Haemostasis
The role of platelets
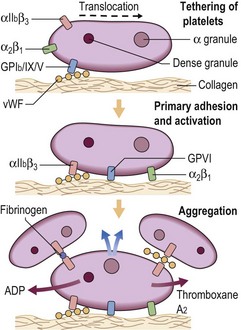
Following damage to a blood vessel there is immediate vasoconstriction to slow blood flow and reduce the risk of exsanguination. The break in the endothelial cell barrier leads to the recruitment of platelets from the circulation to form an occlusive plug. Platelets interact both with the vessel subendothelial matrix (platelet ‘adhesion’) and with each other (platelet ‘aggregation’) (Fig 6.1). The first step in this process, adhesion, does not require platelet metabolic activity. It does, however, lead to the ‘activation’ of platelets.



Coagulation
The coagulation cascade, leading to the generation of thrombin and the formation of a fibrin thrombus, is classically divided into two parts: the intrinsic and extrinsic pathways (Table 6.1).
Table 6.1
The classic coagulation cascade
| Intrinsic pathway | |
| Factor XIIa + Kallikrein → XIa → IXa → Xa | Final common pathway |
| Extrinsic pathway | Factor Xa → Thrombin → Fibrin |
| Factor VIIa – Tissue factor → Xa |
In the intrinsic pathway factor XII is activated by exposed collagen and other negatively charged components of the subendothelium. Activation of factor XII leads to the sequential activation of factors XI, IX, VIII (as cofactor), X and prothrombin. In the extrinsic pathway tissue factor complexes with factor VII with sequential activation of factors VII, X and prothrombin. Both intrinsic and extrinsic pathways terminate in the final common pathway where activated factor X, in association with the cofactor factor Va in the presence of phospholipid and calcium, converts prothrombin into thrombin. Thrombin in turn converts fibrinogen to fibrin by splitting the fibrinopeptides A and B from the centre domain to form fibrin monomers. These monomers combine spontaneously into dimers which assemble to form the fibrin polymer. Factor XIII crosslinks the fibrin polymer to consolidate the thrombus. The conventional division into two pathways is useful in the interpretation of in vitro laboratory tests of haemostasis. The prothrombin time (PT) is a simple measure of the function of the extrinsic pathway and the activated partial thromboplastin time (APTT) monitors the intrinsic pathway (p. 20). However, the physiological pathways at work in vivo are not so simply defined (see Fig 6.2). It seems that the intrinsic pathway is rarely relevant to coagulation in vivo – patients with hereditary deficiency of factor XII have a prolonged APTT but no bleeding disorder. The crucial protein in the initiation of blood coagulation is tissue factor, an integral membrane protein expressed on non-vascular cells. When a blood vessel is damaged, circulating factor VII comes into contact with tissue factor. The tissue factor/factor VIIa complex activates not only factor X (the extrinsic pathway) but also factor IX.
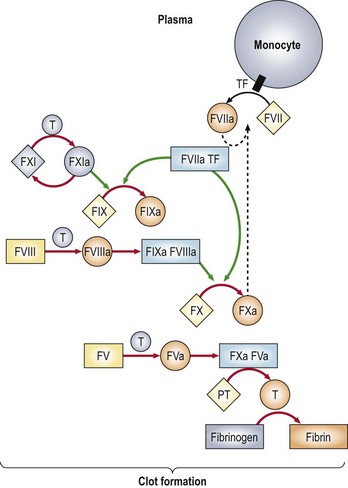
Fig 6.2 Physiological pathways of blood coagulation.
Green arrows indicate the action of enzymes on substrates; red arrows indicate the conversion of a protein from one functional state to another after the cleavage of one or more peptide bonds. F, factor; TF, tissue factor; T, thrombin; PT, prothrombin. Reprinted with permission from Furie B, Furie BC 2004 Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends in Molecular Medicine 10(4):171–178.
Regulation of coagulation
Blood coagulation is modulated by three major inhibitory systems:



Fibrinolysis
Once damaged endothelium is repaired the fibrin thrombus must be removed to restore normal blood flow. Thrombus removal is facilitated by a fibrin-splitting serine protease, plasmin. The fibrinolytic system is shown schematically in Figure 6.3. Release of tissue plasminogen activator (t-PA) from endothelial cells leads to conversion of the proenzyme plasminogen into plasmin. t-PA is most active when bound to fibrin, thus maximising its action at the site of the thrombus. Plasmin has the capacity to digest fibrin in addition to fibrinogen and a number of other proteins. Digestion of a cross-linked thrombus by plasmin leads to the formation of ‘degradation products’ which themselves act as anticoagulants. Fibrinolysis is under strict control; circulating plasmin is inactivated by the protease inhibitor α2-antiplasmin.
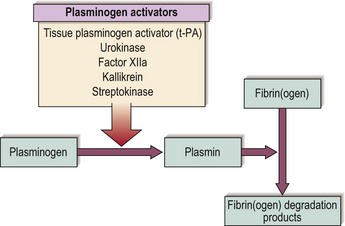
Fig 6.3 The fibrinolytic system.
Note that, unlike the other activators listed, streptokinase is an exogenous activator derived from β-haemolytic streptococci.





