[level-membership-for-anesthesiology-category]
Haematological Disorders and Blood Transfusion
THE PHYSIOLOGY OF BLOOD
Blood Coagulation
The physiology of haemostasis involves a complex interaction between the endothelium, clotting factors and platelets. Normally, the subendothelial matrix and tissue factor are separated from platelets and clotting factors by an intact endothelium. However, when a blood vessel is damaged, vasospasm occurs, which reduces initial bleeding and slows blood flow, increasing contact time between the blood and the area of injury. Initial haemostasis occurs through the action of platelets. Circulating platelets bind directly to exposed collagen with specific glycoprotein Ia/IIa receptors. Von Willebrand factor released from both endothelium and activated platelets strengthens this adhesion. Platelet activation results in a shape change, increasing platelet surface area, allowing the development of extensions which can connect to other platelets (pseudopods). Activated platelets secrete a variety of substances from storage granules, including calcium ions, ADP, platelet activating factor, von Willebrand factor, serotonin, factor V and protein S. Activated platelets also undergo a change in a surface receptor, glycoprotein GIIb/IIIa, which allows them to cross-link with fibrinogen. In parallel with all these changes, the coagulation pathway is activated and further platelets adhere and aggregate (Fig. 13.1).
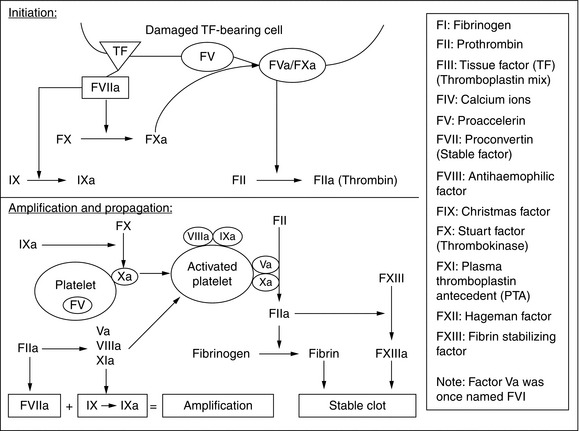
FIGURE 13.1 Clotting processes. From Curry ANG, Pierce JMT. Continuing Education in Anaesthesia, Critical Care & Pain 2007; 2(2): 45–50.
The classical description of coagulation pathways includes an intrinsic pathway and an extrinsic pathway in which clotting factors are designated with Roman numerals (Fig. 13.1). Each pathway consists of a cascade in which a clotting factor is activated and in turn catalyses the activation of another pathway. The intrinsic pathway involves the sequential activation of factors XII, XI and IX. The extrinsic pathway involves the activation of factor VII by tissue factor, and is sometimes called the tissue factor pathway. Of the two pathways, the extrinsic pathway is considered to be the more important because abnormal expression of the intrinsic pathway does not necessarily result in abnormal clotting. The intrinsic pathway may have an additional role in the inflammatory response.
The following steps can be conceptualized (Fig. 13.1):
Common laboratory tests used to investigate coagulation include:




HAEMATOLOGICAL DISORDERS AND THEIR IMPACT ON ANAESTHESIA
Anaemia occurs as a result of decreased red cell production or increased loss due to bleeding or destruction. A number of congenital or acquired conditions can result in anaemia (Table 13.1). Anaemia is defined as a haemoglobin less than 13 g dL− 1 (men) or 12 g dL− 1 (women), but the level of anaemia at which physiological dysfunction occurs in everyday life, or under the stress of surgery, is unclear.
