CHAPTER 10 HAEMATOLOGICAL PROBLEMS
ANAEMIA IN THE CRITICALLY ILL
Anaemia is common in critically ill patients and often necessitates repeated blood transfusion. Anaemia may be the direct result of an underlying disease process, but more commonly is multifactorial. Common factors that may contribute to anaemia are listed in Box 10.1.
INDICATIONS FOR BLOOD TRANSFUSION
 Oxygen carriage and delivery. The oxygen content of blood is given by Hb × SaO2 × 1.34. Raising haemoglobin is an effective way of improving oxygen content and delivery. (See Oxygen delivery and oxygen consumption, p. 68.)
Oxygen carriage and delivery. The oxygen content of blood is given by Hb × SaO2 × 1.34. Raising haemoglobin is an effective way of improving oxygen content and delivery. (See Oxygen delivery and oxygen consumption, p. 68.)



BLOOD PRODUCTS IN THE UK
Recently, potential transmission of new variant Creutzfeldt–Jakob disease (vCJD) has become a concern. It is likely that in the near future screening of donors for vCJD will become available. Currently in the UK all blood products have white cells removed (leucodepletion) as a precaution against vCJD transmission (white cell count < 5 × 106). Continuing concerns regarding the potential for carriage and transmission of vCJD by the UK blood donor pool has resulted in some plasma products being sourced from outside the UK, principally from the USA.
The following component blood products are available.
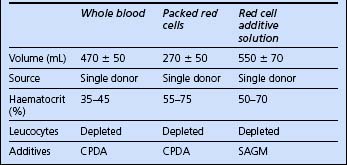
Red cells


The blood products potentially available for red cell replacement are shown in Table 10.1.
Fresh frozen plasma
Recent concerns regarding virus transmission have resulted in treated plasma products becoming available. There are currently two: methylene blue treated and solvent detergent treated. The characteristics of available plasma products are compared in Table 10.2.

Cryoprecipitate
Cryoprecipitate is provided as one to six single donations per pack, suspended in 10–20 mL plasma. It contains fibrinogen and factor VIII. It is used to correct coagulopathy where fibrinogen levels are depleted. Six units generally raise fibrinogen levels by approximately 1 g/L.
ADMINISTRATION OF BLOOD PRODUCTS



Check recipient identity
 When possible, ask the patient to confirm his or her identity and that the details on the identification band are correct.
When possible, ask the patient to confirm his or her identity and that the details on the identification band are correct.

 In some hospitals, a form accompanies blood products, identifying the units issued; this is intended as a record to be placed in the patient’s notes. These forms are not intended to be used as part of the checking procedure, and do not help to ensure that the correct unit of blood is given to the correct patient. The only acceptable checking process is to confirm that the patient details on the product label and those on the patient’s wrist band are the same.
In some hospitals, a form accompanies blood products, identifying the units issued; this is intended as a record to be placed in the patient’s notes. These forms are not intended to be used as part of the checking procedure, and do not help to ensure that the correct unit of blood is given to the correct patient. The only acceptable checking process is to confirm that the patient details on the product label and those on the patient’s wrist band are the same.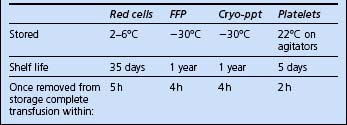









RISKS AND COMPLICATIONS OF BLOOD TRANSFUSION
Complications of blood transfusion include fluid overload, hypothermia, hypocalcaemia, acidosis and dilutional coagulopathy. ARDS and multiple organ failure are also considered to be complications of massive transfusion. Bacterial contamination of blood occurs rarely and is usually fatal (platelet transfusion carries the greatest risk because of the need to store at room temperature).
Acute transfusion reactions are relatively uncommon. They include:





