Haematological disorders
Haemoglobin production in the fetus and newborn
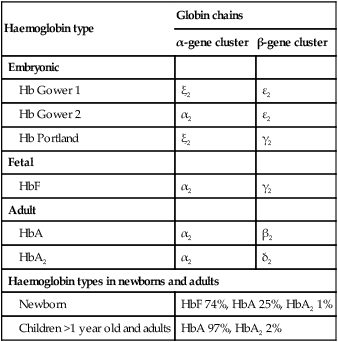
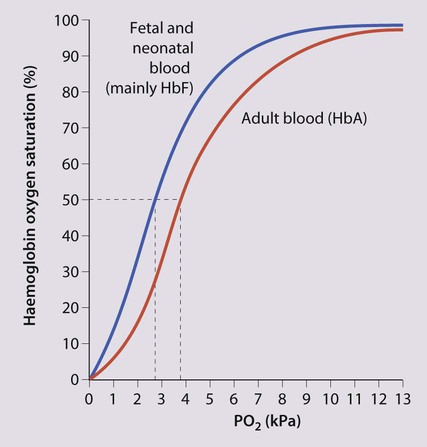
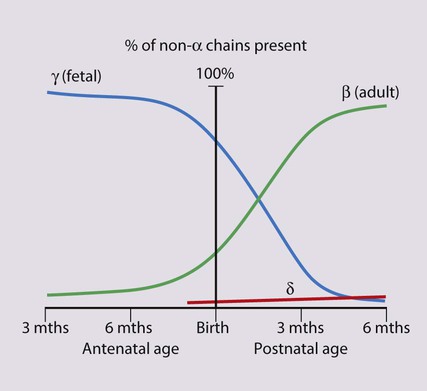
The most important difference between haemopoiesis in the fetus compared with postnatal life is the changing pattern of haemoglobin (Hb) production at each stage of development. The composition and names of these haemoglobins are shown in Table 22.1. Understanding the developmental changes in haemoglobin helps to explain the patterns of abnormal haemoglobin production in some inherited childhood anaemias. Embryonic haemoglobins (Hb Gower 1, Hb Gower 2 and Hb Portland) are produced between 4 and 8 weeks’ gestation, after which haemoglobin production switches to fetal haemoglobin (HbF). HbF is made up of 2 α chains and 2 γ chains (α2γ2) and is the main Hb during fetal life. HbF has a higher affinity for oxygen than adult Hb (HbA), and is therefore better able to hold on to oxygen, an advantage in the relatively hypoxic environment of the fetus (Fig. 22.1). At birth, the types of Hb are: HbF, HbA and HbA2. HbF is gradually replaced by HbA and HbA2 during the first year of life. By 1 year of age, the percentage of HbF is very low in healthy children and increased proportions of HbF are a sensitive indicator of some inherited disorders of haemoglobin production (haemoglobinopathies) .
Table 22.1
Embryonic, fetal and adult haemoglobins
| Haemoglobin type | Globin chains | |
| α-gene cluster | β-gene cluster | |
| Embryonic | ||
| Hb Gower 1 | ξ2 | ε2 |
| Hb Gower 2 | α2 | ε2 |
| Hb Portland | ξ2 | γ2 |
| Fetal | ||
| HbF | α2 | γ2 |
| Adult | ||
| HbA | α2 | β2 |
| HbA2 | α2 | δ2 |
| Haemoglobin types in newborns and adults | ||
| Newborn | HbF 74%, HbA 25%, HbA2 1% | |
| Children >1 year old and adults | HbA 97%, HbA2 2% | |
Haematological values at birth and the first few weeks of life
• At birth, the Hb in term infants is high, 14–21.5 g/dl, to compensate for the low oxygen concentration in the fetus. The Hb falls over the first few weeks, mainly due to reduced red cell production, reaching a nadir of around 10 g/dl at 2 months of age (Fig. 22.2). Normal haematological values at birth and during childhood are shown in the Appendix.
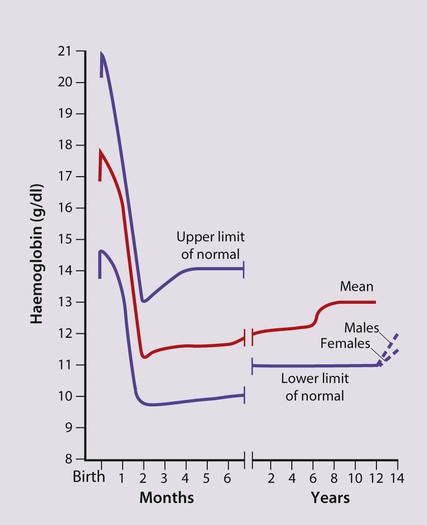
• Preterm babies have a steeper fall in Hb to a mean of 6.5–9 g/dl at 4–8 weeks chronological age.
• Normal blood volume at birth varies with gestational age. In healthy term infants the average blood volume is 80 ml/kg; in preterm infants the average blood volume is 100 ml/kg.
• Stores of iron, folic acid and vitamin B12 in term and preterm babies are adequate at birth. However, in preterm infants, stores of iron and folic acid are lower and are depleted more quickly, leading to deficiency after 2–4 months if the recommended daily intakes are not maintained by supplements.