TABLE 13.1

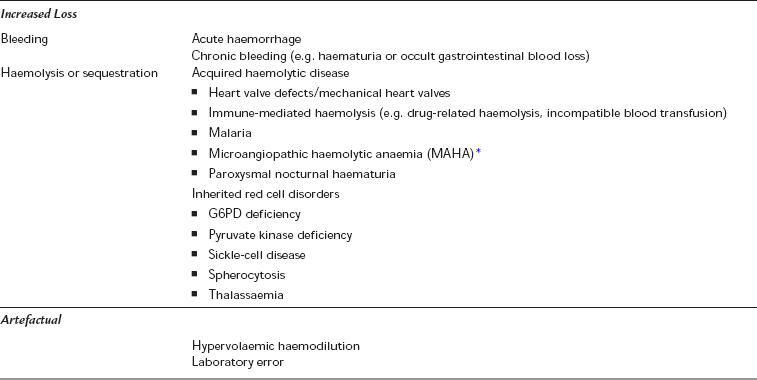
*Causes of MAHA include vasculitis, disseminated intravascular coagulation (DIC), HELLP syndrome (Haemolysis, Elevated Liver enzymes, Low Platelets) and thrombotic thrombocytopaenic purpura/haemolytic uraemic syndrome (TTP/HUS) (see Table 13.2).



Haemoglobinopathies
Inherited and Other Coagulopathies
Various conditions and drugs are known to be associated with increased blood loss during surgery (Tables 13.2 and 13.3). If a patient presents with a known condition or with a history of abnormal bleeding (e.g. menorrhagia or excessive bleeding after previous minor injuries), blood should be sent for a coagulation profile including a platelet count, prothrombin time (PT), thrombin time (TT) and activated partial thromboplastin time (APTT). A platelet count is often done as part of the full blood count, which is considered routine before major surgery. In contrast, coagulation screens should not be considered routine. They are designed for specific investigation of patients with bleeding disorders, not as screening tests. They have a low probability of detecting an abnormality in the absence of any relevant history.
TABLE 13.2
Conditions Which are Known to Increase Blood Loss
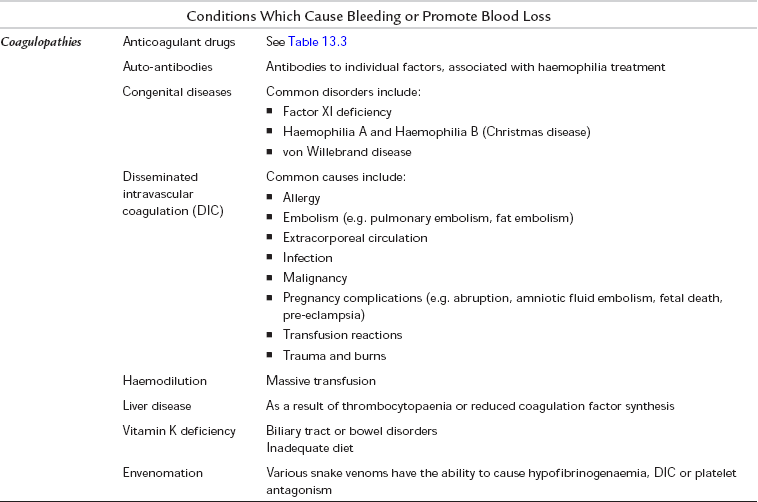


*Heparin-induced thrombocytopaenia (HIT)
**HELLP syndrome (Haemolysis, Elevated Liver enzymes, Low Platelets)
†Thrombotic thrombocytopaenic purpura/haemolytic uraemic syndrome (TTP/HUS)
TABLE 13.3
Drugs which are Known to Increase or Reduce Blood Loss
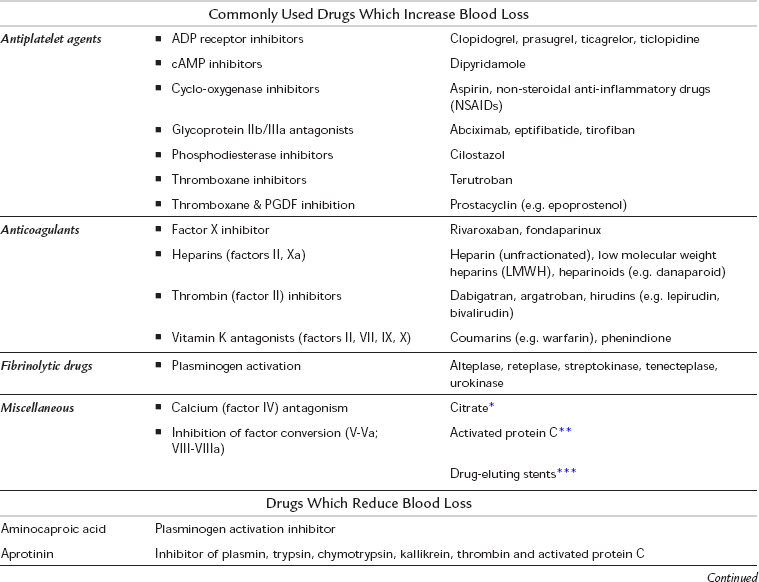

*Citrate may be used to anticoagulate some dialysis machines, and may be used to ‘lock’ central lines (i.e. keep them from becoming blocked with blood clot)
**Activated Protein C has been used in the treatment of severe sepsis
***Used in various angioplasty procedures; mechanism of action depends on drug being released by the stent
Reproduced from Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. British Journal of Haematology 2009; 145(1): 24–33. © Blackwell Publishing.
