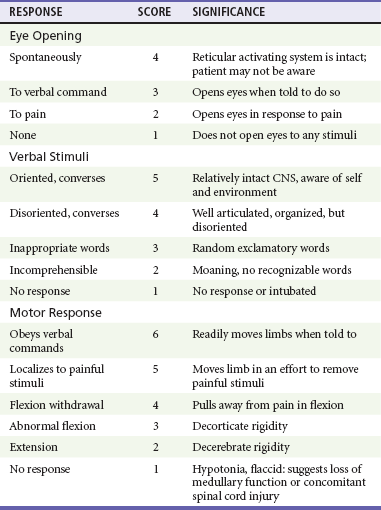
The acute application (within 6 hours) of the GCS in head-injured patients has limitations. Hypoxia, hypotension, and intoxication can falsely lower the initial GCS score.26 Intubation lowers the patient’s GCS score by automatically assigning a score of 1 for verbal response, regardless of the actual contribution of head injury to the clinical examination. Periorbital edema from direct eye trauma may make assessment of spontaneous eye opening difficult. Extremity fractures or occult spinal cord injuries may interfere with the motor examination. Children and patients with difficulty communicating are challenging to assess with the GCS. The GCS may miss subtle mental status changes and does not assess brainstem reflexes or pupillary reflexes. Decisions regarding continued resuscitation of severely head-injured patients should not be based on the initial GCS score because of these limitations. Patients should be fully resuscitated, with evacuation of all surgical lesions, and remain hemodynamically stable, and not be intoxicated before the GCS can be used to predict their prognosis.26
Motor Examination: Posturing
The patient’s acute motor examination assesses for strength and symmetry. Paralysis obscures involuntary reflexes; attempts should be made to perform the motor examination before paralytic agents are given. Hemiparesis contralateral to a fixed and dilated pupil suggests herniation syndrome. A false-localizing motor examination can be caused by contralateral cerebral parenchymal injury occurring simultaneously with the expanding mass lesion or by Kernohan’s notch syndrome (compression of the contralateral cerebral peduncle). False-localizing motor signs can also be caused by extremity trauma, spinal cord, or nerve root injury that makes the examination painful or difficult. If the patient is not cooperative or is comatose, motor movement should be elicited by application of noxious stimuli. Any movement should be recorded. Voluntary purposeful movement must be distinguished from abnormal motor posturing. Decorticate posturing is abnormal flexion of the upper extremity and extension of the lower extremity. The arm, wrist, and elbow slowly flex, and the arm is adducted. The leg extends and internally rotates, with plantar flexion of the foot. Decorticate posturing implies injury above the midbrain. Decerebrate posturing is the result of a more caudal injury and therefore is associated with a worse prognosis.19 The arms extend abnormally and become adducted. The wrist and fingers are flexed, and the entire arm is internally rotated at the shoulder. The neck undergoes abnormal extension, and the teeth may become clenched. The leg is internally rotated and extended, and the feet and toes are plantar flexed.
Brainstem Function
In the acute setting, brainstem activity is assessed by the patient’s respiratory pattern, pupillary size, and eye movements. The oculocephalic response (doll’s eyes maneuver) tests the integrity of the pontine gaze centers. This response cannot be tested until cervical spine fractures have been ruled out. The oculovestibular response (cold water calorics) also permits assessment of the brainstem. Comatose patients no longer demonstrate nystagmus when cold water is placed in the ear canal; the only response is tonic deviation of the eyes toward the instilled cold water.24 This response is dampened by cerumen or blood in the patient’s ear canal, and the tympanic membrane needs to be intact for this test to be performed.
Other Examination Findings
The head and neck should be carefully examined for external signs of trauma that may have also produced underlying TBI. A scalp laceration, contusion, abrasion, or avulsion may overlie a depressed skull fracture. Basilar skull fractures are usually diagnosed by the clinical examination (Box 41-1). Although not always related to severe brain injury, their presence implies that a significant impact force was sustained during head trauma. Carotid artery dissections caused by a hyperflexion-extension neck injury can occasionally be detected by auscultation of a carotid bruit.24 In these patients, a careful neurologic examination should assess for subtle asymmetry between the carotid arteries. The percentage of concurrent cervical spine injury in patients with severe head trauma may be as high as 10.2%.27 Often, other spinal regions are also injured.
Management
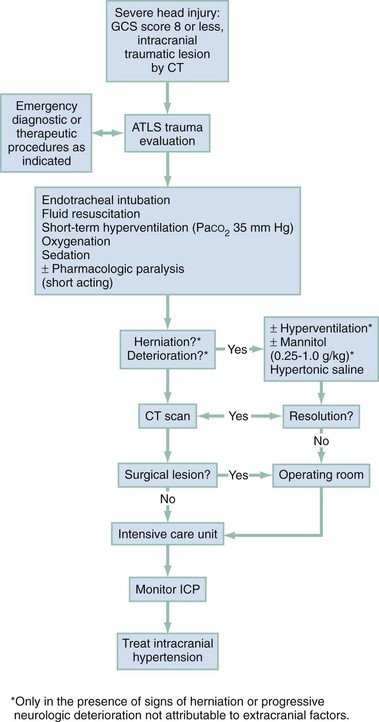
The neurosurgical literature defines severe head injury as TBI manifested by a postresuscitation GCS score of 8 or lower within 48 hours. In the emergency setting, however, this definition is not practical because the outcome for the patient beyond the initial resuscitation is not known. Most emergency medicine research defines severe head injury by a GCS score of 8 or lower at the acute presentation after injury. The presence of any intracranial contusion, hematoma, or laceration is also considered severe injury (see Fig. 41-5).
Approximately 10% of all head-injured patients who reach the ED alive have severe head trauma.1,30 The clinical prognostic indicators in the acute setting are initial motor activity, pupillary responsiveness, the patient’s age and premorbid condition, and the occurrence of secondary systemic insult during the acute period.23,28,31 Up to 25% of these patients have lesions requiring neurosurgical evacuation.18 The prognosis cannot be reliably predicted by the initial GCS score or initial CT scan.
The overall mortality in severe head trauma has improved over the years and now remains stable at approximately 35%.32,33 Mortality for children is lower. For nonsurvivors of head injury who reach the hospital alive, the average time to death is 2 days after trauma. Adult survivors of severe head trauma are usually severely disabled; currently, only 7% have moderate disability or a good outcome. Children older than 2 years who survive a severe closed head injury have a better outcome than adults.
Out-of-Hospital Care
Head trauma can produce profound effects on the cardiovascular system if compression of the brainstem and medulla occurs. Any cardiac dysrhythmia can occur and produce cardiac instability.23,34 All head-injured patients should be placed on a cardiac monitor during transport from the accident scene.
The secondary survey of the head-injured patient should include a search for external signs of head trauma. Scalp lacerations may bleed a large volume into a bulky dressing. A less bulky dressing should be used with firm constant manual pressure applied to avoid excessive blood loss. Many severely head-injured patients are initially combative or agitated. Transporting an agitated patient who is fighting against physical restraints may exacerbate physical injury, cause an increase in ICP, and interfere with appropriate stabilization and management. It may be necessary to use out-of-hospital sedation or neuromuscular blockade for control. The use of sedatives or neuromuscular blockade may influence the initial ED evaluation of the neurotrauma patient. Therefore the risks and benefits of this acute intervention are carefully considered and decisions made on a case-by-case basis. Out-of-hospital protocols allowing the use of sedative agents for selected agitated head-injured patients should be established. Currently used agents include lorazepam (Ativan), diazepam (Valium), midazolam (Versed), and certain butyrophenones (e.g., haloperidol, droperidol). Some investigators are looking at the use of ketamine for prehospital sedation in combative head-injured patients. Recent research questions whether the widely held belief that ketamine increases ICP and CBF is accurate.35 Currently, the existing prehospital data are insufficient to recommend ketamine use in combative prehospital head-injured patients.
Severe head injury is the most common reason for helicopter transfers in trauma care. Although the decision to transport by helicopter should be made on a case-by-case basis, considerations for helicopter use from an accident scene include a long extrication time, ground transport of longer than 30 minutes to an appropriate ED and trauma care facility, two or more severely injured patients at a scene, and assistance in performing expedient lifesaving procedures, especially airway management. Several recent studies have supported that the use of helicopter emergency services improves outcomes in severely head-injured patients.24,36–38
Controversy exists regarding the benefits of out-of-hospital intubations in patients with severe and moderate head injuries. It is unclear whether field intubations truly improve neurologic outcome or survival. Unsuccessful attempts at field intubations may add to out-of-hospital time and increase the risk of aspiration or hypoxia.39–41
In 1997 Winchell and Hoyt showed that patients who had sustained severe head injuries and who were intubated in the out-of-hospital setting had an improved survival compared with those who were not intubated.29 Since that time, others have challenged this finding. In the San Diego paramedic rapid sequence intubation (RSI) trial, Davis and colleagues42 found an increase in mortality and morbidity in patients who sustained severe head injuries and underwent out-of-hospital RSI compared with matched historical controls.42 Potential explanations included frequent hypoxic episodes with associated bradycardia, unintentional hyperventilation, and prolonged scene times for those undergoing out-of-hospital RSI. Wang and colleagues found a fourfold increase in mortality among patients who sustained severe TBI and received ground ambulance intubation compared with ED intubation.24 Flight clinician out-of-hospital intubation has been associated with decreased mortality and improved neurologic outcome, likely because of the higher airway management training requirements of air medical programs.24,37–40 These studies are somewhat limited by lack of generalizability24,42–46 (e.g., most emergency medical service systems do not have RSI capabilities) or by the use of nonvalidated outcome measures of neurologic function.24 Out-of-hospital intubation carries the following risks: frequent hypoxic episodes during intubation attempts, with or without concurrent bradycardia; unintentional hyperventilation of intubated patients; prolonged scene times because of the time demands associated with the intubation process; and persistent hypotension, likely from other injuries that go unattended while the airway management occurs. However, hypoxia must be avoided in head-injured patients, and out-of-hospital airway protocols balance the risks of emergency intubation in an uncontrolled setting with the need to secure an airway at risk.
The key to a successful out-of-hospital RSI program involves well-trained clinicians with specific RSI protocols, involved medical control, frequent continuing education, and consistent quality assurance and improvement. Fakhry and colleagues47 reported that their helicopter clinicians had a 96.6% RSI success rate with few complications and no esophageal intubations.
Emergency Department (Fig. 41-6)
Airway.: RSI is the preferred method for securing the airway in combative or agitated patients. If possible, a brief neurologic examination should be performed before the patient is given any sedative or neuromuscular blocking agents. In general, the agents used for RSI in the head-injured patient are the same as those used for other patients, although attention is given to the increased ICP that can potentially occur with any physical stimulation of the respiratory tract. Lidocaine (1.5-2 mg/kg intravenous push) may attenuate the cough reflex, hypertensive response, and increased ICP associated with intubation, although this is vigorously debated.47–49 If succinylcholine is used, premedication with a subparalytic dose of a nondepolarizing agent can be considered if time permits because fasciculations produced by succinylcholine may increase ICP. The degree of ICP elevation and its clinical significance are unclear, however, and are balanced against the need for rapid establishment of an airway. Etomidate (0.3 mg/kg intravenously [IV]), a short-acting sedative-hypnotic agent, has beneficial effects on ICP by reducing CBF and metabolism.50 In addition, etomidate has minimal adverse effects on blood pressure and cardiac output and fewer respiratory depressant effects than other agents. Concern has been raised that single-dose etomidate used during RSI may adversely affect patient outcome by modulating the inflammatory response and transient adrenal suppression.51 However, research both supporting and refuting this position exists, and no definitive answer has yet been established.51–56
Hypotension.: Hypotension is rarely caused by head injury except as a terminal event. If hypotension is detected at any time in the emergent management of a head-injured patient, a cause other than the head injury should be sought. Some important exceptions occur. Profound blood loss from scalp lacerations can cause hypovolemic hypotension. In infants, hemorrhage into an epidural or subgaleal hematoma can produce profound hypovolemic shock. In the presence of a concomitant high spinal cord injury, neurogenic hypotension may occur. This is rare, and usually the cord injury is apparent on physical examination. In less obvious cases, neurogenic hypotension can be differentiated from hypovolemic hypotension by its nonresponsiveness to fluid administration and by the presence of inappropriate bradycardia in the face of hypotension.
Systemic hypotension cannot be tolerated in the head-injured patient without profound worsening of neurologic outcome; fluids or blood transfusion should therefore be delivered to maintain a systolic blood pressure of at least 90 mm Hg.22 The delivery of large amounts of fluid to severely head-injured patients who are hypotensive from other injuries does not produce clinically significant increases in ICP; fluids should never be withheld in the head trauma patient with hypovolemic hypotension for fear of increasing cerebral edema and ICP. The resuscitation goal is to keep the CPP at 50 or above. Hypotension may interfere with the accurate neurologic assessment of the brain-injured patient. Often, when blood pressure is restored, an improved neurologic status is observed.
Traditionally, normal saline or lactated Ringer’s solution has been used for resuscitation of trauma patients with hypovolemic hypotension. Some research suggests that fluid resuscitation with hypertonic saline (HTS) rather than normal saline may improve neurologic outcome after TBI. However, recent data from the Resuscitation Outcomes Consortium showed no improvement in neurologic outcome when HTS was administered in the prehospital setting in severely head-injured patients not in hemorrhagic shock.57
As many as 50% of patients with severe head injury are victims of multiple trauma.22 The dramatic presentation of the head injury should not distract the clinician from a thorough search for other life threats.
Hyperventilation.: Acute hyperventilation is a lifesaving intervention that can prevent or delay herniation in the patient with severe TBI while more definitive resources are mobilized. The goal is to reduce the PCO2 to the range of 30 to 35 mm Hg. Hyperventilation will reduce ICP by causing cerebral vasoconstriction; the onset of effect is within 30 seconds23 and probably peaks within 8 minutes after the PCO2 drops to the desired range. In most patients, hyperventilation lowers the ICP by 25%; if the patient does not rapidly respond, the prognosis for survival is generally poor.
Prolonged hyperventilation is not recommended because it may cause profound vasoconstriction and ischemia. This vasoconstriction worsens CBF that is already severely compromised during the first 24 hours after TBI.12 Hyperventilation should be viewed as a short-term lifesaving intervention and should be used only when a patient experiences an acute neurologic decline or demonstrates signs consistent with herniation.
Osmotic Agents.: Additional therapy for increased ICP includes the use of osmotic diuretics, such as mannitol and HTS. With deepening coma, pupil inequality, or other deterioration of the neurologic examination, osmotic agents may be lifesaving.
Mannitol is the mainstay for control of elevated ICP in acute severe TBI. The Brain Trauma Foundation and the European Brain Injury Consortium recommend mannitol as the osmotic drug of choice.40,41 However, few comparative data exist on mannitol and other ICP-lowering medications.58 A Cochrane database review concluded that mannitol may have a small beneficial effect compared with pentobarbital.59 ICP-directed therapy based on neurologic signs may also be beneficial. However, other evidence suggests that mannitol may be detrimental compared with HTS.59,60 Further research is needed on optimal osmotic therapy in severe head trauma.