Transfusion-related acute lung injury (TRALI)
Antibodies in the transfused blood product cause activation of the recipient’s white cells, leading to an inflammatory response. The onset is typically within a few hours of transfusion and the clinical features are that of non-cardiogenic pulmonary oedema, which may lead on to the development of ARDS. If TRALI is suspected, the blood transfusion service should be advised so that donors can be screened for white cell antibodies. Treatment is supportive, as for any acute lung injury/ARDS. (See acute lung injury, p. 154.)
For more information on serious hazards of transfusion, see www.shotuk.org. SHOT (Serious Hazards Of Transfusion) is an independent professionally led organization that collects information relating to serious adverse transfusion reactions in the UK.
PATIENTS WHO REFUSE TRANSFUSION
 Blood (red cells, whole blood), FFP, platelets may not be given to Jehovah’s Witnesses under any circumstances.
Blood (red cells, whole blood), FFP, platelets may not be given to Jehovah’s Witnesses under any circumstances.




Management
 Patients with an anticipated major haemorrhage should receive supplementation of iron and other haematinics preoperatively.
Patients with an anticipated major haemorrhage should receive supplementation of iron and other haematinics preoperatively.




NORMAL HAEMOSTATIC MECHANISMS



The classical coagulation cascade is shown in Fig. 10.1. This represents the situation as present in vitro. While it is useful for understanding the serine protease cascade, and for working out in the laboratory the nature of coagulation disorders it does not represent the situation in vivo.
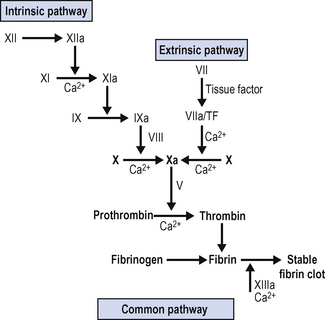
Factor VII forms a complex with tissue factor (TF) on the surface of cells at the site of injury, including platelets. This complex then initiates the coagulation cascade by activating factors X and IX, generating the so-called ‘thrombin burst’ and accelerating clot formation. This is shown in Fig. 10.2.
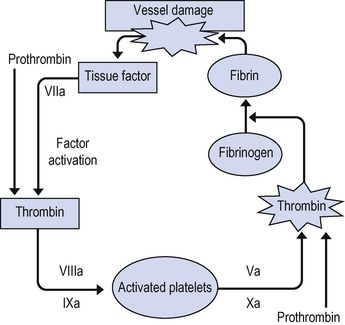
There are also mechanisms within the body to prevent clot formation in healthy vessels and to dissolve established clots. These fibrinolytic pathways are shown in Fig. 10.3.
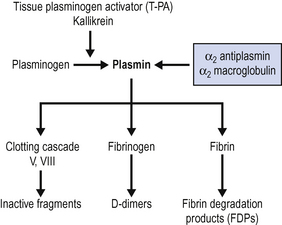
Under normal circumstances, therefore, there is a constant balance maintained between procoagulant mechanisms and anticoagulant mechanisms. If this balance becomes disturbed, bleeding or thrombosis may result.
COAGULOPATHY
Causes
Coagulopathy may result from failure of clot formation, failure of clot stabilization or excessive activation of fibrinolysis. Often more than one process is involved, and the early involvement of a haematologist is advisable. Typical causes of coagulopathy are shown in Box 10.2.
Box 10.2 Typical causes of coagulopathy
| Congenital | Acquired |
|---|---|
| Haemophilia A (factor VIII) | Acquired / functional factor deficiency |
| Haemophilia B (factor IX) | Dilutional coagulopathy |
| Von Willebrand’s disease | Thrombocytopenia |
| Other factor deficiencies | Sepsis |
| Hypothermia | |
| Hepatic dysfunction | |
| Vitamin K deficiency / malabsorption | |
| Renal failure | |
| Drugs |
 Sepsis may produce bone marrow suppression (thrombocytopenia) and triggers inflammatory cascades, which activate both coagulation and fibrinolysis.
Sepsis may produce bone marrow suppression (thrombocytopenia) and triggers inflammatory cascades, which activate both coagulation and fibrinolysis.