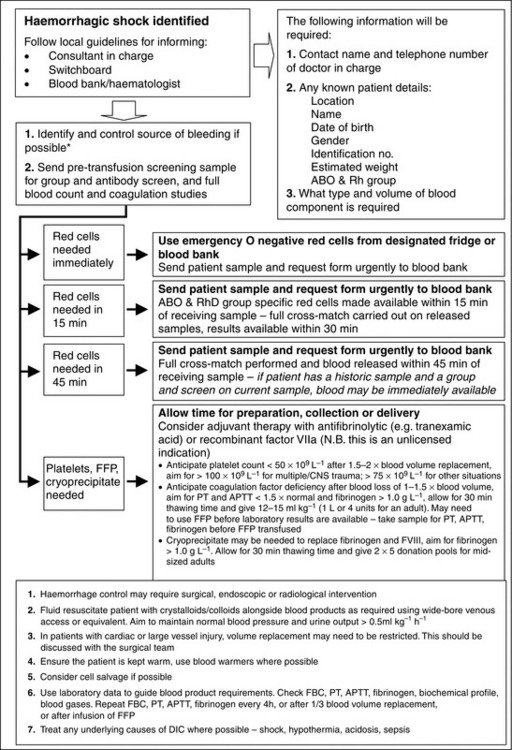
Acquired coagulopathy can also occur as an acute event, for example: following major trauma, during major haemorrhage or in the presence of disseminated intravascular coagulopathy (DIC). In major haemorrhage, clotting factors can become depleted if not replaced promptly. The management of coagulopathy in major haemorrhage should be guided by clinical urgency and laboratory tests (Fig. 13.2).
Coagulopathy of Trauma: Patients who have suffered major trauma have a high incidence of coagulopathy. This is multifactorial and complex but certainly appears before administration of intravenous fluids or blood products, so is not solely an iatrogenic haemodilution effect. The factors associated with coagulopathy are listed in Table 13.4. These factors interact in particular in the ‘lethal triad’ of hypothermia, acidosis and coagulopathy. The effect of hypothermia is not seen in routine coagulation testing because these are performed at 37°C.
TABLE 13.4
Factors Associated with Coagulopathy in Trauma
Physiological dilution of clotting factors
Hypothermia
Acidosis
Red cell loss
Trauma-induced fibrinolysis
Injury-related inflammation
Hypoperfusion
Hypocalcaemia
Genetic predispositions
Iatrogenic – dilution by fluids, anticoagulant effects of intravenous fluids
Disseminated Intravascular Coagulation (DIC): In DIC, the microcirculation of different organs becomes damaged by fibrin clots generated by coagulation pathways which become hyperactive. The pathophysiological production of so many fibrin clots results in a consumptive coagulopathy rendering the patient susceptible to haemorrhage as a result of surgery or other invasive procedures. The causes of DIC are shown in Table 13.2. If DIC is suspected, the cause should be identified and corrected wherever possible. The diagnosis of DIC can be difficult to make and relies upon evaluating the results of several aspects of a coagulation profile. A scoring system exists to evaluate the likelihood of DIC (Table 13.5). Patients who develop DIC in the perioperative period, or who require surgery to treat the cause of DIC (e.g. patients with intra-abdominal sepsis), are at increased risk of major haemorrhage. They are likely to need replacement of consumed coagulation factors in the form of platelets, fresh frozen plasma and cryoprecipitate. Haematological advice should be sought whenever DIC is suspected.