Mannitol (0.25-1 g/kg) can effectively reduce cerebral edema by producing an osmotic gradient that prevents the movement of water from the vascular space into the cells during membrane pump failure and draws tissue water into the vascular space. This reduces brain volume and provides increased space for an expanding hematoma or brain swelling. The osmotic effects of mannitol occur within minutes and peak approximately 60 minutes after bolus administration. The ICP-lowering effects of a single bolus may last for 6 to 8 hours.61 Mannitol has many other neuroprotective properties. It is an effective volume expander in the presence of hypovolemic hypotension and therefore may maintain systemic blood pressure required for adequate cerebral perfusion. It also promotes CBF by reducing blood viscosity and microcirculatory resistance. It is an effective free radical scavenger, reducing the concentration of oxygen free radicals that may promote cell membrane lipid peroxidation. However, mannitol can produce renal failure or hypotension if given in large doses. It may also induce a paradoxical effect of increased bleeding into a traumatic lesion by decompressing the tamponade effect of a hematoma. Because of these and other potential problems, the use of mannitol should be reserved for head-injured patients with evidence of increasing ICP and neurologic deterioration.61
HTS has been used for severe TBI since 1919.62 Numerous clinical studies have demonstrated that HTS can significantly reduce ICP; however, the total number of enrolled patients in these trials is small.58,59,62,63 The interpretation of these clinical studies is complicated by variation in protocols, HTS concentration, and administration. Potential adverse events associated with HTS include renal failure, central pontine myelinolysis, and rebound ICP elevation.64
Encouraging clinical data are available on hospitalized pediatric TBI patients treated with a continuous infusion of 3% normal saline for control of intracranial hypertension.65,66 The clinical studies using HTS for acute field resuscitation of severe TBI have had conflicting results. Using a post hoc analysis of adult trauma data, Vassar and colleagues67 and Wade and colleagues68 showed beneficial effects of HTS on patients with severe TBI. However, Cooper and colleagues69 found no benefit for out-of-hospital use of HTS in reducing elevated ICP and improving CPP compared with lactated Ringer’s in hypotensive patients who had a head injury. As noted earlier, a large, multicenter, double-blinded, randomized, placebo-controlled clinical trial showed no improvement in neurologic outcome when HTS was administered in the prehospital setting in severely head-injured patients not in hemorrhagic shock.57
Barbiturates.: Barbiturate therapy is occasionally used in severely head-injured patients to reduce cerebral metabolic demands of the injured brain tissue. Barbiturates also affect vascular tone and inhibit free radical–mediated cell membrane lipid peroxidation. The effects of barbiturates are delayed relative to other acute interventions for reducing ICP; therefore they are rarely initiated in the ED. If other methods of reducing ICP have been unsuccessful, barbiturates may be added in the hemodynamically stable patient. Pentobarbital is the barbiturate most often used.70
Steroids.: Despite their popularity in the past, there is no benefit to giving steroids in head-injured patients. They do not lower ICP, and high-dose methylprednisolone in moderate and severe TBI is associated with increased mortality.71
Hypothermia.: Although hypothermia remains a significant area of research and promise for patients with severe and moderate TBI, the available scientific evidence is inconclusive with regard to improved mortality or morbidity with prophylactic hypothermia in patients with severe and moderate TBI. In a meta-analysis performed by the Brain Trauma Foundation, duration of hypothermic treatment for more than 48 hours was associated with a reduction in mortality. This finding is significantly limited owing to small sample sizes.72 A large trial of hypothermia use in moderate and severe TBI has recently begun in Europe. The Eurotherm3235 Trial will enroll 1800 patients over 41 months.73
Cranial Decompression.: Patients with signs of herniation who have not responded to other means of ICP reduction and who are rapidly deteriorating in the ED should be considered for placement of emergency burr holes. Emergency trephination has been described for centuries. It is a blind invasive procedure, and the chances of localizing the expanding lesions are uncertain. In patients who do not respond to hyperventilation and osmotic agents and have a unilateral dilated pupil with a history of “talk and deteriorate” after head trauma, emergency cranial decompression may temporarily reverse or arrest the herniation syndrome.74–77 In this setting, emergency trephination may allow enough time for a patient to undergo a formal craniotomy in the operating room.
There has been renewed interest among neurosurgeons in the role of emergent decompressive craniectomy. Numerous papers have been published promoting more liberal use of decompressive craniectomy in patients with severe brain swelling and high ICPs.21 Outcome measurements have not been consistent, and results have been mixed. If performed, the procedure should be done early in the patient’s course. Two ongoing randomized studies may provide better direction regarding when this treatment is best indicated.21
Seizure Prophylaxis.: Up to 12% of all patients who sustain blunt head trauma and 50% of those with penetrating head injury develop early post-traumatic seizures (PTSs).78 Although the occurrence of seizures in the immediate post-trauma period has no predictive value for future epilepsy, early seizures can cause hypoxia, hypercarbia, release of excitatory neurotransmitters, and increased ICP, which can worsen secondary brain injury. Constantly firing neurons are soon depleted of their energy sources, and in the head trauma patient with compromised cerebral metabolism, uncontrolled seizures exacerbate the neurologic deficit.23
Box 41-2 lists accepted indications for early anticonvulsant therapy after head trauma. If the patient is actively seizing, benzodiazepines are administered as effective, rapid-acting first-line anticonvulsants. Lorazepam (0.05-0.1 mg/kg IV over 2-5 minutes up to a total of 4 mg) has been found to be most effective at aborting status epilepticus.23 Diazepam (0.1 mg/kg, up to 5 mg IV, every 10 minutes up to a total of 20 mg) is an alternative. For long-term anticonvulsant activity, phenytoin (18-20 mg/kg IV) or fosphenytoin (15-18 phenytoin equivalents/kg) can be given. Fosphenytoin has the advantages of rapid administration, a smaller volume of fluid for the dose delivered, and less hypotension than phenytoin. In a Cochrane review, the use of antiepileptic drugs reduced the risk of early seizures by 66%.79 Early seizure prophylaxis does not prevent late PTSs; the goal is to prevent additional insult to the damaged brain.78–80
Antibiotic Prophylaxis.: Infection may occur as a complication of penetrating head injury, open skull fractures, and complicated scalp lacerations. Prophylactic antibiotics may be used in these circumstances but are not recommended in patients with only otorrhea or rhinorrhea from a basilar skull fracture.81
Recombinant factor VIIa (rFVIIa) is a hemostatic agent that was originally developed to treat bleeding in hemophiliacs. Considerable interest has arisen regarding its potential use in intracerebral hemorrhage.82,83 rFVIIa costs approximately $1000/mg. Experience from the Iraq war and civilian use in multitrauma patients has produced conflicting results regarding its benefits in traumatic intracranial hemorrhage.82–85 Use of rFVIIa for traumatic head bleeds should be individualized and undertaken in concert with an institution’s treatment protocol. A recent review concluded that although off-label use of rFVIIa increased 140-fold from 2000 to 2008, no evidence exists that rFVIIa improves outcomes in five off-label indications. Specifically, the data regarding the use of rFVIIa in TBI are too limited for determination of the risks and benefits.86 Head-injured patients taking anticoagulants who require an emergent craniotomy may benefit from rFVIIa administration because it decreases the time to operating room intervention.82,83,85,86
Laboratory Tests.: The acute management of the severely head-injured patient is directed by physical examination and diagnostic imaging. Ancillary laboratory tests that may provide useful information in the subsequent management of the patient include a urine toxicology screen, blood alcohol level, complete blood count, electrolytes, glucose, and coagulation studies.
Neuroimaging.: In the acute phase the most useful imaging technique is a non–contrast-enhanced head CT scan. This scan delineates acute intra-axial and extra-axial bleeding, subarachnoid blood, cerebral swelling, ischemic infarction caused by hypoxia after trauma, evidence of increased ICP, and pneumocephalus. Emergency management decisions are strongly influenced by these acute CT scan findings. The bone windows of the CT scan can detect skull fractures (including basilar fractures); plain skull radiographs are not necessary in patients who undergo CT scanning.
Consultation.: All patients with severe head trauma require an imaging modality to determine the extent and nature of the brain injury and the necessity of neurosurgical intervention. Neurosurgical consultation should be obtained as soon as possible to help direct the patient’s subsequent management.
Transfer.: Severely head-injured patients require admission to an institution capable of intensive neurosurgical care and acute neurosurgical intervention. If this is not available at the receiving hospital, the patient should be transferred to an appropriate institution by the most expedient transport method available.
Priority Management.: The hemodynamically unstable patient with multiple trauma that includes head injury presents difficult emergency management decisions. The emergency physician should decide on the sequence that best addresses the most life-threatening pathologic conditions while still preventing morbidity and mortality from other serious injury. If the patient requires immediate surgical intervention for a life-threatening chest or abdominal injury, complete evaluation of the head injury may be curtailed. Moreover, these patients are anesthetized for surgery, and any neurologic deterioration is not detected. Some patients may be too unstable for even an abbreviated head CT scan to be obtained before emergent surgical intervention for other life threats. In this circumstance, early neurosurgical and general surgical consultation should be coordinated by the emergency physician. Intraoperative ventriculostomies or bilateral trephinations may provide some temporary protection from increasing ICP while the patient undergoes surgical correction of the life-threatening injury. A CT scan is performed after the primary life threats have been corrected.
Moderate Head Trauma
Clinical Features and Acute Management
An important clinical scenario in the spectrum of moderate head injury is that of the talk and deteriorate patient. These patients have a GCS score of 13 or greater on presentation but deteriorate to a status of a severe head injury (GCS score 8) within 48 hours.87 Although this description can include patients with minor head trauma, most patients who talk and deteriorate initially have GCS scores suggesting moderate head trauma. When the syndrome was first described by Reilly more than 30 years ago,88 the mortality in head-injured patients who were talking at presentation was estimated to be as high as 38%.87–90 However, CT scans were not readily available at that time, and the GCS was not in widespread use. With the current availability of early CT scanning, as well as rapid transport via emergency medical services, injuries are detected earlier, and the mortality of talk and deteriorate is lower—estimated to be 2.5 to 12%.87,91 In early descriptions of talk and deteriorate, most patients were found to have sustained subdural or epidural hematomas (EDHs). More recently, an equal number have been found to have contusions with subsequent edema.87,91 Patients who talk and deteriorate are generally elders with higher injury severity scores.87,91,92 In addition, anticoagulation puts patients at increased risk.
Because of the varied presentation of patients with moderate head trauma, the initial examination alone cannot accurately predict who will have surgically correctable intracranial lesions. Traumatic intracranial lesions are frequent in moderately head-injured patients. Even patients with a GCS score of 14 have a reported rate of 14% traumatic intracranial lesions.21 A CT scan is essential in patients with moderate head trauma to avoid delayed diagnosis of traumatic mass lesions or diffuse injury. This is especially true for elders or patients on anticoagulation therapy.93,94 Skull radiographs may be useful if the patient has sustained a depressed skull fracture or a penetrating injury but are otherwise rarely helpful.
Disposition
All patients with moderate head injury should be admitted for observation, even with an initial apparently normal CT scan. Most patients improve over the first few days after injury.23 Frequent neurologic checks should be initiated, and a repeated CT scan is indicated if the patient’s condition deteriorates or fails to improve over the first 48 hours after trauma.
Complications
The overall mortality of patients with isolated moderate TBI is approximately 20%, but the morbidity is substantial. Most patients with moderate TBI remain symptomatic for extended periods after head injury. At 3 months after trauma, up to 70% are unable to return to work, 90% have memory difficulties, and more than 90% complain of persistent headaches.95–98 TBI increases long-term mortality. One research group found that with regard to life expectancy, on average, TBI shortens the life span compared with non-TBI patients.99,100 Almost 50% are left with a long-term disability that interferes with their previous daily activities.98 In patients with persistent symptoms of headache, confusion, or memory difficulties, delayed MRI may define lesions in the regions related to cognition that cannot be seen on CT. Although not useful in the acute setting, MRI has prognostic value during subsequent care and assists in directing the future rehabilitation of these patients.
Minor Head Trauma
Minor or minor head trauma (MHT) is an injury to the brain resulting in a temporary and brief interruption of neurologic function after head trauma, which may involve a loss of consciousness (LOC).56,101 The neuropathology involved in producing signs and symptoms of minor TBI may remain at the neurobiochemical level without damage to the microstructure. Heightened ionic flux, surges in levels of glutamate transmitters, disruption of enzymatic pathways, and accumulation of lactate and nitric oxide have been reported in brain tissue after experimental minor TBI.17 Axonal stretching or twisting may occur with some mechanisms producing minor TBI. This injury pattern may promote glutamine-induced neurotoxic cascades that lead to the axonal damage typically described as DAI.102 Experimental models of blast injury also produce these same changes.103
Most authorities, clinical policies, and decision rules now classify minor TBI as that producing a GCS score of 14 or 15.17,101,102,104–106 In fact, the GCS is not sensitive enough to be of prognostic usefulness in minor TBI; a perfect score of 15 in the ED does not take into account the level of alertness or neurologic status immediately after trauma or the presence or absence of focal neurologic injury.31,102,107
From a practical standpoint, minor TBI is a clinical diagnosis. The diagnosis requires a credible mechanism of injury. In civilian life, most mechanisms that do not involve direct craniofacial impact cannot produce minor TBI. For example, belted drivers in low-velocity rear end–impact MVCs are not subjected to high enough acceleration-deceleration or rotary forces to the head to reach the force threshold needed to produce cerebral injury unless there is also direct impact to the head against a stationary object.102,108 It is therefore unlikely that a patient with a whiplash or “jolt” injury would also have minor TBI. Collision sports–related minor TBI can be caused by acceleration-deceleration. Although the extent of this mechanism is not yet known, forces generated in blast injuries in wounded service personnel are the most common cause of minor TBI without direct head impact. Mild TBIs from blast injuries have been called the signature injury of the wars in Iraq and Afghanistan.103
Clinical and Historical Features
Clinical signs and symptoms of minor TBI include balance deficits, impaired verbal memory, delayed language comprehension, slowed speech, amnesia regarding the impact, a period of disordered awareness (with or without LOC), and a finite period of post-trauma amnesia (PTA).91,102,108 Retrograde amnesia is impaired information retrieval and begins with and includes the traumatic event. In minor TBI, it generally lasts up to several minutes and then very rapidly resolves. PTA is impairment of information encoding and therefore does not resolve. PTA is the period of time from the injury to the return of conscious recall: events during this interval are essentially lost to the patient. PTA is a better predictor of injury severity and eventual outcome than the duration of retrograde amnesia or the GCS score.102
Approximately 5 to 15% of patients with minor TBI in the ED with a GCS score of 15 have abnormal CT scans.102,104,109 This estimate is probably inflated, because at least 25% of patients with minor TBI never seek medical care. The incidence of life-threatening lesions that require neurosurgical intervention after minor TBI is variably reported but likely less than 1%104; the goal of emergency evaluation and management of patients with minor TBI is to identify these high-risk patients. Further diagnostic workup hinges on risk stratification in patients with minor TBI (Box 41-3). The criteria are based on several large studies, but because of inconsistent methodology and reporting, they have limitations. For example, LOC in minor head trauma has historically been considered a risk factor for significant injury, but the negative predictive value of LOC has not been determined, and many patients with minor TBI may have sustained only brief or no LOC.110,111 Animal studies and anecdotal reports on humans suggest that the longer the duration of LOC, the more likely that an intracranial lesion exists, but the actual correlation between the duration of the LOC and the incidence of intracranial lesions or injury severity has not been determined.108–112 Other high-risk minor head trauma criteria have also been proposed. The key for the emergency physician, however, is to determine the low-risk patient, and these criteria are less controversial.