• White blood cell counts in neonates are higher than in older children (10–25 × 109/L).
• Platelet counts at birth are within the normal adult range (150–400 × 109/L).
Anaemia
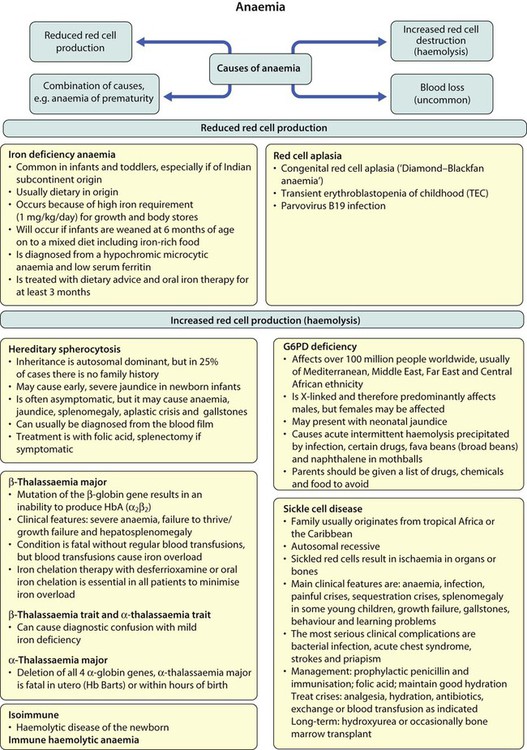
Anaemia results from one or more of the following mechanisms:
• Reduced red cell production – either due to ineffective erythropoiesis (e.g. iron deficiency, the commonest cause of anaemia) or due to red cell aplasia
There may be a combination of these three mechanisms, e.g. anaemia of prematurity.
Using this approach, the principal causes of anaemia are shown in Figure 22.3 and a diagnostic approach to identifying their causes is shown in Figure 22.4.
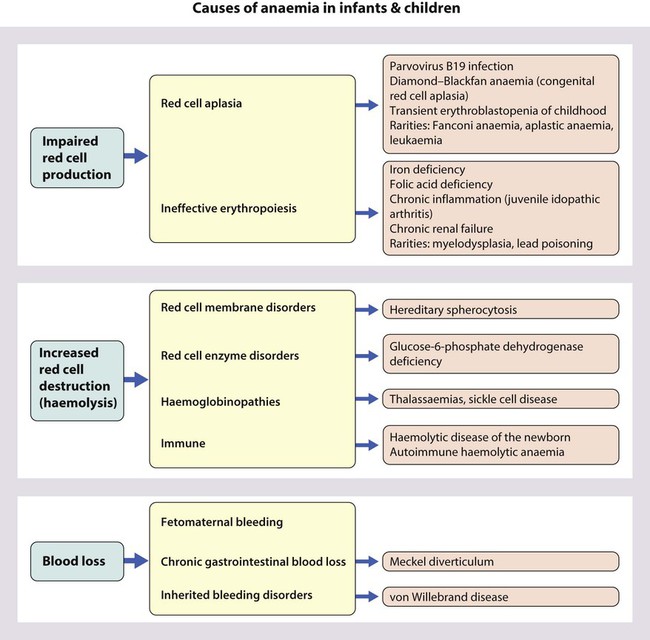
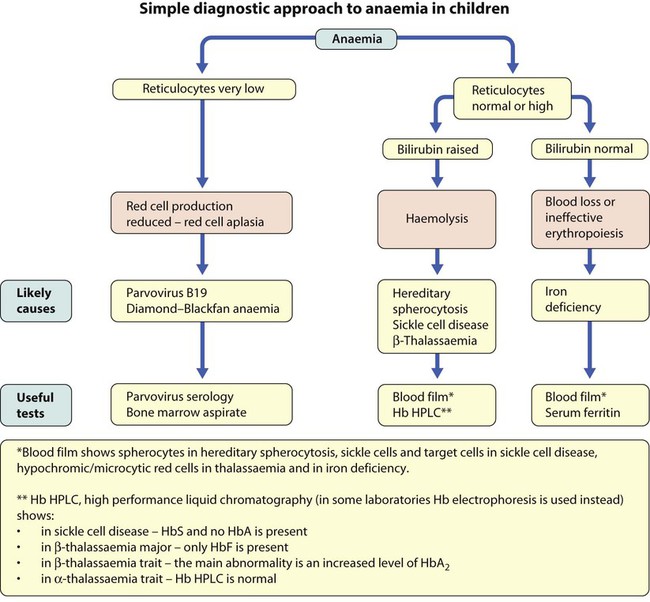
 The definition of anaemia varies with age: Hb <10 g/dl in infants (post-neonatal), Hb <11 g/dl from 1 to 12 years old.
The definition of anaemia varies with age: Hb <10 g/dl in infants (post-neonatal), Hb <11 g/dl from 1 to 12 years old.Causes of anaemia in infants and children
Diagnostic clues to ineffective erythropoiesis are:
• Abnormal mean cell volume (MCV) of the red cells: low in iron deficiency and raised in folic acid deficiency.
Iron deficiency
The main causes of iron deficiency are:
Inadequate intake of iron is common in infants because additional iron is required for the increase in blood volume accompanying growth and to build up the child’s iron stores (Fig. 22.5). A 1-year-old infant requires an intake of iron of about 8 mg/day, which is about the same as his father (9 mg/day) but only half that of his mother (15 mg/day).