TABLE 13.5
Diagnostic Scoring System for Disseminated Intravascular Coagulation (DIC)
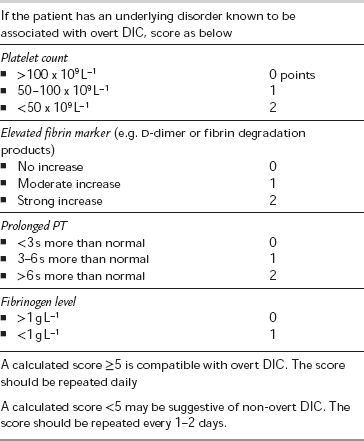
Adapted from the ISTH Diagnostic scoring system for DIC
Drug-Induced Coagulopathies: A list of drugs known to increase blood loss is shown in Table 13.3. If possible, provision should be made to discontinue these preoperatively, taking into account the amount of ‘wash-out’ time which may be required. For example, vitamin K inhibitors such as warfarin can take up to 5 or 6 days, and irreversible platelet inhibitors such as aspirin and clopidogrel may take up to 7 days (the time it takes to generate new platelets). In patients in whom continued anticoagulation is considered essential, for instance those at high risk of venous thromboembolic disease, ‘bridging’ therapy during the wash-out period with shorter-acting anticoagulants such as an unfractionated heparin infusion or low molecular weight heparins (LMWHs) may be considered. These shorter acting agents can be stopped closer to the time of surgery in order to minimize the amount of time the patient is without anticoagulants. In the case of LMWHs, this should be 24 h before surgery, with the last dose being reduced to half normal. In the case of an unfractionated heparin infusion, this can be stopped 2–6 h prior to surgery and the level of anticoagulation monitored by measuring the APTT of the patient’s blood.
Thrombocytopaenia: Thrombocytopenia is usually defined as a platelet count less than 100 × 109 L− 1, but the point at which thrombocytopaenia becomes clinically important depends upon the clinical scenario. Conditions which can result in thrombocytopaenia are shown in Table 13.2. In patients whose platelet count is low, or platelet function is thought to be impaired, a perioperative platelet transfusion may be required.
 ≥ 20 × 109 L− 1 – for a minor intervention (e.g. insertion of a urinary catheter or nasogastric tube)
≥ 20 × 109 L− 1 – for a minor intervention (e.g. insertion of a urinary catheter or nasogastric tube)
 ≥ 30 × 109 L− 1 – for the insertion of a central venous catheter under ultrasound guidance
≥ 30 × 109 L− 1 – for the insertion of a central venous catheter under ultrasound guidance
 ≥ 60–80 × 109 L− 1 – for epidural insertion or neuraxial blockade
≥ 60–80 × 109 L− 1 – for epidural insertion or neuraxial blockade
 ≥ 80 × 109 L− 1 – for uncomplicated surgery
≥ 80 × 109 L− 1 – for uncomplicated surgery
 ≥ 100 × 109 L− 1 – major haemorrhage, major trauma or CNS trauma
≥ 100 × 109 L− 1 – major haemorrhage, major trauma or CNS trauma
INTERVENTIONAL PROCEDURES AND REGIONAL ANAESTHESIA IN COAGULOPATHIC PATIENTS
 LMWH (thromboprophylaxis dose): needle insertion should be delayed until 12 h after last dose; epidural catheters should be removed at least 12 h after the last dose and at least 4 h before the next dose
LMWH (thromboprophylaxis dose): needle insertion should be delayed until 12 h after last dose; epidural catheters should be removed at least 12 h after the last dose and at least 4 h before the next dose


 Ticlopidine: should be discontinued 14 days prior to neuraxial blockade
Ticlopidine: should be discontinued 14 days prior to neuraxial blockade
 Clopidogrel: should be discontinued 7 days prior to neuraxial blockade
Clopidogrel: should be discontinued 7 days prior to neuraxial blockade
 GIIb/IIIa inhibitors: should be discontinued 9–48 h prior to neuraxial blockade.