Imaging Studies
A major decision regarding the emergency management of minor TBI is whether imaging studies should be performed. Several approaches have recently been described, but the research behind these suggestions remains confusing primarily because of differences in study populations, definitions, methods, risk factors included in analysis, and selection of acceptable outcomes. Based on critical assessment of the literature designed to address this question, the American College of Emergency Physicians (ACEP) has developed a clinical policy on neuroimaging of adult ED patients with mild TBI.104 The recommendations from the ACEP clinical policy are summarized in Box 41-4.
A critical comparison of six common decision-making strategies for neuroimaging in mild TBI has been made.113 Although there are differences in the number of scans performed if each of the rules were applied to the same population, the sensitivity in detection of surgically important hematomas was not different among the rules. The final choice of which clinical decision rule to apply, and therefore the level of acceptable risk, rests on the clinician’s attitude toward uncertainty.
The most practical approach regarding imaging in the ED patient is probably selective CT scanning or observation based on risk stratification of the patient with minor TBI. If the low-risk patient is fully awake and not intoxicated, has no focal neurologic findings, has no clinical evidence of skull fracture, and can be kept under competent observation for 12 to 24 hours, neuroimaging is usually not indicated. Patients with moderate-risk minor head trauma (see Box 41-3) should probably undergo CT scanning or prolonged ED observation.
Although CT scanning is extremely sensitive for acute blood, MRI is more sensitive than CT for detecting DAI, ischemia after TBI, and some hemorrhagic lesions, especially those located at the base of the skull or in the posterior fossa.114,115 However, there is no current evidence favoring the use of MRI over CT for MHT in the ED setting.104 Many studies suggest that significant long-term neuropsychiatric sequelae after minor head trauma can occur despite an initial negative head CT scan, and these may be related to lesions seen initially only through MRI or functional imaging.116 Functional imaging is not currently within the scope of emergency assessment of patients with minor TBI but can direct rehabilitation strategies for the small subset of patients with significant morbidity after minor TBI.
Ancillary Studies
No routine laboratory tests are needed for patients with isolated minor head trauma. A urine toxicology screen and blood alcohol level may be useful in interpreting the patient’s mental status. Alcohol can affect the GCS, but this effect is not observed until the blood alcohol concentration is greater than 200 mg/dL; until that level, changes in mental status cannot be explained solely by acute alcohol intoxication.102
When shear injury occurs, proteins are released from neuronal axons. These proteins cross the BBB and can be detected in the peripheral circulation. A number of CNS biomarkers, such as S-100B, neuron-specific enolase, myelin basic protein, cleaved tau, and creatine kinase isoenzyme BB, have been investigated in minor TBI. None have been shown to strongly correlate with long-term outcome, and only S-100B predicts abnormal CT findings in minor TBI if measured within 4 hours of injury.18 However, S-100B appears to lack CNS specificity and is found in adipose, skin, and cartilage; it is therefore often elevated in multiple-trauma patients with no head injury.117 To date, serum biomarkers have lacked the precision needed for meaningful application in the emergency setting.118
Disposition
If a patient with minor head trauma returns to the ED because of persistent symptoms, delayed complications of minor head injury should be sought. If a CT scan was not initially performed, the intensity of symptoms may guide the decision to obtain a CT scan at the second visit. If a negative scan was initially obtained, the likelihood of the subsequent development of an intracranial lesion is small.104 The decision to rescan is more complex in patients from certain subgroups who may be considered more likely to develop delayed complications. These include patients on anticoagulation, those with preexisting neurologic injuries that may obscure an examination, and those with previous neurosurgical procedures (such as ventriculoperitoneal [VP] shunts). The literature about repeat CT scanning in mild traumatic brain injury (MTBI) is scant; however, in one systematic review of severe and moderate TBI, progression of lesions most commonly correlated with overall injury severity and the patients’ use of anticoagulants.119
Patients on Anticoagulation.: All current minor head injury decision-making rules exclude patients who are taking Coumadin or antiplatelet medications. Most current practice guidelines promote that patients who sustain head trauma and are on anticoagulation treatment should have the international normalized ratio (INR) determined and should undergo a CT scan without contrast.
Concussion.: Almost all patients with minor TBI will have rapid and complete resolution of their symptoms. However, a subset of patients with mild TBI report symptoms that persist for long periods after trauma. A concussion (or complicated minor TBI) is a type of minor TBI usually caused by acceleration-deceleration or rotational injury to a freely mobile head and is most commonly associated with collision sports.101,120 It is a complex pathophysiologic state resulting in short-lived distortion of axons, vasculature, and brain neuroanatomy. As with other types of minor TBI, acute CT or MRI abnormalities are not usually found108,120 but functional imaging (i.e., positron emission tomography [PET]) reveals abnormalities with glucose uptake and blood flow.116 Levels of neurotransmitters remain elevated, and a hypermetabolic state may persist in the brain for several days to weeks after the initial injury. This period is vulnerable to cumulative dysfunction if a second injury occurs, especially in children.121
The persistent symptoms after MHT are called the postconcussive syndrome (PCS) and generally include the domains of cognitive, psychomotor, and behavioral performance. The severity and duration of postconcussive symptoms may correlate with the abnormalities found with early functional imaging.116,120 The incidence of PCS after MHT is reported to be 10 to 58% at 1 month after injury,122 but these estimates are based on inpatient studies and are therefore likely to be high.
The most common delayed or persistent postconcussive complaints are headache, sensory sensitivity, memory or concentration difficulties, irritability, sleep disturbances, dizziness, depression, and amnesia regarding the traumatic event. In young children, acute symptoms of concussion differ from those in adults and may include restlessness, lethargy, confusion, or irritability.120 The appearance of symptoms may occur long after the concussion, and they persist long after the acute symptoms of uncomplicated MHT.120 It was once believed that persistent postconcussive symptoms were psychosomatic, litigious, or factitious; however, the presence of abnormalities on functional imaging and with sophisticated neuropsychometric testing suggests a pathophysiologic basis for PCS.17,122 Studies of concussed athletes (for whom preinjury baseline data are available) show that the cognitive domain most frequently involved in PCS is memory.17 Dizziness, headache, and nausea occurring early after trauma are associated with prolonged PCS; patients with all three symptoms have a 50% chance of persistent symptoms at 6 months, compared with a 28% chance for those with MHT with none of these symptoms at presentation.122 Most adult patients with PCS recover within 3 to 12 months after injury.100,104,115,120
Approximately 300,000 sports-related concussions occur annually each year. Among these, about 135,000 are in child athletes aged 5 to 18 years.123 A study of concussed football players showed acute symptoms lasting at least 5 days, cognitive impairments lasting 5 to 7 days, and balance deficits lasting 3 to 5 days after concussion; 91% were back to their preinjury baseline by 7 days, but some had deficits on verbal fluency tests as long as 90 days after injury.124 As a result of these and other data, educational campaigns have been launched for athletes on the signs and symptoms of concussion and steps to take when a concussion has occurred during play. The CDC has developed a number of “Heads Up” tool kits aimed at players, parents, and coaches to address these goals.123 The CDC, National Football League (NFL), NFL Players Association, Professional Football Athletic Trainers Society, and NFL Physician’s Society initiated a joint effort in 2010 with similar aims.
The demonstrated period of neurodysfunction and the delayed return to cognitive and physical baseline that follows concussive impact have lead to the development of several scoring systems to grade severity of concussions with the goal of determining when it is safe for an athlete to return to play. No single set of guidelines has emerged that has been universally accepted, but all are predicated on the concern about a period of vulnerability after impact. Football players sustaining concussion appear susceptible to an additional concussion, with the majority of reinjury occurring within 10 days after the first injury. This is variably attributed to balance defects, delayed reflexes, delayed speed of information processing, or simply continued exposure to collision sports.123 Recent consensus of sports medicine experts emphasizes physical and cognitive rest after sports-related concussion until complete symptom resolution.123,125 Although some organizations have published guidelines for rapid return to play (RTP) after a concussion (including same-day return), most are applicable only to professional or elite athletes and only when adequate sideline resources are available to detect subtle symptoms (e.g., on-site sports doctors, formal sideline neuropsychometric testing). For high school and collegiate athletes, a stepwise strategy has been developed to clear the athlete for RTP107 (Table 41-3). Progression from one step to the next requires that the athlete be asymptomatic; if symptoms recur, the patient drops back one step in the progression. This graduated RTP protocol cannot be achieved without referral and long-term care; in the ED management of concussed athletes, no RTP should be allowed until such follow-up and progress have occurred. The second impact syndrome (SIS) occurs when an athlete sustains a second concussion before being completely asymptomatic from the first and then experiences a rapid, usually fatal, neurologic decline. It is postulated that persistent neurochemical disruptions and altered autoregulatory mechanisms after a first injury make the brain particularly vulnerable to marked brain swelling and subsequent herniation after a seemingly minor second impact. Although the existence and frequency of the SIS are controversial and hotly debated in the sports medicine literature, its serious implications affect subsequent management decisions regarding head-injured athletes and others with concussion.126 All current recommendations for RTP after a sports-related concussion, including the graduated RTP protocol described earlier, state that players with concussion should not return to play for at least 1 week after they have become asymptomatic.
Table 41-3
Graduated Return-to-Play Protocol
| REHABILITATION STAGE | FUNCTIONAL EXERCISE AT EACH STAGE OF REHABILITATION | OBJECTIVE OF EACH STAGE |
| 1. No activity | Complete physical and cognitive rest | Recovery |
| 2. Light aerobic exercise | Walking, swimming, or stationary cycling, keep intensity to <70% of maximum predicted heart rate; no resistance training | Increase heart rate |
| 3. Sport-specific exercise | Skating drills in ice hockey, running drills in soccer; no head impact activities | Add movement |
| 4. Noncontact training drills | Progression to more complex training drills, for example, passing drills in football and ice hockey; may start progressive resistance training | Exercise, coordination, and cognitive load |
| 5. Full-contact practice | With medical clearance, participate in normal training activities | Restore athlete’s confidence; coaching staff assesses functional skills |
| 6. Return to play | Normal game play |
Adapted from McCrory P, et al: Consensus statement on concussion in sport: The 3rd International Conference on Concussion in Sport held in Zurich, November 2008. J Athl Train 44:434-448, 2009.
Pediatric Head Injuries
Approximately 650,000 to 1 million children are emergently evaluated yearly in the United States for head trauma, with 80 to 90% classified as mild.127,128 However, TBI accounts for the largest source of childhood mortality and morbidity after trauma. The etiology of TBI is age related; in infants and the very young, abusive injury is a major cause of TBI. In toddlers, abusive injury must be considered, but injury from falls appears more common. Once children enter school, injuries from physical activities (such as bicycle riding) and MVCs (especially pedestrians stuck by cars) are the most common causes of TBI.129–131
Pathophysiology
Until the cranial sutures close, children’s skulls are more distensible than those of adults. As a result, young children often sustain less TBI after head trauma than adults with comparable nonfatal mechanisms of injury.132 However, children appear to have an age-dependent brain vulnerability. Very young children (younger than 1 year) have higher mortality after head trauma than older children with injuries with the same severity of injury. Many factors contribute to this. Medical attention is often delayed in children with nonaccidental injuries. Because of limited language and comprehension, an accurate formal neurologic examination in young children is sometimes difficult. Medical personnel tend to underestimate the extent of the injuries in small children and are often reluctant to initiate invasive procedures that may be necessary to aid in the diagnostic workup, such as intravenous access for sedation in CT scanning.
The types of TBI sustained after head trauma in children differ from those in adults. Children have fewer traumatic mass lesions (with the exception of SDHs in the very young), fewer hemorrhagic contusions, more diffuse brain swelling, and more diffuse axonal injury. Of head-injured patients younger than 20 years who talk and deteriorate, 39% have brain swelling only (i.e., no mass lesions), whereas 87% of patients older than 40 who talk and deteriorate have mass lesions.89
Clinical Features
The initial physical presentation of a child with TBI may have minimal predictive value in terms of the severity of trauma sustained.133 Therefore repeated examinations to detect changes in the neurologic status are paramount.
In principle, the acute neurologic assessment of head-injured children is the same as that of adults. Because of its reliance on developed language skills and the patient’s attention and cooperation, the GCS is difficult to apply to very young children. A modified scale (see Table 38-4) has been developed134 and has been shown to accurately predict the need for neurosurgical intervention in head-injured preverbal children.135
Children with severe or moderate head injury are clinically similar to adults with these degrees of injury, although children have an increased incidence of early (i.e., within the first week after trauma) PTSs after severe head injury.23 Low GCS score and young age are associated with an increased early PTS risk. Even after adjustment for GCS score, children younger than 2 years have up to a threefold increased risk compared with older (i.e., older than 12 years) children.66 Overall, up to 6% of all head-injured children and 20% of severely head-injured children have early PTS. Most PTSs occur within the first 24 hours and do not predict seizures later in the post-traumatic period. However, early PTS can exacerbate secondary brain injury by creating hypoxia and increasing cerebral metabolism and ICP. Although prophylactic use of anticonvulsants is not recommended to prevent late PTSs, acute anticonvulsant prophylaxis is a treatment option in severely head-injured infants and young children to prevent early PTSs.66
The clinical presentation of post-traumatic intracranial lesions in infants who have sustained moderate or severe TBI can be extremely subtle, especially in those younger than age 6 months. Often these lesions are associated with scalp injuries and skull fractures but no other symptoms. Toddlers are also difficult to assess; the only symptom of an intracranial injury may be irritability.138,139 Recommendations are also offered regarding the ED workup in these very young children.
Inflicted head injury, also described as abusive head injury or nonaccidental head injury, is the most common cause of head injury deaths in infants. Child abuse is considered in all children with unexplained head injuries or injuries not consistent with the history provided. The shaken baby syndrome is most often seen in children younger than 1 year and is inflicted by violently shaking an infant while holding him or her by the torso or extremities. The uncontrolled head movement causes an acceleration-deceleration brain injury, with a rotary component, as the moving brain strikes against the interior of the skull. The resultant shearing injury causes widespread damage. Patients have a broad range of symptoms, from nonspecific complaints such as vomiting or sleepiness, to seizures and coma. Classically described findings include SDHs, and subarachnoid hemorrhage (SAH) without signs of external trauma. Diffuse retinal hemorrhages are seen in 50 to 100% of all patients with shaken baby syndrome; the extent of hemorrhage appears related to injury severity and may predict the prognosis.130
Children with concussions tend to be symptomatic longer after mild TBI than adults. As in adults, postconcussive symptoms in children include neurocognitive changes. The severity and duration of postconcussive symptoms is related to the severity of initial injury, as well as personal and environmental factors. Children who have sustained LOC, post-traumatic amnesia, a GCS score less than 15 at presentation, or mental status changes are more likely to have prolonged postconcussive symptoms.140,141 Thus children should not return to sports or play activities until completely symptom free, and then only at the discretion of their primary physician.120 Concussive injuries in children also produce two unique clinical circumstances. Many children experience a brief impact seizure at the time of relatively minor head injury. By the time the child is evaluated, he or she is at baseline neurologic function. Impact seizures do not appear to predict subsequent early PTSs. However, a post-traumatic impact seizure may confuse the initial assessment of the severity of the head injury and prompt a more aggressive workup than needed. Postconcussive blindness, another serious complication of concussive injuries in children, is usually associated with impact to the back of the head. Children experience a temporary loss of vision that can persist from minutes to hours before normal vision returns.