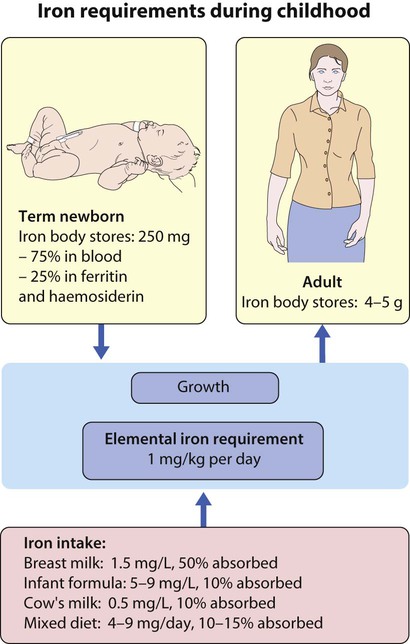
• Breast milk (low iron content but 50% of the iron is absorbed)
• Infant formula (supplemented with adequate amounts of iron)
• Cow’s milk (higher iron content than breast milk but only 10% is absorbed)
• Solids introduced at weaning, e.g. cereals (cereals are supplemented with iron but only 1% is absorbed).
Iron deficiency may develop because of a delay in the introduction of mixed feeding beyond 6 months of age or to a diet with insufficient iron-rich foods, especially if it contains a large amount of cow’s milk (Box 22.1). Iron absorption is markedly increased when eaten with food rich in vitamin C (fresh fruit and vegetables) and is inhibited by tannin in tea.
 Infants should not be fed unmodified cow’s milk as its iron content is low and poorly absorbed.
Infants should not be fed unmodified cow’s milk as its iron content is low and poorly absorbed.Clinical features
Most infants and children are asymptomatic until the Hb drops below 6–7 g/dl. As the anaemia worsens, children tire easily and young infants feed more slowly than usual. The history should include asking about blood loss and symptoms or signs suggesting malabsorption. They may appear pale but pallor is an unreliable sign unless confirmed by pallor of the conjunctivae, tongue or palmar creases. Some children have ‘pica’, a term which describes the inappropriate eating of non-food materials such as soil, chalk, gravel or foam rubber (see Case History 22.1). There is evidence that iron deficiency anaemia may be detrimental to behaviour and intellectual function.
 Treatment of iron deficiency anaemia is with dietary advice and oral iron therapy for several months.
Treatment of iron deficiency anaemia is with dietary advice and oral iron therapy for several months.Red cell aplasia
There are three main causes of red cell aplasia in children:
• Congenital red cell aplasia (‘Diamond–Blackfan anaemia’)
• Transient erythroblastopenia of childhood
• Parvovirus B19 infection (this infection only causes red cell aplasia in children with inherited haemolytic anaemias and not in healthy children).
Increased red cell destruction (haemolytic anaemia)
• Red cell membrane disorders (e.g. hereditary spherocytosis)
• Red cell enzyme disorders (e.g. glucose-6-phosphate dehydrogenase deficiency)
• Haemoglobinopathies (abnormal haemoglobins, e.g. β-thalassaemia major, sickle cell disease).
Haemolysis from increased red cell breakdown leads to:
The diagnostic clues to haemolysis are:
• Raised reticulocyte count (on the blood film this is called ‘polychromasia’ as the reticulocytes have a characteristic lilac colour)
• Unconjugated bilirubinaemia and increased urinary urobilinogen
• Abnormal appearance of the red cells on a blood film (e.g. spherocytes, sickle shaped or very hypochromic) (Fig. 22.6)
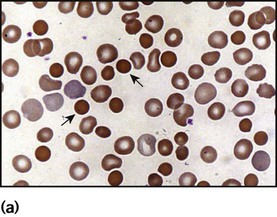
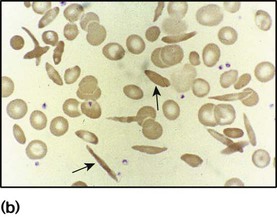
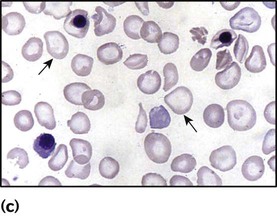
• Positive direct antiglobulin test (only if an immune cause, as this test identifies antibody-coated red blood cells)
Hereditary spherocytosis
Clinical features
• Jaundice – usually develops during childhood but may be intermittent; may cause severe haemolytic jaundice in the first few days of life
• Anaemia – presents in childhood with mild anaemia (haemoglobin 9–11 g/dl), but the haemoglobin level may transiently fall during infections
• Mild to moderate splenomegaly – depends on the rate of haemolysis
• Aplastic crisis – uncommon, transient (2–4 weeks), caused by parvovirus B19 infection
Haemoglobinopathies
These are red blood cell disorders which cause haemolytic anaemia because of reduced or absent production of HbA (α- and β-thalassaemias) or because of the production of an abnormal Hb (e.g. sickle cell disease). α-Thalassaemias are caused by deletions (occasionally mutations) in the α-globin gene. β-Thalassaemia and sickle cell disease are caused by mutations in the β-globin gene. Clinical manifestations of the haemoglobinopathies affecting the β-chain are delayed until after 6 months of age when most of the HbF present at birth has been replaced by adult HbA (Fig. 22.7, Table 22.2).