GIIb/IIIa inhibitors: should be discontinued 9–48 h prior to neuraxial blockade.
THROMBOSIS AND ACUTE ISCHAEMIC EVENTS
All hospital patients should undergo an assessment of their risk of developing venous thromboembolism (VTE) in order to ensure that appropriate prophylactic measures are taken. Reassessment should be undertaken after 24 h, and at any time that the patient’s clinical condition changes. Any assessment should weigh the risk of developing VTE against the risk of bleeding which might occur when pharmacological prophylaxis is prescribed (Tables 13.6 and 13.7). Methods of pharmacological prophylaxis include subcutaneous low molecular weight heparins, subcutaneous unfractionated heparin and newer anticoagulants such as fondaparinux, dabigatran and rivaroxaban. Antiplatelet agents such as aspirin are not considered to provide adequate protection against VTE when used in isolation.
TABLE 13.6
Conditions which are Known to Increase Thrombosis Risk
| Acquired | Antiphospholipid syndrome Cardiac failure Diabetes Heparin-induced thrombocytopaenia Hyperlipidaemia Malignancy Myeloproliferative disorders Nephrotic syndrome Oral contraceptive pill (oestrogen therapies) Paroxysmal nocturnal haemoglobinuria Polycythaemia TTP/HUS* |
| Congenital | Antithrombin deficiency Dysfibrinogenaemia Factor V Leiden variant (activated protein C resistance) Hyperhomocysteinaemia Protein C deficiency Protein S deficiency Prothrombin genetic variant |
*Thrombotic thrombocytopaenic purpura/haemolytic uraemic syndrome (TTP/HUS)
TABLE 13.7
Risk Assessment for Venous Thromboembolism (VTE)
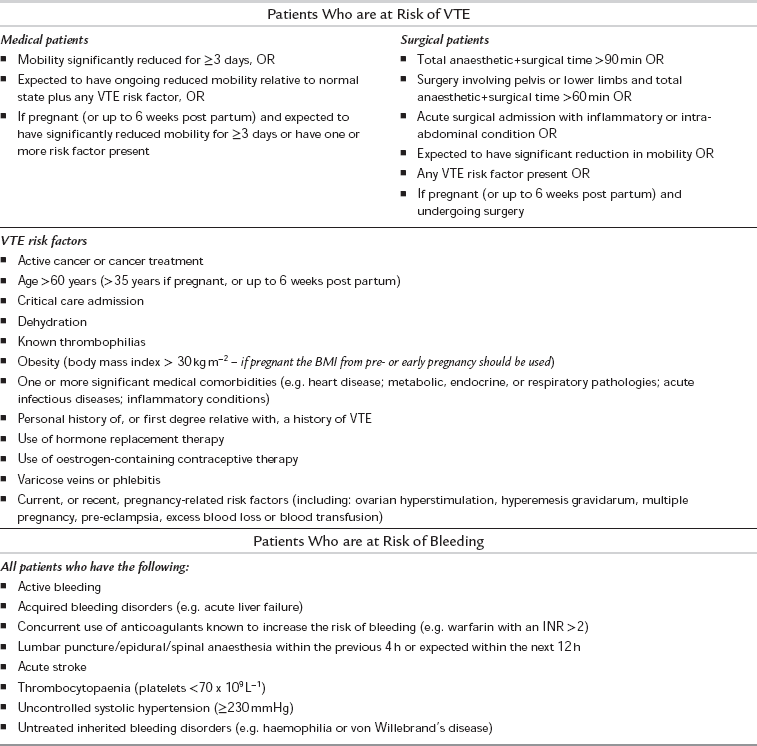
Adapted from the NICE guidelines, Venous thromboembolism: reducing the risk, 2010
BLOOD PRODUCTS AND BLOOD TRANSFUSION
The transfusion of whole blood is relatively uncommon and donated blood is usually separated into its constituent components, which are then available for transfusion. A wide range of blood products are available, the most common of which are listed in Table 13.8, along with their indications. Units of packed red cells are most commonly transfused during the resuscitation of acute haemorrhage, or as a treatment of symptomatic anaemia.