Diagnosis and Management
Children with severe TBI who are transported directly to a pediatric trauma center or an adult trauma center with pediatric capabilities have better outcomes than those transported to nontrauma hospitals.66 Hypoxia and hypoventilation are common in pediatric head-injured patients, and they develop more rapidly in children than in adults. In children with a GCS score below 8, airway management should be aggressive to prevent hypoxia, aspiration, and hypercarbia and to allow mild hyperventilation if the patient has signs of herniation. There is no evidence to support the advantage of out-of-hospital intubation over bag-valve-mask ventilation of pediatric patients with TBI66,142; scene time should not be prolonged with multiple attempts at endotracheal intubation.
Hypotension is a late finding in children in shock; poor perfusion should be clinically identified and corrected as soon as possible to ensure adequate CPP. There is no evidence to suggest that aggressive fluid resuscitation exacerbates cerebral edema in head-injured children. In children, unlike in adults, hypovolemic hypotension can occur because of head trauma. Hypotension from intracranial bleeding can occur in children younger than 1 year with a large linear skull fracture and an underlying large EDH.132 The intracranial blood can seep through the fracture and produce a large galeal or subperiosteal hematoma. Hypotension from intracranial bleeding can also occur in a child with hydrocephalus and a functioning shunt. Blood may accumulate without much evidence of increased ICP. Scalp lacerations can also produce significant hemorrhage and subsequent hypotension.
Up to 80% of children with severe head trauma have elevated ICP.132 In infants, a bulging fontanel suggests elevated ICP. Other signs of elevated ICP include bradycardia, papilledema, declining level of consciousness, and seizures. When increased ICP is indicated by physical examination, methods to reduce ICP should be initiated. As with adults, acute hyperventilation has immediate effects but is never indicated for prophylaxis or for prolonged management of increased ICP.66 Hyperosmolar therapy is effective at reducing ICP. There have been no studies describing the superiority of one osmotic agent over another in the treatment of TBI, and no direct comparisons have been done. Although efficacy studies were based only on adults, mannitol at doses of 0.25 to 1.0 g/kg has become a mainstay in the treatment of elevated ICP in children with severe TBI. Bolus therapy has a theoretic benefit compared with continuous treatment; over prolonged periods, mannitol may accumulate in injured areas of the brain where the BBB is no longer intact, thus creating a reverse osmotic shift.143,144 Several studies support the effectiveness of HTS in lieu of mannitol for elevated ICP in pediatric head-injured patients. Treatment is with a continuous infusion of 3% normal saline at 0.1 to 1.0 mL/kg body weight per hour, titrated to effect.66
When minor head injury is considered in children, age is an important consideration in interpreting clinical and historical findings. Children younger than age 2 years are often difficult to assess and may have subtle clinical findings. In general, the literature supports the conclusion that younger patients are at higher risk for intracranial injury. One clinical sign of potential brain injury in children younger than age 2 years is the presence of a scalp hematoma, especially a large parietal scalp hematoma.143–145 In an observational cohort study involving children with low risk for brain injuries, scalp hematomas were present in 93% of children 2 years old or younger who had brain injuries.144
More than 50% of children with head injury of all severities in the United States undergo emergent cranial CT scanning.127,146 Cranial CT is the diagnostic imaging modality of choice in the evaluation of moderate or severe pediatric TBI and should also be strongly considered in pediatric patients with high-risk minor TBI. However, potential risks are associated with childhood exposure to radiation. Children are more sensitive to radiation than adults because of rapidly dividing cells during normal growth. The younger the child, the more potential risk. Increased rates of solid tumor cancers146 and decreased cognitive performance have been associated with radiation doses similar to that of a single head CT scan.147 An additional safety concern is the frequent need to sedate young children in order to perform an adequate imaging study. In these circumstances, the risks of radiation exposure and sedation should be weighed against the likelihood of an intracranial lesion in the child with minor head trauma.
It is estimated that up to 98% of cranial CT scans done to assess head injury in children are negative.145,148 Several clinical prediction rules have been developed to assist in decision-making for emergent neuroimaging of children with minor TBI. Comparison of these various rules is difficult because of variable methods and inclusion criteria. Most have not been validated in a separate population.148,149 Unless both sensitivity and specificity are high, such rules may actually result in an increased number of CT scans being performed. Kupperman and colleagues,145 with a research network including 25 EDs and 4200 patients, have derived and validated a clinical prediction rule for minor head trauma that includes subsets of children younger than 2 years and from 2 to 18 years old. The rule performs well with a very low risk of missed clinically important TBI in both age subgroups. An algorithm suggested by this study is presented in Figure 41-7. The CATCH study derived a decision rule that identified two levels of risk for children with minor head injury.150 The large cohort of 3800 patients was identified from 10 Canadian pediatric teaching institutions. Children with a GCS score below 15 at 2 hours after trauma, a suspected open or depressed skull fracture, a worsening headache, or persistent irritability were found to be at increased risk for the need for a neurosurgical intervention and considered high risk; moderate risk (brain injury on CT with no need for neurosurgical intervention) was associated with any sign of basal skull fracture, a large boggy scalp hematoma, or a dangerous mechanism of injury (e.g., MVC or fall greater than 3 feet).
The use of skull radiographs in the diagnostic workup of head-injured children is controversial but may be appropriate in some circumstances. As with adults, when a CT scan is indicated, skull radiographs are not necessary. Up to 11% of children younger than age 2 years will sustain a skull fracture associated with head trauma, and 15 to 30% of these will have TBI. Therefore in children younger than age 2 years, a skull fracture is a predictor of TBI.151 The presence of a skull fracture in children significantly increases the likelihood of intracranial pathology; conversely, a negative radiographic skull screen does not guarantee the absence of TBI. Parietal skull fractures are the most common. Often, fractures occur in infants who sustain relatively minor head injury. Skull films may be useful as a screening tool in determining the need for a CT scan, especially in children 2 years old or younger whose neurologic examination is difficult to obtain and interpret. In alert children younger than age 2 years with minor head injury, a low-risk history, normal findings on physical examination that includes a neurologic and mental status examination appropriate for age, and a scalp hematoma, skull radiographs may be a useful screen.139 If the skull radiograph is negative for a fracture, a CT scan may be unnecessary. If the skull radiograph shows a fracture, CT imaging is indicated.
Post-traumatic leptomeningeal cysts or growing skull fractures are delayed complications of linear skull fractures in infancy. If a tear in the dura accompanies the linear fracture, the meninges may fill with CSF and prolapse through the fracture margins, thus preventing fracture healing.23,139 These cysts can grow in size and have the potential to cause a mass effect. If a linear fracture is found by skull radiography, close follow-up is indicated to assess for this rare delayed complication.
Overall, children who sustain severe head injury have lower mortality and a better neurologic outcome than comparably injured adults. This is probably because of the neuroplasticity of the young brain; however, in children younger than age 2 years, the prognosis after severe head injury is poor.152 Very young children have immature cerebrovascular autoregulation, which increases the risk of cerebral edema formation. The immature brain has increased susceptibility to permanent injury because of incomplete myelination.
The emergency evaluation of children with minor head injury is especially challenging, given their potentially dramatic presentation and the added difficulty of obtaining an accurate neurologic assessment. Evidence-based consensus guidelines have been proposed for the evaluation and management of minor head injury in children.23,127,138,139 Parents should be educated about the warning signs and symptoms of delayed complications of minor head trauma.
Penetrating Head Injuries
Penetrating brain injury (PBI) occurs at a rate of 12 per 100,000 population and can be caused by missile injuries or impalement.153 The United States has the highest penetrating head injury rate among developed countries in the world, with the most common cause being gunshot wounds (GSWs). These dramatic injuries are increasing in frequency, and the neuroscientific understanding of the complicated cerebral events that occur with penetrating head injury does not yet equal our understanding of the pathophysiology of blunt injuries.
Civilian GSWs to the head account for approximately 31,000 deaths per year, and up to 70% of all patients who sustain a GSW to the head are dead at the scene.154,155 Overall, the mortality caused by a GSW to the head is estimated to be 90%.156 If the patient is hemodynamically stable, has not sustained secondary systemic insults such as hypoxia or hypotension, has no expanding mass lesions from the missile injury, and has not ingested intoxicants that may interfere with assessment, prognosis after a GSW to the head can be predicted by the GCS score and pupillary responsiveness at presentation.157 If the GCS score at presentation is less than 5, mortality approaches 100%. If the GCS score at presentation is greater than 8 and the pupils are reactive, survival approaches 75%. Survivors of GSWs to the head tend to do well, with up to 60% returning to their former employment.157
Pathophysiology
Missile injuries to the head can result in several different patterns of damage. Tangential wounds are caused by an impact that occurs at an oblique angle to the skull. If the missile has high velocity but low energy, it can travel around the skull under the scalp without passing through the skull. Intracranial damage, primarily cortical contusions, can occur at the initial site of impact because of pressure waves generated by the impact. In one study, 24% of patients with tangential GSWs also had intracranial hemorrhage, and 16% sustained skull fractures.158,159 Many patients with tangential GSWs have a GCS score of 15 on presentation.
Penetrating missile wounds are produced with moderate- to high-velocity projectiles discharged at close range. The majority of the civilian PBI literature deals with penetrating missile wounds.156 The penetrating object may travel through the entire skull, bounce off the opposite inner table of the skull and ricochet within the brain, or stop somewhere within the cranial cavity. Bullets that penetrate the skull do not travel in a straight path. The wounding capacity of a firearm is related to the kinetic energy of its missile on impact and how much energy is dissipated in the tissues. Low-velocity missiles tend to be deflected by intracranial structures. The final track is therefore erratic and occasionally bears no relation to the exit or entrance site of the missile.160,161 High-velocity missiles can project straight through the tissues and easily fracture bones. Flight stability and the angle at which the bullet strikes its target affect the path through the brain. Within tissue, destabilizing motions include deviation of the longitudinal axis of the bullet from a straight line (yaw), forward rotation of the bullet around its center of mass (tumbling), and oscillatory motion of the bullet axis around its center of mass (rotation). As the bullet passes through the brain, a tissue cavity is created. This cavity can be as much as 10 times the diameter of the missile. A percussion shock wave is also created, lasting 2 msec but causing little tissue destruction.156,160,161
The morbidity and mortality from missile injuries to the head depend on the intracranial path, speed of entry, and the size and type of the penetrating object. Projectiles that cross the midline or the geographic center of the brain, pass through the ventricles, or come to rest in the posterior fossa are associated with extremely high mortality.157,160,161 High-velocity wounds are associated with greater mortality than low-velocity injuries. Large missiles or missiles that fragment within the cranial vault are usually fatal. The design of the bullet and its fragmentation potential (capacity to deform or fragment) also contribute to final tissue destruction and patients’ morbidity and mortality.
Many GSWs to the head are intentionally self-inflicted injuries. The percentage of penetrating head injuries caused by self-inflicted GSWs ranges from 13 to 88%.157 Characteristics of self-inflicted GSWs include injury on the dominant side, powder burns at the entrance site, and large stellate scalp lacerations caused by dissection of the subgaleal layer by exploding gases released close to the scalp. In suicide attempts, GSWs to the head tend to traverse the midline in the coronal plane and often involve major vascular structures. If the self-inflicted GSW has an entrance through the mouth, injury to the hard palate may occur with potential upper airway compromise. The careful aim and close range of self-inflicted GSWs to the head make these injuries particularly devastating; mortality is higher than with non–self-inflicted penetrating injuries, and odds ratios of death vary from 1.63 to 5.83.157
Management
Emergency treatment should include intravenous antibiotics because penetrating missiles are contaminated with skin, bone, and hair. Tissue contamination may be widespread because of the cavitation caused by the missile as it passes through the brain.162 Approximately 90% of all CNS infections associated with penetrating TBI injuries occur within 6 weeks. Most neurosurgeons do not automatically débride bone and missile fragments owing to a growing body of literature that shows no evidence that removal of these foreign bodies decreases infection rate.18,156
Between 30 and 50% of patients with PBI develop seizures; 10% of these occur in the first week.18,156 Anticonvulsants should be given in the acute setting to prevent early PTSs in the patient with PBI, especially if the patient is to be transported to another institution after acute stabilization. Anticonvulsants should not be given beyond the first week after PBI because this has not been shown to prevent the development of late seizures.
Skull radiographs may be useful in determining the number of penetrating fragments and their track. A CT scan defines the precise location of the missile, its intracranial path, the presence of bone or missile fragments, extra-axial or intracerebral blood collections or other traumatic lesions, and pneumocephalus. CT scanning is the radiologic test of choice for PBI.163 Pneumocephalus is often associated with missile wounds that penetrate the sinuses but can be caused by free air sucked into the penetration cavity behind the projectile.
All tangential GSWs should be evaluated with a head CT scan secondary to the high incidence of associated intracranial injury.159 When a penetrating head injury is caused by impalement, the penetrating object should be left in place to be removed at surgery. A skull radiograph shows the size of the object, the angle of impalement, and the depth of penetration. Angiography may be indicated to better discern location referable to key vascular structures.