Table 22.2
Haemoglobins in haemoglobinopathies
| HbA | HbA2 | HbF | HbS | |
| Newborn | 25% | 1% | 74% | − |
| Adult | 97% | 2% | − | − |
| β-Thalassaemia trait | >90% | ↑ | + ↑ | − |
| β-Thalassaemia major | − | ↑ | ↑ | − |
| Sickle cell trait | 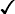 |
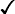 |
+ ↑ | 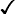 |
| Sickle cell disease | − | 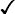 |
+ ↑ |  |
Sickle cell disease
There are three main forms of sickle cell disease and the sickle trait:
• Sickle cell anaemia (HbSS) – patients are homozygous for HbS, i.e. virtually all their Hb is HbS; they have small amounts of HbF and no HbA because they have the sickle mutation in both β-globin genes.
• HbSC disease (HbSC) – affected children inherit HbS from one parent and HbC from the other parent (HbC is formed as a result of a different point mutation in β-globin), so they also have no HbA because they have no normal β-globin genes.
• Sickle β-thalassaemia – affected children inherit HbS from one parent and β-thalassaemia trait from the other. They have no normal β-globin genes and most patients can make no HbA and therefore have similar symptoms to those with sickle cell anaemia.
• Sickle trait – inheritance of HbS from one parent and a normal β-globin gene from the other parent, so approximately 40% of the haemoglobin is HbS. They do not have sickle cell disease but are carriers of HbS, so can transmit HbS to their offspring. They are asymptomatic and are only identified as a result of blood tests.
Management
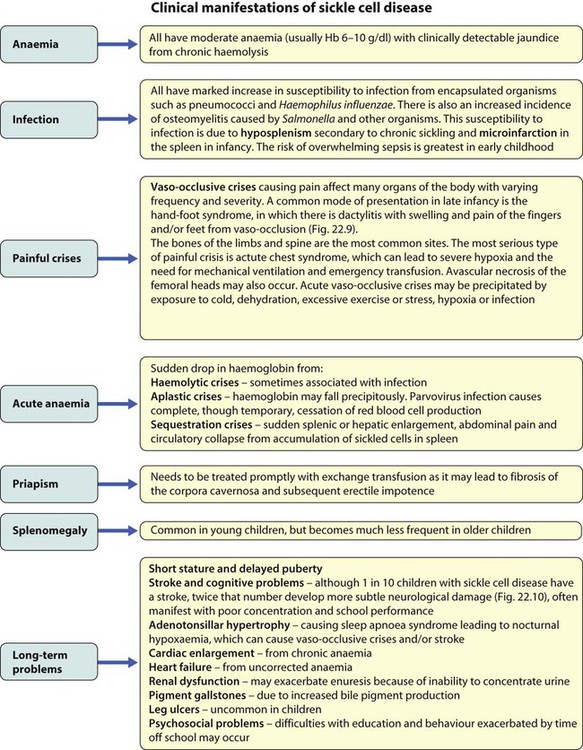
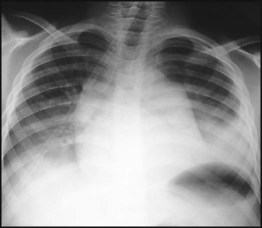
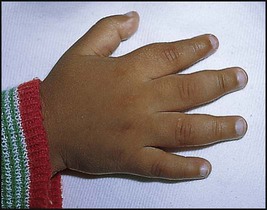
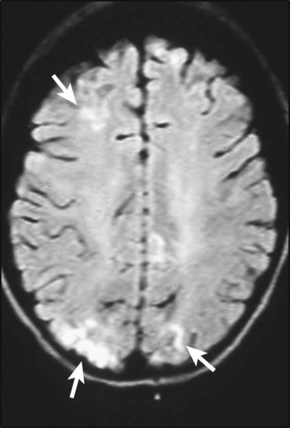
Treatment of chronic problems – Children who have recurrent hospital admissions for painful vaso-occlusive crises or acute chest syndrome (see Case History 22.2) may benefit from hydroxyurea, a drug which increases their HbF production and helps protect against further crises. It requires monitoring for side-effects, especially white blood cell suppression. The most severely affected children (1–5%) who have had a stroke or who do not respond to hydroxyurea may be offered a bone marrow transplant. This is the only cure for sickle cell disease but can only be safely carried out if the child has an HLA-identical sibling who can donate their bone marrow – the cure rate is 90% but there is a 5% risk of fatal transplant-related complications.
β-Thalassaemias
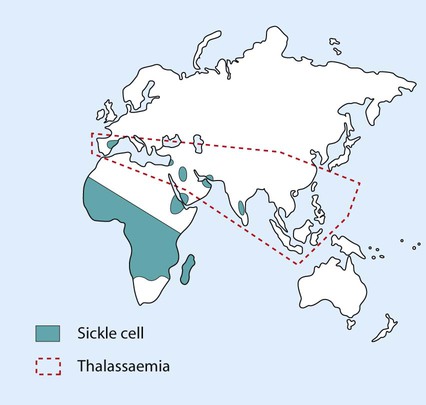
The β-thalassaemias occur most often in people from the Indian subcontinent, Mediterranean and Middle East (Fig. 22.12). In the UK, most affected children are born to parents from the Indian subcontinent; in the past, many were born to Greek Cypriots, but this has become uncommon through active genetic counselling within their community.