TABLE 13.8
Indications and Uses of Blood Products
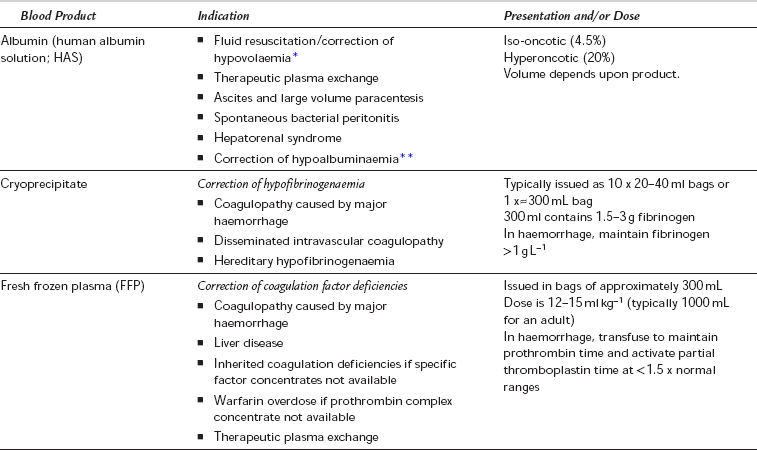

*Albumin may be used safely for volume resuscitation, but there is little evidence to suggest that it is associated with improved outcomes when compared with other resuscitation fluids. It may be associated with worse outcomes when used in certain conditions such as traumatic brain injury.
**The use of human albumin solution to correct hypoalbuminaemia is contentious. Hypoalbuminaemia is associated with increased mortality in critically ill patients, but actively correcting it is not clearly associated with improved outcome. Hyperoncotic albumin has also been used in the management of adult respiratory distress syndrome (ARDS).
Red Cell Storage Lesion
 Patients of blood group O can receive only group O donated blood
Patients of blood group O can receive only group O donated blood
 Patients of blood group A can receive group O or group A donated blood
Patients of blood group A can receive group O or group A donated blood
 Patients of blood group B can receive group O or group B donated blood
Patients of blood group B can receive group O or group B donated blood
 Patients of blood group AB can receive groups O, A, B or AB donated blood
Patients of blood group AB can receive groups O, A, B or AB donated blood
 Patients with RhD positive blood can receive RhD positive or RhD negative blood
Patients with RhD positive blood can receive RhD positive or RhD negative blood

Other, less common, red blood cell antibody/antigen reactions may also occur, and if time allows, a full cross-match should be undertaken. In more urgent situations a more limited approach may be necessary (Fig. 13.2).