Complications after Head Injury
Seizures
PTSs are relatively common in the acute or subacute period. Acute PTSs are usually brief and are probably caused by transient mechanical and neurochemical changes within the brain. After the acute seizure, the patient often has no additional seizure activity. In the subacute period, 24 to 48 hours after trauma, seizures are caused by worsening cerebral edema, small hemorrhages, or penetrating injuries. PTSs are common in children and can be precipitated by relatively minor head injury.164 Acute PTS prophylaxis in the ED is recommended for some head-injured patients even if they have not had a seizure (see Box 41-2).78,80,164 This is especially important in patients who will have neuromuscular blockade to facilitate management or transfer because the clinical manifestations of seizures are lost in these patients. Phenytoin and fosphenytoin are used as first-line agents for prophylaxis. The decision to maintain the head-injured patient on long-term anticonvulsant therapy during the recovery period depends on the patient’s subsequent course. Long-term seizure prophylaxis is not indicated for all patients who have had PTSs in the acute or subacute period. The utility of prophylactic anticonvulsants to prevent late PTSs has not been proved, and their use is not recommended.78 However, if a seizure occurs after the acute or subacute period, anticonvulsants are indicated. TBI has been estimated to account for 20% of all epilepsy in the general population.165,166 It is the leading cause of epilepsy in young adults. In a longitudinal cohort study following individuals with moderate and severe head injury, Yasseen and colleagues167 found that 9% of this population was being treated for epilepsy. Estimates of late PTSs in patients with moderate and severe TBI range from 4.2 to 25.3% and manifest themselves as long as 12 years postinjury.168
Central Nervous System Infections
Meningitis after Basilar Fractures.: Post-traumatic meningitis is caused by a variety of microbes, depending on the portal of bacterial entry. Patients have typical signs and symptoms of meningitis, including fever, altered mental status, and occasional focal neurologic signs. In patients with a CSF leak after basilar fracture, early meningitis (i.e., within 3 days of injury) is usually caused by pneumococci. Ceftriaxone or cefotaxime is a reasonable antibiotic choice with the addition of vancomycin. Gram-negative organisms often cause meningitis that develops more than 3 days after trauma.169 A third-generation cephalosporin, with nafcillin or vancomycin added to ensure coverage of Staphylococcus aureus, should be started. In children, post-traumatic meningitis may be caused by Haemophilus influenzae. Prophylactic antibiotics are not consistently recommended in the acute setting in patients with CSF leaks caused by basilar skull fractures during the first 7 days.169 However, Friedman and colleagues found that prophylactic antibiotics administered in the first week reduced meningitis risk by one half.170 The risk of meningitis does increase after 7 days of CSF leakage, and prophylactic antibiotics may be warranted.171
Brain Abscess.: Brain abscesses develop infrequently after penetrating missile injuries to the head. Abscesses can also develop after open depressed skull fractures if bone fragments are not removed or as a postoperative complication. Post-traumatic CSF fistulae and fractures that disrupt air-filled sinuses predispose to the formation of brain abscesses. Clinical manifestations include headaches, nausea, vomiting, declining mental status, signs of increased ICP, or new focal neurologic findings in patients who had been improving after trauma. Occasionally, nuchal rigidity, hemiparesis, or seizures may be present. Systemic signs are often subtle, and CSF leukocytosis may be absent.
The treatment of brain abscess is usually operative drainage. The patient with cerebritis may respond to intravenous antibiotics but requires close monitoring with repeated CT scans. Common organisms isolated from post-traumatic abscesses are S. aureus and gram-negative aerobes.169
Cranial Osteomyelitis.: Cranial osteomyelitis can occur after penetrating injury to the skull. The clinical manifestations include pain, tenderness, swelling, and warmth at the infected site. More than 50% of cases are obvious on plain skull radiographs.169 Technetium bone scans can help in the diagnosis when the skull radiographs are negative, but false-positive bone scans occur in patients with previous trauma or craniotomy. Adding a gallium scan helps to differentiate infection from other causes of a positive technetium scan. Patients with post-traumatic cranial osteomyelitis require surgical débridement and removal of the infected bone. Antibiotic choice is determined by culture results. If systemic symptoms are present, an underlying subdural or epidural empyema is often present.
Medical Complications
Disseminated Intravascular Coagulation
The injured brain is a source of tissue thromboplastin that activates the extrinsic clotting system. Disseminated intravascular coagulation (DIC) can develop within hours after any injury disrupting brain tissue. It has been detected in nearly all patients with severe TBI.93,172 DIC increases morbidity and mortality after severe head trauma as well as the risk of delayed intracranial hemorrhage. If a stable patient with DIC suddenly deteriorates, a repeat CT scan should be obtained to rule out hemorrhage.
The extent of tissue destruction determines the degree of DIC that develops. The diagnosis is based on abnormalities in INR, prothrombin and partial thromboplastin time, platelets, plasma fibrinogen levels, and fibrin degradation products. Patients with coagulopathy or abnormal platelet function require interventions to correct these. Patients with moderate or severe head trauma are at extremely high risk to experience venous thromboembolic events (VTEs) after admission to the hospital. Recent evidence suggests that patients with moderate or severe head trauma can be safely treated with low-molecular-weight heparin to reduce the risk of VTE without significantly increasing the risk of intracerebral hematoma expansion.172
Neurogenic Pulmonary Edema
Neurogenic pulmonary edema can develop minutes to days after head trauma. Two theories exist on its pathophysiology: catecholamine surge or blast with elevated hydrostatic forces and increased capillary permeability directly caused by brain injury versus a systematic inflammatory reaction.173–175 Lowering the ICP appears to reverse the neurogenic stimulation that causes this edema.176 Close ventilator management is essential to improved outcomes in patients with acute lung injury secondary to TBI.175
Cardiac Dysfunction
A variety of cardiac rhythm, rate, and conduction abnormalities are detected after head injury. Harvey Cushing noted a connection between cardiac dysrhythmias and intracranial bleeding in the beginning of the 20th century.177 These abnormalities can be life-threatening and require aggressive therapy. In addition, adequate cardiac output is essential in head-injured patients to ensure cerebral perfusion. Many head-injured patients with cardiac dysfunction have concurrent myocardial injury from underlying disease or from chest injury. However, brain injury can cause primary cardiac dysfunction. Cardiac rhythm abnormalities have been reported in up to 70% of all patients with SAH and more than 50% of all patients with intracranial hemorrhage.34,174 In SAH the cardiac dysrhythmias may result from autonomic nervous system dysfunction that subsequently affects ventricular polarization. High levels of circulating catecholamines have been measured in head-injured patients, with increased sympathetic nervous system activation.34
The most common cardiac dysrhythmia after head injury is supraventricular tachycardia, but many other rhythms have been observed. Findings on the electrocardiogram include diffuse large upright or inverted T waves, prolonged QT intervals, ST segment depression or elevation, and U waves. The primary goal in the emergency management of cardiac dysfunction after head trauma is ensuring adequate tissue perfusion and avoiding hypoxia. Dysrhythmias in head-injured patients often resolve as ICP is reduced.34,174 Standard advanced cardiac life support protocols should then be used because concurrent cardiac injury may also be present in multiply traumatized patients.
Specific Injuries
Disruption of the galea results in a gaping scalp laceration. Large lacerations of the galea are closed to prevent the edges of the wound from pulling apart as the muscles within the galea contract. The skin, dermis, and galea can usually be repaired in a single layer with interrupted or vertical mattress sutures of 0-3.0 nylon or polypropylene.178 Several recent studies have evaluated the use of staples versus sutures to close scalp lacerations that do not involve the galea. For both adult and pediatric scalp lacerations that begin beyond the hairline, staples have been shown to be cheaper, take less time, and have the same outcome than sutures if used in the appropriate manner.179–181 Staples cannot be used to close the galea and may not be effective alone when hemostasis is a problem.181 Recently an alternative method of scalp laceration closure has been described in which bundles of the patient’s hair on each side of the wound are twisted together and then secured with tissue glue.182,183 This may provide another method of effective repair and prevent the need to remove the closure material after the wound has healed.
Because of the rich blood supply of the scalp, even very large scalp avulsions can survive. If the avulsion remains attached to the rest of the scalp by a tissue bridge, it should be reattached to the surrounding tissue. If the avulsion is completely detached from the scalp, it should be treated as any other amputated part and reimplanted as soon as possible.178 Scalp abrasions are often contaminated with pieces of dirt or other debris. The wound should be cleaned as thoroughly as possible and inspected for puncture wounds or other areas that penetrate beyond the superficial layers of the skin to ensure the removal of unsuspected foreign bodies. A careful inspection often reveals a small scalp laceration within the abraded area. Antibiotics are usually not needed for carefully managed scalp wounds because rapid healing is facilitated by the rich blood supply of the scalp.
Skull Fractures
Clinical Assessment and Significance
Skull fractures are local injuries caused by direct impact to the skull. The presence of a skull fracture does not always indicate underlying brain injury. However, the force required to fracture the skull is substantial, and all patients with skull fractures are carefully evaluated to ensure that no additional injury is present. With increasing severity of head injury, the likelihood of skull fracture increases, and the presence of a skull fracture after trauma increases the likelihood of a TBI.184–186 It is often difficult to predict the presence of a skull fracture by clinical examination, and if this can be done, it is likely that substantial underlying brain injury is also present. The pattern, extent, and type of skull fracture depend on the force of the impact applied and the ratio of the impact force to the impact area. The fracture usually starts at the point of maximum impact.
Clinically significant skull fractures result in intracranial air and pass through an air-filled space (e.g., sinus), are associated with an overlying scalp laceration (open skull fracture), are depressed below the level of the skull’s inner table, or overlie a major dural venous sinus or the middle meningeal artery.166,187
A noncontrast head CT scan with bone windows has become the imaging modality of choice for patients with suspected skull fractures.186,187 Plain radiographs can be useful when CT scan is not available; if an intracranial foreign body or a linear or depressed fracture is detected, a noncontrast head CT is indicated to rule out intracranial traumatic pathology.187
Linear Fractures
Patients with nonsignificant linear fractures can usually be discharged from the ED with competent home observation. In patients with significant linear fractures, admission for observation is warranted.186
Depressed Fractures
Depressed skull fractures are clinically important because they predispose to significant underlying brain injury and to complications of head trauma, such as infection and seizures. When a depressed fracture occurs, traumatic impact drives the bone piece below the plane of the skull. The edges of the depressed portion of skull may become locked underneath the adjacent intact bone and fail to rebound into their previous position. As a result, the depressed piece of bone can penetrate tissue and lacerate the dura. Depressed skull fractures are usually caused by direct impact injury with small blunt objects, such as a hammer or a baseball bat. Most depressed skull fractures occur over the parietal or temporal regions. If the free piece of bone is depressed deeper than the adjacent inner table of the skull, most neurosurgeons consider this injury significant enough to require surgical elevation.23,186
Depressed skull fractures are most often open fractures with disruption of the galea. They can often be felt with palpation of the skull beneath a scalp laceration. This examination should be done cautiously to avoid driving a depressed bone fragment deeper into the cranial tissue. The clinical examination for a depressed skull fracture may be misleading. The mobility of the scalp can result in nonalignment of the fracture with an overlying scalp laceration. As a result, the skull underlying the laceration may be normal, with the depressed area several centimeters away. Scalp swelling may also interfere with physical examination findings and hide any palpable bone defects. The signs and symptoms of a depressed skull fracture depend on the depth of depression of the free bone piece. About 25% of patients sustaining a depressed skull fracture report LOC.186 Neurologic deficits may be present, depending on the extent of underlying brain tissue injury.
Depressed skull fractures, as well as any type of penetrating skull injury, may increase the risk for developing seizures.188 ED patients suspected of having a depressed skull fracture warrant prophylaxis for PTSs, especially if they have an altered level of consciousness or require chemical paralysis. Depressed skull fractures may also increase the risk for intracranial and meningeal infections; patients with depressed skull fractures should receive prophylactic antibiotics.23,166,186,188
Basilar Fractures
Basilar fractures are linear fractures at the base of the skull. The fracture usually occurs through the temporal bone, with bleeding into the middle ear producing hemotympanum. Patients with basilar fractures are at risk for extra-axial hematomas because of the proximity of the fracture to the middle cerebral artery. Often, the fracture has caused a dural tear, which produces a communication among the subarachnoid space, the paranasal sinuses, and the middle ear. This offers a route for the introduction of infection into the cranial cavity and is suggested by a CSF leak.23 As with linear skull fractures, a basilar fracture is not always associated with significant underlying brain injury; these fractures are the result of considerable impact force, however, and TBI must be ruled out.
Basilar fractures can compress and entrap the CNs that pass through the basal foramina, can dislocate the bones of the auricular chain, and can disrupt the otic canal or cavernous sinuses, with subsequent injury to CNs III, IV, and V. Fractures of the sphenoid bone can disrupt the intracavernous internal carotid artery, creating the potential for the formation of pseudoaneurysms or carotid venous fistulae. The diagnosis of a basilar skull fracture is based on associated clinical signs and symptoms (see Box 41-1).
Patients with clinical evidence suggesting a basilar skull fracture should have a CT scan, to define the fracture and to rule out concurrent intracranial pathology, and should be admitted for observation. Because the basilar skull fracture may afford an entrance for bacteria, antibiotics are often considered. However, most CSF leaks resolve spontaneously in about 1 week with no complications, and, in general, antibiotics are not given prophylactically during the first week of CSF rhinorrhea.23 Counter to this approach, Friedman and colleagues found prophylactic antibiotics to reduce the risk of meningitis by half.170 If a patient with a previously diagnosed CSF leak returns to the ED later with fever, the diagnosis of meningitis should be strongly suspected and appropriate workup (i.e., lumbar puncture) and antibiotic treatment initiated immediately.
Diffuse Axonal Injury
Prolonged traumatic coma not caused by mass lesions, ischemic insult, or nontraumatic causes of coma is thought to result from DAI. DAI or diffuse traumatic axonal injury is a pathologic process in which axons sustain a primary insult in which they are torn (axotomy) and secondary insults that can result in progressive cellular pathologic changes and eventual axon death (Fig. 41-8).155 Affected axons are dispersed between areas of undamaged cells (Fig. 41-9). Badly damaged axons become edematous and eventually begin to separate from one another, causing widespread disruption of cortical physiology and microanatomy in the white matter of the brain and brainstem. Complete separation of axons does not always cause cell death; acute uncoupling of CBF and metabolism and apoptosis are thought to be the primary factors linked to axonal cell death after DAI.21,155 Experimental laboratory data have indicated that neurons can partially repair and regenerate damaged axons. Recovery depends on the reversal or correction of structural and physiologic abnormalities.17
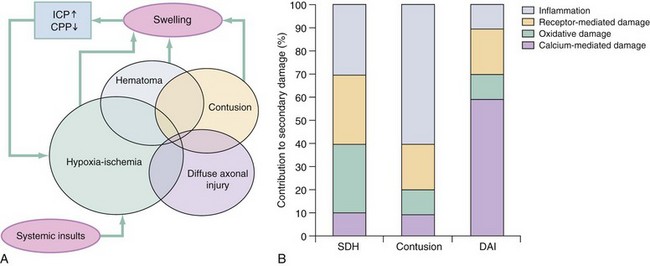
DAI is thought to be the cause of persistent traumatic coma that begins immediately at the time of trauma; however, some patients with DAI may recover consciousness briefly before lapsing into prolonged coma. No specific acute focal traumatic lesions are noted on a head CT scan. MRI is more sensitive in detecting subtle injury in DAI, but it is often not practical to perform MRI on critically injured patients. Occasionally, small petechial hemorrhages in proximity to the third ventricle and within the white matter of the corpus callosum or within the internal capsule of the brainstem are detected. DAI is the most common CT finding after severe head trauma, estimated to occur in 50% of all comatose head-injured patients.166
Because diagnostic studies cannot predict the extent of the axonal damage, the severity of the injury is determined by the clinical course. Patients with mild DAI are in coma for 6 to 24 hours. Approximately one third of patients with mild DAI demonstrate decorticate or decerebrate posturing, but by 24 hours they are following commands.166 The mortality in this group is 15% and is associated with infectious complications or concurrent intracranial injuries. Most patients who recover have mild or no permanent disabilities.