• β-Thalassaemia major – This is the most severe form of the disease. HbA (α2β2) cannot be produced because of the abnormal β-globin gene.
• β-Thalassaemia intermedia – This form of the disease is milder and of variable severity. The β-globin mutations allow a small amount of HbA and/or a large amount of HbF to be produced.
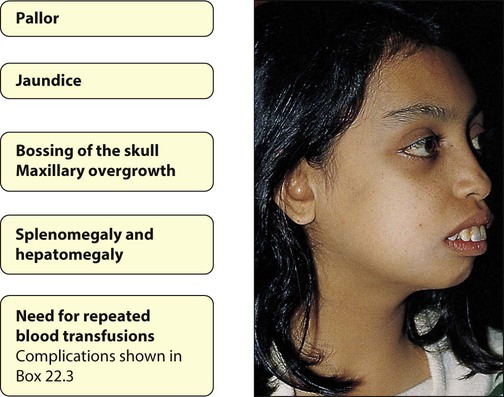
Clinical features (Fig. 22.13)
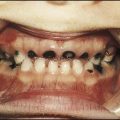
• Severe anaemia, which is transfusion dependent, from 3–6 months of age and jaundice
• Failure to thrive/growth failure
• Extramedullary haemopoiesis, prevented by regular blood transfusions. In the absence of regular blood transfusion, develop hepatosplenomegaly and bone marrow expansion; the latter leads to the classical facies with maxillary overgrowth and skull bossing (very rare in the UK and developed countries).
Management
The condition is uniformly fatal without regular blood transfusions, so all patients are given lifelong monthly transfusions of red blood cells. The aim is to maintain the haemoglobin concentration above 10 g/dl in order to reduce growth failure and prevent bone deformation. Repeated blood transfusion causes chronic iron overload, which causes cardiac failure, liver cirrhosis, diabetes, infertility and growth failure. For this reason, all patients are treated with iron chelation with subcutaneous desferrioxamine, or with an oral iron chelator drug, such as deferasirox, starting from 2 to 3 years of age. Patients who comply well with transfusion and chelation have a 90% chance of living into their forties and beyond. However, compliance is difficult. Those who cannot comply have a high mortality in early adulthood from iron overload. The complications of multiple transfusions are shown in Box 22.3. An alternative treatment for β-thalassaemia major is bone marrow transplantation, which is currently the only cure. It is generally reserved for children with an HLA-identical sibling as there is then a 90–95% chance of success (i.e. transfusion independence and long-term cure) but a 5% chance of transplant-related mortality.
Anaemia in the newborn
Reduced red blood cell production
There are two main but rare causes in the newborn and both cause red cell aplasia:
Increased red cell destruction (haemolytic anaemia)
• Immune (e.g. haemolytic disease of the newborn)
• Red cell membrane disorders (e.g. hereditary spherocytosis)
• Red cell enzyme disorders (e.g. glucose-6-phosphate dehydrogenase deficiency)
Haemolytic disease of the newborn (immune haemolytic anaemia of the newborn) is due to antibodies against blood group antigens. The most important are: anti-D (a ‘rhesus’ antigen), anti-A or anti-B (ABO blood group antigens) and anti-Kell. The mother is always negative for the relevant antigen (e.g. rhesus D-negative) and the baby is always positive; the mother then makes antibodies against the baby’s blood group and these antibodies cross the placenta into the baby’s circulation causing fetal or neonatal haemolytic anaemia. The diagnostic clue to this type of haemolytic anaemia is a positive direct anti-globulin test (Coombs test). This test is only positive in antibody-mediated anaemias and so is negative in all the other types of haemolytic anaemia. (These conditions are considered further in Chapter 10.)
Blood loss
• Feto-maternal haemorrhage (occult bleeding into the mother)
• Twin-to-twin transfusion (bleeding from one twin into the other one)
• Blood loss around the time of delivery (e.g. placental abruption).
The main diagnostic clue is severe anaemia with a raised reticulocyte count and normal bilirubin.
