[level-membership-for-hematology-oncology-and-palliative-medicine-category]14
Haemolytic anaemia I – General features and inherited disorders
General features of haemolysis
In classification of the haemolytic anaemias there are three main considerations:
 The mode of acquisition of the disease: is it an inherited disorder or a disorder acquired in later life?
The mode of acquisition of the disease: is it an inherited disorder or a disorder acquired in later life?


The simple classification in Table 14.1 relies upon division of the main clinical disorders into inherited and acquired types. In general, it can be seen that inherited disorders are intrinsic to the red cell and acquired disorders extrinsic. The inherited disorders can be subdivided depending on the site of the defect within the cell – in the membrane, in haemoglobin, or in metabolic pathways. Acquired disorders (discussed in the next section) are broadly divided depending on whether the aetiology has an immune basis.
Table 14.1
Classification of the haemolytic anaemias
| Inherited disorders | |
| Red cell membrane | Hereditary spherocytosis and hereditary elliptocytosis |
| Haemoglobin | Thalassaemia syndromes and sickling disorders |
| Metabolic pathways | Glucose-6-phosphate dehydrogenase and pyruvate kinase deficiency |
| Acquired disorders | |
| Immune | Warm and cold autoimmune haemolytic anaemia |
| Isoimmune | Rhesus or ABO incompatibility (e.g. haemolytic disease of newborn, haemolytic transfusion reaction) |
| Non-immune and trauma | Valve prostheses, microangiopathy, infection, drugs or chemicals, hypersplenism |
Diagnosis of a haemolytic anaemia
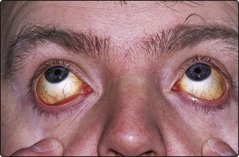
Recognition of the general clinical and laboratory features of haemolysis usually precedes diagnosis of a particular clinical syndrome. Where haemolysis leads to significant anaemia the resultant symptoms are as for other causes of anaemia. However, the increased red cell breakdown of the haemolytic anaemias causes an additional set of problems. Accelerated catabolism of haemoglobin releases increased amounts of bilirubin into the plasma such that patients may present with jaundice (Fig 14.1). Where the spleen is a major site of red cell destruction there may be palpable splenomegaly. Severe prolonged haemolytic anaemia in childhood can lead to expansion of the marrow cavity and associated skeletal abnormalities including frontal bossing of the skull.
Inherited disorders
Disorders of the red cell membrane
Hereditary spherocytosis
This is the most common cause of inherited haemolytic disease in northern Europeans. The disease is heterogeneous with a variable mode of inheritance. There are many possible gene mutations with alterations in spectrin, ankyrin and other membrane proteins. In a blood film the red cells are spheroidal (‘spherocytes’) with a reduced diameter and more intense staining than normal red cells (Fig 14.2). These abnormal red cells are prone to premature destruction in the microvasculature of the spleen.
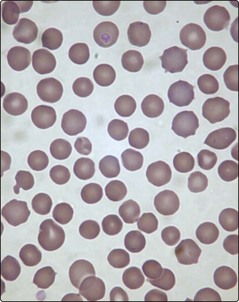
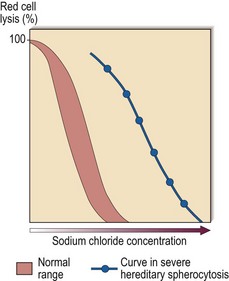
Diagnosis is facilitated by the presence of a family history. The combination of general features of haemolysis and spherocytes in the blood is suggestive of hereditary spherocytosis but not diagnostic as spherocytes may also be seen in autoimmune haemolysis. The two haemolytic disorders are distinguished by the direct antiglobulin test, which is negative in hereditary spherocytosis and nearly always positive in immune haemolysis. Useful screening tests for hereditary spherocytosis include measurement of osmotic fragility (Fig 14.3) and flow cytometric analysis of eosin-5-maleimide binding. In difficult cases, gel electrophoretic analysis of red cell membranes is helpful.
Abnormalities of red cell metabolism
The red cell has metabolic pathways to generate energy and also to protect it from oxidant stress (Fig 14.4). Loss of activity of key enzymes may lead to premature destruction; there are two common examples.
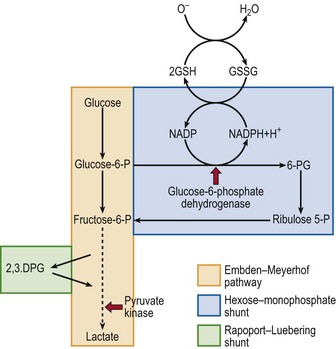
Fig 14.4 Schematic diagram of red cell metabolism.
This shows the key roles of pyruvate kinase in the Embden–Meyerhof pathway (the cell’s source of ATP) and glucose-6-phosphate dehydrogenase in the hexose-monophosphate shunt (the cell’s protection from oxidant stress). The broken line represents several intermediate steps.





[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]14
Haemolytic anaemia I – General features and inherited disorders
General features of haemolysis
In classification of the haemolytic anaemias there are three main considerations:
The mode of acquisition of the disease: is it an inherited disorder or a disorder acquired in later life?
The simple classification in Table 14.1 relies upon division of the main clinical disorders into inherited and acquired types. In general, it can be seen that inherited disorders are intrinsic to the red cell and acquired disorders extrinsic. The inherited disorders can be subdivided depending on the site of the defect within the cell – in the membrane, in haemoglobin, or in metabolic pathways. Acquired disorders (discussed in the next section) are broadly divided depending on whether the aetiology has an immune basis.
Table 14.1
Classification of the haemolytic anaemias
| Inherited disorders | |
| Red cell membrane | Hereditary spherocytosis and hereditary elliptocytosis |
| Haemoglobin | Thalassaemia syndromes and sickling disorders |
| Metabolic pathways | Glucose-6-phosphate dehydrogenase and pyruvate kinase deficiency |
| Acquired disorders | |
| Immune | Warm and cold autoimmune haemolytic anaemia |
| Isoimmune | Rhesus or ABO incompatibility (e.g. haemolytic disease of newborn, haemolytic transfusion reaction) |
| Non-immune and trauma | Valve prostheses, microangiopathy, infection, drugs or chemicals, hypersplenism |
Diagnosis of a haemolytic anaemia
Recognition of the general clinical and laboratory features of haemolysis usually precedes diagnosis of a particular clinical syndrome. Where haemolysis leads to significant anaemia the resultant symptoms are as for other causes of anaemia. However, the increased red cell breakdown of the haemolytic anaemias causes an additional set of problems. Accelerated catabolism of haemoglobin releases increased amounts of bilirubin into the plasma such that patients may present with jaundice (Fig 14.1). Where the spleen is a major site of red cell destruction there may be palpable splenomegaly. Severe prolonged haemolytic anaemia in childhood can lead to expansion of the marrow cavity and associated skeletal abnormalities including frontal bossing of the skull.
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