The transfusion of blood products is not without risk, and, if possible, should not be done without the informed consent of the recipient. A list of the most common complications of transfusion is shown in Table 13.9. Of particular note are the risks associated with the transfusion of incompatible blood products, which are considered to be ‘Never Events’ within the UK (the equivalent to ‘No Pay’ events within the US system). A ‘zero-tolerance’ approach to pre-transfusion sampling, and blood product checking and administration is recommended, which includes:
TABLE 13.9
Errors and Complications of Blood Transfusion (as Classified and Defined by the SHOT Report 2009)
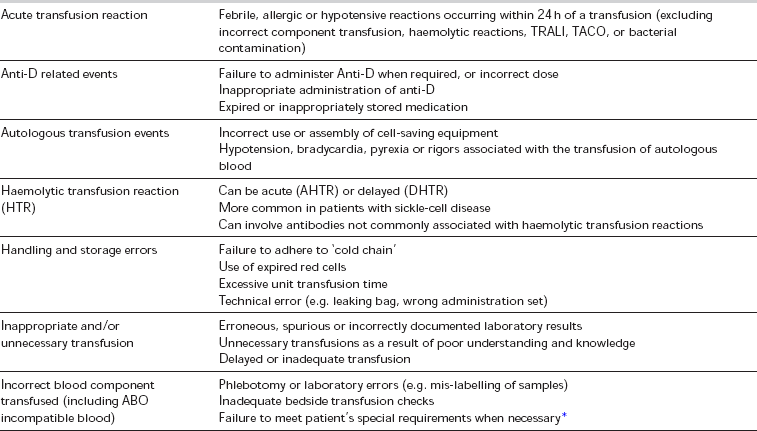
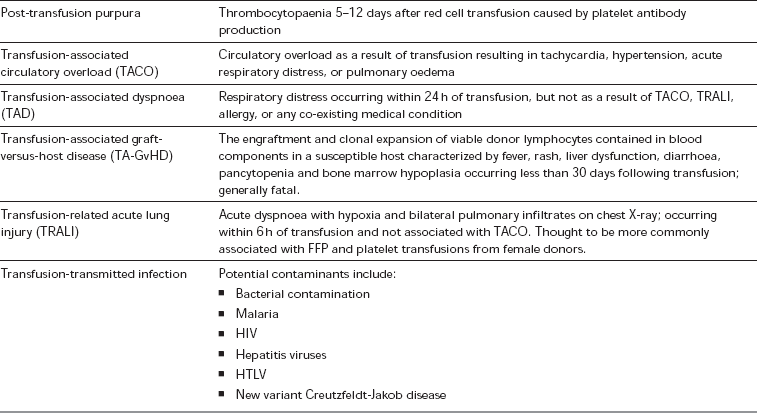
*Special requirements include irradiated blood, or blood where cytomegalovirus is absent (CMV negative blood) – for indications see text







MAJOR HAEMORRHAGE
When major haemorrhage occurs, it is often necessary to transfuse large volumes of both packed red cells and products which promote clotting, such as fresh frozen plasma (FFP), cryoprecipitate and platelets. Major haemorrhage protocols have been developed in order to aid this process, an example of which is shown in Figure 13.2. If possible, the transfusion of red cells and clotting products should be guided by laboratory results, or by near-patient testing (for example near-patient haemoglobinometers or thromboelastography – TEG). However, waiting for confirmation of coagulopathy before administering appropriate therapy is likely to lead to greater blood loss, use of more blood products and worse outcome. Major trauma is well recognized as causing early coagulopathy even before any fluids or blood products are given.
Predictable Blood Loss
Blood conservation strategies should be employed whenever possible. These can involve the proactive or reactive use of pharmacological treatments, for example the administration of tranexamic acid, or topical fibrin sealants (Table 13.3). Alternatively, mechanical methods of blood conservation may be employed, for example the use of tourniquets for lower limb surgery.
Association of Anaesthetists of Great Britain and Ireland. Management of anaesthesia for Jehovah’s Witnesses. AAGBI; 1999.
Beed, M., Levitt, M., Bokhari, S.W. Intensive care management of patients with haematological malignancy. Continuing Education in Anaesthesia, Critical Care & Pain. 2010;10:167–171.
Chee, Y.L., Crawford, J.C., Watson, H.G., et al. Guidelines on the assessment of bleeding risk prior to surgery or invasive procedures. Br. J. Haematol. 2008;140:496–504.
Curry, A.N.G., Pierce, J.M.T. Conventional and near-patient tests of coagulation. Continuing Education in Anaesthesia, Critical Care & Pain. 2007;2:45–50.