Bleeding disorders
Normal haemostasis
There are five main components:
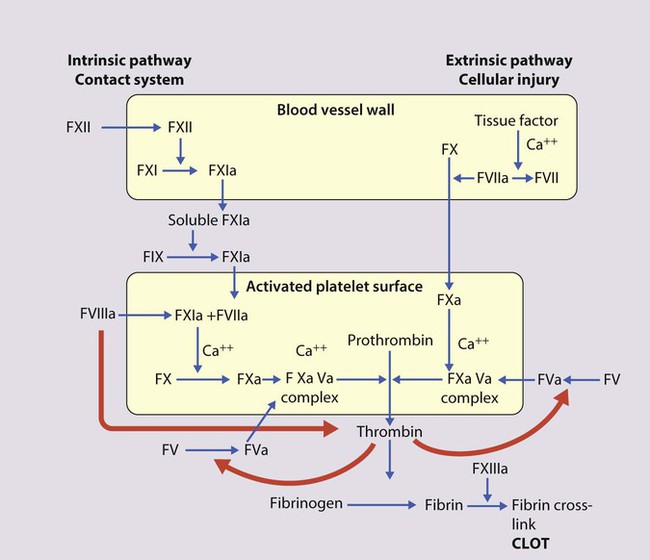
1. Coagulation factors – are produced (mainly by the liver) in an inactive form and are activated when coagulation is initiated (usually by tissue factor (TF), which is released by vessel injury; see Fig. 22.14)
2. Coagulation inhibitors – these either circulate in plasma or are bound to endothelium and are necessary to prevent widespread coagulation throughout the body once coagulation has been initiated
3. Fibrinolysis – this process limits fibrin deposition at the site of injury due to activity of the key enzyme plasmin
4. Platelets – are vital for haemostasis as they aggregate at sites of vessel injury to form the primary haemostatic plug which is then stabilised by fibrin
5. Blood vessels – both initiate and limit coagulation. Intact vascular endothelium secretes prostaglandin I2 and nitric oxide (which promote vasodilatation and inhibit platelet aggregation). Damaged endothelium releases TF and procoagulants (e.g. collagen and von Willebrand factor) and there are inhibitors of coagulation on the endothelial surface (thrombomodulin, antithrombin and protein S) to modulate coagulation.
The endpoint of the coagulation cascade is generation of thrombin. A simplified model is shown in Figure 22.14. The two main pathways for thrombin generation were identified many years ago as the intrinsic and extrinsic pathways. Important components of these pathways are still being discovered. In recent years, the crucial role of tissue factor (TF) in haemostasis has been recognised and it is now thought that the extrinsic pathway is the one primarily responsible for initiating both normal haemostasis and thrombotic disease.
Diagnostic approach
The diagnostic evaluation of an infant or child for a possible bleeding disorder includes:
• Identifying features in the clinical presentation that suggest the underlying diagnosis, as indicated in Box 22.4
• Initial laboratory screening tests to determine the most likely diagnosis (Table 22.3)
Table 22.3
Investigations in haemophilia A and von Willebrand disease
| Haemophilia A | von Willebrand disease | |
| PT | Normal | Normal |
| APTT | ↑↑ | ↑ or normal |
| Factor VIII:C | ↓↓ | ↓ or normal |
| vWF Antigen | Normal | ↓ |
| RiCoF (activity) | Normal | ↓ |
| Ristocetin-induced platelet aggregation | Normal | Abnormal |
| vWF multimers | Normal | Variable |
PT, prothrombin time; APTT, activated partial thromboplastin time; RiCoF, ristocetin co-factor, measures vWD activity.
• Specialist investigation to characterise a deficiency or exclude important conditions that can present with normal initial investigations, e.g. mild von Willebrand disease, factor XIII deficiency and platelet function disorders.
The most useful initial screening tests are:
• Full blood count and blood film
• Prothrombin time (PT) – measures the activity of factors II, V, VII and X
• Activated partial thromboplastin time (APTT) – measures the activity of factors II, V, VIII, IX, X, XI and XII
• If PT or APTT is prolonged, a 50 : 50 mix with normal plasma will distinguish between possible factor deficiency or presence of inhibitor
• Thrombin time – tests for deficiency or dysfunction of fibrinogen
• Quantitative fibrinogen assay
• D-dimers – to test for fibrin degradation products
• Biochemical screen, including renal and liver function tests.
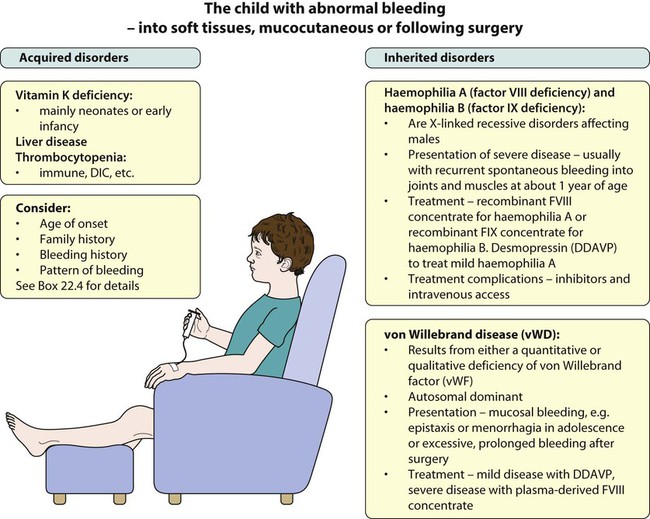
Haemophilia
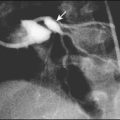
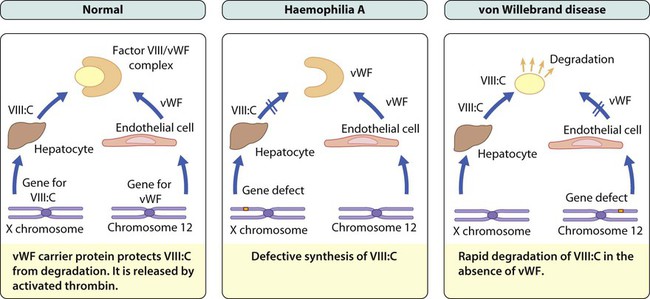
The commonest severe inherited coagulation disorders are haemophilia A and haemophilia B. Both have X-linked recessive inheritance. In haemophilia A, there is FVIII deficiency (Fig. 22.15); it has a frequency of 1 in 5000 male births. Haemophilia B (FIX deficiency) has a frequency of 1 in 30 000 male births. Two-thirds of newly diagnosed infants have a family history of haemophilia, whereas one-third are sporadic. Identifying female carriers requires a detailed family history, analysis of coagulation factors and DNA analysis. Prenatal diagnosis is available using DNA analysis.