Moderate DAI is the most common clinical picture. Patients with moderate DAI are in coma for longer than 24 hours. Often, they are victims of falls or vehicular crashes and have associated basilar skull fractures. Patients may exhibit transient decortication or decerebration but eventually recover purposeful movements. On awakening, patients have prolonged severe post-traumatic amnesia and moderate to severe persistent cognitive deficits. Almost 25% die of complications of prolonged coma.166
Severe DAI is almost always caused by vehicular crashes. Patients remain in coma for prolonged periods and demonstrate persistent brainstem dysfunction (posturing) and autonomic dysfunction (e.g., hypertension and hyperpyrexia). Diffuse brain swelling subsequent to injury causes intracranial hypertension. Herniation syndrome can occur if elevated ICP does not respond to medical or surgical intervention. Some patients with severe DAI eventually awaken but are severely disabled. Some patients remain in a persistent vegetative state, but most with severe DAI die from their head injury. All patients with DAI have an identical presentation of coma. Recent research has identified several biomarkers that may have clinical relevance in the future.155 However, currently no early clinical or biomarker predictor exists that differentiates patients with mild, moderate, or severe DAI.
Cerebral Contusions
The clinical presentation of patients with contusions is frequently delayed. They may have sustained only a brief LOC, but the duration of post-traumatic confusion and obtundation may be prolonged. If contusions occur near the sensorimotor cortex, focal neurologic deficits may be present. Many patients with significant contusions make uneventful recoveries, but contusions may cause significant neurologic problems, including increased ICP, PTSs, and focal deficits. Agrawal and colleagues189 showed that significant progression of cerebral contusions is not an infrequent occurrence. Patients with lower GCS scores and larger cerebral contusions are at higher risk for needing delayed surgical intervention.189
Epidural Hematoma
EDHs are blood clots that form between the inner table of the skull and the dura. Most EDHs are caused by direct impact injury that causes a forceful deformity of the skull. Often, a fracture occurs across the middle meningeal artery, vein, or a dural sinus. The temporoparietal region is the most likely site for an EDH.159 The high arterial pressure of the bleeding vessel dissects the dura away from the skull, permitting the formation of the hematoma.
An EDH is usually unilateral, and 20% of patients have other intracranial lesions, usually SDHs or contusions.23 The deterioration of a patient who has an EDH from arterial bleeding can be rapid and dramatic. Because of their rapid formation, EDHs from arterial bleeding are usually detected within hours after injury and often earlier in children. EDHs that develop from a dural sinus tear develop more slowly, and clinical manifestations may be delayed, with resultant delays in detection.
EDH is primarily a disease of the young and accounts for 2.7 to 4% of all patients who have experienced TBI.23,190 EDHs are rare in elders and children younger than 2 years of age because of the close attachment of the dura to the skull in both patient populations. The classic presentation of an EDH is described as head trauma producing a decreased level of consciousness followed by a “lucid” interval. Although the patient’s consciousness is less decreased during the lucid interval, a completely normal mental status may not return before a second episode of decreased consciousness occurs. The lucid interval is not pathognomonic for an EDH and occurs in patients who sustain other expanding mass lesions. Approximately 47% of patients with EDHs present classically.190 The development of symptoms and signs of EDH is entirely dependent on how quickly the EDH is developing within the cranial vault. Patients with an EDH often complain of a severe headache, sleepiness, dizziness, nausea, and vomiting. A small EDH may remain asymptomatic, but this is rare.
If the patient is not in coma when the diagnosis of EDH is established and if the condition is rapidly treated, the mortality is 5 to 10%. If the patient is in coma, the mortality from EDH is approximately 20%. The mortality in pediatric patients undergoing surgical evacuation is 5%.190 If the EDH is rapidly detected and evacuated, the functional outcome is excellent.
On CT scan, an EDH appears hyperdense, biconvex, ovoid, and lenticular. The EDH does not usually extend beyond the dural attachments at the suture lines. The margins are sharply defined, and the hematoma usually bulges inward toward the brain (see Fig. 41-9). EDHs of mixed density on CT may be actively bleeding.
A posterior fossa EDH is the most common traumatic mass lesion of the posterior fossa and accounts for 5% of EDHs.23 Direct occipital trauma resulting in a skull fracture that disrupts a venous sinus is the usual cause, and most patients have external evidence of occipital injury. Most patients become symptomatic within 24 hours after injury, with complaints of headache, nausea, vomiting, and nuchal rigidity. Most patients eventually have a decreased level of consciousness. On CT scan, a posterior fossa EDH looks similar to other EDHs, but it may cross the midline and extend above the tentorium to the supratentorial compartment (Fig. 41-10). Expert consensus guidelines support rapid surgical evacuation for any patient who has mass effect on CT or progressive neurologic deterioration.175 Mortality approaches 26%.23
Recent studies have investigated the indications for immediate operative intervention for EDHs. Epidurals greater than 30 cm3 in volume should be evacuated surgically, regardless of the patient’s GCS score. Furthermore, it is strongly recommended that comatose patients with an acute EDH and anisocoria on pupillary examination undergo surgical evacuation as soon as possible.190
Subdural Hematoma
SDHs are more common than EDHs, occurring in up to 30% of patients with severe head trauma.159 The slow bleeding of venous structures delays the development of clinical signs and symptoms. As a result, the hematoma compresses the underlying brain tissue for prolonged periods and can cause significant tissue ischemia and damage.
SDHs are classified by the time to clinical presentation. Acute SDHs are symptomatic within 24 hours after trauma. Patients with acute SDHs often have a decreased level of consciousness. Most patients with an SDH have a GCS score less than 8. Approximately 12 to 38% of patients will have a lucid period at some point in their presentation. The overall mortality of patients who have an SDH and require surgical intervention is 40 to 60%.159
If the SDH is very small (only a few millimeters thick at its widest point on CT scan), some neurosurgeons may choose careful observation for these patients. Even a small SDH may be accompanied by extensive brain tissue damage that can cause an increase in ICP sufficient to precipitate a herniation syndrome. Current consensus guidelines recommend that acute SDHs with a thickness greater than 10 mm or a midline shift of more than 5 mm on CT should be evacuated surgically, regardless of the patient’s GCS score.159 Research indicates that the longer the time between clinical worsening and operative treatment, the worse the patient’s clinical outcome. In these cases, surgical evacuation should be performed as soon as possible.159
Unlike EDHs, SDHs often extend beyond the suture lines (Fig. 41-11). An SDH may follow the contour of the tentorium and be detected within the interhemispheric fissure (Fig. 41-12). Many patients with an acute SDH also show CT evidence of intracerebral lesions contralateral to the SDH.
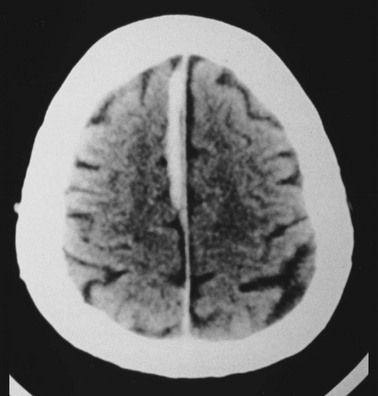
Figure 41-12 Non–contrast-enhanced computed tomography scan of intrahemispheric acute subdural hematoma.
A chronic SDH becomes symptomatic 2 weeks or more after trauma. The signs and symptoms may be very subtle or nonspecific, but many patients demonstrate unilateral weakness or hemiparesis.23 Most report an altered level of consciousness, but some patients are unable to recall their head injury or describe only a minor injury. In 20% of cases, chronic subdurals are bilateral. A chronic SDH may have initially been a small asymptomatic SDH that eventually expanded owing to a combination of recurrent hemorrhage and escape of plasma into the hematoma. At some point, a critical mass is reached and the chronic SDH becomes symptomatic. On CT scan, a chronic SDH may appear isodense or hypodense to brain parenchyma. In these cases, indirect evidence of the lesion includes a midline shift, effacement of the ipsilateral cortical sulci, and ventricular compression. Contrast may increase the likelihood of identifying a chronic SDH that has become isodense. On CT scan, blood of various ages is seen as a mixed-density lesion. On MRI, a chronic SDH appears hyperdense. The treatment of chronic SDHs is controversial. If they become symptomatic, chronic SDHs require surgical evacuation. Most patients have a good outcome after surgery. Overall, the mortality from surgically drained chronic SDH approaches 4%, with decreased survival in elders.23
In children the presence of an SDH should prompt consideration of child abuse. Many types of injury can produce SDH in children, but the infant who is repeatedly and forcibly shaken is especially susceptible.130 Infants may have SDH because of birth trauma. In these cases the initial clinical manifestation may be a generalized seizure within the first 6 months of life. On examination the infant may have a bulging fontanel or an enlarged head circumference. A careful history may elicit long-standing constitutional symptoms, such as failure to thrive or lethargy.
Subdural Hygroma
On CT scans, SDHGs appear crescent shaped in the extra-axial space. The CT density is the same as that of CSF. Bilateral SDHGs are common. If SDHGs are asymptomatic, observation is reasonable management. Otherwise, they are surgically evacuated. Mortality approaches 20% and appears to depend on the severity of other intracranial injury.23
Traumatic Subarachnoid Hemorrhage
Traumatic subarachnoid hemorrhage (TSAH) is blood within the CSF and meningeal intima and probably results from tears of small subarachnoid vessels. TSAH is detected on the first CT scan in up to 33% of patients with severe TBI and has an incidence of 44% in all cases of severe head trauma. It is therefore the most common CT scan abnormality seen after head injury. Data from the National Traumatic Coma Data Bank demonstrate a 60% unfavorable outcome in severely brain-injured patients in the presence of TSAH compared with a 30% unfavorable outcome if no TSAH occurs.191 An increased incidence of skull fractures and contusions is found in patients with TSAH compared with patients with no TSAH. The amount of blood within the TSAH correlates directly with the outcome and inversely with the presenting GCS score.
Intracerebral Hematoma
Intracerebral hematomas (ICHs) are formed deep within the brain tissue and are usually caused by shearing or tensile forces that mechanically stretch and tear deep small-caliber arterioles as the brain is propelled against irregular surfaces in the cranial vault. Resulting small petechial hemorrhages subsequently coalesce to form ICHs. Approximately 85% are in the frontal and temporal lobes. They are often found in the presence of extra-axial hematomas, and in many patients multiple ICHs are present.192,193 Isolated ICHs may be detected in as many as 12% of all patients with severe head trauma.
The clinical effects of ICH depend on size, location, and whether the bleeding is continuing. ICHs have been reported with all degrees of severity of head trauma. More than 50% of patients with ICH sustain LOC at the time of impact. The patient’s subsequent level of consciousness depends on the severity of the impact and coexisting lesions. Combined with contusions, other concurrent lesions, and subsequent perilesion edema, an ICH can produce substantial mass effects and precipitate a herniation syndrome (Fig. 41-13).
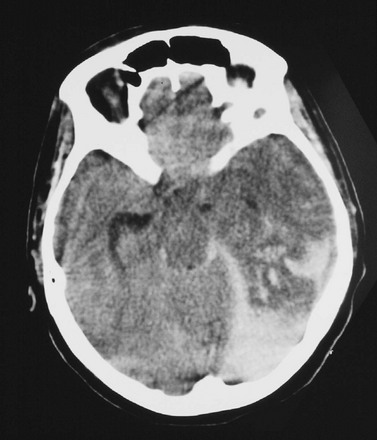
An ICH may be detected on the first CT scan immediately after injury but often is not seen for several hours or days. Unlike contusions, ICHs are usually deep in the brain tissue and often become well demarcated over time. On CT scan, an ICH appears as a well-defined hyperdense homogeneous area of hemorrhage (Fig. 41-14).
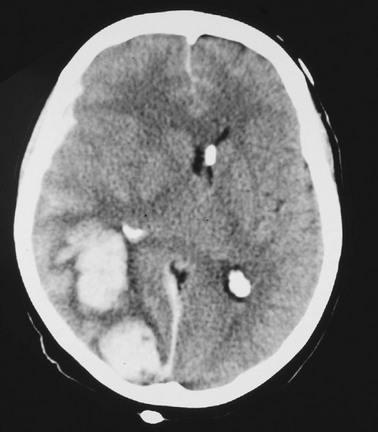
Many patients with an ICH require emergent intervention or surgery to control elevated ICP. Mortality is low in patients who are conscious before surgery; in unconscious patients, mortality approaches 45%.192,193 ICHs that bleed into the ventricles or cerebellum also carry a high mortality rate.
Traumatic Intracerebellar Hematoma
Key Concepts















 Children with severe head trauma have fewer intracranial lesions than adults but more edema. In children, increasing edema alone can cause talk and deteriorate or other significant neurologic decline.
Children with severe head trauma have fewer intracranial lesions than adults but more edema. In children, increasing edema alone can cause talk and deteriorate or other significant neurologic decline.
 Clinical decision rules such as Kupperman and colleagues145 algorithm can significantly reduce the number of head CT scans performed on children who experience head trauma.
Clinical decision rules such as Kupperman and colleagues145 algorithm can significantly reduce the number of head CT scans performed on children who experience head trauma.
 Skull fractures have more clinical significance in children than in adults.
Skull fractures have more clinical significance in children than in adults.




References
1. Faul, M, Xu, L, Wald, MM, Coronado, VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths, 2002-2006. Atlanta: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010.
2. Brenner, LA, et al. Traumatic brain injury, posttraumatic stress disorder, and postconcussive symptom reporting among troops returning from Iraq. J Head Trauma Rehabil. 2010;25:307–312.
3. Hoge, CW, et al. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N Engl J Med. 2008;358:4534–4563.
4. Jaffee, MS, et al. Acute clinical care and care coordination for traumatic brain injury within Department of Defense. J Rehabil Res Dev. 2009;46:655–666.
5. Greenwald, BD, Burnett, DM, Miller, MA. Congenital and acquired brain injury. 1. Brain injury: Epidemiology and pathophysiology. Arch Phys Med Rehabil. 2003;84:S3–S7.
6. Leventhal, JM, Martin, KD, Asnes, AG. Fractures and traumatic brain injuries: Abuse versus accidents in a U.S. database of hospitalized children. Pediatrics. 2010;126:e104–e115.
7. Carpenter, M. Gross anatomy of the brain. In Carpenter MB, ed.: Core Text of Neuroanatomy, 4th ed, Baltimore: Williams & Wilkins, 1991.
8. Rockswold, G. Head injury. In: Tintanelli J, ed. Emergency Medicine. New York: McGraw-Hill, 1996.
9. Lenzlinger, P. Overview of basic mechanisms underlying neuropathological consequences of head trauma. In: Miller LP, Hayes RL, Newcomb JK, eds. Head Trauma: Basic, Preclinical, and Clinical Directions. New York: Wiley-Liss; 2001:4–5.
10. Zwienenberg, M, Muizellar, J. Vascular aspects of severe head injury. In: Miller LP, Hayes RL, Newcomb JK, eds. Head Trauma: Basic, Preclinical, and Clinical Directions. New York: Wiley-Liss; 2001:303–326.
11. Reivich, M. Arterial PCO2 and cerebral hemodynamics. Am J Physiol. 1964;206:25–35.
12. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. XIV. Hyperventilation. J Neurotrauma. 2007;24(Suppl 1):S87–S90.