Investigations
Basic investigations include platelet count, prothrombin time (PT), activated partial thromboplastin time (APTT), thrombin time (TT), fibrinogen, and fibrinogen breakdown products (FDPs / D dimers). If a specific factor deficiency is considered likely, then individual factor assay may be appropriate. Seek haematological advice. Normal ranges are shown in Table 10.4.
| Normal range | Significance | |
|---|---|---|
| Platelets | 150–450 × 109/L | See thrombocytopenia below |
| PT | 12–14 s (INR = PT/control; normal INR = 1) | Extrinsic and common pathway Marker of hepatic dysfunction / vitamin K deficiency Used to monitor warfarin therapy |
| APTT | 30–40 s | Intrinsic and common pathway Used to monitor heparin therapy |
| TT | 10–12 s | Tests conversion of fibrinogen to fibrin Prolonged by heparin/FDPs/D dimers |
| Fibrinogen | >2 g/L | Reduced in dilutional coagulopathy, liver failure, fibrinolysis (DIC) |
| D dimers | <0.2 g/L | Increased in presence of fibrinolysis (DIC) |
Thromboelastography (TEG)
While the in-vitro tests listed above can be useful diagnostically to determine the likely cause of a coagulopathy, they test individual aspects of the coagulation process rather than reflect the overall process of clot formation. The thromboelastograph can be used to provide a dynamic test of coagulation and fibrinolysis. This can be used to help identify the need for FFP, cryoprecipitate, platelets or antifibrinolytic therapy. An increasing number of hospitals have access to TEG devices in theatre and some critical care units. If available, you should familiarize yourself with the device and interpretation of the information that it gives you before you have to use it in an emergency situation. There is now a body of evidence to suggest that the management of coagulopathy based on interpretation thromboelastogram and related technologies not only helps in the management of haemostasis, but also reduces the quantities of clotting products required. A typical TEG trace is shown in Fig. 10.4.
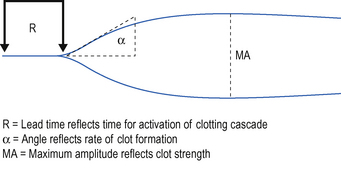
Management of coagulopathy
 Ensure that the patient is adequately resuscitated. Oxygen, i.v. access and adequate volume or blood replacement. Correct hypothermia.
Ensure that the patient is adequately resuscitated. Oxygen, i.v. access and adequate volume or blood replacement. Correct hypothermia.








Activated factor VII (VIIa)
There has been a great deal of interest recently in the role of activated factor VII in the management of coagulopathy and uncontrolled bleeding. Activated factor VII binds to exposed tissue factor on damaged endothelial surfaces and activates the coagulation process, leading to localized fibrin production and clot formation. There is increasing evidence that VIIa is effective in reducing bleeding when other measures have failed. (See Normal haemostatic mechanisms, p. 255.)
THROMBOCYTOPENIA
Thrombocytopenia is a common finding in critically ill patients. Common causes are shown in Box 10.3.
Box 10.3 Causes of thrombocytopenia
| Reduced production | Increased destruction / sequestration |
|---|---|
| Bone marrow failure Drugs, toxins Viral infections |
Infection Disseminated intravascular coagulation (DIC) Mechanical devices (balloon pump/CVVHD) Clot / mechanical destruction Heparin-induced thrombocytopenia (HIT) Immune thrombocytopenia purpura (ITP) Thrombotic thrombocytopenia purpura (TTP) Sequestration (e.g. splenomegaly) |
 In some cases, thrombocytopenia is associated with increased microvascular thrombotic processes. Under these circumstances, giving platelets may increase the risk of clinically significant thrombosis. Do not give platelets unless there is active bleeding, or the platelet count is less than 20 × 109/L and the risk of thrombosis has been excluded (see below).
In some cases, thrombocytopenia is associated with increased microvascular thrombotic processes. Under these circumstances, giving platelets may increase the risk of clinically significant thrombosis. Do not give platelets unless there is active bleeding, or the platelet count is less than 20 × 109/L and the risk of thrombosis has been excluded (see below).