Hoffbrand, A.V., Moss, P. Essential haematology. Oxford: Wiley-Blackwell; 2010.
Horlocker, T.T., Wedel, D.J., Benzon, H., et al. Regional anesthesia in the anticoagulated patient: defining the risks (The Second ASRA Consensus Conference on Neuraxial Anesthesia and Anticoagulation). Reg. Anesth. Pain Med. 2003;28:172–197.
Levi, M., Toh, C.H., Thachil, J., et al. Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br. J. Haematol. 2009;145:24–33.
Mason, R. Anaesthesia databook, third ed. Greenwich Medical Media; 2001.
McClelland, D.B.L., Handbook of transfusion medicine. fourth ed. United Kingdom Blood Services, 2007.
National Institute for Health and Clinical Excellence (UK). Venous thromboembolism: reducing the risk. NICE; 2010.
Wilson, M., Forsyth, P., Whiteside, J. Haemoglobinopathy and sickle-cell disease. Continuing Education in Anaesthesia, Critical Care & Pain. 2010;10:24–28.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Haematological Disorders and Blood Transfusion
THE PHYSIOLOGY OF BLOOD
Blood Coagulation
The physiology of haemostasis involves a complex interaction between the endothelium, clotting factors and platelets. Normally, the subendothelial matrix and tissue factor are separated from platelets and clotting factors by an intact endothelium. However, when a blood vessel is damaged, vasospasm occurs, which reduces initial bleeding and slows blood flow, increasing contact time between the blood and the area of injury. Initial haemostasis occurs through the action of platelets. Circulating platelets bind directly to exposed collagen with specific glycoprotein Ia/IIa receptors. Von Willebrand factor released from both endothelium and activated platelets strengthens this adhesion. Platelet activation results in a shape change, increasing platelet surface area, allowing the development of extensions which can connect to other platelets (pseudopods). Activated platelets secrete a variety of substances from storage granules, including calcium ions, ADP, platelet activating factor, von Willebrand factor, serotonin, factor V and protein S. Activated platelets also undergo a change in a surface receptor, glycoprotein GIIb/IIIa, which allows them to cross-link with fibrinogen. In parallel with all these changes, the coagulation pathway is activated and further platelets adhere and aggregate (Fig. 13.1).
FIGURE 13.1 Clotting processes. From Curry ANG, Pierce JMT. Continuing Education in Anaesthesia, Critical Care & Pain 2007; 2(2): 45–50.
The classical description of coagulation pathways includes an intrinsic pathway and an extrinsic pathway in which clotting factors are designated with Roman numerals (Fig. 13.1). Each pathway consists of a cascade in which a clotting factor is activated and in turn catalyses the activation of another pathway. The intrinsic pathway involves the sequential activation of factors XII, XI and IX. The extrinsic pathway involves the activation of factor VII by tissue factor, and is sometimes called the tissue factor pathway. Of the two pathways, the extrinsic pathway is considered to be the more important because abnormal expression of the intrinsic pathway does not necessarily result in abnormal clotting. The intrinsic pathway may have an additional role in the inflammatory response.
The following steps can be conceptualized (Fig. 13.1):
Common laboratory tests used to investigate coagulation include:
HAEMATOLOGICAL DISORDERS AND THEIR IMPACT ON ANAESTHESIA
Anaemia occurs as a result of decreased red cell production or increased loss due to bleeding or destruction. A number of congenital or acquired conditions can result in anaemia (Table 13.1). Anaemia is defined as a haemoglobin less than 13 g dL− 1 (men) or 12 g dL− 1 (women), but the level of anaemia at which physiological dysfunction occurs in everyday life, or under the stress of surgery, is unclear.
TABLE 13.1
*Causes of MAHA include vasculitis, disseminated intravascular coagulation (DIC), HELLP syndrome (Haemolysis, Elevated Liver enzymes, Low Platelets) and thrombotic thrombocytopaenic purpura/haemolytic uraemic syndrome (TTP/HUS) (see Table 13.2).