Clinical features
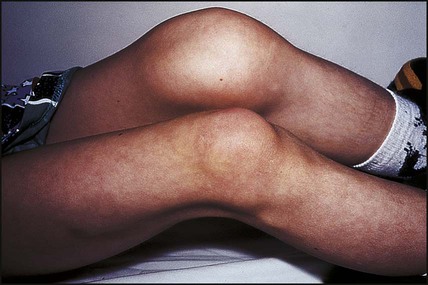
The disorder is graded as severe, moderate or mild, depending on the FVIII:C (or IX:C in haemophilia B) level (Table 22.4). The hallmark of severe disease is recurrent spontaneous bleeding into joints and muscles, which can lead to crippling arthritis if not properly treated (Fig. 22.16). Most children present towards the end of the first year of life, when they start to crawl or walk (and fall over). Bleeding episodes are most frequent in joints and muscles. Where there is no family history, non-accidental injury may initially be suspected. Almost 40% of cases present in the neonatal period, particularly with intracranial haemorrhage, bleeding post-circumcision or prolonged oozing from heel stick and venepuncture sites. The severity usually remains constant within a family.
Table 22.4
| Factor VIII:C | Severity | Bleeding tendency |
| <1% | Severe | Spontaneous joint/muscle bleeds |
| 1–5% | Moderate | Bleed after minor trauma |
| >5–40% | Mild | Bleed after surgery |
von Willebrand disease (vWD)
Von Willebrand factor (vWF) has two major roles:
• It facilitates platelet adhesion to damaged endothelium
• It acts as the carrier protein for FVIII:C, protecting it from inactivation and clearance.
Von Willebrand disease (vWD) results from either a quantitative or qualitative deficiency of von Willebrand factor (vWF). This causes defective platelet plug formation and, since vWF is a carrier protein for FVIII:C, patients with vWD also are deficient in FVIII:C (see Fig. 22.15).
Acquired disorders of coagulation
The main acquired disorders of coagulation affecting children are those secondary to:
• Haemorrhagic disease of the newborn due to vitamin K deficiency (see Ch. 10)
Thrombocytopenia
• Severe thrombocytopenia (platelets <20 × 109/L) – risk of spontaneous bleeding
• Moderate thrombocytopenia (platelets 20–50 × 109/L) – at risk of excess bleeding during operations or trauma but low risk of spontaneous bleeding
• Mild thrombocytopenia (platelets 50–150 × 109/L) – low risk of bleeding unless there is a major operation or severe trauma.
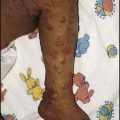
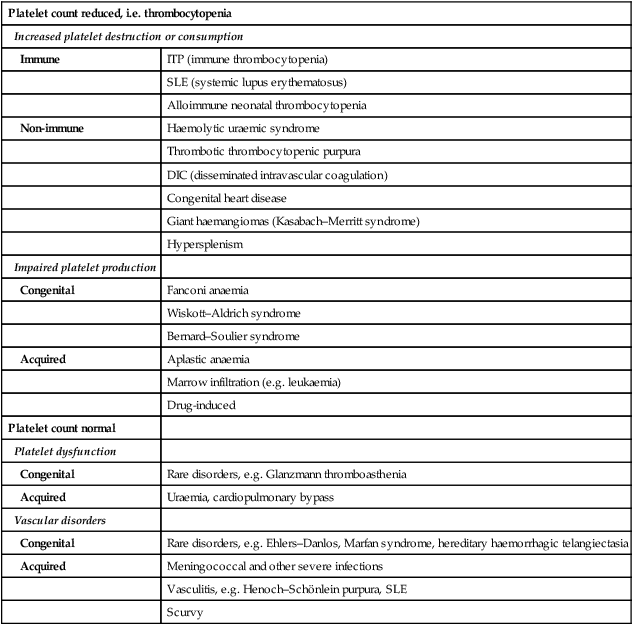
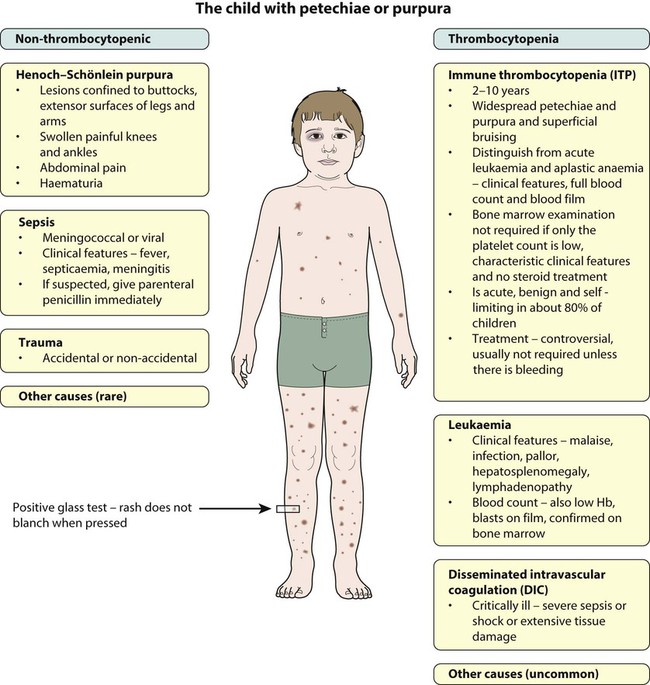
Thrombocytopenia may result in bruising, petechiae, purpura and mucosal bleeding (e.g. epistaxis, bleeding from gums when brushing teeth). Major haemorrhage in the form of severe gastrointestinal haemorrhage, haematuria and intracranial bleeding is much less common. The causes of easy bruising and purpura are listed in Table 22.5. While purpura may signify thrombocytopenia, it also occurs with a normal platelet count from platelet dysfunction and vascular disorders.