DISSEMINATED INTRAVASCULAR COAGULATION
Disseminated intravascular coagulation (DIC) is a complex process arising as a result of generalized activation of the inflammatory cascade. It involves activation of clotting within the microvasculature, with consequent tissue damage. There is a consumptive coagulopathy, where normal clotting fails to take place because of depletion of circulating factors. The process is generally accompanied by activated fibrinolysis, with clot instability. The breakdown products of fibrinogen (D-dimer) and fibrin (FDPs) are in themselves anticoagulant, thus adding an extra level of complexity.
PURPURIC DISORDERS
Purpura are small purple spots in the skin resulting from microvascular haemorrhage. Although these are not always pathological, e.g. ‘senile purpura’ is secondary to vessel fragility in old age, they should raise suspicion of underlying pathology. Typically, purpura are associated with thrombocytopenia (see above), but they are also seen in other conditions in which there is inflammation or damage to the microvasculature. Important causes are shown in Box 10.4.
THROMBOTIC DISORDERS
A number of factors predispose to thrombosis in ICU patients (Table 10.5).
| Vascular endothelial damage | Trauma / surgery Central venous catheters |
| Altered blood flow | Vascular disease Immobilization Shock states Central venous catheters Effects of vasoactive drugs |
| Altered platelet activity and coagulation state | Underlying disease processes Activation of inflammatory cascades Stress response to surgery, trauma, sepsis |
Some patients are at particular at risk of thrombosis. These include the pregnant, the obese and those with carcinomatosis, pelvic / hip injuries or surgery, myeloproliferative disease, systemic lupus erythematosus (lupus anticoagulant) and conditions such as TTP–HUS (see TTP–HUS, p. 263).
Arterial thrombosis / embolization
Arterial thrombosis / embolization commonly follows vascular surgery in arteriopathies, but can also be seen following trauma or accompanying other prothrombotic conditions such as heparin-induced thrombocytopenia (HIT) (see p. 263). Peripheral small vessel occlusion (e.g. trash foot) is common after aortic surgery, but may also accompany other conditions such as bacterial endocarditis and mycotic aneurysm. This usually improves over time. Embolization of a peripheral (limb) artery results in a pale, weak, numb and pulseless limb with loss of Doppler signals. Central arterial thrombosis of the distal aorta also occurs rarely (saddle embolus).





THE IMMUNOCOMPROMISED PATIENT
All critically ill patients on the intensive care unit should be considered to have some degree of altered immune function (immunocompromise). The causes are multifactorial as shown in Box 10.5.
Box 10.5 Causes of altered immune function in critically ill patients
Impaired barrier defences (breaches in skin / impaired cough reflexes / impaired gut perfusion)
Some specific patient groups are particularly at risk of relative immunocompromise. These include those with alcoholic liver disease and those suffering from malignancy. Such immunocompromise may be difficult to diagnose, as bone marrow function and other measures of response to infection may be apparently normal.
Patients with significant immunocompromise are, however, increasingly common in the ICU. Immune deficiency may be inherited (e.g. severe combined immune deficiency, SCID) or acquired. Most commonly, it is seen in patients with depressed bone marrow function, either as a result of an underlying disease process or as a result of treatment (e.g. following chemotherapy or immunosuppressive treatment following transplantation). Typical causes of significant immunocompromise are shown in Box 10.6.
Management
The principles of management are largely the same as for the immunocompetent patient. Resuscitation and stabilization are the initial priorities.










AIDS
Patients newly presenting with a disease characteristic of an immunosuppressed state, and who are subsequently diagnosed as HIV positive during the course of critical care admission, would not normally be commenced on antiretroviral therapy until after discharge from the intensive care unit, or recovery from the acute infective process. This is because acute institution of antiretroviral agents is unlikely significantly to affect the prognosis during the acute illness.