13. Laffey, JG, Kavanagh, BP. Hypocapnia. N Engl J Med. 2002;347:43–53.
14. Biros, MH, Heegaard, W. Prehospital and resuscitative care of the head-injured patient. Curr Opin Crit Care. 2001;7:444–449.
15. Gordon, KE. Pediatric minor traumatic brain injury. Semin Pediatr Neurol. 2006;13:243–255.
16. Gordon, KE, Dooley, JM, Fitzpatrick, EA, Wren, P, Wood, EP. Concussion or mild traumatic brain injury: Parents appreciate the nuances of nosology. Pediatr Neurol. 2010;43:253–257.
17. Bazarian, JJ, Blyth, B, Cimpello, L. Bench to bedside: Evidence for brain injury after concussion—looking beyond the computed tomography scan. Acad Emerg Med. 2006;13:199–214.
18. Chesnut, RM. Care of central nervous system injuries. Surg Clin North Am. 2007;87:119–156.
19. Greve, MW, Zink, BJ. Pathophysiology of traumatic brain injury. Mt Sinai J Med. 2009;76:97–104.
20. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. Introduction. J Neurotrauma. 2007;24(Suppl 1):S1–S2.
21. Maas, AI, Stocchetti, N, Bullock, R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741.
22. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. I. Blood pressure and oxygenation. J Neurotrauma. 2007;24(Suppl 1):S7–S13.
23. Greenberg, M. Handbook of Neurosurgery, 7th ed. New York: Thieme; 2010.
24. Wang, HE, Peitzman, AB, Cassidy, LD, Adelson, PD, Yealy, DM. Out-of-hospital endotracheal intubation and outcome after traumatic brain injury. Ann Emerg Med. 2004;44:439–450.
25. Kellie, G. An account of the appearances observed in the dissection of two of the three individuals presumed to have perished in the storm of the third, and his bodies were discovered in the vicinity of Leith on the morning of the 4th November 1821 with some reflections on the pathology of the brain. Trans Med Chir Sci (Edinburgh). 1824;1:84–169.
26. Monroe, A. Observation on the Structure and Function of the Nervous System. Edinburgh: Creek and Johnson; 1783.
27. Graham, D, Gennarelli, T. Pathology of brain damage after head injury. In: Cooper P, Golfinos J, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:133–154.
28. Marmarou, A, et al. Predominance of cellular edema in traumatic brain swelling in patients with severe head injuries. J Neurosurg. 2006;104:720–730.
29. Fodstad, H, Kelly, PJ, Buchfelder, M. History of the Cushing reflex. Neurosurgery. 2006;59:1132–1137.
30. Langlois, JA, Rutland-Brown, W, Thomas, KE. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths. Atlanta: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2006.
31. Teasdale, G, Jennett, B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81–84.
32. Marmarou, A, et al. IMPACT database of traumatic brain injury: Design and description. J Neurotrauma. 2007;24:239–250.
33. Stein, SC, Georgoff, P, Meghan, S, Mizra, K, Sonnad, SS. 150 years of treating severe traumatic brain injury: A systematic review of progress in mortality. J Neurotrauma. 2010;27:1343–1353.
34. Provencio, J, Bleck, T. Cardiovascular disorders related to neuroemergencies. In: Cruz J, ed. Neurologic and Neurosurgical Emergencies. Philadelphia: WB Saunders; 1998:39–50.
35. Bar-Joseph, G, Guilburd, Y, Tamir, A, Guilburd, JN. Effectiveness of ketamine in decreasing intracranial pressure in children with intracranial hypertension. J Neurosurg Pediatr. 2009;4:40–46.
36. Davis, DP, et al. The impact of aeromedical response to patients with moderate to severe traumatic brain injury. Ann Emerg Med. 2005;46:115–122.
37. McVey, J, Petrie, DA, Tallon, JM. Air versus ground transport of the major trauma patient: A natural experiment. Prehosp Emerg Care. 2010;14:45–50.
38. Thomas, SH. Helicopter EMS transport outcomes literature: Annotated review of articles published 2004-2006. Prehosp Emerg Care. 2007;11:477–488.
39. Bernard, SA, et al. Prehospital rapid sequence intubation improves functional outcome for patients with severe traumatic brain injury: A randomized controlled trial. Ann Surg. 2010;252:959–965.
40. Davis, DP, et al. Prehospital airway and ventilation management: A trauma score and injury severity score–based analysis. J Trauma. 2010;69:294–301.
41. Dumont, TM, Visioni, AJ, Rughani, AI, Tranmer, BI, Crookes, B. Inappropriate prehospital ventilation in severe traumatic brain injury increases in-hospital mortality. J Neurotrauma. 2010;27:1233–1241.
42. Davis, DP, et al. The effect of paramedic rapid sequence intubation on outcome in patients with severe traumatic brain injury. J Trauma. 2003;54:444–453.
43. Davis, DP, et al. Paramedic rapid sequence intubation for severe traumatic brain injury: Perspectives from an expert panel. Prehosp Emerg Care. 2007;11:1–8.
44. Davis, DP, Kimbro, TA, Vilke, GM. The use of midazolam for prehospital rapid-sequence intubation may be associated with a dose-related increase in hypotension. Prehosp Emerg Care. 2001;5:163–168.
45. Davis, DP, et al. Paramedic-administered neuromuscular blockade improves prehospital intubation success in severely head-injured patients. J Trauma. 2003;55:713–719.
46. Davis, DP, et al. The impact of prehospital endotracheal intubation on outcome in moderate to severe traumatic brain injury. J Trauma. 2005;58:933–939.
47. Fakhry, SM, et al. Prehospital rapid sequence intubation for head trauma: Conditions for a successful program. J Trauma. 2006;60:997–1001.
48. Salhi, B, Stettner, E. In defense of the use of lidocaine in rapid sequence intubation. Ann Emerg Med. 2007;49:84–86.
49. Vaillancourt, C, Kapur, AK. Opposition to the use of lidocaine in rapid sequence intubation. Ann Emerg Med. 2007;49:86–87.
50. Modica, PA, Tempelhoff, R. Intracranial pressure during induction of anaesthesia and tracheal intubation with etomidate-induced EEG burst suppression. Can J Anaesth. 1992;39:236–241.
51. Yeung, JK, Zed, PJ. A review of etomidate for rapid sequence intubation in the emergency department. CJEM. 2002;4:194–198.
52. Albert, SG, Ariyan, S, Rather, A. The effect of etomidate on adrenal function in critical illness: A systematic review. Intensive Care Med. 2011;37:901–910.
53. Dmello, D, Taylor, S, O’Brien, J, Matuschak, GM. Outcomes of etomidate in severe sepsis and septic shock. Chest. 2010;138:1327–1332.
54. Warner, KJ, Cuschieri, J, Jurkovich, GJ, Bulger, EM. Single-dose etomidate for rapid sequence intubation may impact outcome after severe injury. J Trauma. 2009;67:45–50.
55. Jabre, P, et al. Etomidate versus ketamine for rapid sequence intubation in acutely ill patients: A multicentre randomised controlled trial. Lancet. 2009;374:293–300.
56. Hohl, CM, et al. The effect of a bolus dose of etomidate on cortisol levels, mortality, and health services utilization: A systematic review. Ann Emerg Med. 2010;56:105–113.
57. Bulger, EM, et al. Out-of-hospital hypertonic resuscitation following severe traumatic brain injury: A randomized controlled trial. JAMA. 2010;304:1455–1464.
58. Oddo, M, et al. Effect of mannitol and hypertonic saline on cerebral oxygenation in patients with severe traumatic brain injury and refractory intracranial hypertension. J Neurol Neurosurg Psychiatry. 2009;80:916–920.
59. Wakai, A, Roberts, I, Schierhout, G. Mannitol for acute traumatic brain injury. Cochrane Database Syst Rev. 1, 2007.
60. Wakai, A, Roberts, I, Schierhout, G. Mannitol for acute traumatic brain injury. Cochrane Database Syst Rev. 4, 2005.
61. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. II. Hyperosmolar therapy. J Neurotrauma. 2007;24(Suppl 1):S14–S20.
62. Weed, L, McKibben, P. Experimental alteration of brain bulk. Am J Physiol. 1919;48:531–558.
63. Rockswold, GL, et al. Hypertonic saline and its effect on intracranial pressure, cerebral perfusion pressure, and brain tissue oxygen. Neurosurgery. 2009;65:1035–1042.
64. White, H, Cook, D, Venkatesh, B. The use of hypertonic saline for treating intracranial hypertension after traumatic brain injury. Anesth Analg. 2006;102:1836–1846.
65. Walker, PA, et al. Modern approaches to pediatric brain injury therapy. J Trauma. 2009;67:S120–S127.
66. Adelson, PD, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents. Chapter 11. Use of hyperosmolar therapy in the management of severe pediatric traumatic brain injury. Pediatr Crit Care Med. 2003;4:S40–S44.
67. Vassar, MJ, Perry, CA, Gannaway, WL, Holcroft, JW. 7.5% sodium chloride/dextran for resuscitation of trauma patients undergoing helicopter transport. Arch Surg. 1991;126:1065–1072.
68. Wade, CE, et al. Individual patient cohort analysis of the efficacy of hypertonic saline/dextran in patients with traumatic brain injury and hypotension. J Trauma. 1997;42:S61–S65.
69. Cooper, DJ, et al. Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: A randomized controlled trial. JAMA. 2004;291:1350–1357.
70. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. XI. Anesthetics, analgesics, and sedatives. J Neurotrauma. 2007;24(Suppl 1):S71–S76.
71. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. XV. Steroids. J Neurotrauma. 2007;24(Suppl 1):S91–S95.
72. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. III. Prophylactic hypothermia. J Neurotrauma. 2007;24(Suppl 1):S21–S25.
73. Sinclair, HL, Andrews, PJ. Bench-to-bedside review: Hypothermia in traumatic brain injury. Crit Care. 2010;14:204.
74. Nelson, JA. Local skull trephination before transfer is associated with favorable outcomes in cerebral herniation from epidural hematoma. Acad Emerg Med. 2011;18:78–85.
75. Smith, SW, et al. Emergency department skull trephination for epidural hematoma in patients who are awake but deteriorate rapidly. J Emerg Med. 2010;39:377–383.
76. Nelson, JA. Local skull trephination before transfer is associated with favorable outcomes in cerebral herniation from epidural hematoma. Acad Emerg Med. 2011;18:78–85.
77. Smith, SW, et al. Emergency department skull trephination for epidural hematoma in patients who are awake but deteriorate rapidly. J Emerg Med. 2010;39:377–383.
78. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. XIII. Antiseizure prophylaxis. J Neurotrauma. 2007;24(Suppl 1):S83–S86.
79. Schierhout, G, Roberts, I. Anti-epileptic drugs for preventing seizures following acute traumatic brain injury. Cochrane Database Syst Rev. 2001;4:CD000173.
80. Chang, BS, Lowenstein, DH. Practice parameter: Antiepileptic drug prophylaxis in severe traumatic brain injury: Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;60:10–16.
81. Brain Trauma Foundation, et al. Guidelines for the management of severe traumatic brain injury. IV. Infection prophylaxis. J Neurotrauma. 2007;24(Suppl 1):S26–S31.
82. McQuay, N, Jr., Cipolla, J, Franges, EZ, Thompson, GE. The use of recombinant activated factor VIIa in coagulopathic traumatic brain injuries requiring emergent craniotomy: Is it beneficial? J Neurosurg. 2009;111:666–671.
83. Stein, DM, Dutton, RP, Kramer, ME, Handley, C, Scalea, TM. Recombinant factor VIIa: Decreasing time to intervention in coagulopathic patients with severe traumatic brain injury. J Trauma. 2008;64:620–628.
84. Hauser, CJ, et al. Results of the CONTROL trial: Efficacy and safety of recombinant activated factor VII in the management of refractory traumatic hemorrhage. J Trauma. 2010;69:489–500.
85. Brown, CV, et al. Recombinant factor VIIa for the correction of coagulopathy before emergent craniotomy in blunt trauma patients. J Trauma. 2010;68:348–352.
86. Yank, V, et al. Systematic review: Benefits and harms of in-hospital use of recombinant factor VIIa for off-label indications. Ann Intern Med. 2011;154:529–540.
87. Tan, JE, Ng, I, Lim, J, Wong, HB, Yeo, TT. Patients who talk and deteriorate: A new look at an old problem. Ann Acad Med Singapore. 2004;33:489–493.
88. Reilly, PL, Graham, DI, Adams, JH, Jennett, B. Patients with head injury who talk and die. Lancet. 1975;2:375–377.
89. Lobato, RD, et al. Head-injured patients who talk and deteriorate into coma. Analysis of 211 cases studied with computerized tomography. J Neurosurg. 1991;75:256–261.
90. Rockswold, GL, Pheley, PJ. Patients who talk and deteriorate. Ann Emerg Med. 1993;22:1004–1007.
91. Goldschlager, T, Rosenfeld, JV, Winter, CD. ‘Talk and die’ patients presenting to a major trauma centre over a 10 year period: A critical review. J Clin Neurosci. 2007;14:618–624.
92. Dunn, LT, Fitzpatrick, MO, Beard, D, Henry, JM. Patients with a head injury who “talk and die” in the 1990s. J Trauma. 2003;54:497–502.
93. Stein, SC, Chen, XH, Sinson, GP, Smith, DH. Intravascular coagulation: A major secondary insult in nonfatal traumatic brain injury. J Neurosurg. 2002;97:1373–1377.
94. Stein, SC, Smith, DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479–488.
95. Colohan, AR, Oyesiku, NM. Moderate head injury: An overview. J Neurotrauma. 1992;9(Suppl 1):S259–S264.
96. Ruttan, L, Martin, K, Liu, A, Colella, B, Green, RE. Long-term cognitive outcome in moderate to severe traumatic brain injury: A meta-analysis examining timed and untimed tests at 1 and 4.5 or more years after injury. Arch Phys Med Rehabil. 2008;89:S69–S76.
97. Stein, SC, Ross, SE. Moderate head injury: A guide to initial management. J Neurosurg. 1992;77:562–564.
98. van Velzen, JM, van Bennekom, CA, Edelaar, MJ, Sluiter, JK, Frings-Dresen, MH. How many people return to work after acquired brain injury? A systematic review. Brain Inj. 2009;23:473–488.
99. Schievink, WI. Spontaneous spinal cerebrospinal fluid leaks: A review. Neurosurg Focus. 2000;9:e8.
100. Schlosser, RJ, Woodworth, BA, Wilensky, EM, Grady, MS, Bolger, WE. Spontaneous cerebrospinal fluid leaks: A variant of benign intracranial hypertension. Ann Otol Rhinol Laryngol. 2006;115:495–500.
101. Kulstad, EB, Kalimullah, EA, Tekwani, KL, Courtney, DM. Etomidate as an induction agent in septic patients: Red flags or false alarms? West J Emerg Med. 2010;11:161–172.
102. Ching, KY, Baum, CR. Newer agents for rapid sequence intubation: Etomidate and rocuronium. Pediatr Emerg Care. 2009;25:200–210.
103. Elder, GA, Cristian, A. Blast-related mild traumatic brain injury: Mechanisms of injury and impact on clinical care. Mt Sinai J Med. 2009;76:111–118.