Table 22.5
Causes of purpura or easy bruising
| Platelet count reduced, i.e. thrombocytopenia | |
| Increased platelet destruction or consumption | |
| Immune | ITP (immune thrombocytopenia) |
| SLE (systemic lupus erythematosus) | |
| Alloimmune neonatal thrombocytopenia | |
| Non-immune | Haemolytic uraemic syndrome |
| Thrombotic thrombocytopenic purpura | |
| DIC (disseminated intravascular coagulation) | |
| Congenital heart disease | |
| Giant haemangiomas (Kasabach–Merritt syndrome) | |
| Hypersplenism | |
| Impaired platelet production | |
| Congenital | Fanconi anaemia |
| Wiskott–Aldrich syndrome | |
| Bernard–Soulier syndrome | |
| Acquired | Aplastic anaemia |
| Marrow infiltration (e.g. leukaemia) | |
| Drug-induced | |
| Platelet count normal | |
| Platelet dysfunction | |
| Congenital | Rare disorders, e.g. Glanzmann thromboasthenia |
| Acquired | Uraemia, cardiopulmonary bypass |
| Vascular disorders | |
| Congenital | Rare disorders, e.g. Ehlers–Danlos, Marfan syndrome, hereditary haemorrhagic telangiectasia |
| Acquired | Meningococcal and other severe infections |
| Vasculitis, e.g. Henoch–Schönlein purpura, SLE | |
| Scurvy | |
Immune thrombocytopenia (ITP)
Clinical features
Most children present between the ages of 2 and 10 years, with onset often 1–2 weeks after a viral infection. In the majority of children, there is a short history of days or weeks. Affected children develop petechiae, purpura and/or superficial bruising (see Case History 22.3). It can cause epistaxis and other mucosal bleeding but profuse bleeding is uncommon, despite the fact that the platelet count often falls to <10 × 109/L. Intracranial bleeding is a serious but rare complication, occurring in 0.1–0.5%, mainly in those with a long period of severe thrombocytopenia.
Management
Case History
22.3 Immune thrombocytopenic purpura (ITP)
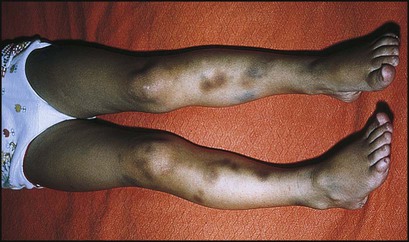
Sian, aged 5 years, developed bruising and a skin rash over 24 h. She had had an upper respiratory tract infection the previous week. On examination she appeared well but had a purpuric skin rash with some bruises on the trunk and legs (Fig. 22.17). There were three blood blisters on her tongue and buccal mucosa, but no fundal haemorrhages, lymphadenopathy or hepatosplenomegaly. Urine was normal on dipsticks testing. A full blood count showed Hb 11.5 g/dl with normal indices, WBC and differential normal, platelet count 17 × 109/L. The platelets on the blood film were large; the film was otherwise normal. A diagnosis of ITP was made and she was discharged home. Her parents were counselled and given emergency contact names and telephone numbers. They were also given literature on the condition and advised that she should avoid contact sports but should continue to attend school. Over the next 2 weeks she continued to develop bruising and purpura but was asymptomatic. By the third week, she had no new bruises, and her platelet count was 25 × 109/L; the blood count and film showed no new abnormalities. The following week, the platelet count was 74 × 109/L and a week later it was 200 × 109/L. She was discharged from follow-up.
Thrombosis in children
Thrombosis is uncommon in children and about 95% of venous thromboembolic events are secondary to underlying disorders associated with hypercoagulable states (see below). Thrombosis of cerebral vessels usually presents with signs of a stroke. (The condition is considered further in Chapters 10 and 27.) Rarely, children may inherit abnormalities in the coagulation and fibrinolytic pathway that increase their risk of developing clots even in the absence of underlying predisposing factors. These conditions are termed congenital prothrombotic disorders (thrombophilias). They are:
• DIC (disseminated intravascular coagulation)
• Polycythaemia (e.g. due to congenital heart disease)
• SLE (systemic lupus erythematosus) and persistent antiphospholipid antibody syndrome.