104. Wilbur, K, Zed, PJ. Is propofol an optimal agent for procedural sedation and rapid sequence intubation in the emergency department? CJEM. 2001;3:302–310.
105. Matis, G, Birbilis, T. The Glasgow Coma Scale—a brief review. Past, present, future. Acta Neurol Belg. 2008;108:75–89.
106. Smits, M, et al. Predicting intracranial traumatic findings on computed tomography in patients with minor head injury: The CHIP prediction rule. Ann Intern Med. 2007;146:397–405.
107. Johnston, KM, et al. Current concepts in concussion rehabilitation. Curr Sports Med Rep. 2004;3:316–323.
108. Sheedy, J, Geffen, G, Donnelly, J, Faux, S. Emergency department assessment of mild traumatic brain injury and prediction of post-concussion symptoms at one month post injury. J Clin Exp Neuropsychol. 2006;28:755–772.
109. Iverson, GL. Outcome from mild traumatic brain injury. Curr Opin Psychiatry. 2005;18:301–317.
110. Fabbri, A, et al. Clinical performance of NICE recommendations versus NCWFNS proposal in patients with mild head injury. J Neurotrauma. 2005;22:1419–1427.
111. Smits, M, et al. A history of loss of consciousness or post-traumatic amnesia in minor head injury: “conditio sine qua non” or one of the risk factors? J Neurol Neurosurg Psychiatry. 2007;78:1359–1364.
112. Haydel, MJ, et al. Indications for computed tomography in patients with minor head injury. N Engl J Med. 2000;343:100–105.
113. Stein, SC, Fabbri, A, Servadei, F, Glick, HA. A critical comparison of clinical decision instruments for computed tomographic scanning in mild closed traumatic brain injury in adolescents and adults. Ann Emerg Med. 2009;53:180–188.
114. Mittl, RL, et al. Prevalence of MR evidence of diffuse axonal injury in patients with mild head injury and normal head CT findings. AJNR Am J Neuroradiol. 1994;15:1583–1589.
115. Hijaz, TA, Cento, EA, Walker, MT. Imaging of head trauma. Radiol Clin North Am. 2011;49:81–103.
116. Wilde, EA, et al. Diffusion tensor imaging of acute mild traumatic brain injury in adolescents. Neurology. 2008;70:948–955.
117. Townend, W, et al. Rapid elimination of protein S-100B from serum after minor head trauma. J Neurotrauma. 2006;23:149–155.
118. Siman, R, et al. A panel of neuron-enriched proteins as markers for traumatic brain injury in humans. J Neurotrauma. 2009;26:1867–1877.
119. Wang, MC, Linnau, KF, Tirschwell, DL, Hollingworth, W. Utility of repeat head computed tomography after blunt head trauma: A systematic review. J Trauma. 2006;61:226–233.
120. McCrory, P, et al. Consensus statement on concussion in sport: The 3rd International Conference on Concussion in Sport held in Zurich, November 2008. J Athl Train. 2009;44:434–448.
121. Thomas, DG, et al. Identifying neurocognitive deficits in adolescents following concussion. Acad Emerg Med. 2011;18:246–254.
122. Bazarian, JJ, Atabaki, S. Predicting postconcussion syndrome after minor traumatic brain injury. Acad Emerg Med. 2001;8:788–795.
123. Centers for Disease Control and Prevention (CDC). Nonfatal traumatic brain injuries from sports and recreation activities—United States, 2001-2005. MMWR Morb Mortal Wkly Rep. 2007;56:733–737.
124. McCrory, P. Does second impact syndrome exist? Clin J Sport Med. 2001;11:144–149.
125. McCrory, P, et al. Consensus statement on concussion in sport—the Third International Conference on Concussion in Sport held in Zurich, November 2008. Phys Sportsmed. 2009;37:141–159.
126. McCrory, P, Johnston, KM, Mohtadi, NG, Meeuwisse, W. Evidence-based review of sport-related concussion: Basic science. Clin J Sport Med. 2001;11:160–165.
127. Kuppermann, N. Pediatric head trauma: The evidence regarding indications for emergent neuroimaging. Pediatr Radiol. 2008;38(Suppl 4):S670–S674.
128. Atabaki, SM, et al. A clinical decision rule for cranial computed tomography in minor pediatric head trauma. Arch Pediatr Adolesc Med. 2008;162:439–445.
129. Duhaime, AC, Christian, CW, Rorke, LB, Zimmerman, RA. Nonaccidental head injury in infants—the “shaken-baby syndrome.”. N Engl J Med. 1998;338:1822–1829.
130. Matschke, J, et al. Shaken baby syndrome: A common variant of non-accidental head injury in infants. Dtsch Arztebl Int. 2009;106:211–217.
131. Huh, JW, Raghupathi, R. New concepts in treatment of pediatric traumatic brain injury. Anesthesiol Clin. 2009;27:213–240.
132. Weiner, H, Weinberg, J. Head injury in the pediatric age group. In Cooper P, Golfinos J, eds.: Head Injury, 4th ed, New York: McGraw-Hill, 2000.
133. Falk, AC, Cederfjall, C, von Wendt, L, Klang Soderkvist, B. Management and classification of children with head injury. Childs Nerv Syst. 2005;21:430–436.
134. James, HE. Neurologic evaluation and support in the child with an acute brain insult. Pediatr Ann. 1986;15:16–22.
135. Holmes, JF, Palchak, MJ, MacFarlane, T, Kuppermann, N. Performance of the pediatric Glasgow Coma Scale in children with blunt head trauma. Acad Emerg Med. 2005;12:814–819.
136. Reference deleted in proofs.
137. Fink, EL, Kochanek, PM, Clark, RS, Bell, MJ. How I cool children in neurocritical care. Neurocrit Care. 2010;12:414–420.
138. Schutzman, SA, et al. Evaluation and management of children younger than two years old with apparently minor head trauma: Proposed guidelines. Pediatrics. 2001;107:983–993.
139. Schutzman, SA, Greenes, DS. Pediatric minor head trauma. Ann Emerg Med. 2001;37:65–74.
140. Kirkwood, MW, et al. Management of pediatric mild traumatic brain injury: A neuropsychological review from injury through recovery. Clin Neuropsychol. 2008;22:769–800.
141. Yeates, KO, et al. Longitudinal trajectories of postconcussive symptoms in children with mild traumatic brain injuries and their relationship to acute clinical status. Pediatrics. 2009;123:735–743.
142. Gausche, M, et al. Effect of out-of-hospital pediatric endotracheal intubation on survival and neurological outcome: A controlled clinical trial. JAMA. 2000;283:783–790.
143. Palchak, MJ, Holmes, JF, Kuppermann, N. Clinician judgment versus a decision rule for identifying children at risk of traumatic brain injury on computed tomography after blunt head trauma. Pediatr Emerg Care. 2009;25:61–65.
144. Palchak, MJ, et al. A decision rule for identifying children at low risk for brain injuries after blunt head trauma. Ann Emerg Med. 2003;42:492–506.
145. Kuppermann, N, et al. Identification of children at very low risk of clinically important brain injuries after head trauma: A prospective cohort study. Lancet. 2009;374:1160–1170.
146. Klemetti, S, Uhari, M, Pokka, T, Rantala, H. Evaluation of decision rules for identifying serious consequences of traumatic head injuries in pediatric patients. Pediatr Emerg Care. 2009;25:811–815.
147. Hall, P, et al. Effect of low doses of ionising radiation in infancy on cognitive function in adulthood: Swedish population based cohort study. BMJ. 2004;328:19.
148. Maguire, JL, Boutis, K, Uleryk, EM, Laupacis, A, Parkin, PC. Should a head-injured child receive a head CT scan? A systematic review of clinical prediction rules. Pediatrics. 2009;124:e145–154.
149. Klig, JE, Kaplan, CP. Minor head injury in children. Curr Opin Pediatr. 2010;22:257–261.
150. Osmond, MH, et al. CATCH: A clinical decision rule for the use of computed tomography in children with minor head injury. CMAJ. 2010;182:341–348.
151. Dunning, J, et al. Derivation of the children’s head injury algorithm for the prediction of important clinical events decision rule for head injury in children. Arch Dis Child. 2006;91:885–891.
152. Beaudin, M, Saint-Vil, D, Ouimet, A, Mercier, C, Crevier, L. Clinical algorithm and resource use in the management of children with minor head trauma. J Pediatr Surg. 2007;42:849–852.
153. Kaufman, HH. Civilian gunshot wounds to the head. Neurosurgery. 1993;32:962–964.
154. Adekoya, N, Thurman, DJ, White, DD, Webb, KW. Surveillance for traumatic brain injury deaths—United States, 1989-1998. MMWR Surveill Summ. 2002;51:1–14.
155. Beaman, V, Annest, JL, Mercy, JA, Kresnow, M, Pollock, DA. Lethality of firearm-related injuries in the United States population. Ann Emerg Med. 2000;35:258–266.
156. Part 1. Guidelines for the management of penetrating brain injury. Introduction and methodology. J Trauma. 2001;51:S3–S6.
157. Part 2. Prognosis in penetrating brain injury. J Trauma. 2001;51:S44–S86.
158. Anglin, D, Hutson, HR, Luftman, J, Qualls, S, Moradzadeh, D. Intracranial hemorrhage associated with tangential gunshot wounds to the head. Acad Emerg Med. 1998;5:672–678.
159. Farhat, HI, Hood, B, Bullock, MR. A tangential gunshot wound to the head: Case report and review of the literature. J Emerg Med. 2009.
160. Maiden, N. Ballistics reviews: Mechanisms of bullet wound trauma. Forensic Sci Med Pathol. 2009;5:204–209.
161. Maiden, N. Historical overview of wound ballistics research. Forensic Sci Med Pathol. 2009;5:85–89.
162. Antibiotic prophylaxis for penetrating brain injury. J Trauma. 2001;51:S34–S40.
163. Neuroimaging in the management of penetrating brain injury. J Trauma. 2001;51:S7–S11.
164. Mazzola, CA, Adelson, PD. Critical care management of head trauma in children. Crit Care Med. 2002;30:S393–S401.
165. Hauser, WA, Annegers, JF, Kurland, LT. Prevalence of epilepsy in Rochester, Minnesota: 1940-1980. Epilepsia. 1991;32:429–445.
166. Lowenstein, DH. Epilepsy after head injury: An overview. Epilepsia. 2009;50(Suppl 2):4–9.
167. Yasseen, B, Colantonio, A, Ratcliff, G. Prescription medication use in persons many years following traumatic brain injury. Brain Inj. 2008;22:752–757.
168. Masel, BE, DeWitt, DS. Traumatic brain injury: A disease process, not an event. J Neurotrauma. 2010;27:1529–1540.
169. Lapointe, M. Basic principles of antimicrobial therapy of CNS infections. In: Cooper P, Golfinos J, eds. Head Injury. 4th ed. New York: McGraw-Hill; 2000:483.
170. Friedman, JA, Ebersold, MJ, Quast, LM. Persistent posttraumatic cerebrospinal fluid leakage. Neurosurg Focus. 2000;9:e1.
171. Brodie, HA, Thompson, TC. Management of complications from 820 temporal bone fractures. Am J Otol. 1997;18:188–197.
172. Dudley, RR, et al. Early venous thromboembolic event prophylaxis in traumatic brain injury with low-molecular-weight heparin: Risks and benefits. J Neurotrauma. 2010;27:2165–2172.
173. Rogers, FB, et al. Neurogenic pulmonary edema in fatal and nonfatal head injuries. J Trauma. 1995;39:860–868.
174. Szerlip, NJ, Bholat, O, McCunn, MM, Aarabi, B, Scalea, TM. Extracorporeal life support as a treatment for neurogenic pulmonary edema and cardiac failure secondary to intractable intracranial hypertension: A case report and review of the literature. J Trauma. 2009;67:E69–E71.
175. Mascia, L. Acute lung injury in patients with severe brain injury: A double hit model. Neurocrit Care. 2009;11:417–426.
176. Hanson, W. Acute respiratory failure in neuroemergencies. In: Cruz J, ed. Neurologic and Neurosurgical Emergencies. Philadelphia: WB Saunders; 1998:28–29.
177. Cushing, H. The blood pressure reaction of acute cerebral compression illustrated by cases of intracranial hemorrhage. Am J Med Sci. 1903;125:1017–1044.
178. Lammers, R. Principles of wound management. In: Roberts J, Hedges J, eds. Clinical Procedures in Emergency Medicine. Philadelphia: WB Saunders; 2004:623–655.
179. Hogg, K, Carley, S. Towards evidence based emergency medicine: Best BETs from the Manchester Royal Infirmary. Staples or sutures in children with scalp lacerations. Emerg Med J. 2002;19:328–329.
180. Hogg, K, Carley, S. Towards evidence based emergency medicine: Best BETs from the Manchester Royal Infirmary. Staples or sutures for repair of scalp laceration in adults. Emerg Med J. 2002;19:327–328.
181. Singer, A, McBride, M. Surgical staples. In: Singer A, Hollander J, Blumm R, eds. Skin and Soft Tissue Infection: A Practical Evidence Based Guide. Shelton, Conn: Peoples Medical Publishing House; 2011:79–82.
182. Karaduman, S, Yuruktumen, A, Guryay, SM, Bengi, F, Fowler, JR, Jr. Modified hair apposition technique as the primary closure method for scalp lacerations. Am J Emerg Med. 2009;27:1050–1055.
183. Ong, ME, et al. Hair apposition technique for scalp laceration repair: A randomized controlled trial comparing physicians and nurses (HAT 2 study). Am J Emerg Med. 2008;26:433–438.
184. Carson, HJ. Brain trauma in head injuries presenting with and without concurrent skull fractures. J Forensic Leg Med. 2009;16:115–120.
185. Muñoz-Sánchez, MA, et al. Skull fracture, with or without clinical signs, in mTBI is an independent risk marker for neurosurgically relevant intracranial lesion: A cohort study. Brain Inj. 2009;23:39–44.
186. Bullock, MR, et al. Surgical management of depressed cranial fractures. Neurosurgery. 2006;58:S56–S60.
187. Heegaard, W, Biros, M. Skull fractures in adults. Up to Date. 2012.
188. Chen, JW, Ruff, RL, Eavey, R, Wasterlain, CG. Posttraumatic epilepsy and treatment. J Rehabil Res Dev. 2009;46:685–696.
189. Agrawal, A, Timothy, J, Pandit, L, Manju, M. Post-traumatic epilepsy: An overview. Clin Neurol Neurosurg. 2006;108:433–439.
190. Daele, JJ, Goffart, Y, Machiels, S. Traumatic, iatrogenic, and spontaneous cerebrospinal fluid (CSF) leak: Endoscopic repair. B-ENT. 2011;7(Suppl 17):47–60.
191. Chan, EK, Yan, B, Ryan, MM. Spontaneous intracranial hypotension in childhood: A case report and review of the literature. J Child Neurol. 2011;26:761–766.
192. Schievink, WI. Spontaneous spinal cerebrospinal fluid leaks. Cephalalgia. 2008;28:1345–1356.
193. Cheuret, E, et al. Intracranial hypotension in a girl with Marfan syndrome: Case report and review of the literature. Childs Nerv Syst. 2008;24:509–513.